Ревматичні хвороби
Содержание
- Глава 13. Системний червоний вовчак
- Глава 14. Антифосфоліпідний синдром
- Глава 15. Системна склеродермія
- Глава 16. Ідіопатичні запальні міопатії
- Глава 17. Хвороба Шегрена
- Глава 18. Змішане захворювання сполучної тканини
- Глава 19. Ревматична поліміалгія
- Глава 20. Рецидивуючий поліхондрит
- Глава 21. Системні васкуліти. Васкулопатії. Синдром Гудпасчера
- Глава 22. Ревматоїдний артрит
- Глава 23. Ювенільний артрит
- Глава 24. Спондилоартропатії
- Глава 25. Артрити, пов’язані з інфекцією
- Глава 26. Гостра ревматична лихоманка та хронічна ревматична хвороба серця
- Глава 27. Кристалічні артропатії
- Глава 28. Остеоартроз
- Глава 29. Остеопороз
- Глава 30. Захворювання кісток
- Глава 31. Фіброміалгії
- Глава 32. Хвороба Педжета
- Глава 33. Інфекційний ендокардит
- Глава 34. Хвороби міокарда
- Глава 35. Вроджені вади серця
Глава 13. Системний червоний вовчак
Існує декілька визначень СЧВ. Найбільш розповсюдженими є наступні.
СЧВ — хронічне полісиндромне захворювання переважно молодих жінок та дівчат, що розвивається на фоні генетично зумовленої неповноцінності імунорегуляторних процесів, яка призводить до безконтрольної продукції антитіл до власних клітин та їх компонентів із розвитком аутоімунного та імунокомплексного хронічного запалення.
СЧВ — системне аутоімунне захворювання невідомої етіології, що характеризується гіперпродукцією органоспецифічних аутоантитіл до різноманітних компонентів клітинного ядра з розвитком імунозапального пошкодження тканин та внутрішніх органів.
Код за МКХ-10
М32 Системний червоний вовчак.
Епідеміологія
Кількість випадків СЧВ коливається від 4 до 250 на 100 тис. населення. Пік захворюваності припадає на вік 15–25 років. Жінки хворіють у 8–10 разів частіше, ніж чоловіки. У дітей співвідношення дівчаток та хлопців меньше — 3:1. У препубертатному віці частота захворюваності хлопчиків та дівчаток приблизно однакова. СЧВ найбільш часто розвивається в репродуктивному віці, під час вагітності та в післяпологовий період. Відзначено підвищення частоти та тяжкості перебігу захворювання серед представників негроїдної раси, пуерториканців, китайців. Смертність при СЧВ в 3 вища, ніж у популяції.
Етіологія та патогенез
Аутоімунний характер ураження при СЧВ вважається доведеним, проте конкретний етіологічний фактор захворювання не встановлено. Обговорюють роль різноманітних інфекційних агентів, токсичних речовин, деяких медикаментозних засобів, але прямих доказів участі будь-якого визначеного фактора досі не отримано. Існують непрямі підтвердження етіологічної («тригерної») ролі вірусної або бактеріальної інфекції, спадкової схильності, порушення гормональної регуляції. Вірогідність участі вірусної інфекції в розвитку СЧВ на сьогодні обговорюється у зв’язку з підвищеним титром антитіл до ряду РНК- та ДНК-вмісних вірусів (кору, краснухи, паротиту, грипу, вірусу Епштейна — Барр, цитомегаловіруса, вірусу простого герпесу та ін.), а також виявленням у хворих на СЧВ маркерів персистуючої вірусної інфекції (ретровірусів), таких як лімфоцитотоксичні антитіла та антитіла до вірусної двоспіральної РНК. Незважаючи на наявність непрямих доказів ролі вірусної інфекції при СЧВ, прямі на сьогодні — відсутні.
Важлива роль у розвитку СЧВ належить генетичній схильності, на користь якої свідчать сімейна розповсюдженість захворювання, яка у багато разів перевищує популяційну, діагностування СЧВ у обох монозиготних близнюків більш ніж у 70% хворих, а також доволі висока частота (більш ніж у ⅓ випадків) виявлення у практично здорових членів сімей хворих на СЧВ таких імунологічних зсувів, як: гіпергаммаглобулінемія, антиядерні та антилімфоцитарні антитіла, відкладання імуноглобулінів у дермоепітеліальному стику.
СЧВ — клінічно гетерогенне аутоімунне захворювання. Виявлено багато генетичних локусів, які підвищують ризик виникнення СЧВ. Особливий інтерес становлять дані про вивчення антигенів гістосумісності. Найбільше значення надають антигенам НLА A11, B8, B35, DR2 і DR3 та відсутності алеля О (близько 80% хворих). Відзначено, що при гострому і підгострому перебігу частіше, ніж у контролі, виявляють носійство антигенів НLА A11, B7 і B35, а при первинно-хронічному — B8 і B35, DR2 і DR3 (при зменшенні кількості DR1). Існує різниця в генетичній характеристиці хворих з ураженням нирок. Так, при люпус-нефриті відзначають збільшення кількості антигенів A9 і B16, а при нефротичному синдромі — DR3. Ураження ЦНС, а також люпус-нефрит супроводжуються носійством A9 та зменшенням кількості DR7.
Виявлено чіткі взаємозв’язки між клінічними проявами СЧВ і FCGR2A, ITGAM, STAT4, TNSF4 та генами IL21. Наприклад, при ниркових порушеннях визначено виражену кореляцію з алелем ризику розвитку СЧВ в ITGAM, а також дискоїдними висипами.
Вивчення генетичної характеристики загальної популяції та хворих на СЧВ показало, що носійство антигенів НLА A11 у 14,9 раза підвищувало ризик виникнення СЧВ, B18 — у 4,9 раза, B8 — у 4,3 раза, а поєднання A11 i B8, A11 i B18, A1 i A11 — у 24 рази. Не тільки наявність додаткових НLА-антигенів сприяє розвитку захворювання, але й висока частота нульових (DR1, DR7, C4) антигенів може відіграти роль у виникненні СЧВ. Присутність різних антигенів, їх поєднання, а також асоціація з нульовими антигенами можуть визначати клінічний поліморфізм СЧВ та тяжкість перебігу захворювання. Сьогодні проводять дослідження антигенів НLА з метою визначення їх ролі у прогнозуванні перебігу захворювання, виявленні факторів ризику розвитку тяжких форм СЧВ.
Слід зазначити, що при СЧВ, як і при інших аутоімунних захворюваннях, виявляють підвищення НLA B8 i DR3, які зумовлюють гіпофункцію Т-супресорів, послаблення елімінації імунних комплексів у зв’язку з дефектами макрофагальної системи печінки та селезінки, тобто ведуть до порушення регуляторних механізмів імунної системи, що певною мірою пояснює можливість розвитку аутоімунних порушень.
При СЧВ виявляють також генетично детермінований дефект системи комплементу, дефіцит С2 і С4 компонентів комплементу, дефіцит натуральних кілерів. Відзначено зв’язок СЧВ з негенетично зумовленим дефіцитом окремих компонентів комплементу (С1q, С4, С2), поліморфізмом генів рецепторів FcyR II (порушення кліренсу імунних комплексів) і ФНП-α, підвищенням частоти носійства НLА-В8 і НLА-DR3.
Важливе значення при СЧВ, як і при інших аутоімунних захворюваннях, належить гормональному фактору, зокрема статі (у жінок частота виникнення СЧВ у 9 разів вища). Так, роль статі пов’язана з локалізацією низки генів, що регулюють імунну відповідь у Х-хромосомі, та з високим рівнем естрогенів, які підвищують активність Т-хелперів, продукцію ІЛ, які послаблюють елімінацію імунних комплексів. Крім того, відомо, що статеві гормони впливають також на активність мікросомальних ферментів (цитохром Р450), які є стабілізаторами мембран. Так, андрогени збільшують їх активність, у той час як естрогени — пригнічують, і таким чином активізують процеси перекисного окиснення ліпідів, призводячи до дестабілізації мембран. У чоловіків, хворих на СЧВ, відзначено тенденцію до гіпоандрогенемії та гіперпродукції пролактину.
У процесі розвитку СЧВ як аутоімунного захворювання має відбуватися порушення функції тимуса (центрального органа імунної системи), відповідального за дозрівання і диференціацію всіх субпопуляцій Т-лімфоцитів. При СЧВ вже на ранньому етапі захворювання у 90% хворих зареєстровано появу аутоантитіл до тканини тимуса, проте роль змін у тимусі в аутоімунізації до кінця не розкрита.
Таким чином, вищеописані фактори призводять до розвитку імунорегуляторних порушень, зокрема до дефіциту супресорної функції імунної системи. Розвиток дефіциту супресорних механізмів може бути також наслідком порушень у системі ідіотип-антиідіотипових антитіл, підтвердженням чому є зниження в крові рівня антиідіотипових антитіл до аутоантитіл перед загостренням аутоімунного захворювання.
Серед факторів, що ініціюють СЧВ, є надлишкова інсоляція, охолодження, стресові ситуації, фізичні перевантаження, лікарські препарати та ін.
У результаті впливу ініціюючих факторів за наявності мультифакторної схильності, пов’язаної з генетично детермінованим порушенням імунітету, статтю, а також, можливо, вірусною інфекцією, формуються аутоімунні механізми розвитку СЧВ.
Зниження імуносупресивних функцій при СЧВ, що веде до порушення імунологічної толерантності, надмірної активності В-лімфоцитів, характеризується підвищеною продукцією антитіл до нативної (двоспіральної) ДНК, а також інших ядерних (дезоксинуклеопротеїну, гістонів, Sm-антигену тощо) і цитоплазматичних антигенів (Rо/SSА, Lа/SSВ, мітохондрій, рибосом та ін.). Патогенетичне значення антинуклеарних антитіл полягає в їх здатності формувати ЦІК, які, відкладаючись у структурах різних органів, можуть викликати їх пошкодження (наприклад комплекси антитіл нативної ДНК і нативної ДНК-комплементу у патогенезі люпус-нефриту). Роль імунокомплексних процесів, особливо їх наявність у ЦІК нативної ДНК, є провідним у патогенезі СЧВ (СЧВ — класичний приклад імунокомплексної хвороби людини). Механізми формування імунокомплексного процесу пов’язані також зі змінами в системі комплементу, зокрема з генетично детермінованим дефіцитом С2 і С4 компонентів комплементу, ослабленням елімінації імунних комплексів у зв’язку з дефектом макрофагальної системи. Підтвердженням ролі імунних комплексів у патогенезі СЧВ є їх виявлення в клубочках нирок, судинах, базальній мембрані шкіри, хоріоїдальних сплетеннях мозку; загальна гіпокомплементемія, зниження окремих компонентів комплементу (С1, С3, С4), а також кореляція між рівнем ЦІК, гіпокомплементемією та активністю хвороби.
Крім того, важливе значення в патогенезі СЧВ мають два взаємопов’язані процеси:
- на ранній стадії захворювання переважає поліклональна (В-клітинна) активація імунітету, надалі — антигенспецифічні (Т-клітинні) імунні реакції;
- фундаментальне імунне порушення, що лежить в основі СЧВ, — вроджені або індуковані дефекти програмованої загибелі клітин (апоптозу).
При СЧВ продукуються аутоантитіла приблизно до 40 із більш ніж 2000 потенційно аутоантигенних клітинних компонентів, найбільш важливі з яких — ДНК і мультивалентні внутрішньоклітинні нуклеопротеїнові комплекси (нуклеосома, рибонуклеопротеїни, Rо/Lа та ін.). Висока імуногенність останніх відзначається здатністю перехресно зв’язувати В-клітинні рецептори і накопичуватися на поверхні «апоптотичних» клітин. Характерні різноманітні дефекти клітинного імунітету, що мають властивість гіперпродукувати Тh2-цитокіни (ІЛ-4, ІЛ-6, ІЛ-10). Останні — аутокринні фактори активації В-лімфоцитів, що синтезують антиядерні аутоантитіла. Естрогени можуть стимулювати синтез Тh2-цитокінів.
На тлі зниження загального вмісту В-клітин у периферичній крові при СЧВ спостерігають характерний перерозподіл субпопуляцій В-клітин у бік збільшення кількості «наївних» В-клітин і плазмобластів. При СЧВ, як і при інших аутоімунних захворюваннях, В-клітини відіграють важливу роль не тільки в синтезі аутоантитіл, а й презентації аутоантигенів Т-клітинам. В експериментальних дослідженнях продемонстровано, що у трансгенних МRL/lpr мишей, у яких кількість В-лімфоцитів — в нормі, але синтез аутоантитіл відсутній, можливий розвиток важкого нефриту, який морфологічно характеризується інфільтрацією тканини нирок активованими Т-клітинами. І навпаки, у мишей ліній JHD/lpr і МRL /+, у яких на фоні генетичної схильності до СЧВ відсутні В-клітини, розвитку захворювання не виявлено. Встановлено, що активацію й диференціювання В-клітин регулює стимулятор В-лімфоцитів (B-lymphocyte stimulator), який також називають фактором активації В-клітин (B-cell-activating factor). Вважають, що взаємодія між стимулятором В-лімфоцитів і відповідним рецептором, що належать до суперсімейства ФНП, відіграє важливу роль в імунопатогенезі захворювання. У трансгенних мишей з гіперекспресією стимулятора В-лімфоцитів розвивається вовчакоподібний синдром, що нагадує СЧВ у людини, з гіперпродукцією імунних комплексів, РФ, антитіл до нативної та денатурованої ДНК. При цьому концентрація стимулятора В-лімфоцитів в сироватці корелює з активністю патологічного процесу. У мишей лінії NZВ/NZW F1 (класична лабораторна модель СЧВ людини) в нирках виявлено збільшення вмісту В-клітинних цитокінів і самих В-клітин. Подібні дані отримано при обстеженні хворих на СЧВ.
Ефекторні механізми, що визначають пошкодження внутрішніх органів при СЧВ, пов’язані, в першу чергу, з гуморальними імунними реакціями (синтез антиядерних антитіл). На відміну від деяких форм системних васкулітів, розвиток яких викликаний відкладенням в судинах ЦІК, виникнення вовчакового нефриту зумовлено локальним (in situ) формуванням імунних комплексів в ниркових клубочках. Спочатку ядерні антигени (ДНК, нуклеосоми та ін.) зв’язуються з компонентами клубочків нирки, а потім взаємодіють із відповідними антитілами. Інший можливий механізм — перехресна взаємодія антитіл до ДНК з компонентами клубочка.
Системне імунне запалення може бути пов’язано з цитокінзалежним (ІЛ-1 і ФНП-α) пошкодженням ендотелію, активацією лейкоцитів і системи комплементу. Припускають, що останній механізм має особливо велике значення в ураженні органів, недоступних для імунних комплексів (наприклад ЦНС).
Таким чином, схематично патогенез СЧВ можна представити як комплексний вплив генетичних, гормональних й імунорегуляторних факторів, спрямований на продукцію антитіл В-лімфоцитами.
Психоневрологічні симптоми при СЧВ, як правило, виникають непередбачувано, але в більшості випадків асоціюються з певними біомаркерами. При вивченні таких біомаркерів, як вовчаковий антикоагулянт (ВА), антитіла до кардіоліпіну (АКЛ), анти-р2-глікопротеїд I, антирибосомний Р-рецептор, встановлено, що у пацієнтів із СЧВ наявність ВА підвищує ризик майбутнього внутрішньочерепного тромбозу, а антирибосомних антитіл Р — вовчакового психозу.
Крім того, встановлено, що пацієнти з СЧВ мають підвищений ризик атеросклерозу. Так, високі рівні лептину та низькі адипонектину асоціюються з атеросклерозом й імуномодулюючими функціями в загальній популяції. До того ж встановлено, що високі рівні лептину значно підвищують ризик субклінічного атеросклерозу при СЧВ і також асоціюються зі збільшенням кількості запальних біомаркерів атеросклерозу.
Високі рівні лептину можуть допомогти виявити пацієнтів із СЧВ з ризиком розвитку атеросклерозу.
Дослідження під час лікування хворих на СЧВ із серцево-судинними захворюваннями аторвастатином не показало зменшення вираженості субклінічних проявів атеросклерозу або активності захворювання протягом 2 років.
Дефіцит вітаміну D є поширеним та асоціюється з багатьма хронічними хворобами, включаючи аутоімунні.
У дослідженні дефіциту вітаміну D встановлено, що його значно частіше виявляють у пацієнтів із СЧВ та наявністю антиядерних антитіл порівняно з хворими з відсутністю антиядерних антитіл і контролем. Крім того, пацієнти з високою В-клітинною активацією мали більш низькі рівні вітаміну D, ніж хворі з низькою В-клітинною активацією.
У пацієнтів із дефіцитом вітаміну D сироваткова активність інтерферону альфа вища, ніж у хворих без дефіциту цього вітаміну.
Підсумовано, що вітамін D відіграє важливу роль у синтезі аутоантитіл в патогенезі СЧВ.
Патоморфологія
При СЧВ можливе ураження більшості органів і тканин організму. Патологічний процес характеризується 4 основними видами гістологічних змін, які в різних поєднаннях відзначають в більшості уражених органів. До них відносять фібриноїдні зміни, склероз, гематоксилінові тільця, судинні зміни.
1. Фібриноїдні зміни характеризуються наявністю безклітинного матеріалу з вираженою еозинофілією, який має форму ниток або пучків та зовні нагадує фібрин. Вважають, що виникнення цих змін зумовлене ураженням основної субстанції сполучної тканини внаслідок відкладення плазми. Навколо ділянок, які зазнали фібриноїдних змін, формуються слабко виражені запальні інфільтрати, що складаються в основному з лімфоцитів і плазматичних клітин. Така запальна реакція більш виражена в серозних оболонках після відкладення фібрину.
2. У результаті хронічної запальної реакції навколо відкладення фібрину відзначають потовщення колагенових волокон, збільшення кількості фібробластів, розростання сполучної тканини. Зміни найбільш виражені в селезінці, де фіброзна тканина, що формується навколо селезінкових артерій у вигляді концентричних кіл, зумовлює виникнення феномену «цибулинного лушпиння», однієї з двох ознак, майже патогномонічних для СЧВ.
3. Іншою характерною ознакою СЧВ є гематоксилінові тільця (за даними електронної мікроскопії — продукт деградації клітинних ядер), які за класичним описом мають приблизно розмір ядра, є округло-овальної форми, безструктурні, щільність їх менша, ніж звичайного ядра, при забарвленні гематоксилін-еозином набувають від пурпурного до рожево-блакитного кольору. Ймовірно, вони ідентичні включенням, виявленим в LЕ-клітинах.
4. Поширене ураження артеріол і капілярів більше, ніж будь-які інші зміни, сприяє появі клінічної картини СЧВ. В інтимі розвиваються фібриноїдні зміни, що супроводжуються звуженням просвіту судини, чому сприяє також утворення колагену. Майже завжди відзначають потовщення ендотеліального шару невеликих артеріол та інших судин. Незважаючи на ці зміни, тромбози зустрічаються порівняно рідко.
Нирки. Найбільш характерною патологією при СЧВ є вовчакова нефропатія, в основі якої лежить імунокомплексний гломерулонефрит. При найбільш легкій формі ураження нирок патологічний процес обмежується змінами ендотеліальних клітин і потовщенням базальних мембран капілярів. При цьому можлива невелика проліферація клітин клубочків. Такі зміни можуть виникати лише в небагатьох клубочках або частині кожного клубочка. У цій стадії перебіг патологічного процесу може бути безсимптомним, захворювання виявляють тільки при біопсії нирок. Описані зміни зворотні. Залежно від стадії патологічного процесу, в якій проводиться дослідження нирок, можуть бути виявлені всі види морфологічних змін. Відзначають ураження клубочків, канальців, інтерстицію. Гістологічні ознаки вовчакового нефриту — вогнищевий некроз з облітерацією капілярів, фібриноїдні зміни, проліферація клітин і гематоксилінові тільця. Характерною ознакою є феномен «дротяної петлі», названий так тому, що фібриноїдні зміни стінок капілярів клубочків зумовлюють чіткість контурів потовщених капілярних петель. При СЧВ розрізняють гломерулонефрит без характерних морфологічних ознак вовчака, який може бути мембранозним або мембранозно-проліферативним і мати вогнищевий або дифузний характер, та гломерулонефрит із характерними морфологічними ознаками СЧВ: вовчаковий нефрит, нефросклероз, що трактується як результат гломерулонефриту. Найбільш несприятливим в прогностичному відношенні є дифузний проліферативний гломерулонефрит.
Серце. При СЧВ можливе ураження всіх оболонок серця, його частота перевищує частоту, про яку свідчать клінічні дані. Відкладення фібриноїду в перикарді супроводжується запальними змінами навколишньої тканини, що становлять морфологічну основу вовчакового перикардиту. Відкладення фібриноїду в міокарді призводить до осередкового міокардиту з фіброзом й атрофією волокон серцевого м’яза, що іноді зумовлює розвиток серцевої недостатності. Типовим для СЧВ є розвиток ендокардиту Лібмана — Сакса, який характеризується наявністю тромботичних вегетацій на стулках клапанів, особливо мітрального, іноді з переходом на пристінковий ендокардит.
Шкіра. Гістологічно ураження шкіри характеризується гіперкератозом із закупоркою волосяних фолікул, атрофією епідермісу, вакуольною дегенерацією базальної мембрани, судинними змінами та лімфоцитарною інфільтрацією дерми. Характерною ознакою ураження шкіри при СЧВ є відкладення IgG i IgM в ділянці дермоепідермального з’єднання в ураженій та неураженій шкірі.
Легені. У легенях при СЧВ розвиваються судинні зміни та пневмоніт із потовщенням альвеолярних перетинок. Часто приєднується вторинна інфекція. Як прояв серозиту — плеврит.
ЦНС. Ураження ЦНС і периферичної нервової системи характеризується фібриноїдними змінами судин мозку і мозкових оболонок, а також судин, що забезпечують кровопостачання периферичних нервів. Крім того, в ЦНС часто виявляють неспецифічні зміни, включно з демієлінізацією та гліозом.
Лімфатичні вузли при СЧВ збільшуються, можлива наявність атрофії фолікулів, іноді вогнищ некрозу з нечіткими контурами без клітинної реакції. У патологічний процес може залучатися печінка, що морфологічно проявляється інфільтрацією строми лімфоїдними, плазматичними клітинами, макрофагами, а також жировою дистрофією паренхіми, коагуляційним некрозом гепатоцитів. Специфічні зміни для СЧВ виявляють в селезінці. Це атрофія лімфоїдних фолікулів, виражена плазматизація, феномен «цибулинного лушпиння», а також іноді відкладення гомогенних білкових преципітатів, які не дають позитивної реакції на амілоїд.
Класифікація
Характерна особливість СЧВ — різноманіття клінічних проявів і варіантів перебігу хвороби. Класифікація СЧВ включає визначення варіанта перебігу хвороби залежно від характеру початку (гострий, підгострий або первинно-хронічний) (табл. 13.1).
Таблиця 13.1
Клінічна класифікація
|
Характер перебігу хвороби |
Гострий Підгострий Хронічний Рецидивуючий поліартрит Синдром дискоїдного вовчака Синдром Рейно Синдром Верльгофа Синдром Шегрена АФС |
|
|
Ступінь активності процесу |
Відсутній (0) Мінімальний (I) Помірний (II) Високий (III) |
|
|
Клініко-морфологічна характеристика уражень |
шкіри |
Симптом метелика Капілярити Ексудативна еритема, пурпура Дискоїдний вовчак Ретикулярне ліведо та ін. |
|
суглобів |
Артралгії Поліартрит (гострий, підгострий, хронічний) |
|
|
серозних оболонок |
Плеврит, перикардит (випітний, сухий, адгезивний), перигепатит, периспленіт, полісерозит |
|
|
серця |
Міокардит, ендокардит, недостатність мітрального клапана, міокардіофіброз, міокардіодистрофія |
|
|
легень |
Гострий, хронічний пневмоніт Пневмосклероз |
|
|
нирок |
Люпус-нефрит нефротичного або змішаного типу, пієлонефротичний синдром, сечовий синдром |
|
|
нервової системи |
Менінгоенцефалополірадикулоневрит, поліневрит, інсульти й інфаркт мозку Васкуліт судин головного мозку |
|
Гострий перебіг захворювання. Характерний швидкий розвиток мультиорганних проявів включно з ураженням нирок і ЦНС та висока імунологічна активність.
Підгострий перебіг захворювання. Перебіг захворювання — хвилеподібний, з періодичним виникненням загострень і розвитком поліорганної симптоматики протягом 2–3 років з моменту появи перших симптомів.
Первинно-хронічний варіант перебігу. Властиве тривале превалювання одного або декількох симптомів: дискоїдних висипань, синдрому Рейно, артриту, судомного синдрому, гематологічних порушень, синдрому Шегрена. Множинні органні ураження з’являються до 5–10-го року хвороби. Первинно-хронічний варіант перебігу найбільш часто відзначають при поєднанні СЧВ і вторинного АФС.
Крім того, виділяють клініко-імунологічні варіанти.
СЧВ з дебютом у дитячому та підлітковому віці. Характерний гострий перебіг. В якості перших симптомів можливі поліартрит, лейкоцитоз, лімфаденопатія, гепато- і спленомегалія. Розвиток активних форм вовчакового нефриту у підлітків — вкрай несприятлива прогностична ознака.
Дебют СЧВ у віці старше 50 років («СЧВ у літніх») діагностують в 15–22% випадків. Характерна висока частота ураження серцево-судинної системи, серозитів, вторинного синдрому Шегрена, а також підвищена продукція різних аутоантитіл. Тяжкі форми ураження нирок і ЦНС виявляють рідко. Прогноз більш сприятливий, ніж при «класичному» СЧВ. Висока частота летальних випадків, зумовлених супутніми захворюваннями (ішемічна хвороба серця, гіпертонічна хвороба, злоякісні новоутворення, інфекції) і ускладненнями лікарської терапії (наприклад шлунково-кишковими кровотечами).
СЧВ у чоловіків. На частку чоловіків припадає 6–20% хворих. Захворювання починається в більш старшому віці, ніж у жінок; характеризується підвищенням частоти ураження нирок і деякими іншими прогностично несприятливими ознаками (судомні напади, тромбоцитопенія, наявність АФЛ), а також нетиповим суглобовим синдромом із залученням суглобів нижніх кінцівок і розвитком сакроілеїту у 25% хворих, більш високою частотою дискоїдного ураження шкіри. Прогноз у чоловіків, на відміну від жінок, більш серйозний.
Підгострий шкірний червоний вовчак — особливий варіант СЧВ з поширеними фоточутливими папулосквамозними (псоріазиформними) або кільцеподібними поліциклічними шкірними висипами. Своєрідність цієї форми полягає в рідкісності тяжкого ураження нирок і ЦНС, високій частоті виявлення анти-Rо антитіл та переважанні хворих чоловічої статі (співвідношення чоловіків і жінок — 4:1).
Вторинний АФС розвивається у 20–30% хворих на СЧВ. Характерні рецидивуючі судинні тромбози, акушерська патологія, рідше — тромбоцитопенія та інші прояви (серцево-судинні, неврологічні, шкірні та ін.). У ряді випадків АФС може передувати симптомам СЧВ, переважати в клінічній картині та бути фактором у визначенні лікувальної тактики.
Синдром неонатального вовчака — модель пасивно набутого аутоімунного захворювання, пов’язаного з трансплацентарною передачею материнських аутоантитіл до розчинних антигенів тканинних рибонуклеопротеїнів, у першу чергу Rо/SS-А і Lа/SS-В (виявляють у 20–30% хворих на СЧВ та деякі інші ревматичні захворювання). Прояви неонатального вовчака включають ураження шкіри, пневмоніти, гепатолієнальний синдром, ураження серця (міокардити, перикардити, уроджена повна поперечна блокада) і цитопенію. Ознаки носять транзиторний характер, як правило, повністю зникають без будь-якого лікування через 4–12 тиж в міру елімінації материнських імуноглобулінів з організму дитини.
Антифосфоліпідний синдром
АФС — симптомокомплекс, що характеризується венозними та/або артеріальними тромбозами, акушерською патологією (невиношування в I і II триместр вагітності, передчасні пологи), рідше тромбоцитопенією, а також іншими (серцево-судинні, неврологічні, шкірні та ін.) проявами, пов’язаними з гіперпродукцією АФЛ.
АФС може розвиватися у 20–30% пацієнтів із СЧВ.
Клінічна картина
Клінічна картина СЧВ відзначається надзвичайним поліморфізмом. Хворіють переважно жінки репродуктивного віку (20–30 років), нерідко підлітки. Поширене ураження тканин при СЧВ зумовлює можливість численних клінічних варіантів захворювання, у зв’язку з чим важко описати його типову картину. Поряд з випадками гострого тяжкого перебігу хвороби з септичною лихоманкою, полісерозитом і коматозним станом можна діагностувати моносимптомні та малохарактерні форми (артралгія, гемолітична анемія, гломерулонефрит, неврит). Слід пам’ятати про те, що лихоманка — типовий прояв СЧВ, може випереджати інші симптоми на кілька місяців. Необхідно також враховувати анамнестичні вказівки на алергію на ліки та зменшення кількості лейкоцитів у крові, особливо лімфопенію, зареєстровану в минулому.
Усе більшу увагу на сьогодні привертають такі досить часті прояви СЧВ, як епілепсія, нейропсихічні порушення й артрит, які можуть випереджати виникнення розгорнутої картини захворювання на місяці та навіть роки.
Захворювання найчастіше починається непомітно, з неспецифічних симптомів: підвищення температури тіла, виникнення болю в суглобах, нездужання та слабкості, трофічних розладів, зменшення маси тіла, поява різних шкірних висипів. Спочатку нерідко діагностують РА, поки не виявляють LЕ-клітини і АНФ. Ознаки вісцеральних уражень у багатьох випадках з’являються лише через кілька років.
Майже у кожного четвертого хворого процес починається гостро з високої температури і швидкого зменшення маси тіла, а також змін шкірних покривів.
Суглобовий синдром відзначають рідше, ніж при поступовому початку хвороби, зміни в нирках на ранньому етапі захворювання розглядають як прогностично несприятливу ознаку. Ранні симптоми СЧВ: артрит (50%), зміни шкіри (26%), підвищення температури (7%), епілептичні напади (4%), плеврит (3%), а також психози, пурпура й анемія. Подальший перебіг СЧВ може бути рецидивуючим з відносно тривалими ремісіями або хронічно прогресуючим. Характер клінічних проявів змінюється при кожному рецидиві хвороби. Так, якщо в дебюті захворювання відзначають клінічні ознаки гемолітичної анемії, то в подальшому вона може змінитися серозитом або люпус-нефритом та ін. Для перебігу СЧВ, як правило, характерні загальні симптоми — підвищення температури тіла в 85% випадків, зменшення маси тіла в 60% хворих, поява слабкості. Найбільш характерними клінічними проявами СЧВ є наступні симптоми: артрит і артралгії — 91,6%; лихоманка — 83,6%, LЕ-клітини — 75,6%; зміни шкірних покривів — 71,5%; лімфаденопатія — 58,6%; анемія — 56,5%; анорексія, нудота, блювання — 53,2%; міалгія — 48,2%; диспротеїнемія — 53%; ураження нирок — 46,1; плеврит — 45,0; лейкопенія — 42,6%; перикардит — 30,5%; ексудат у плевральну порожнину — 30,5; ураження ЦНС — 25,5%.
Суглобовий синдром — ознака, яку найчастіше виявляють більш ніж у 90% хворих, в основному у вигляді рецидивуючих артралгій чи артритів, рідше — стійкого больового синдрому з больовими контрактурами. Особливо характерні помірно виражені артралгії або тендовагініти, які нерідко помилково трактують як ранній прояв ревматоїдного поліартриту. Поряд з підвищеною чутливістю до сонячних променів і лікарських препаратів протягом багатьох років може проявлятися також біль в суглобах, який розцінюють як прояв росту організму («біль зростання») або ревматизму. Досить часто артралгії не відповідають вираженості синовіту. Справжній синовіт, типовий симптом СЧВ, звичайно буває нестійким і рідко викликає деструктивні зміни хряща. При СЧВ можливе ураження будь-яких суглобів, особливо дрібних суглобів кистей, а також променезап’ясткових, гомілковостопних, рідше великих. У багатьох хворих може розвиватися деформуючий неерозивний артрит кистей (синдром Жакку), особливо в осіб із високими титрами антигістонових антитіл. Рідкість виникнення ерозивних змін є істотною відмінністю між синовітом при СЧВ і РА.
Своєрідною формою ураження суглобів при СЧВ є асептичний остеонекроз, який може розвиватися у будь-якому суглобі, але найчастіше в голівці стегнової кістки, колінних суглобах. При цьому нерідко основне захворювання тривалий час перебуває в стадії ремісії, а в клінічній картині хвороби переважають симптоми асептичного некрозу голівки стегнової кістки, через що хворі стають частково або повністю знерухомленими. Порушення функції суглобів, викликане остеонекрозом, вимагає корекції, у зв’язку з чим у таких хворих застосовують ендопротезування.
У чоловіків, хворих на СЧВ, останнім часом досить часто (½ випадків) діагностують сакроілеїт (НLА В27-негативний). При суглобовому синдромі з ураженням періартикулярних тканин виявляють також розрив зв’язок, що призводить до значних порушень функції суглоба. Відновлення функції суглоба можливе лише оперативним шляхом. Хірургічне втручання проводять не скасовуючи ГК якомога раніше, до розвитку рубців.
У деяких хворих хронічний тендовагініт сухожиль згиначів призводить до формування контрактур, особливо великих пальців кистей (характерний вовчаковий великий палець), інших пальців кистей і стоп. СЧВ асоційований з підвищеним ризиком розвитку ОП. Частота спонтанних переломів у хворих на СЧВ приблизно в 5 разів вища, ніж у популяції. Поряд із традиційними факторами ризику розвитку ОП у хворих на СЧВ сприяє системне запалення; порушення ліпідного спектра; патологія яєчників (у жінок) і гіпогонадизм (у чоловіків); вторинний гіперпаратиреоз, обмеження рухової діяльності та інсоляції, медикаментозна терапія. Негативно впливають на кісткову масу ГК, циклофосфамід, антикоагулянти, гонадотропін-рилізинг-гормон.
Ураження скелетних м’язів (міалгії, підвищена чутливість м’язів при пальпації, м’язова слабкість, атрофія) у ряді випадків не відрізняється від такого при класичних ІЗМ. Є дані про нерідкісний (до 30%) розвиток синдрому фіброміалгії.
Рентгенологічним методом виявляють епіфізарний ОП переважно суглобів кистей і променезап’ясткових суглобів. При хронічному поліартриті (з довгостроковим перебігом) з деформаціями можливе звуження суглобових щілин в основному міжфалангових суглобів кисті, рідше променезап’ясткових, а також стоншення субхондральних пластинок, поява дрібних узур суглобових кінців кісток з підвивихами. При біопсії синовіальної оболонки діагностують гострий або підгострий синовіт з ядерною патологією та наявністю гематоксилінових тілець.
Частота шкірних проявів у хворих на СЧВ за Дюбуа:
1. Судинний метелик — 36,7%
2. Фотосенсибілізація — 32,7%
3. Хронічні дискоїдні зміни — 28,6%
4. Нерубцева алопеція — 21,3%
5. «Метелик» за типом відцентрової еритеми — 20,9%
6. Петехії, пурпура, екхімози — 19,8%
7. Неспецифічні макулопапульозні зміни — 19,0%
8. Синдром Рейно — 18,4%
9. Кон’юнктивіт — 10,0%
10. Макулопапульозні зміни на відкритих ділянках тіла — 9,1%
11. Зміни слизових оболонок — 9,1%
12. Дифузна гіперпігментація — 8,4%
13. Кропив’янка — 6,9%
14. Еритема в ділянці суглобів — 6,5%
15. Ламкість волосся — 5,6%
16. Виразки гомілок — 5,6%
17. Підшкірні вузлики — 5,0%
18. Періорбітальний набряк — 4,8%
19. Тромбофлебіт — 4,6%
20. Набряклість обличчя — 4,6%
21. Жовтяниця — 3,8%
22. Рубцева алопеція — 3,6%
23. Сильний свербіж — 2,8%
24. Гангрена пальців — 1,3%
25. Навколонігтьова еритема — 1,1%
26. Бульозні зміни — 0,4%
27. Еритема в ділянці піднебіння — 1,3%
Ураження шкіри
Ураження шкіри — одна з найбільш важливих з діагностичної точки зору ознак, зустрічається в 55–90% хворих. В 20–30% випадків шкірний синдром — перший прояв хвороби, а в 60–70% хворих він розвивається на різних етапах захворювання. Шкірні прояви включають еритему обличчя у вигляді метелика, підгострий шкірний червоний вовчак, дискоїдні елементи, алопецію, панікуліт, дифузну пальмарну еритему, еритему навколонігтьового валика, сітчасте ліведо, геморагії та ін.
Фотосенсибілізацію (підвищену чутливість до інсоляції) відзначають в 45–70% випадків, у половини хворих вона призводить до загострення хвороби. Фотоіндукуючі зміни шкірних покривів розвиваються на відкритих ділянках тіла (обличчя, зона декольте, верхні кінцівки) та представлені різноманітними макулярними, папульозними і бульозними ураженнями, а також класичною еритемою. Найбільш часто у хворих реєструють ураження шкіри у вигляді ізольованих або зливних еритематозних плям різної величини, різко відмежованих від оточуючої здорової тканини, які поширюються до периферії (відцентрова еритема Бієтта). Типову локалізацію на відкритих ділянках, у першу чергу на обличчі, особливо на носі та щоках, з утворенням фігури метелика вважають патогномонічною ознакою захворювання. Судинний (васкулітний) метелик у вигляді нестійкого, пульсуючого розлитого почервоніння з ціанотичним відтінком, що посилюється під впливом сонця, вітру, морозу, психоемоційних навантажень, відзначають рідше. Рідко виявляють ураження шкіри у вигляді численних еритематозних, різко окреслених кільцеподібних висипів, що нагадують багатоформну ексудативну еритему — синдром Роуелла.
Типові вогнища дискоїдного червоного вовчака з характерними ознаками — еритемою, інфільтрацією, гіперкератозом й атрофією — відзначають у 20–25% хворих. Улюблена локалізація дискоїдних елементів — обличчя, вушні раковини, шия, волосиста частина голови, рідше — верхні кінцівки. Можливе розташування вогнищ на слизовій оболонці порожнини рота з виразкою. У результаті ураження на місці вогнищ залишаються ділянки рубцевої атрофії. Дисемінований червоний вовчак — ще один вид шкірного ураження, утворений множинними, розсіяними по різних ділянках шкірного покриву вогнищами дискоїдного вовчака. Діагностично значущі капілярити — набрякла еритема з дрібнокрапковими геморагіями на подушечках пальців рук, долонях та підошвах (рис. 13.1).
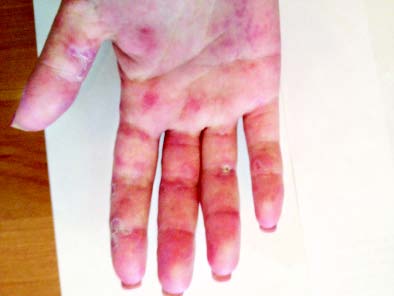
Рис. 13.1. Капілярити долонь при хронічному перебігу СЧВ
Шкірні геморагії спостерігають у 9–20% хворих; як правило, вони пов’язані з тромбоцитопенією або порушенням функції тромбоцитів; можуть бути зумовлені прийомом лікарських препаратів (ГК, НПЗП, саліцилатів), а також шкірним васкулітом. Активний шкірний васкуліт може бути представлений некротичними виразками кінцівок, гангреною пальців, інфарктами шкіри. Хронічні виразки нижніх кінцівок розвиваються у 3–5% хворих. Вищеперелічені зміни можуть бути проявами вторинного АФС.
Дифузна гіперпігментація шкіри зустрічається у 5–8% хворих на СЧВ, зазвичай на відкритих ділянках тіла, розгинальних поверхнях кінцівок. Можлива депігментація, яка в разі ураження великих ділянок шкіри нерідко створює косметичні проблеми.
Алопеція — часта (24–70%), але неспецифічна ознака СЧВ. Уражаються волосиста частина голови, брови, вії, пахвові западини та інші ділянки. Виділяють кілька форм алопеції: вогнищева і дифузна, рубцева і нерубцева (рис. 13.2).
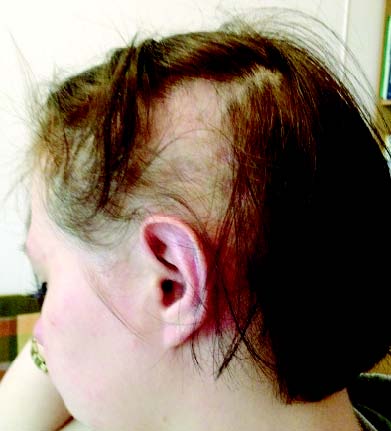
Рис. 13.2. Нерубцева алопеція при хронічному перебігу СЧВ
Ураження нігтів виявляють у ¼ хворих на СЧВ, частіше при активному процесі. Характерні дифузне почервоніння півмісяців, поздовжня і поперечна смугастість, атрофія навколонігтьового валика, оніхолізис, лейконіхія.
Ураження слизових оболонок
Ураження слизових оболонок виявляють у 7–40% хворих, зазвичай у період загострення захворювання. Зміни торкаються головним чином порожнини рота і носа, рідше — залучаються кон’юнктива і геніталії. У порожнині рота відзначають еритематозні ділянки з геморагічними вкрапленнями й ерозіями слизової оболонки (енантема), помірно болісні виразки (афтозний стоматит), білясті бляшки з неправильними контурами. Залучення в патологічний процес червоної облямівки губ (люпус-хейліт) характеризується вираженим запаленням, набряком, тріщинами, утворенням ерозій і виразок, покритих серозно-кров’яними кірками. Ерозивно-виразковий процес може супроводжуватися печінням і вираженою хворобливістю. Результат ураження — атрофія. Червона облямівка стоншується, на поверхні з’являються телеангіектазії. Тривало існуючі виразки в порожнині носа, зумовлені лейкоцитокластичним ангіїтом, іноді можуть призводити до перфорації носової перегородки (0,5–1% хворих).
Ураження дихальної системи
Ураження дихальної системи відзначають на різних етапах захворювання більш ніж у половини хворих на СЧВ. У патологічний процес залучені практично всі відділи, включаючи верхні дихальні шляхи, паренхіму легенів, плевру, судинну систему та дихальні м’язи.
Гортань піддається ураженню в 2–4% випадків. Клінічні прояви включають неспецифічне запалення слизової оболонки гортані, рухові розлади (параліч або парез голосових зв’язок), підглотковий стеноз, запальний набряк гортані, різні інфекційні ураження. Може розвинутися некротизуючий васкуліт з обструкцією повітроносних шляхів.
Полісерозит зустрічається досить часто при СЧВ (у 40–90% хворих) і разом з ураженням шкіри та суглобовим синдромом становить класичну діагностичну тріаду. Найбільш часто відзначають ураження плеври і перикарда, набагато рідше — очеревини. Серозит частіше буває сухий фібринозний, рідше — випітний. Випіт, як правило, мінімальний, швидко зникає, залишаючи спайки. В ексудаті можна виявити LЕ-клітини. Клінічна картина плевриту, перикардиту, периспленіту, перигепатиту відповідає загальновідомій: біль, шум тертя плеври, перикарда, очеревини над ділянкою селезінки і печінки. У діагностиці перикардиту допомагає ехокардіографія, при проведенні якої виявляють невеликий випіт у порожнині перикарда майже у половини хворих. При рентгенологічному дослідженні діагностують плевроперикардіальні спайки, потовщення костальної, міжчасткової, медіастинальної плеври, що вказують на перенесений в минулому серозит. Останнім часом завдяки ранній діагностиці та проведенню активної адекватної терапії полісерозити вкрай рідко закінчуються облітерацією плеври і перикарда, а також вираженими периспленітом і перигепатитом.
Ураження легень при СЧВ за даними (Насонова В.А., 1989) розвивається на 2–4-му році хвороби. Пневмоніт у хворих на СЧВ клінічно характеризується задишкою, кровохарканням, аускультативно — ослабленим диханням, наявністю незвучних вологих хрипів у нижніх відділах, а рентгенологічно — високим стоянням діафрагми, стійким посиленням легеневого малюнка і його деформацією (вогнищевосітчастою) переважно в нижніх і середніх відділах легень, симетричністю ураження, іноді наявністю дископодібних ателектазів. Крім пневмоніту, при СЧВ можливі й інші легеневі зміни, такі як бактеріальна пневмонія, туберкульоз, кандидоз та ін.
Легенева гіпертензія розвивається у 5–14% хворих. Опосередковується різними механізмами: артеріальним вазоспазмом, васкулітом великих судин, тромбозом, гострим і хронічним ураженням паренхіми, ендотеліальною дисфункцією. Легенева гіпертензія може бути зумовлена тромбоемболією гілок легеневої артерії, що виявляють у 5–12% хворих на СЧВ в межах вторинного АФС. У більшості випадків ознаки легеневої гіпертензії з’являються через кілька років після початку СЧВ, розвиваються повільно і мають тенденцію до поступового прогресування. Прогноз при розвитку легеневої гіпертензії залежить від ступеня її вираженості та швидкості підвищення тиску в легеневій артерії.
Гострий вовчаковий пневмоніт — рідкісний варіант вовчакового ураження легень (1–4%). Клінічними проявами є лихоманка, задишка, продуктивний кашель, можливі кровохаркання і плевральний больовий синдром. За допомогою рентгенологічних методів виявляють одно- або двобічні інфільтрати в нижніх легеневих зонах. Легеневі (альвеолярні) геморагії діагностують відносно рідко (2%), проте їх перебіг, як правило, тяжкий, від них може залежати прогноз хвороби. Клінічна симптоматика нагадує гострий вовчаковий пневмоніт з прогресуючим зниженням гемоглобіну і гематокриту, розвитком артеріальної гіпоксемії та гострого респіраторного дистрес-синдрому. Тяжкі обструктивні зміни нижніх дихальних шляхів розвиваються рідко. Порушення функціональних дихальних тестів реєструють приблизно в 10% хворих, в яких немає виражених змін на рентгенограмах. Клінічна картина облітеруючого бронхіоліту неспецифічна: лихоманка, кашель, задишка; рентгенологічно виявляють вогнищеві або дифузні інфільтрати. Іноді спостерігається дисфункція дихальних м’язів, що призводить до виникнення задишки за відсутності якого-небудь органічного ураження легень та дихальних шляхів. Слабкість дихальних м’язів зазвичай не асоційована з генералізованою м’язовою слабкістю.
Ураження серця при СЧВ вельми характерне та зустрічається в ⅓ хворих. При люпус-кардиті можуть бути уражені всі оболонки серця. За даними інших авторів (Насонова В.А., Бунчук Н.В. (ред.), 1997), частота ураження серця при СЧВ може становити від 88,8 до 100%. Найбільш характерними ознаками ураження серця при СЧВ є перикардит, перебіг якого частіше латентний, який клінічно діагностують рідко. При ехокардіографічному дослідженні, при своєчасному його проведенні, досить часто реєструють присутність випоту в перикарді.
Класичний вовчаковий ендокардит, відомий як ендокардит Лібмана — Сакса, анатомічно характеризується дрібними бородавками по вільному краю мітрального клапана з наступним рубцюванням і формуванням клапанних вад, частіше — недостатності клапанів. Важливою складовою діагностики є ехокардіографія і фонокардіографія. Можливий також розвиток вовчакового міокардиту, проте досі він залишається більше анатомічним, ніж клінічним діагнозом, оскільки проходить із мізерними клінічними проявами. Іноді відзначають розширення меж серця, ритм галопу, порушення ритму і провідності. У ряді випадків нерідко при СЧВ виявляють коронарний артеріїт. Зумовлена вовчаковим ураженням нирок гіпертензія істотно обтяжує як клінічну картину, так і прогноз СЧВ відносно кардіальної патології. Іноді у хворих на СЧВ розвивається легенева гіпертензія. Етіологія її невідома, вона може бути зумовлена тромбозом, асоційованим з АФС.
Ураження судин. При СЧВ в патологічний процес залучаються судини дрібного й середнього калібру. Хоча багато вісцеральних проявів СЧВ зумовлені васкулітом, можливі й більш очевидні ознаки ураження судинного русла. Найбільш характерні симптоми васкуліту — сітчасте ліведо, дигітальні інфаркти, рецидивуючий тромбофлебіт і хронічні виразки гомілок — асоціюються з АФС. Крім того, системне ураження судинного русла поєднується також з високим рівнем кріоглобулінів у хворих на СЧВ, клінічно проявляється виразковим ураженням шкіри, кріоглобулінемічною пурпурою, капіляритом, цереброваскулітом, нефритом, ліведо, стійким синдромом Рейно та ін. (рис. 13.3).
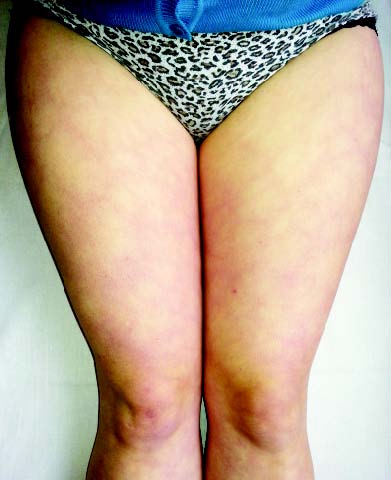
Рис. 13.3. Сітчасте ліведо
У 6–18% хворих на СЧВ діагностують синдром Рейно, який виступає продромальним, або раннім, симптомом СЧВ. Патогенез синдрому Рейно нез’ясований. Тільки в деяких випадках в його основі — кріоглобулінемія.
Загалом наявність синдрому Рейно при СЧВ є показником доброякісного, повільно прогресуючого перебігу хвороби, особливо, якщо в дебюті СЧВ синдром Рейно набуває рецидивуючого характеру. Для такої форми СЧВ притаманний розвиток міалгічного синдрому, телеангіектазій. Однак, якщо СЧВ починається зі стійкого синдрому Рейно, то це прогностично несприятливий симптом. У таких хворих відзначають схильність до розвитку полісиндромності за типом системного васкуліту з ураженням нирок, ЦНС, у ряду пацієнтів — легень з тенденцією до незворотних порушень кровообігу в уражених органах (розвиток некрозів шкіри, кісток, дифузного гломерулонефриту з ангіопатією сітківки, важкої гіпертензії, цереброваскуліту, легеневої гіпертензії). Прогноз при вовчаковому системному васкуліті несприятливий.
Ураження нирок
Ураження нирок вкрай різноманітне, його виявляють у 35–90% хворих. У переважної більшості розвиток вовчакового нефриту проявляється протягом перших 5 років хвороби, у 3–10% хворих ураження нирок може бути першим проявом СЧВ. Активні форми вовчакового нефриту розвиваються звичайно в перші роки захворювання на тлі вираженого імунозапального процесу, частіше в молодому віці. Хворим старшого віку властивий менш агресивний перебіг нефриту (як клінічних, так і морфологічних варіантів).
На сьогодні використовують морфологічну класифікацію вовчакового нефриту, запропоновану ВООЗ у 1982 р. і переглянуту в 1995 р. Встановлено взаємозв’язок між особливостями морфологічної картини гломерулонефриту та його клінічними проявами, характером перебігу і прогнозом (табл. 13.2).
Таблиця 13.2
Зіставлення клініко-морфологічних характеристик при вовчаковому гломерулонефриті
|
Клас нефриту (за класифікацією ВООЗ) |
Морфологічні особливості |
Клінічні прояви |
|
Клас I: мінімальні зміни |
Нормальні клубочки при світловій мікроскопії та наявність депозитів у мезангію при імунофлуоресценції |
Відсутні зміни в сечі, функція нирок нормальна. Прогноз сприятливий, проте можлива трансформація в більш серйозний тип нефриту |
|
Клас II (8–30%): мезангіальний проліферативний гломерулонефрит |
Різний ступінь мезангіальної гіперклітинності з наявністю імунних депозитів в мезангію |
Протеїнурія <1 г/добу, гематурія. Функція нирок збережена. Розвиток нефротичного синдрому і ниркової недостатності нехарактерний. Прогноз сприятливий за відсутності трансформації в більш тяжкий морфологічний тип нефриту |
|
Клас III (10–25%): фокальний гломерулонефрит |
Може бути активне або хронічне, сегментарне або тотальне, ендо- або екстракапілярне ушкодження із залученням < 50% клубочків |
Протеїнурія > 1 г/добу, у 20–30% випадків розвивається нефротичний синдром. Зміни сечового осаду мають помірний характер. Перебіг характеризується неухильним прогресуванням. Високий ризик розвитку хронічної ниркової недостатності, резистентність до імуносупресивної терапії. Прогноз відносно сприятливий у разі адекватної терапії |
|
Клас IV (20–60%): дифузний проліферативний гломерулонефрит |
Морфологія та сама, що при класі III, але в процес залучено > 50% клубочків |
Протеїнурія > 3 г/добу, нефротичний синдром з активним сечовим осадом. Ниркова недостатність і артеріальна гіпертензія відносно рідкі, імунологічні порушення виражені помірно. Прогноз сприятливий для хворих із помірною протеїнурією, серйозніший — при нефротичному синдромі (особливо тривалому). У частини хворих можлива спонтанна ремісія |
|
Клас V (10–20%): мембранозний гломерулонефрит |
Рівномірне потовщення базальної мембрани клубочків внаслідок субепітеліального і внутрішньомембранного відкладення імунних депозитів |
Виражена протеїнурія, гематурія, циліндрурія. Висока частота артеріальної гіпертензії і ниркової недостатності різного ступеня вираженості, низький рівень комплементу. Прогноз за неадекватної терапії несприятливий |
|
Класс VI: нефросклероз |
Дифузні зміни: дифузний і сегментарний гломерулосклероз, атрофія канальців, інтерстиціальний фіброз, артеріолосклероз |
Клінічні та лабораторні ознаки хронічної ниркової недостатності різної вираженості. Прогноз несприятливий |
У клінічній практиці для вибору тактики лікування використовують клініко-лабораторні та морфологічні критерії тяжкості нефриту (табл. 13.3).
Таблиця 13.3
Критерії тяжкості вовчакового нефриту
|
Ступінь тяжкості |
Характеристика |
|
Фокальний вовчаковий нефрит (III клас) і дифузний проліферативний вовчаковий нефрит (IV клас) |
|
|
Низький |
Фокальний нефрит без несприятливих гістологічних ознак (півмісяці, фібриноїдний некроз або індекс хронізації >3) і без несприятливих клінічних ознак (нормальна функція нирок, протеїнурія <3 г/добу) |
|
Помірний |
Часткова відповідь або відсутність відповіді після індукційної терапії фокального нефриту або фокальний нефрит з несприятливими гістологічними ознаками, або підвищення рівнів сироваткового креатиніну щонайменше на 30%, або дифузний гломерулонефрит без несприятливих гістологічних ознак |
|
Високий |
Відсутність ремісії через 6–12 міс від початку терапії або порушення функції нирок, фібриноїдний некроз та півмісяці в більш ніж 25% клубочків, або змішаний мембранозний і проліферативний нефрит, або високий індекс хронізації ізольовано або в комбінації з високим індексом активності, або швидкопрогресуючий вовчаковий нефрит (подвоєння рівня сироваткового креатиніну протягом 2–3 міс) |
|
Мембранозний нефрит (клас V) |
|
|
Низький |
Протеїнурія <3 г/добу, нормальна функція нирок |
|
Помірний |
Нефротичний синдром, нормальна функція нирок Нефротичний синдром, функція нирок порушена |
Перебіг вовчакового нефриту характеризується досить частими загостреннями, при цьому кожне загострення посилює тяжкість ураження ниркових структур, розвиток або прогресування гломерулярного склерозу та інтерстиціального фіброзу.
У 10–27% хворих на вовчаковий нефрит розвивається ниркова недостатність, що прогресує до термінальної стадії.
Розвиток і прогресування ниркової недостатності часто супроводжується зменшенням вираженості або навіть повним зникненням екстраренальних проявів СЧВ і зниженням лабораторних показників активності захворювання. Факторами ризику прогресування вовчакового нефриту вважають чоловічу стать, дитячий і молодий вік хворих в період дебюту хвороби, «ранній» початок нефриту, приналежність до негроїдної раси і погані соціально-економічні умови.
Перебіг вовчакового нефриту може ускладнюватися розвитком інфекційного ураження нирок, в першу чергу пієлонефриту. Описано також ускладнення, пов’язані з лікарською терапією: геморагічний цистит (циклофосфамід), анальгетична нефропатія (НПЗП). Розвиток АА-амілоїдозу при СЧВ — казуїстичний.
Ураження органів травлення
Патологія ШКТ зустрічається в 50% хворих на СЧВ. Ураження слизової оболонки рота відзначають у 7–40% випадків. Виразковий стоматит зазвичай реєструють на тлі високої активності захворювання. Хворобливі виразки можуть порушувати прийом їжі та ковтання. Фарингіт частіше діагностують у дітей. У третини хворих з активним СЧВ виникають анорексія, нудота, блювання, діарея. Розвиток пептичних виразок (від 4 до 12%) пов’язують із застосуванням НПЗП і меншою мірою — ГК.
Асцит виявляють у 8–10% хворих. Основна причина його виникнення — нефротичний синдром. Асцит може бути викликаний застійною серцевою недостатністю (при цьому відсутній больовий синдром, а асцитична рідина є транссудатом), перитонітом (больовий синдром виражений, наявний ексудат) і цирозом печінки (рідко).
Украй несприятлива прогностична ознака — розвиток панкреатиту, зумовленого активністю захворювання або васкулітом в судинах підшлункової залози. Крім того, панкреатит може виникати внаслідок гіповолемії, холециститу, прийому алкоголю і ряду лікарських препаратів (ГК, азатіоприн, тіазидні діуретики). Частота даного прояву — 1–8%.
Ураження кишечнику пов’язано із залученням серозних оболонок і ураженням судин брижі. Васкуліт мезентеріальних артерій з розвитком кишкових інфарктів і виразок відносять до найбільш важких проявів.
Патологія печінки варіює від незначного збільшення її в розмірах (у 10–31% хворих) до важкого гепатиту. Найбільш часті причини жовтяниці при СЧВ (1–4% пацієнтів) — геморагічна анемія та вірусний гепатит, рідше обструкція жовчовивідних шляхів і цироз печінки. Рідко розвивається васкуліт печінки, що призводить до інфарктів і спонтанних розривів органа з картиною гострого живота. Підвищення рівнів печінкових ферментів (як правило, не більше ніж у 3 рази), за відсутності інфекції гепатотропними вірусами, зазвичай зумовлено прийомом лікарських препаратів (ацетилсаліцилова кислота, преднізолон, циклофосфамід) та тромбозом дрібних судин печінки.
Ураження нервової системи
Симптоми ураження нервової системи відзначено у більшості хворих на різних стадіях захворювання і включають практично весь спектр неврологічної симптоматики. Можуть бути першою ознакою хвороби, яка виникає задовго до появи розгорнутої картини СЧВ. Характерний головний біль (часто за типом мігрені), рефрактерний до ерготаміну, блокаторів β-адренорецепторів, анальгетиків і трициклічних антидепресантів.
Найбільш частий варіант цереброваскулярних порушень — транзиторні ішемічні атаки в каротидному (джексонівські епілептичні напади, пірамідні симптоми, порушення мови) та/або вертебробазилярному (запаморочення, ністагм, диплопія, нудота, блювання) басейні. Інсульти виникають рідше і пов’язані, як правило, із вторинними патогенетичними механізмами (наявність АФЛ, уремія, артеріальна гіпертензія).
При високій активності СЧВ розвивається синдром псевдопухлини мозку: внутрішньочерепна гіпертензія без вогнищевого неврологічного дефіциту з дифузними головними болями, нудотою, блюванням, запамороченням, загальмованістю. При обстеженні виявляють застійні диски зорових нервів. Епілептичні напади (генералізовані судомні, генералізовані без судом, фокальні) спостерігають у 20–50% хворих, зазвичай у період загострення. Хорея — руховий синдром екстрапірамідного генезу, розвивається в 1–4% випадків у осіб молодого віку на ранніх етапах захворювання. Церебральна атаксія спостерігається рідко (менш ніж у 1% хворих), проявляється порушенням рівноваги та координації, дизартрією, зниженням тонусу м’язів.
Мієлопатія розвивається менш ніж в 1% випадків. Зумовлена активним захворюванням і наявністю АФЛ (за рахунок ішемічного некрозу і демієлінізації волокон спинного мозку), може мати вторинний характер (компресійні переломи хребців, туберкульоз, вірусна інфекція). Початок мієлопатії найчастіше гострий: з’являються прогресуюча м’язова слабкість й оніміння нижніх кінцівок, що мають висхідний характер. Надалі виникають сегментарні порушення чутливості та рухової діяльності на рівні ураження спинного мозку, провідникові порушення чутливості, центральні парапарези, тетрапарези, пара- і тетраплегії, порушення функції тазових органів. Прогноз вовчакової мієлопатії вкрай несприятливий, що пов’язано з розвитком у більшості випадків сепсису. Енцефаломієлопатія — багатовогнищеве демієлінізуюче ураження головного і спинного мозку з клінічною картиною, яка нагадує розсіяний склероз. Синдром Девіса — поєднання невропатії зорового нерва з синдромом поперечної мієлопатії.
Периферичні невропатії при СЧВ діагностують у 2–21% хворих, при цьому краніальні невропатії виявляють у 16% випадків. Клінічні ознаки невропатії зазвичай наростають поступово на тлі активних проявів захворювання, рідко діагностуються. Неврит зорового нерва — не часте, але важке ураження. Його слід запідозрити при різкому одно- або двосторонньому зниженні гостроти зору, повній сліпоті, болю у ділянці очного яблука. Вторинні ураження нервової системи розвиваються на тлі артеріальної гіпертензії, уремії, гіпоксемії (внаслідок анемії, серцевої та легеневої недостатності), інтеркурентних інфекцій; прийому ГК (психоорганічні симптоми), амінохінолінових похідних (дратівливість, афективні стани, судомні напади), НПЗП (головний біль, асептичний менінгіт). Психічні, нервово-психічні та поведінкові проблеми властиві 10–80% хворих на СЧВ. Психотичні стани (30–50% пацієнтів) включають дезорієнтацію, зорові та слухові галюцинації, явища аутизму і параноїдну симптоматику. Маніакально-депресивний психоз спостерігають рідко, частіше виникають маніакальноподібні та депресивні розлади. Є поодинокі випадки кататонії. Депресії у хворих на СЧВ мають помірний характер, проявляються соматичними та психопатологічними симптомами. У деяких хворих депресія може тривати роками, можливі суїцидальні спроби.
Офтальмологічні прояви включають сухий кератокон’юнктивіт у рамках вторинного синдрому Шегрена, епісклерит, іридоцикліт, ішемічну невропатію, неврит зорового нерва, а також зміни, зумовлені прийомом лікарських препаратів (задня субкапсулярна катаракта). Залучення органа слуху в патологічний процес відзначають рідко (0–3% випадків) у вигляді середнього отиту і невриту слухового нерва з виникненням приглухуватості різного ступеня.
Ендокринні порушення при СЧВ недостатньо вивчені, хоча й виявляють їх більш високу поширеність у даної категорії хворих порівняно з популяцією. Розвиток цукрового діабету відзначено у 7–9% пацієнтів, який в ряді випадків зумовлений ГК-терапією. Частота гіпотиреозу становить 1–10%, гіпертиреозу — 3–11%, аутоімунного тиреоїдиту Хашимото — 0,5–4%.
Лімфоаденопатія — ознака СЧВ, яку досить часто виявляють — у 37–68% випадків. Нерідко збільшення лімфатичних вузлів буває настільки значним, що може викликати у лікаря підозру на лімфопроліферативне захворювання. Збільшення селезінки відзначають у 9–41% хворих.
Хворі на СЧВ із синдромами Рейно і Шегрена являють собою особливу групу, у них переважно відзначають хронічний перебіг, лімфаденопатію, неспецифічні ураження шкіри, артралгії, міалгії, сечовий синдром у вигляді незначної протеїнурії, більш сприятливий прогноз.
Гематологічні зміни характерні для СЧВ і мають діагностичне значення. Як і клінічні симптоми вони дуже різні та в деяких випадках тривалий час можуть бути єдиним чи одним із провідних проявів СЧВ.
Анемію діагностують досить часто, як правило, вона нормохромна, помірно виражена. При шлунково-кишкових кровотечах (у результаті основного захворювання або стероїдних виразок) розвивається гіпохромна анемія. Значно рідше відзначають аутоімунну гемолітичну анемію у результаті появи в крові антитіл до власних еритроцитів, що визначають за допомогою реакції Кумбса. У деяких випадках СЧВ починається з гемолітичної анемії, тривалий час хворі перебувають під спостереженням і одержують лікування з приводу цього захворювання, поки не приєднаються інші, вже класичні, прояви СЧВ (люпус-нефрит, симптом метелика та ін.).
Лейкопенію виявляють більш ніж у половини хворих на СЧВ, нерідко зі зменшенням кількості лімфоцитів (стійким або минущим). Практично у кожного пацієнта з СЧВ протягом хвороби при динамічному спостереженні діагностують лейкопенію (іноді одноразово). Однак лікування ГК та інфекція, яка часто виникає, можуть призводити до збільшення кількості лейкоцитів.
Тромбоцитопенія зустрічається рідше, але може бути вираженою, а іноді тромбоцитопенічна пурпура може бути провідною ознакою, особливо в дебюті хвороби. Лейкопенія і тромбоцитопенія при СЧВ можуть бути викликані аутоантитілами, як і аутоімунна гемолітична анемія.
У патогенезі геморагічного синдрому хворих на СЧВ має значення порушення як мікроциркуляторної, так і гемокоагуляційної ланки гомеостазу. Важливе значення в патогенезі геморагічного синдрому при СЧВ має активація фібринолізу. При поєднанні з тромбоцитопенією, відносною або абсолютною гіпофібриногенемією, зниженою резистентністю капілярів, подовженням часу й підвищенням інтенсивності кровотечі навіть помірне зростання активності фібринолізу збільшує ймовірність кровоточивості.
При СЧВ відзначають порушення здатності крові згортатися, зумовлене появою АФЛ (АКЛ, ВА, хибнопозитивна реакція Вассермана) і розвитком АФС. Висловлюється припущення, що ВА може зв’язуватися з фосфоліпідами мембран ендотеліальних клітин, блокуючи вивільнення арахідонової кислоти і порушуючи тим самим вироблення простацикліну, що сприяє агрегації тромбоцитів та розвитку тромбозу. Поява антитіл до фосфоліпідів при СЧВ веде до розвитку патології судинної стінки і тромбозів. Клінічно АФС характеризується схильністю до поширених тромбозів (тромби формуються як у венозному, так і артеріальному руслі), внутрішньоутробною загибеллю плоду (повторні аборти), ураженням ЦНС (інсульти, епісиндром, психоз), сітчастим ліведо, ураженням ендокарда, тромбоцитопенією (зумовленою порушенням агрегаційних властивостей тромбоцитів й імунологічних параметрів), асептичним некрозом кісток, розвитком хронічних виразок гомілок. У хворих на СЧВ із АФС рідше виявляють LЕ-клітини, антитіла до ДНК та антинуклеарний фактор, але частіше — кріоглобулінемію.
Лабораторні дослідження
Клінічний аналіз крові:
- Збільшення ШОЕ часто має місце, але погано корелює з клінічною активністю захворювання (ШОЕ може бути в межах норми у хворих під час загострення та зростати в період ремісії). За відсутності видимих причин для збільшення ШОЕ слід виключити супутню інфекцію.
- Лейкопенія (зазвичай лімфопенія) корелює з клінічною активністю захворювання.
- Гіпохромна анемія може бути наслідком хронічного запального процесу, прихованої шлункової кровотечі, прийому деяких лікарських препаратів. Кумбс-позитивна аутоімунна гемолітична анемія розвивається рідко, в основному при активних формах хвороби.
- Тромбоцитопенію зазвичай діагностують у пацієнтів із вторинним АФС. Дуже рідко виникає аутоімунна тромбоцитопенія, пов’язана з синтезом антитіл до тромбоцитів.
Аналіз сечі. Виявляють протеїнурію, гематурію, лейкоцитурію, циліндрурію, ступінь вираженості яких залежить від клініко-морфологічного варіанта вовчакового нефриту.
Біохімічний аналіз крові. Відхилення біохімічних показників неспецифічні, залежать від характеру ураження внутрішніх органів у різні періоди хвороби. Підвищення рівня СРБ не характерно, у більшості випадків відображає розвиток супутньої інфекції.
Імунологічні дослідження
- АНФ або антиядерний фактор у високому титрі виявляють у 95% хворих на СЧВ. Відсутність АНФ ставить під сумнів діагноз СЧВ.
- Антитіла до двоспіральної ДНК виявляють у 20–60% хворих. Відносно специфічні. Підвищення рівня корелює з активністю захворювання і розвитком вовчакового нефриту.
- Антитіла до Sm (Smith) виявляють у 10–30% хворих. Високоспецифічні. Антитіла до малого ядерного рибонуклеопротеїну частіше визначають у хворих із картиною ЗЗСТ (феномен Рейно, міозит, щільний набряк кистей). Антитіла до Rо/SS-А поєднуються з лімфопенією, тромбоцитопенією, фотодерматитом, легеневим фіброзом, синдромом Шегрена. Антитіла до Lа/SS-В часто виявляють разом з антитілами до Rо, але їх клінічне значення не з’ясовано.
- АФЛ — АКЛ (підвищення рівня IgG та/або IgМ), ВА, хибнопозитивна реакція Вассермана — характерні для АФС.
- Зниження загальної гемолітичної активності комплементу (СН50) і його окремих компонентів (СЗ і С4) відзначають у хворих на вовчаковий нефрит, що корелює з активністю нефриту (особливо СЗ). Зниження СН50 і рівня окремих білків системи комплементу може бути зумовлено їх генетично детермінованим дефіцитом.
Інструментальні методи дослідження
За наявності показань: рентгенографія кісток і суглобів, УЗД суглобів і м’яких тканин, МРТ, денситометрія (визначення МЩКТ) — до початку лікування, потім щорічно.
Рентгенографія органів грудної клітки (не рідше 1 разу на рік), за наявності показань — функціональні тести, бронхоскопія, КТ легень, ехокардіографія (для діагностики легеневої гіпертензії).
ЕКГ, ехоКГ, УЗД судин з вимірюванням товщини комплексу «інтима-медіа» та діаметра сонних артерій (виявлення субклінічних ознак атеросклерозу). За наявності показань — моніторування ЕКГ за методом Холтера, ангіографія та ін.
Езофагогастродуоденоскопія (не рідше 1 разу на рік), УЗД органів черевної порожнини, за наявності показань — КТ та МРТ. МРТ, КТ, електроенцефалографія за показаннями.
Діагностичні критерії системного червоного вовчака (ACR, 1997)
1. Еритема-метелик
Фіксована еритема, плоска або що піднімається над поверхнею шкіри, на вилицях, з тенденцією до поширення на назолабіальні складки.
2. Дискоїдний вовчак
Еритематозні плями, що піднімаються, зі щільно прилягаючими роговими лусочками і закупореними волосяними фолікулами; згодом на місці висипів формується рубцева атрофія.
3. Фотосенсибілізація
Поява висипів після надмірної інсоляції (дані анамнезу або спостереження лікаря).
4. Виразки порожнини рота
Ульцерація ротової, носової порожнин, ковтки.
5. Артрит
Неерозивний артрит, що уражає 2 і більше периферичних суглобів, що характеризується хворобливістю, припухлістю і випотом.
6. Серозит
а) плеврит: переконливі дані анамнезу про плевритичний біль або шум тертя плеври, зафіксований лікарем, або наявність плеврального випоту
або
б) перикардит: зафіксовані на ЕКГ ознаки перикардиту або шум тертя перикарда чи наявність перикардіального випоту.
7. Ураження нирок
а) персистуюча протеїнурія: >0,5 г/добу або більше +++, якщо підрахунок не проводиться;
б) циліндрурія: можуть бути еритроцитарні, гемоглобінові, зернисті, воскоподібні, змішані циліндри.
8. Ураження нервової системи
а) судоми
або
б) психоз
За відсутності провокуючих ліків або метаболічних порушень, таких як уремія, кетоацидоз або електролітний дисбаланс.
9. Гематологічні зміни
а) гемолітична анемія з ретикулоцитозом
або
б) лейкопенія <4000/мм3 у 2 і більше дослідженнях
або
в) лімфопенія <1500/мм3 у 2 і більше дослідженнях
або
г) тромбоцитопенія <100 000/мм3 за відсутності провокуючих ліків.
10. Імунологічні порушення
а) анти-ДНК: антитіла до нативної ДНК у високому титрі
або
б) анти-Sm: присутність антитіл до Sm ядерного антигену
або
в) виявлення АФЛ на підставі:
- високого рівня IgM або IgG АКЛ;
- виявлення ВА з використанням стандартної методики;
- хибнопозитивна серологічна реакція на сифіліс протягом не менше 6 міс, підтверджена реакцією іммобілізації блідих трепонем або реакцією імунної флуоресценції.
11. Антиядерні антитіла
Високі титри антиядерних антитіл в реакції імунної флуоресценції або еквівалентній реакції в будь-який момент за відсутності ліків, здатних викликати медикаментозний червоний вовчак.
За наявності 4 або більше з 11 вищеперерахованих критеріїв можна поставити діагноз СЧВ.
Чутливість становить 96%, специфічність — 96%.
Приклади формулювання діагнозів
СЧВ: гострий перебіг, активна фаза, активність III ступеня, з ураженням шкіри — симптом метелика; суглобів — поліартрит; серозних оболонок — ексудативний плеврит, перикардит; нирок — люпус-нефрит нефротичного типу; нервової системи — церебральний васкуліт з епілептиформним синдромом.
СЧВ: підгострий перебіг, активність II ступеня, АФС з ураженням шкіри — ретикулярне ліведо, капілярити з явищами дигітального некрозу; суглобів — поліартрит, асептичний некроз голівки правої стегнової кістки; серця — ендокардит Лібмана — Сакса, недостатність мітрального клапана, серцева недостатність I ступеня; нервової системи — дисциркуляторна енцефалопатія, астеновегетативний синдром.
СЧВ: хронічний перебіг, активна фаза, активність I ступеня, синдром дискоїдного вовчака, рецидивуючий поліартрит (синдром Жакку), серозних оболонок — адгезивний плеврит.
Диференційна діагностика
Диференційну діагностику проводять з гематологічними захворюваннями (гемолітичні анемії, тромбоцитопенічна пурпура), системними васкулітами, лімфопроліферативними захворюваннями, іншими ревматичними захворюваннями (хвороба Шегрена, первинний лімфогранулематоз, фіброміалгія, ранній РА, ювенільний хронічний артрит), змішаною кріоглобулінемією при гепатиті С, сироватковою хворобою, синдромом медикаментозного вовчака, паранеопластичним синдромом, саркоїдозом, запальними захворюваннями кишечнику, інфекційними захворюваннями (бореліоз Лайма, туберкульоз, вторинний сифіліс, інфекційний мононуклеоз, вірусні артрити, ВІЛ-інфекція, гепатит В та ін.).
Показання до консультації спеціалістів
Невролог: уточнення характеру і ступеня ураження нервової системи, підбір симптоматичної терапії у випадку розвитку неврологічної симптоматики.
Психіатр: призначення симптоматичної терапії і вирішення питання про необхідність лікування в спеціалізованому стаціонарі за наявності психічних розладів (особливо психозу і депресії, які супроводжуються суїцидальними думками).
Окуліст: уточнення генезу зорових порушень (патологія судин сітківки, прояв побічної дії ГК, наявність синдрому Шегрена).
Акушер-гінеколог: спільне спостереження в період вагітності.
Нефролог: вирішення питання про проведення біопсії нирки і гемодіалізу. З метою оцінки активності процесу та ураження систем та органів при СЧВ в клінічній практиці використовують індекси SLAM, SLEDAI, ECLAM.
Лікування
Лікування має бути максимально індивідуальним. Для вибору тактики ведення хворого необхідно визначити провідний синдром, оцінити активність хвороби, характер і ступінь вираженості супутньої патології. Виключно важливе розмежування загострення СЧВ і гострого інфекційного процесу. Динаміка лабораторних (особливо імунологічних) показників у більшості випадків не є підставою для коригування терапії. Слід пам’ятати, що хворі на СЧВ нерідко схильні до розвитку алергічних реакцій на багато лікарських засобів, у першу чергу, на антибактеріальні (особливо сульфаніламіди).
Мета лікування:
- Досягнення клініко-лабораторної ремісії захворювання.
- Запобігання ураженням життєво важливих органів і систем, у першу чергу нирок і ЦНС.
- Покращання якості життя.
Показання до госпіталізації:
- Лихоманка неясного генезу (інфекційні ускладнення — одна з найбільш частих причин смерті хворих на СЧВ).
- Біль в грудній клітці.
- Наявність симптомів дифузного ураження ЦНС.
- Виражені цитопенії.
- Активні форми вовчакового нефриту і швидкопрогресуюча ниркова недостатність.
- Гострий пневмоніт або легенева кровотеча.
- Загострення СЧВ (при неможливості коригування лікування на амбулаторному етапі).
Немедикаментозне лікування
Загальні рекомендації: зменшення психоемоційного навантаження, тривалості перебування на сонці, активне лікування супутніх захворювань. У період загострення захворювання і на тлі лікування цитотоксичними препаратами показана ефективна контрацепція. Протипоказані пероральні контрацептивні препарати з високим вмістом естрогенів, оскільки вони можуть викликати загострення СЧВ. Слід уникати введення вакцин і лікувальних сироваток. З метою профілактики ОП рекомендовано припинення паління, прийому їжі з високим вмісту кальцію, фізичні вправи. Для профілактики атеросклерозу показано дієту з низьким вмістом жирів і холестерину (ХС), припинення паління, контроль маси тіла, фізичні вправи.
Медикаментозне лікування
Основна терапія СЧВ до сьогодні — це неспецифічна імуносупресія.
У механізмах розвитку СЧВ важлива роль належить Toll-подібним рецепторам, які розташовані на дендритних клітинах і В-клітинах і можуть активуватися вірусами, бактеріями РНК і ДНК, які містяться в імунних комплексах при СЧВ.
Високі рівні цитокінів, хемокінів, факторів росту, які синтезуються моноцитами/макрофагами, дендритними та ендотеліальними клітинами, викликають активацію захворювання і пошкодження органів.
У цей процес включаються: В-активуючий фактор В-лімфоцитів, ФНП-α, інтерферон γ, ІЛ-12, -6, -10.
Пригнічення цих факторів і клітин можливе різними засобами, наприклад імуносупресорами, ГК, біологічними агентами.
Глюкокортикоїди
ГК короткої дії (преднізолон і метилпреднізолон) — найбільш ефективні лікарські засоби для лікування СЧВ. Доза ГК залежить від активності захворювання.
- Хворим з низькою активністю захворювання призначають невеликі дози ГК (преднізолон <10 мг/добу).
- При помірній активності — середні дози (20–40 мг/добу, або бетаметазон 1–2 мл глибоко внутрішньом’язово одноразово) протягом 4 тиж з поступовим зниженням до підтримувальної дози.
- При високій активності хвороби і важкому ураженні ЦНС, нирок, системи крові (тромбоцитопенія, гемолітична анемія) призначають високі дози ГК у поєднанні з цитотоксичними препаратами.
Абсолютним показанням для призначення високих доз ГК (1 мг/кг/добу і більше) є швидко прогресуюче ураження життєво важливих органів. Тривалість прийому високих доз ГК варіює від 4 до 12 тиж залежно від вираженості клінічного ефекту. Зниження дози слід проводити поступово, під ретельним клініко-лабораторним контролем, а підтримувальні дози (5–10 мг/добу) необхідно приймати протягом багатьох років.
Пульс-терапія (500–1000 мг метилпреднізолону внутрішньовенно (в/в) крапельно і протягом як мінімум 30 хв 3 дні поспіль) показана хворим з високою активністю СЧВ з метою досягнення швидкого ефекту, а також зниження доз пероральних ГК. Показання до включення в комплексне лікування хворих на СЧВ цитотоксичних препаратів: активний вовчаковий нефрит; висока загальна активність хвороби; резистентність до ГК, розвиток побічних реакцій на ГК на ранніх етапах лікування (наприклад явищ гіперкортицизму у підлітків), необхідність швидкого зниження підтримувальної дози преднізолону, що перевищує 15–20 мг/добу. Цитотоксичні препарати слід застосовувати тривало (до декількох років), при цьому регулярне спостереження та індивідуальний підбір дози дозволяють значно зменшити кількість побічних реакцій і ускладнень.
Циклофосфамід — препарат вибору при вовчаковому нефриті та тяжкому ураженні ЦНС. Призначення циклофосфаміду щомісячно по 0,5–1,0 г/м2 в/в крапельно протягом 6 міс, а потім кожні 3 міс протягом 2 років у поєднанні з пульс-терапією метилпреднізолоном (по 1,0 г/добу послідовно протягом 3 днів) і пероральним прийомом ГК (преднізолон 40–60 мг/добу) дозволяє підвищити виживаність хворих на проліферативний вовчаковий нефрит більшою мірою, ніж монотерапія ГК (у тому числі пульс-терапія) або комбінування ГК з азатіоприном. У більшості випадків застосування циклофосфаміду дозволяє контролювати клінічні прояви СЧВ, рефрактерні до монотерапії, високими дозами ГК (тромбоцитопенія, ураження ЦНС, легеневі геморагії, системний васкуліт).
Азатіоприн слід застосовувати для підтримки індукованої циклофосфамідом ремісії вовчакового нефриту, лікування резистентних до ГК форм аутоімунної гемолітичної анемії, тромбоцитопенії та торпідних уражень шкіри. Комбінована терапія азатіоприном та ГК сприяє підвищенню загальної виживаності хворих на вовчаковий нефрит. Стандартна терапевтична доза азатіоприну становить 2–3 мг/кг/добу. Максимальний ефект на тлі лікування цим препаратом проявляється не раніше 6–9 міс.
Мікофенолова кислота завдяки наявності цитостатичної (а не цитотоксичної) активності викликає побічні ефекти рідше, ніж азатіоприн. Терапевтична доза становить 2–3 г/добу, її розділяють на 2 прийоми з інтервалом 12 год, підтримувальна доза — 1 г/добу.
Метотрексат призначають при рефрактерному до монотерапії ГК вовчаковому артриті й ураженнях шкіри.
Циклоспорин в дозі <5 мг/кг/добу — препарат другого ряду при нефротичному синдромі, пов’язаному з мембранозним вовчаковим нефритом і тромбоцитопенією.
Нестероїдні протизапальні препарати
НПЗП в стандартних терапевтичних дозах можна застосовувати для лікування м’язово-скелетних проявів СЧВ, лихоманки і помірно вираженого серозиту. У пацієнтів із вторинним АФС НПЗП слід використовувати з обережністю, оскільки вони можуть сприяти розвитку тромбозів у хворих зі схильністю до гіперкоагуляції.
Амінохінолінові похідні
Гідроксихлорохін використовують при ураженнях шкіри, суглобів і конституціональних порушеннях. Його застосування дозволяє запобігти розвитку загострень СЧВ. Крім того, гідроксихлорохін знижує рівень ліпідів і ризик тромботичних ускладнень. Необхідне проведення повного офтальмологічного обстеження 1 раз на рік у зв’язку з ризиком розвитку ретинопатії (1:5000).
Белімумаб
Белімумаб — людське рекомбінантне моноклональне антитіло — IgG1. Розроблений спеціально для терапії СЧВ і є першим (за період більш ніж 50 років) препаратом, схваленим регуляторними органами Європи та США, а тепер й України, для терапії цього захворювання. Механізм дії белімумабу полягає в попередженні зв’язування цитокіну BlyS (B-lymphocytes stimulator — В-лімфоцитарний стимулятор) з клітинними рецепторами аутореактивних «перехідних» (transitional) і наївних В-клітин (роль В-лімфоцитів та цитокінів у патогенезі СЧВ висвітлено вище). Це призводить до пригнічення характерної для СЧВ В-клітинної гіперреактивності, зокрема синтезу аутоантитіл. Крім того, блокада BLyS може призводити до зниження виживаності В-клітин у центрах росту лімфоїдних органів, диференціювання В-клітин пам’яті в аутоантитіло-продукуючі клітини і зменшення синтезу прозапальних цитокінів (Shum K., Askanase A., 2012; Насонов Е.Л. и соавт., 2012).
Белімумаб показаний для зниження активності захворювання у дорослих пацієнтів з позитивною реакцією антитіл до СЧВ, які отримують стандартну терапію. Белімумаб вводять шляхом в/в інфузії в дозі 10 мг/кг маси тіла. Після першого введення (1-й день) наступне застосування необхідне через 14 та 28 днів; у подальшому препарат призначають кожні 4 тиж. Протипоказанням до застосування белімумабу є гіперчутливість до будь-якого компонента препарату (Інструкція для медичного застосування препарату белімумаб).
У дослідженнях ІІІ фази у серопозитивних пацієнтів, які отримували белімумаб у дозі 10 мг/кг, продемонстровано зниження активності захворювання і ризику серйозних загострень СЧВ. У хворих цієї групи відзначали нормалізацію рівня аутоантитіл, підвищення концентрації С3- і С4-компонентів комплементу. При зменшенні загальної кількості В-клітин і деяких субпопуляцій В-лімфоцитів (наївних й активованих В-клітин і плазматичних клітин) незмінним залишався рівень В-клітин пам’яті (CD20+/CD27+), а також CD4+ і CD8+ Т-клітин.
У пацієнтів з високими значеннями індексу SELENA-SLEDAI (≥10), низьким рівнем комплементу та підвищеними рівнями антитіл до двоспіральної ДНК (порівняно з плацебо) ефективність белімумабу була вищою, ніж у загальній популяції хворих, які одержували белімумаб. Також в цій підгрупі відмічено поліпшення віддалених результатів (зниження частоти загострень, зменшення потреби в терапії ГКС, покращання якості життя) (Stohl W. et al., 2012; van Vollenhoven R.F. et al., 2012).
Екстракорпоральні методи лікування
Плазмаферез
Метод використовують для лікування тяжко хворих зі швидкопрогресуючим ураженням життєво важливих органів (пневмоніт, ураження ЦНС, швидкопрогресуючий люпус-нефрит з нирковою недостатністю) у поєднанні з активною терапією циклофосфамідом та ГК.
Внутрішньовенний імуноглобулін
В/в імуноглобулін застосовують у хворих на СЧВ з тяжким ураженням ЦНС, при вираженій тромбоцитопенії, приєднанні бактеріальної інфекції. Схеми застосування препарату на сьогодні нестандартизовані: зазвичай доза в/в імуноглобуліну варіює від 0,4 до 2,0 г/кг/добу і протягом 4–5 днів.
Терапія системного червоного вовчака залежно від ураження органів і тканин
Ураження шкіри
- При шкірних ураженнях показані: низькі дози ГК (преднізолон 7,5–10,0 мг/добу); гідроксихлорохін (400 мг/добу); місцеві ГК, але необхідно уникати застосування фторованих препаратів, особливо на обличчі, у зв’язку з ризиком розвитку атрофії шкіри; сонцезахисні креми (проти α- і β-ультрафіолетових променів).
- При генералізованому ураженні шкіри, резистентному до комбінованої терапії низькими дозами ГК і гідроксихлорохіну, використовують азатіоприн в дозі 2–3 мг/кг/добу або метотрексат 7,5–15,0 мг/тиж.
- При генералізованому шкірному васкуліті або бульозному ураженні шкіри показано болюсне введення циклофосфаміду (0,5–1,0 г/м2) у поєднанні з метилпреднізолоном (1 г). При неефективності — синхронна інтенсивна терапія.
Артралгії та артрити
- Низькі дози ГК (<10 мг/добу преднізолону).
- Гідроксихлорохін (200–400 мг/добу).
- НПЗП в стандартних дозах. У пацієнтів із АФС необхідно з обережністю застосовувати селективні НПЗП.
- За наявності резистентності до терапії ГК, гідроксихлорохіном і НПЗП — метотрексат 7,5–15,0 г/тиж.
Полісерозит (плеврит/перикардит)
- Преднізолон у дозі 0,25–0,5 мг/кг/добу.
- НПЗП у стандартних дозах.
- При неефективності — пульс-терапія метилпреднізолоном у поєднанні з азатіоприном.
- При частих рецидивах серозиту — в/в імуноглобулін (0,5 г/кг і протягом 5 останніх днів).
Пневмоніт
- Пульс-терапія метилпреднізолоном (1 г) у поєднанні з циклофосфамідом (0,75–1,0 г/м2) з подальшим призначенням ГК реr оs: преднізолон у дозі 1 мг/кг/добу.
- При альвеолярній кровотечі показаний плазмаферез з наступним введенням метилпреднізолону (1 г) і циклофосфаміду (1 г/м2).
Аутоімунна гемолітична анемія
- Преднізолон 1 мг/кг/добу.
- У випадку вираженого (< 70 г/л) швидко прогресуючого зниження рівня гемоглобіну необхідне проведення пульс-терапії метилпреднізолоном (1 г послідовно протягом 3 днів).
- При неефективності ГК-терапії — азатіоприн у дозі 2–3 мг/кг/добу.
- У пацієнтів з тяжкою швидкопрогресуючою гемолітичної анемією, рефрактерною до ГК-терапії, — циклофосфамід (0,75–1,0 г/м2). Тромбоцитопенія (рівень тромбоцитів <50×109/л).
- Преднізолон у дозі 1 мг/кг/добу.
- При швидкому зниженні рівня тромбоцитів — пульс-терапія метилпреднізолоном (1 г послідовно протягом 3 днів).
- За відсутності протягом 1 тиж ефекту на тлі ГК-терапії — пульс-терапія циклофосфамідом або азатіоприном у дозі 2–3 мг/кг/добу. Вибір імуносупресивного препарату залежить від вираженості та швидкості прогресування тромбоцитопенії, а також наявності інших проявів СЧВ (при нефриті показаний циклофосфамід).
- При неефективності імуносупресивної терапії, вираженому зниженні рівня тромбоцитів (<25×109/л) і наявності геморагічного синдрому показано застосування в/в імуноглобуліну (0,5 г/кг/добу протягом 3–5 послідовних днів).
- При тромботичній тромбоцитопенічній пурпурі показаний плазмаферез у поєднанні з введенням свіжозамороженої плазми і пульс-терапією ГК.
Лейкопенія
- Не вимагає спеціального лікування.
- При агранулоцитозі показана пульс-терапія метилпреднізолоном (не менше 1,0 г).
Нервово-психічні порушення
- Преднізолон 1 мг/кг/добу у поєднанні з циклофосфамідом (щомісячне болюсне введення 0,5–1,0 г/м2). При розвитку побічних реакцій на тлі лікування циклофосфамідом або наявності протипоказань до його застосування використовують азатіоприн у дозі 2–3 мг/кг/добу.
- При розвитку загрозливих для життя станів (кома, поперековий мієліт, епістатус) показаний плазмаферез у поєднанні з пульс-терапією метилпреднізолоном (не менше 1 г) і циклофосфамідом (не менше 1 г/м2).
Вовчаковий нефрит
Найбільш численні дослідження проводили при вовчаковому нефриті. Сучасна стандартна терапія — в/в введення циклофосфаміду 1000 мг 1 раз на місяць — переглянута Національним інститутом охорони здоров’я США (National Institutes of Health — NIH).
За останніх 15 років розроблено альтернативні способи введення циклофосфаміду в набагато менших дозах і протягом більш короткого часу, ніж за схемою NIH, а саме, протягом 6 міс циклофосфамід вводили по 500 мг 1 раз на 2 тиж. Ця схема за ефективністю не поступалася NIH, але спостерігалася тенденція до зниження ризику розвитку інфекцій та токсичної дії циклофосфаміду.
Вибір схеми лікування і його тривалості базується на оцінці результатів гістологічного дослідження (морфологічний клас вовчакового нефриту, індекси активності та хронізації) і лабораторних показників (рівні протеїнурії та сироваткового креатиніну). Виділяють індукційну (тривалістю 3–6 міс) і підтримувальну фази лікування.
При резистентності до терапії циклофосфамідом і метилпреднізолоном відповідно до рекомендацій ЕULAR застосовують при люпус-нефриті та нейролюпусі 1000 мг циклофосфаміду в/в 1 раз на 1 міс протягом 6 міс, а потім по 1000 мг в/в кожні 3 міс протягом 1,5 року.
Найбільш успішним вважається застосування мікофенолової кислоти, яка блокує проліферацію Т- і В-лімфоцитів, знижує синтез антитіл, а також інгібує NO-синтетазу, чим зменшує пошкодження.
У пацієнтів із СЧВ з активним люпус-нефритом результати терапії мікофеноловою кислотою в дозі 2–3 г/добу порівняно з терапією циклофосфамідом протягом 6 міс (500 мг/м2) були аналогічні.
Але у африканців, іспанців і латинських американців відповідь на мікофенолову кислоту була краща порівняно з азатіоприном, а також показала кращий результат у підтримувальній терапії протягом 36 міс та у попередженні смертності, розвитку термінальної стадії захворювань нирок, загостренні нефриту. Поєднання мікофенолової кислоти з ГК — метод вибору ведення тяжких форм СЧВ. ГК + імуносупресори — стандарт для лікування тяжкого нефриту.
У хворих на люпус-нефрит, рефрактерний до циклофосфаміду та інших цитостатиків, застосовували ритуксимаб у дозі 1000 мг 2 рази на рік. Основою для призначення ритуксимабу є дані про те,що у хворих на СЧВ розподіл субпопуляцій B-клітин у процесі їх розвитку і дозрівання порівняно з нормальним станом, змінено. Також відзначають підвищення рівнів преплазмоцитів, клітин пам’яті та преклітин зародкового центру, а також значно знижені рівні «наївних» B-клітин. Встановлено, що ритуксимаб забезпечував клінічний ефект у 159 із 208 (79%) пролікованих хворих на СЧВ (у яких антималярійні, імуносупресивні препарати, а також ГК неефективні в якості терапії першої лінії) з покращенням середніх показників шкал оцінки СЧВ (BILAG, SLEDAI, SLAM), ниркової функції, а також зниженням дози ГК. Крім того, особливо виражене покращення відзначали у хворих із психоневрологічними симптомами.
Основні принципи лікування хворих на системний червоний вовчак під час вагітності
Перед призначенням лікування необхідно оцінити активність хвороби і співвідношення між користю від лікування для матері і можливим ризиком несприятливого впливу лікарських препаратів на плід.
- Протипоказана терапія циклофосфамідом і метотрексатом у зв’язку з високим ризиком тератогенної дії.
- Слід уникати призначення НПЗП (за винятком низьких доз ацетилсаліцилової кислоти).
- Гідроксихлорохін сприяє зниженню частоти і зменшенню вираженості загострень СЧВ і не супроводжується несприятливим впливом на плід.
- Для профілактики загострення СЧВ можливе призначення низьких доз преднізолону (<10 мг/добу).
- При загостренні СЧВ показано підвищення дози ГК, при необхідності — проведення пульс-терапії ГК; при вираженій тромбоцитопенії, рефрактерної до ГК, — імуноглобулін в/в.
Експертиза непрацездатності
Працездатність хворих I, II або III групи залежить від характеру перебігу захворювання, ступеня загострення процесу, тяжкості ураження внутрішніх органів (у першу чергу нирок і ЦНС). Визначення груп інвалідності проводять відповідно до загальноприйнятої класифікації і критеріїв, використовуваних при здійсненні медико-соціальної експертизи. При наданні хворим трудових рекомендацій необхідно мати на увазі протипоказані їм умови праці, пов’язані з наявністю несприятливих метеорологічних і санітарно-гігієнічних факторів, що провокують рецидив захворювання. Протипоказаними виробничими чинниками для таких хворих слід вважати: фізичну працю, що вимагає вираженого напруження, вимушеного положення тіла, певного темпу; розумову працю, що вимагає вираженого напруження або певного темпу; «очні зміни»; вплив сонячних променів або інших видів опромінення (ультрафіолетове, інфрачервоне, електрозварювання); охолодження, коливання температури, підвищену вологість; високу температуру зовнішнього середовища; запиленість; роботи, пов’язані з забрудненням шкірних покривів. При визначенні групи інвалідності у хворого необхідно керуватися наступним.
I групу інвалідності встановлюють при високому ступені (III ступінь) активності процесу, резистентності до проведеної терапії, різко виражених і стійких порушеннях функцій органів і систем організму.
II групу інвалідності визначають при помірній активності процесу та схильності до тривалих загострень, хронічному перебігу захворювання з ураженням серцево-судинної системи та серцевою недостатністю I–II ступеня, при ураженні легень з дихальною недостатністю II ступеня, ураженні нирок з нирковою недостатністю I–II ступеня, ураженні суглобів з порушенням функції II ступеня, ураженні ЦНС з органічною симптоматикою і церебральною недостатністю I–II ступеня.
III групу інвалідності встановлюють при мінімальній активності процесу, ураженні вісцеральних органів без вираженого порушення їх функції, якщо у виконуваній роботі за фахом мають місце протипоказані фактори праці, а рекомендоване працевлаштування призводить до зниження кваліфікації або пов’язане зі значним зменшенням обсягу виробничої діяльності, а також при обмеженні можливості працевлаштування осіб з низькою кваліфікацією або тих, які раніше не працювали.
Працездатними визнаються хворі зі стійкою мінімальною активністю процесу або в яких захворювання тривалий час знаходиться у неактивній стадії, за відсутності функціональних порушень з боку органів і систем та протипоказаних чинників у виконуваній роботі.
Інвалідам III групи доступні всі професії, в яких використовується інтелектуальна праця, не пов’язані зі значними нервово-психічними навантаженнями (бухгалтер, економіст, юрист, культпрацівник, діловод, адміністративно-господарські працівники), а також професії, де застосовується легка кваліфікована фізична праця (оператор, наладник, майстер з ремонту годинників, оптик), а також некваліфікована (працівник сфер комунального, культурно-побутового обслуговування). Інвалідам III групи (особам середнього віку, чия професія вимагала важкої та середньої тяжкості фізичної праці) показані перенавчання або перекваліфікація на всі професії, пов’язані з легкою фізичною працею в умовах сприятливого мікроклімату з виключенням професійних шкідливих факторів. Для адаптації до нових умов і характеру праці їм доцільно рекомендувати сидячу або зі змінним положенням тіла роботу в зменшеному обсязі або зі скороченим робочим днем. При сприятливому перебігу захворювання обсяг роботи може бути розширеним.
Інвалідам II групи за відсутності вираженої активності може бути рекомендована робота в спеціально створених умовах (спеццех, спецділянки) на підприємствах, де вони працювали до інвалідності, з урахуванням професійних навичок. Інваліди цієї групи при вираженій активності процесу і порушенні функції окремих органів можуть виконувати роботу вдома: дрібне складання, в’язання, їм показані канцелярська праця, творча робота, не пов’язана зі значним нервово-психічним напруженням.
У всіх випадках трудові влаштування мають проводитися з урахуванням різнобічних медичних і соціальних факторів та індивідуального підходу до кожного хворого. Це сприятиме профілактиці інвалідності та залученню більшої кількості кваліфікованих фахівців до суспільно корисної діяльності.
Диспансерне спостереження
Хворі підлягають постійному диспансерному спостереженню з метою виявлення ознак загострення захворювання й ураження внутрішніх органів.
Профілактика СЧВ спрямована на попередження виникнення захворювання і загострення хвороби. При проведенні первинної профілактики необхідно обстежити родичів хворих на СЧВ. При виявленні таких симптомів, як стійка лейкопенія, збільшення ШОЕ, гіпергаммаглобулінемія, антитіла до ДНК, вовчакові клітини, хибнопозитивна реакція Вассермана, кріоглобулінемія та ін., рекомендується такий самий щадний режим, як і хворим на СЧВ. Цим особам необхідно уникати надмірної інсоляції, переохолодження, щеплення, грязелікування та інших фізичних методів терапії. Регулярному спостереженню підлягають хворі на дискоїдний вовчак. В якості вторинної профілактики поряд з адекватною терапією рекомендується: своєчасно звертатися до лікаря при зміні самопочуття, регулярно проходити диспансерне обстеження; приймати гормональні препарати у суворо призначеній дозі; дотримуватися розпорядку дня, що включає 1–2-годинний сон вдень, та дієти з обмеженням кухонної солі та вуглеводів, що включає продукти, багаті білками та вітамінами; не засмагати, не переохолоджуватися; уникати різних оперативних втручань, щеплень, вакцин і введення сироваток (крім життєво необхідних); дотримуватися щадного режиму, не забувати про обережне загартовування (ранкова гімнастика, обтирання теплою водою, тривалі прогулянки на свіжому повітрі, заняття спортом); при загостренні осередкової або інтеркурентної інфекції дотримуватися ліжкового режиму, приймати антибіотики, десенсибілізуючі засоби; лікування осередкової інфекції має бути наполегливим, переважно консервативним, лише за крайньої необхідності можливе хірургічне втручання під прикриттям підвищених доз ГК та антибіотиків; при ураженні шкіри для захисту від сонячних променів застосовувати фотозахисні мазі, при почервонінні обличчя змащувати шкіру ГК-вмісними мазями. У хворих з клінічно вираженим люпус-нефритом прогноз значно гірший і прямо залежить від ступеня вираженості ниркової патології. Прогноз особливо несприятливий у тих випадках, коли люпус-нефрит діагностують протягом 2 років від початку захворювання.
Рекомендації для пацієнта
- Регулярний контакт з медичним персоналом для отримання консультативної допомоги з приводу будь-яких питань, періодичні медичні обстеження.
- Зміна способу життя у зв’язку з хронічним характером захворювання (у тому числі вирішення питання професійної орієнтації та працевлаштування), адекватні дозовані фізичні навантаження.
- Дотримання режиму праці та відпочинку, дієти, рекомендованих схем лікування (у тому числі відмова від нетрадиційних (народних) методів лікування).
- Виключення впливу факторів, які сприяють загостренню захворювання.
Прогноз
На сьогодні прогноз СЧВ значно покращився: 5-річна виживаність досягає 86–95%, 10-річна — 70–80%, 20-річна — 60–70%. Однак смертність при СЧВ в 3 рази вища, ніж у популяції.
До факторів несприятливого прогнозу відносять ураження нирок (особливо дифузний проліферативний гломерулонефрит), артеріальну гіпертензію, приналежність до чоловічої статі, початок захворювання у віці до 20 років, супутній АФС, високу активність захворювання, високі значення індексу ушкодження, приєднання інфекції, ускладнення медикаментозної терапії.
Глава 14. Антифосфоліпідний синдром
АФС — аутоімунне системне захворювання з широким спектром переважно тромботичних клінічних проявів на тлі підвищеної продукції АФЛ. За сучасними уявленнями, основу АФС становить своєрідна васкулопатія, зумовлена незапальним та/або тромботичним ураженням судин і закінчується їх оклюзією. Основні ознаки — венозні та/або артеріальні тромбози, різні форми акушерської патології (у першу чергу звичне невиношування вагітності — більше 2 випадків втрати плоду); тромбоцитопенія. Різні неврологічні, шкірні, серцево-судинні та гематологічні порушення належать до додаткових ознак. Серологічні маркери цього синдрому — АФЛ.
Основні типи АФС — ВА і АКЛ — гетерогенна популяція аутоантитіл, що реагують з широким спектром антигенних детермінант. Хоча вивчення АФЛ фактично почалося ще на початку століття з розробки Вассерманом серологічного методу діагностики сифілісу (реакція Вассермана), інтерес до цих антитіл особливо зріс в останнє десятиліття у зв’язку з описом АФС. Вперше клінічний опис синдрому (ще не названого АФС) представлено E.A. Johansson і співавторами в 1977 р. як «периферичний судинний синдром, який перехрещується з СЧВ», який характеризувався рецидивуючими венозними тромбозами, геморагічною капіляропатією з циркулюючим антикоагулянтом і хибнопозитивною реакцією Вассермана. І нарешті в 1983 р. описано цей синдром спочатку G.R.V. Hughes, а потім E.N. Harris під назвою «антикардіоліпіновий синдром, або синдром антитіл до фосфоліпідів». Проте пізніше ці самі автори запропонували більш широку назву — АФС. У 1994 р. на VI Міжнародному симпозіумі з вивчення АКЛ запропоновано називати АФС синдромом Hughes (Хьюза), за іменем англійського ревматолога, який вперше його описав і зробив найбільший внесок у розробку проблеми. У зв’язку з виявленням кофакторів АФЛ дискутується й інша назва цього синдрому — антифосфоліпідкофакторний, або антифосфоліпідбілковий синдром.
Епідеміологія
Частота виявлення IgG-АКЛ в сироватці здорових людей варіює від 0 до 14%, у середньому — 2–4% (у високому титрі — < 0,2%), і підвищується в осіб похилого віку.
Захворювання у молодих людей розвивається частіше, ніж у літніх; виявляють також у дітей і навіть новонароджених. Як і інші аутоімунні ревматичні хвороби, АФС частіше діагностують у жінок, ніж у чоловіків (співвідношення 5:1), зазвичай у середньому віці (близько 35 років). При вторинному АФС співвідношення жінок і чоловіків становить 7,5:1, а при первинному — 3,5:1. Первинний і вторинний АФС виявляють майже з однаковою частотою.
Клінічні прояви АФС спостерігають у 30–60% хворих із ВА і у 30–50% — з помірним або високим титром IgG-АКЛ.
Етіологія
Причини АФС не встановлено. Відомо, що підвищення титру АФЛ в крові (як правило, транзиторне) спостерігається на тлі широкого спектра бактеріальних і вірусних інфекцій.
- Вірусні інфекції, викликані вірусом гепатиту С, вірусом Епштейна — Барр, ВІЛ, цитомегаловірусом, парвовірусом В19, аденовірусом, а також вірусами Неrpes zoster, кору, краснухи, Т-клітинного лейкозу людини типу I.
- Бактеріальні інфекції: лепра, туберкульоз і захворювання, викликані іншими мікобактеріями; сальмонельози, стафілококові, стрептококові інфекції, Ку-лихоманка.
- Інфекції, викликані спірохетами: сифіліс, лептоспіроз, хвороба Лайма (Borrelia burgdorferi).
- Паразитарні інфекції: малярія, лейшманіоз, токсоплазмоз.
Однак, незважаючи на гіперпродукцію АФЛ, тромботичні ускладнення у хворих з інфекціями розвиваються рідко. Це пов’язують з різницею в імунологічних властивостях АФЛ в осіб з АФС та інфекційними захворюваннями. За результатами 9-річного спостереження за 122 хворими на СЧВ і АФС у 8% випадків приєднання інтеркурентної інфекції було провокуючим фактором розвитку катастрофічного АФС.
Генетична схильність
Синтез АФЛ, як і багатьох інших аутоантитіл, має безсумнівну генетичну основу. За даними сімейно-генетичних досліджень, відзначено підвищення частоти виявлення АФЛ у сім’ях хворих на АФС. Описано випадки АФС (частіше первинного) у членів однієї родини. Аналізу зв’язку між розвитком первинного та вторинного (пов’язаного з СЧВ) АФС (або синтезом АФЛ) і антигенами головного комплексу гістосумісності присвячено дуже велику кількість досліджень. Передбачається, що зв’язок між розвитком АФС і алелями НLА більшою мірою відображає генетичну схильність до синтезу АФЛ, а не до розвитку клінічних проявів власне АФС. До алелей НLА, які найчастіше пов’язані з розвитком АФС, відносять НLА DRВ1 * 04 (DR4), DRВ1 * 07 (DR7), DRВ1 * 301 (ВК6), DRw53, DQA1 * 0102, DQА1 * 0201, DRA1 * 0301, DQB1 * 302 (DQ8), DQB1 * 0604/5/7/9. Не виключено, що вищеперелічені алелі перебувають у неврівноваженому зчепленні з іншими більш важливими генами системи НLА. Наприклад, є дані, що гіперпродукція АФЛ пов’язана з генетично зумовленими дефектами комплементу (С4А/С4В-дефіцит).
Оскільки навіть мінімальні генетично зумовлені зміни білків згортання крові можуть призводити до вираженого підвищення ризику тромбозів, особливий інтерес представляє вивчення генетичного поліморфізму генів, що кодують синтез цих білків при АФС. Є дані про зв’язок між розвитком АФС і генетичним поліморфізмом р2-глікопротеїну I, лейденського фактора V, гена протромбіну, інгібітора активатора плазміногену 1, тромбоцитарного GР IIb/IIIа 1/2 рецептора, Fсу-рецептора (FcP-IIа) та ін.
Патогенез
За сучасними уявленнями, АФЛ — не тільки серологічний маркер, але й важливий «патогенетичний» медіатор, що викликає розвиток основних клінічних проявів АФС (тромбозів, акушерської патології, цитопенії та ін.). У цілому АФЛ мають здатність впливати на більшість процесів, які становлять основу регуляції гемостазу, порушення яких призводить до гіперкоагуляції. Взаємодія АФЛ із фосфоліпідами — складний феномен, у реалізації якого ключову роль відіграють так звані кофактори. Необхідною умовою для зв’язування АКЛ, виділених із сироваток пацієнтів з АФС, з кардіоліпіном служить наявність так званого кофактора АКЛ, який був ідентифікований як бета-2-глікопротеїн I. Поряд з бета-2-глікопротеїном I як «кофактори» (або аутоантигени) можуть виступати інші білки, багато з яких беруть безпосередню участь у регуляції згортання крові. До них відносять протромбін (чинник II), білок С, білок S, анексин V, тромбомодулін, фактори V, VII/VIIа і XII, високомолекулярний і низькомолекулярний кініноген, гепарин та ін. Однак тільки синтез АФЛ у людини не може спровокувати клінічно значущих порушень гемостазу, що призводять до розвитку АФС. Це стало підставою для гіпотези «подвійного удару» (two-hit hypothesis), згідно з якою АФЛ («перший удар») створюють умови для гіперкоагуляції, а формування тромбу індукується додатковими медіаторами («другий удар»), що підсилюють активацію каскаду згортання крові, вже викликану АФЛ. Дійсно, є дані про те, що частота тромбозів у пацієнтів з АФЛ в крові значно зростає за наявності інших факторів ризику гіперкоагуляції, наприклад вагітності, паління, хірургічних втручань і особливо вроджених тромбофілій. Крім того, АФЛ — надзвичайно гетерогенна популяція аутоантитіл, кожне з яких володіє своїм унікальним патогенним потенціалом; деякі хворі з АФС мають підвищений ризик атеросклерозу, який може відображати наявність особливої популяції «атерогенних» АФЛ. Таким чином, розвиток АФС реалізується за рахунок декількох взаємодоповнюючих АФЛ-залежних і АФЛ-незалежних механізмів. Можливі механізми активності АФЛ умовно можуть бути розділені на гуморальні та клітинні.
Класифікація
На сьогодні виділяють наступні основні форми АФС:
- первинний АФС: політромботичний синдром, порушення мозкового кровообігу, особливо в осіб молодого віку; звичне невиношування вагітності та внутрішньоутробна загибель плода (постійно повторювані викидні за відсутності акушерсько-гінекологічної патології); алергія на лікарські препарати (хінідин, гідралазин, фенотіазини, кокаїн, прокаїнамід).
До підтипу первинного АФС належить синдром Снеддона — поєднання судинно-мозкових порушень і ліведо при виявленні в крові АФЛ;
- вторинний АФС: на тлі аутоімунних захворювань (СЧВ, вузликовий періартеріїт, РА, ССД, імунний тиреоїдит); на тлі злоякісних новоутворень (тимома, карциноми, гематологічні пухлини); на тлі інфекційних та інфекційно-імунних захворювань (хвороба Лайма, бронхіальна астма, ВІЛ-інфекція, стафілококова, стрептококова інфекція); «катастрофічний» АФС (гостра дисемінована коагулопатія/васкулопатія) з гострим мультиорганним тромбозом;
- інші мікроангіопатичні синдроми (тромботична тромбоцитопенічна пурпура/гемолітикоуремічний синдром); HELLP-синдром (гемоліз, підвищення активності печінкових ферментів, зниження вмісту тромбоцитів), ДВЗ-синдром; гіпопротромбінемічний синдром;
- «серонегативний» АФС. В останні роки обговорюється можливість існування так званого АФЛ-негативного варіанта. АФС, при якому є клінічні прояви патології, але відсутні класичні серологічні маркери — вовчакові антитіла і АКЛ.
Попередні критерії класифікації катастрофічного АФС:
1. Залучення 3 або більше органів, систем та/або тканин (бажано інструментальне підтвердження оклюзії судин; залучення нирок визначається як підвищення креатиніну на 50%, підвищення артеріального тиску >180/100 мм рт. ст., протеїнурія >0,5 г/добу).
2. Розвиток поліорганних проявів одночасно або в межах <1 тиж.
3. Морфологічне підтвердження оклюзії судин принаймні одного органа або тканини (може бути представлено поряд з наявністю ознак васкуліту).
4. Серологічне підтвердження наявності АФЛ (ВА або АКЛ); якщо у хворого раніше не діагностували АФС, наявність АФЛ має бути підтверджена в двох дослідженнях з інтервалом не менше 6 тиж.
АФС вважають вірогідним катастрофічним за наявності всіх 4 ознак, а вірогідним:
- якщо є всі 4 ознаки, однак залучені тільки 2 органи, системи та/або тканини;
- якщо є всі 4 ознаки, за винятком серологічного дослідження (наявність АФЛ), за 6 тиж до того;
- якщо є 1-ша, 2-га і 4-та ознаки;
- якщо є 1-ша, 3-тя і 4-та ознаки і розвиток третього випадку тромбозу за період >1 тиж, але <1 міс, незважаючи на антикоагулянтну терапію.
Клінічна картина
В основі судинної патології при АФС лежить незапальна тромботична васкулопатія, що зачіпає судини будь-якого калібру і локалізації (від капілярів до великих судин, включаючи аорту). У зв’язку з цим спектр клінічних проявів АФС надзвичайно різноманітний.
Найбільш часті і характерні прояви АФС — венозні та/або артеріальні тромбози (розвиваються приблизно у третини пацієнтів, у сироватці яких виявлено АФЛ) й акушерська патологія.
Основні клінічні прояви антифосфоліпідного синдрому (за частотою):
- Частота більше 30%
- Тромбоз глибоких вен кінцівок
- Спонтанні аборти в ранні терміни вагітності
- Тромбоцитопенія
- Частота більше 20%
- Сітчасте ліведо
- Мігрень
- Інсульт
- Частота більше 10%
- Тромбоемболія легеневих артерій
- Транзиторні ішемічні атаки
- Спонтанні аборти в пізні терміни вагітності
- Потовщення/дисфункція клапанів серця
- Гемолітична анемія
- Частота більше 1%
- Прееклампсія
- Епісиндром
- Виразки ніг
- Минуща сліпота
- Інфаркт міокарда
- Еклампсія
- Тромбоз артерій нижніх кінцівок
- Тромбоз вен верхніх кінцівок
- Тромбоз артерій верхніх кінцівок
- Псевдоваскулітні ураження
- Гангрена пальців верхніх і нижніх кінцівок
- Кардіоміопатія
- Стенокардія
- Вегетації на клапанах
- Ураження нирок (тромбоз клубочків, інфаркт нирок, тромбоз ниркових артерій, тромбоз ниркових вен)
- Мультиінфарктна деменція
- Некрози шкіри
- Аваскулярний некроз кісток
- Легенева гіпертензія
- Тромбоз підключичної вени
- Гостра енцефалопатія
- Рестеноз після аортокоронарного шунтування
- Ураження травного тракту (ішемія стравоходу і кишечнику)
- Тромбоз артерій сітківки
- Інфаркт селезінки
- Легеневий мікротромбоз
- Нейропатія зорового нерва
- Частота менше 1%
- Транзиторна амнезія
- Тромбоз мозкових вен
- Церебральна атаксія
- Внутрішньосерцеві тромбози
- Інфаркт підшлункової залози
- Синдром Аддісона
- Ураження печінки (синдром Бадда — Кіарі, тромбоз дрібних ниркових вен)
- Тромбоз вен сітківки
- Крововиливи в нігтьове ложе
- Післяпологовий кардіопульмональний синдром
- Інші типи ураження легень (гострий респіраторний дистрес-синдром дорослих, легеневі геморагії, тромбоз легеневої артерії)
Венозний тромбоз, особливо тромбоз глибоких вен нижніх кінцівок, — найбільш частий прояв АФС, у тому числі в дебюті захворювання. У загальній популяції АФЛ визначають приблизно у 10% хворих з венозними тромбозами. При АФС частота венозних тромбозів протягом 6 років спостереження коливається від 29 до 55%. У половини хворих розвиваються тромбоемболії легеневих артерій. Тромби зазвичай локалізуються в глибоких венах нижніх кінцівок, але нерідко — і печінкових, портальних, поверхневих венах та ін. Характерні повторні емболії легеневих артерій з глибоких вен нижніх кінцівок, іноді це призводить до розвитку легеневої гіпертензії. АФС (частіше первинний, ніж вторинний) — друга за частотою причина розвитку синдрому Бадда — Кіарі. Тромбоз центральної вени надниркової залози може призводити до наднирковозалозної недостатності.
Артеріальні тромбози в цілому трапляються рідше, ніж венозні. Вони проявляються ішемією та/або інфарктами головного мозку, коронарних артерій, порушеннями периферичного кровообігу. Найбільш частою локалізацією тромбозу (у 50% випадків) служать мозкові, рідше коронарні артерії. До рідкісних проявів АФС належать тромбоз великих артерій, а також висхідного відділу аорти (з розвитком синдрому дуги аорти) і черевної аорти.
Рецидиви тромбозів. Характерна особливість АФС — високий ризик рецидивування венозних й артеріальних тромбозів. При цьому у хворих із першим тромбозом в артеріальному руслі повторні тромбози також частіше розвиваються в артеріях. Якщо ж першим тромбозом був венозний, то повторні тромбози, як правило, відбуваються у венозному руслі. Відзначено, що у пацієнтів з високим рівнем ІgG-АКЛ в сироватці (>100 GPL) повторні порушення мозкового кровообігу розвивалися в більш ранні терміни, ніж у пацієнтів з низьким рівнем АФЛ.
Акушерська патологія
Звичне невиношування вагітності, рецидивуючі спонтанні аборти, внутрішньоутробна загибель плода, прееклампсія/еклампсія — діагностичні ознаки АФС.
Прийнято такі визначення несприятливих акушерських результатів:
- аборт спонтанний, так званий викидень, до 20-го тижня гестації. У I триместр до 13-го тижня, у II триместр — з 14-го до 20-го тижня; рецидивуючі втрати вагітності (рецидивуючі викидні) — 2 і більше послідовних спонтанних аборти;
- преембріонічна втрата плода — від моменту зачаття до 5-го тижня гестації;
- ембріонічна втрата плода — від 5-го до 9-го тижня гестації;
- передчасні пологи — живородіння або народження мертвого плода після 20–37 тиж гестації (після цього періоду пологи вважають строковими);
- загибель плода — внутрішньоутробна загибель у період після 10 тиж гестації, народження мертвого плода після 20 тиж гестації.
Наслідками патології вагітності при АФС можуть бути незрозуміла загибель плода після 10 тиж гестації, рання прееклампсія або еклампсія, внутрішньоутробна затримка розвитку плода, а також тромбоемболічні ускладнення, пов’язані з вагітністю. Не підтверджено вплив АФЛ на неуспішні випадки штучного запліднення.
Неврологічні прояви
Основною причиною ураження ЦНС виступає тромбоз мозкових артерій, що призводить до ішемії мозку, однак виділяють ряд неврологічних і нейропсихічних проявів, пов’язаних з іншими механізмами. Синтез АФЛ асоціюється з надзвичайно широким спектром патології центральної та периферичної нервової системи. Інсульт і транзиторні ішемічні атаки — другі за частотою після венозних тромбозів клінічні прояви АФС. Ураження нервової системи належить до найбільш частих і важких (потенційно смертельних) проявів АФС і включає:
- транзиторні ішемічні атаки;
- ішемічний інсульт;
- гостру ішемічну енцефалопатію;
- тромбоз мозкових вен;
- епісиндром;
- мігрень;
- хорею;
- розсіяний склероз;
- поперечний мієліт;
- ідіопатичну внутрішньочерепну гіпертензію;
- нейросенсорну приглухуватість;
- синдром Гійєна — Барре;
- транзиторну загальну амнезію;
- очні синдроми;
- паркінсонічний гіпертонус;
- когнітивні порушення;
- деменцію;
- депресію;
- психоз;
- інші неврологічні та психіатричні захворювання.
Є пропозиції виключити з критеріїв діагнозу АФС такі ознаки, як судинний головний біль за типом мігрені, поперечний мієліт, прояви, подібні до розсіяного склерозу, епілепсії та хореї, що пов’язано з недостатністю доказів їх зв’язку з синтезом АФЛ. При виявленні АФЛ (у середньо- або високопозитивних титрах) у хворих із перерахованими ознаками за відсутності у них тромбозу або акушерської патології вони підлягають проспективному спостереженню, оскільки в подальшому можливий розвиток АФС.
Ураження легень
У рамках АФС описано такі форми легеневої патології:
- емболію та інфаркт легень (виявляють приблизно у третини пацієнтів із рецидивуючим тромбозом глибоких вен гомілки); нерідко призводять до легеневої гіпертензії, а потім до правошлуночкової недостатності (іноді легенева гіпертензія поєднується з ізольованою недостатністю тристулкового клапана);
- гострий респіраторний дистрес-синдром дорослих;
- внутрішньоальвеолярні легеневі крововиливи;
- тромбоз великих і дрібних легеневих судин;
- фіброзуючий альвеоліт;
- післяпологовий кардіопульмональний синдром, що характеризується лихоманкою, плевритом, задишкою та легеневими інфільтратами;
- рефрактерну незапальну легеневу васкулопатію.
Основний легеневий прояв АФС — легенева тромбоемболічна хвороба — практично не відрізняється від «звичайної» тромбоемболії у хворих без АФЛ і може бути першим проявом захворювання. Характерні рецидиви легеневих тромбозів. При АФС частота легеневої гіпертензії становить 1,8–4,5%, а частота виявлення АФЛ у хворих із хронічною тромбоемболічною легеневою гіпертензією коливається від 10 до 20%.
Ураження серцево-судинної системи
Ураження серця займає особливо важливе місце в спектрі проявів АФС і характеризується різноманітними формами патології.
- Ураження коронарних артерій:
- тромбоз великих гілок коронарних артерій;
- множинний інтраміокардіальний тромбоз;
- рестеноз після аортокоронарного шунтування;
- рестеноз після черезшкірної транслюмінальної коронарної ангіопластики.
- Ураження клапанів серця:
- псевдоінфекційний ендокардит із вегетацією;
- недостатність та/або стеноз мітрального, аортального та/або тристулкового клапана;
- потовщення, фіброз і кальциноз стулок клапанів.
- Ураження міокарда (хронічна ішемічна кардіоміопатія).
- Внутрішньосерцевий тромбоз.
- Артеріальна гіпертензія.
- Легенева гіпертензія.
- Синдром дуги аорти.
- Атеросклероз і дисліпопротеїнемія.
Характеристика АФЛ-асоційованого ураження клапанів серця:
- Наявність в крові АФЛ (лабораторні критерії АФС) та в поєднанні:
- з ехоКГ-пошкодженням;
- та/або з регургітацією клапанів та/або стенозом мітрального, та/або аортального клапана, та/або комбінованих пороків (дослідження клапанів може бути виконано за допомогою черезстравохідної ехоКГ або без неї).
- Визначення пошкодження клапанів.
- Потовщення клапанів більше 3 мм.
- Локальне потовщення стулки клапана в проксимальній або середній частині.
- Нерівні вузлики на передсерцевій поверхні мітрального клапана та/або шлуночковій поверхні аортального клапана.
- Патологію клапанів слід підтверджувати при допплер-ехоКГ.
- Інтерпретувати результати дослідження повинні два фахівці з ехоКГ.
- Функціональний та об’єктивний стан слід оцінювати за міжнародними критеріями діагностування хвороб серця.
- В усіх вищезгаданих випадках за наявності в анамнезі гострої ревматичної лихоманки або інфекційного ендокардиту необхідно їх виключення.
- Пацієнтів з достовірним АФС виключають з цієї групи.
- Констатація СЧВ (за критеріями ACR) обов’язкова.
Артеріальна гіпертензія
Частим ускладненням АФС виступає артеріальна гіпертензія. Вона буває лабільною, нерідко поєднується із сітчастим ліведо і ураженням мозкових артерій у рамках синдрому Снеддона, але також може бути стабільною, злоякісною, що призводить до гіпертонічної енцефалопатії. Розвиток артеріальної гіпертензії при АФС може бути пов’язаний з багатьма причинами, у тому числі тромбозом ниркових судин, інфарктом нирок, тромбозом черевного відділу аорти («псевдокоарктація») або клубочків нирки. Є дані про високу частоту стенозу ниркових артерій (26%) у пацієнтів молодого віку (<50 років) з АФС та артеріальною гіпертензією. При вивченні великої групи пацієнтів (n=600) з АФС встановлено, що поширеність артеріальної гіпертензії при АФС становить 29%, причому у 86% хворих відзначали сітчасте ліведо.
Гематологічні порушення
Тромбоцитопенія — типова гематологічна ознака АФС, проте геморагічні ускладнення проявляються рідко, вони, як правило, пов’язані з супутнім дефектом специфічних факторів згортання крові, патологією нирок або передозуванням антикоагулянтів.
Характеристика АФЛ-асоційованої тромбоцитопенії:
- Визначення у крові АФЛ (лабораторні критерії АФС) у поєднанні з тромбоцитопенією (<100×109/л), підтвердженою двічі в проміжку не менше 12 тиж.
- Виключення хворих із тромботичною тромбоцитопенічною пурпурою, ДВЗ, псевдотромбоцитопенією і гепарин-індукованою тромбоцитопенією.
- Помірна (50–100×109/л) або виражена (<50×109/л) тромбоцитопенія.
- Свідчення наявності або відсутності СЧВ.
- Хворі з достовірним АФС (діагностичні критерії виключаються з цього визначення).
Розвиток кровоточивості може побічно свідчити про наявність антитіл до протромбіну. Нерідко виявляють Кумбс-позитивну гемолітичну анемію (10%); рідше діагностують синдром Еванса (поєднання аутоімунної тромбоцитопенії та аутоімунної гемолітичної анемії).
Ураження нирок
Прояви патології нирок при АФС вельми різноманітні. У більшості пацієнтів діагностують тільки безсимптомну помірну протеїнурію (<2 г/добу) без порушення функції нирок, однак в інших може розвиватися гостра або підгостра ниркова недостатність з вираженою протеїнурією (аж до нефротичного синдрому), активним сечовим осадом та артеріальною гіпертензією. При СЧВ ураження нирок може бути пов’язано як з мікротромбозами, так і імунокомплексним ураженням клубочків, однак ізольований мікроангіотромбоз виявляють порівняно рідко. За даними біопсії нирок, мікротромбоз клубочків виступає головним морфологічним проявом ураження нирок тільки у 10% хворих на СЧВ. Тромбоз ниркових артерій може призводити до інфаркту нирок, який проявляється однобічним або двобічним болем у попереку, гематурією, порушенням функції нирок, тяжкою артеріальною гіпертензією. Навпаки, тромбоз ниркових вен зазвичай протікає безсимптомно і лише іноді супроводжується вираженим болем та порушенням функції нирок.
Характеристика АФЛ-асоційованої нефропатії:
- Наявність в крові АФЛ (лабораторні критерії АФС) у поєднанні з гістологічним виявленням тромботичної мікроангіопатії, яка включає як артеріоли, так і капіляри клубочків та/або одну чи більше наступних ознак:
- фіброзна гіперплазія інтими, залучаються організовані тромби з реканалізацією або без неї;
- фіброз або фіброзно-клітинна оклюзія артерій або артеріол;
- великі зони атрофічних канальців, що містять еозинофільні циліндри.
- Васкуліт, тромботична тромбоцитопенічна пурпура, гемолітико-уремічний синдром, злоякісна гіпертензія та інші причини хронічної ниркової недостатності виключаються.
- Пацієнти з достовірним АФС виключаються.
- При СЧВ перелічені зміни слід відрізняти від асоційованих із вовчаковою нефропатією.
Ураження очей
У пацієнтів з АФС можливі втрата зору, оклюзії артерій і вен сітківки та ішемічний передній неврит очного нерва.
Ураження печінки
У рамках АФС виділяють наступні форми печінкової патології:
- синдром Бадда — Кіарі, пов’язаний з обструкцією великих печінкових вен і розвитком некрозу печінки; портальна гіпертензія іноді поєднується з легеневою гіпертензією;
- обструкція дрібних печінкових вен (печінкова венооклюзивна хвороба) характеризується нетромботичним концентричним звуженням просвіту дрібних центролобулярних вен (нерідко це ускладнення асоціюється з вузловою гіперплазією печінки); вузлова регенераторна гіперплазія зазвичай розвивається на тлі венооклюзивної хвороби; інфаркти печінки розвиваються рідко, головним чином у вагітних в рамках НЕLLР-синдрому або в післяпологовий період; гепатит (виявлено кілька пацієнтів, у яких АФС розвивався на тлі хронічного активного гепатиту).
Ураження травного тракту
У пацієнтів з АФС можливі ішемія та некроз стравоходу, шлунка і кишечнику, які призводять до шлунково-кишкових кровотеч, болю у животі (аж до симптомів гострого живота), некрозу та перфорації стравоходу, гігантських виразок у шлунку і атипових виразок у дванадцятипалій кишці. Є дані про розвиток гострого холециститу й оклюзії селезінкових судин, які спричиняють інфаркт селезінки.
Ураження надниркових залоз
Недостатність надниркових залоз при АФС у дорослих, а також у дітей та підлітків описана багатьма авторами. В основі наднирковозалозної недостатності лежить поєднання тромбозу та надниркових вен і геморагічного інфаркту, звичайно двостороннього. Факторами ризику крововиливу в надниркову залозу при АФС служать тяжка супутня патологія, тромбоемболії в анамнезі та післяопераційний період.
Ураження шкіри
Ураження шкіри при АФС характеризується різноманітними клінічними проявами.
- Сітчасте ліведо (найбільш часто).
- Виразки шкіри:
- виразки, що нагадують ураження шкіри при ліведо-васкуліті;
- великі виразки, схожі на гангренозну піодермію;
- посттромбофлебітичні виразки.
- Псевдоваскулітне і васкулітне ураження:
- пурпура;
- долонна і підошовна еритема;
- вузлики;
- пустули.
- Множинні крововиливи в нігтьове ложе.
- Поверхневий некроз шкіри.
- Гангрена пальців рук і ніг.
- Тромбофлебіт підшкірних вен.
- Злоякісні атрофічні папулоподібні ураження.
- Множинні геморагії в нігтьовому ложі.
Найбільш важливим шкірним проявом АФС є ліведо (від латинського «сітчастий синець»), яке являє собою стійкі синюваті плями, що нагадують мереживо або вічка рибальської сітки. Блідий центр кожного вічка пояснюється спазмом перпендикулярної артеріоли, який перфорує дерму. У деяких випадках ціанотичний або бордово-червоний колір периферії кожної блідої ділянки шкіри зумовлений застоєм крові у венозних сплетіннях, які супроводжують артеріолу.
Визначення АФЛ-асоційованого сітчастого ліведо:
- Наявність в крові АФЛ (лабораторні критерії АФС) разом з оболонкою ліведо.
- Сітчасте ліведо — постійне, необоротне, при зігріванні фіолетове, червоне або синє, сітчасте або плямисте ураження шкіри тулуба, рук і/або ніг; може складатися з правильної форми безперервних кіл (постійне ліведо) або неправильних переривчастих кривих (livedo racemosa — кістоподібне ліведо). Ширина розгалуження може бути >10 мм (велике сітчасте ліведо) або <10 мм (дрібне сітчасте ліведо).
- Виділяють 4 варіанти сітчастого ліведо: дрібне кістоподібне ліведо (fine livedo racemosa), велике кістоподібне ліведо, дрібне правильної форми і велике правильної форми ліведо.
- Гістологічні зміни підтверджують діагноз, але необов’язково. Зміни включають часткову або повну оклюзію просвіту малих або середніх артерій, або артеріол дермо-епідермального стику без периваскулярної інфільтрації чи негативний тест прямої імунофлуоресценції.
Діагностичні критерії (за: Alarcon-Segovia D. et al., 1992)
Клінічні критерії:
- Звичне невиношування вагітності
- Венозні тромбози
- Артеріальна оклюзія
- Трофічні виразки гомілок
- Сітчасте ліведо
- Гемолітична анемія
- Тромбоцитопенія
Біологічні критерії:
- Високий рівень АФЛ
- IgG або IgМ > 5 SD
- Наявність у пацієнта двох і більше клінічних критеріїв і одного біологічного дозволяють встановити діагноз АФС.
Лабораторна діагностика
- АКЛ IgG- або ІgМ-ізотипів, які виявляють в сироватці в середніх або високих титрах хоча б 2 рази протягом 12 тиж за допомогою стандартизованого імуноферментного методу.
- Антитіла до бета-2-глікопротеїну I IgG- та/ або ІgМ-ізотипів, які визначають у сироватці в середніх або високих титрах принаймні 2 рази протягом 12 тиж за допомогою стандартизованого імуноферментного методу.
- ВА в плазмі у 2 або більше випадках дослідження з проміжком не менше 12 тиж, який визначається відповідно до рекомендацій Міжнародного товариства з тромбозів та гемостазу (дослідницька група з вивчення ВА/фосфоліпідзалежних антитіл):
- збільшення часу згортання плазми в фосфоліпідзалежних коагуляційних тестах (активований частковий тромбопластиновий час, каоліновий час згортання, протромбіновий час, тести з отрутами Рассела, текстариновий час);
- відсутність корекції збільшення часу згортання скринінгових тестів у тестах змішування з донорською плазмою;
- скорочення або корекція збільшення часу згортання cкринінгових тестів при додаванні фосфоліпідів;
- виключення інших коагулопатій, наприклад інгібітору VIII фактора згортання крові або гепарину (подовжуючі фосфоліпідзалежні тести згортання крові).
Інструментальні дослідження
Тромбози при АФС мають бути верифіковані за допомогою інструментальних методів дослідження. Їх застосування залежить від передбачуваного тромбування судини або органа. Методи ідентифікації тромбозу включають ультразвукове допплерівське сканування судин, венографію; для ідентифікації ураження ЦНС — КТ та МРТ; в кардіології — від ЕКГ, моніторування ЕКГ, ехоКГ до коронарографії; для підтвердження тромбоемболії легеневої артерії — радіоізотопне сканування легень; в особливих (за показаннями) випадках — ангіографію. Для верифікації тромбозів черевної порожнини, крім УЗД, показано проведення КТ органів черевної порожнини, за показаннями — лапароскопічне дослідження для верифікації тромбозу. При веденні вагітних із АФС показано ультразвукове дослідження плода починаючи з 18–20-го тижня кожні 4–6 тиж, функціональні тести активності плода (кардіотокографія, допплерометрія).
Диференційна діагностика
Диференційна діагностика залежить від клінічних проявів. Відзначають ряд генетично детермінованих і набутих захворювань, які призводять до рецидивуючої втрати вагітності, тромбоемболічних ускладнень або одночасно обох наслідків (табл. 14.1).
Таблиця 14.1
Диференційна діагностика АФС
|
Захворювання |
Клінічні прояви |
|
Системні васкуліти |
|
|
Вузликовий поліартеріїт |
Сітчасте ліведо, дистальна гангрена кінцівок, виразки шкіри, некрози шкіри, ураження нервової системи, нирок |
|
Облітеруючий тромбангіїт (хвороба Вінівартера — Бюргера) |
Рецидивуючий мігруючий флебіт, дистальна гангрена кінцівок, виразки шкіри, некроз шкіри, інфаркт міокарда, тромбоз судин брижі, ураження ЦНС |
|
Геморагічний васкуліт |
Геморагічний висип на шкірі, виразки та некроз шкіри, ураження нирок |
|
Скроневий артеріїт (хвороба Хортона) |
Тромбоз артерій сітківки, головний біль |
|
Неспецифічний аортоартеріїт (хвороба Такаясу) |
Синдром дуги аорти, ураження клапанів серця |
|
Тромботична тромбоцитопенічна пурпура (хвороба Мошковица) |
Рецидивуючі тромбози судин різного калібру, тромбоцитопенія, гемолітична аутоімунна анемія |
|
Гемолітико-уремічний синдром |
Рецидивуючі тромбози судин різного калібру, ураження нирок, гемолітична анемія, геморагії |
|
Шкірний васкуліт |
Виразки та некрози шкіри, ліведо-васкуліт |
|
Ревматичні хвороби |
|
|
ГРЛ |
Розвиток пороків серця, тромбози судин різної локалізації, частіше ЦНС та кінцівок за механізмом кардіогенної тромбоемболії |
|
СЧВ |
Тромбози, гематологічні порушення, ліведо |
|
Склеродермія |
Ліведо, дистальна гангрена кінцівок, виразки шкіри |
|
Тромбофілії |
|
|
Спадкові хвороби в результаті мутації факторів згортання крові, антикоагулянтів плазми |
Рецидивуючі тромбози судин різного калібру та локалізації, шкірні виразки |
|
ДВЗ-синдром |
Тромбоемболічні ускладнення |
|
Інфекційні хвороби |
|
|
Туберкульоз, вірусні гепатити та ін. |
Тромбоемболії, поперечний мієліт, ліведо |
Приклади формулювання діагнозів
Первинний АФС з ураженням шкіри — синдром ліведо, трофічні виразки гомілки; ураженням нервової системи — епілептиформний синдром; акушерською патологією — повторні звичні аборти, тромбоцитопенія.
Лікування
Профілактика і лікування тромбозу при АФС становить складну проблему. Це пов’язано з неоднорідністю патогенезу АФС, поліморфізмом клінічних проявів, відсутністю достовірних клінічних та лабораторних показників, що дозволяють прогнозувати рецидив тромботичних порушень. Фактично досі не розроблено загальноприйнятих на міжнародному рівні стандартів, які стосуються тактики ведення пацієнток з різними формами АФС, а пропоновані на сьогодні рекомендації засновані на результатах «відкритих» випробувань або ретроспективного аналізу результатів захворювання. Слід підкреслити, що ГК, цитостатики і плазмаферез застосовували тільки при АФС на тлі СЧВ або іншого аутоімунного захворювання за необхідності придушити активність основного захворювання (наприклад СЧВ) або при «катастрофічному» АФС. У випадках АФС без ознак іншого захворювання, а також при високих рівнях АФЛ вони неефективні (і навіть протипоказані), оскільки тривала ГК-терапія потенційно може підвищити ризик рецидиву тромбозу, а цитотоксичні препарати (і ГК) збільшують ризик ускладнень антикоагулянтної терапії.
У зв’язку з високим ризиком рецидиву тромбозу при АФС переважна більшість хворих потребують проведення профілактичної антикоагулянтної терапії протягом тривалого часу, іноді довічно. Виняток можуть становити пацієнти зі стійкою (протягом декількох років) нормалізацією рівнів АФЛ за відсутності рецидиву тромбозу та виключення інших чинників ризику розвитку тромбозів. Однак і в цьому випадку ризик рецидиву тромбозу не може бути повністю виключений, тому пацієнти потребують ретельного динамічного спостереження та, ймовірно, прийому низьких доз ацетилсаліцилової кислоти.
Одне з найбільш дискусійних питань — доцільність профілактичного лікування пацієнтів із позитивними результатами визначення АФЛ, але без клінічних проявів АФС. Виділяють потенційно контрольовані і неконтрольовані фактори ризику виникнення тромбозів при АФС.
Вважається, що ризик розвитку і рецидиву тромбозу при АФС можна знизити, виключивши фактори ризику, але справжня ефективність цих рекомендацій залишається невідомою. На думку більшості дослідників, в осіб з високим рівнем АФЛ у крові, але за відсутності клінічних проявів АФС (у тому числі й у вагітних без вказівок на акушерську патологію в анамнезі) слід обмежитися призначенням низьких доз ацетилсаліцилової кислоти. Додатковий «профілактичний» ефект, ймовірно, можуть забезпечити амінохінолінові препарати, які поряд із протизапальною дією мають антитромботичні та гіполіпідемічні властивості.
Стандарт профілактики рецидивуючої втрати плода — низькі дози ацетилсаліцилової кислоти (50–100 мг) протягом періоду від настання вагітності до народження дитини. Під час вагітності низькі дози ацетилсаліцилової кислоти можна комбінувати з підшкірним введенням гепарину натрію або низькомолекулярних гепаринів. Необхідно підкреслити, що ризик ОП у матері може бути зведений до мінімуму введенням низькомолекулярних гепаринів (надропарин кальцію, натрію далтепарин та ін.). Застосування варфарину під час вагітності протипоказано, оскільки призводить до варфаринової ембріопатії з порушенням росту епіфізів кісток і гіпоплазією носової перегородки, а також неврологічних порушень.
Варфарин належить до непрямих антикоагулянтів — похідних кумаринового ряду і характеризується швидким ефектом гіпокоагуляції з короткою післядією. Контроль лікування непрямими антикоагулянтами здійснюють за протромбіновим часом та на сьогодні його не вважають досить надійним. Для визначення протромбінового часу використовують тромбопластин, біологічна активність якого залежить від виду тканини і технології одержання. Для того, щоб стандартизувати результати визначення протромбінового часу з застосуванням різних тромбопластинів ВООЗ (1983) рекомендує використовувати міжнародне нормалізоване відношення (МНВ). Виділяють два рівні гіпокоагуляційної терапії варфарином: помірний, який відповідає значенням МНВ 2,0–3,0, та інтенсивний при МНВ — 2,5–3,5. Більш низьке значення МНВ може бути достатнім за відсутності факторів підвищеного ризику тромбозу або у хворих з ризиком геморагічних ускладнень. У деяких випадках резистентність до непрямих антикоагулянтів у пацієнтів із АФС має генетичну природу (за нашими даними, 6%) і пов’язана з мутацією факторів згортання крові V і II. У носіїв цих мутацій при призначенні непрямих антикоагулянтів необхідно контролювати рівень природного антикоагулянту — протеїну С, зниження якого при лікуванні непрямими антикоагулянтами призводить до рикошетного тромбозу.
Центральне місце в лікуванні гострого тромбозу при АФС займають прямі антикоагулянти: гепарин натрію або низькомолекулярні гепарини, або гепариноїди. Тромболізис, тромбектомію і емболектомію в гострій стадії тромбозу глибоких вен гомілок і легеневої тромбоемболії проводять рідко. Стандартом лікування є гепаринотерапія з одночасним призначенням пероральних антикоагулянтів. Дискусії ведуться лише щодо переваг гепаринів з різною молекулярною масою. На сьогодні у профілактиці та лікуванні хворих з венозними тромбоемболіями перевагу віддають низькомолекулярним гепаринам, враховуючи їх достатню ефективність і менший ризик небажаних реакцій. Лікування гострого венозного тромбозу починають з 4- або 7-денного введення гепарину натрію, непрямі антикоагулянти приєднують одночасно у мінімальних дозах при значенні МНВ до 2,0. Цих показників необхідно досягти принаймні за добу до відміни гепарину натрію. Подальше підтримання гіпокоагуляції на рівні МНВ 2,0–3,0 досить ефективне, щоб попередити рецидив венозної тромбоемболії, при цьому тривалість лікування — обернено пропорційна ризику рецидиву. Пацієнти, які мають спадкові фактори ризику і більше одного епізоду венозного тромбозу, повинні отримувати антикоагулянтну терапію більше 6 міс. Як довго слід проводити лікування антитромботичними препаратами: 1–2 роки або, можливо, довічно, і чи може менш інтенсивна антикоагуляція бути достатньо ефективною, поки нез’ясовано. Лише нові дослідження дозволять відповісти на ці питання.
В окремих клінічних спостереженнях або невеликих відкритих випробуваннях повідомляється про ефективність плазмаферезу, в/в введення імуноглобуліну, простацикліну, фібринолітичних препаратів, препаратів на основі риб’ячого жиру у жінок з акушерською патологією.
Лікування плазмаферезом, високими дозами ГК (у тому числі пульс-терапія) і цитостатиками залишається основним методом при катастрофічному АФС. Хороший ефект відзначали при поєднанні плазмаферезу з в/в введенням імуноглобуліну у розрахунку 2 г/кг протягом 2 діб або по 400 г/кг протягом 5 діб.
При помірній тромбоцитопенії, яку нерідко відзначають у хворих із АФС, спеціального лікування не потрібно. Тромбоцитопенія при вторинному АФС в рамках СЧВ зазвичай добре контролюється ГК, амінохіноліновими препаратами, а в резистентних випадках — низькими дозами ацетилсаліцилової кислоти. Тактику лікування резистентної тяжкої тромбоцитопенії (<50 000/мм3), яка створює загрозу кровотеч, до кінця не розроблено. Цим пацієнтам, поряд із застосуванням ГК у високих дозах, доцільно призначати в/в імуноглобулін.
Прогноз
Прогноз при АФС врешті залежить від ризику рецидивування тромбозів, який, як уже зазначалося, хоча і відрізняється істотною варіабельністю, в цілому досить високий у хворих із вторинним і первинним АФС. У загальній популяції пацієнтів із СЧВ частота тромбозів коливається від 7 до 12%, а частота тромбозів, пов’язана з летальним кінцем, досягає 27%. У багатоцентровому дослідженні (European Working Party on Systemic Lupus Erythematosus), заснованому на тривалому (10 років) спостереженні 1000 пацієнтів із СЧВ, встановлено, що основними причинами летальності при СЧВ протягом перших 5 років хвороби є інфекції, а в наступні 5 років — тромбози (26,1%). Прогноз у хворих на СЧВ із АФС (або з АФЛ) гірший, ніж у пацієнтів із СЧВ без АФЛ.
За відсутності антикоагулянтної терапії частота рецидивування тромбозів при АФС досягає 55%. На тлі антикоагулянтної терапії ризик повторних тромбозів вельми високий (9 на 100 пацієнтів на рік); такі тромбози можуть призводити до летальних наслідків. За даними проспективного дослідження, повторні тромбози відзначено у 9% пацієнтів протягом 4 років спостереження. Комбінований індекс пошкодження (SDI) протягом 5 і 15 років спостереження був вищим у пацієнтів з АФС порівняно з хворими без АФС, 15-річна виживаність пацієнтів із АФС становила 65%, а хворих без АФС — 90% (р=0,03). У третини пацієнтів з первинним АФС розвивається незворотне ураження внутрішніх органів. Повторні тромбози виявлено у 19% хворих (незважаючи на лікування варфарином), а 5 із 39 пацієнтів потребували оперативного лікування з приводу патології клапанів серця протягом 10 років спостереження. За деякими даними, у пацієнтів з АФС має місце суттєве підвищення післяопераційної летальності (20%) при хірургічних втручаннях на клапанах серця. Особливо несприятливий прогноз відзначають у пацієнтів із катастрофічним АФС, при якому летальність становила 46%. Цікаво, що у 36% хворих, що вижили, констатували повторні тромботичні епізоди (незважаючи на антикоагулянтну терапію).
Глава 15. Системна склеродермія
ССД — аутоімунне захворювання сполучної тканини з характерним ураженням шкіри, судин, опорно-рухового апарату і внутрішніх органів (легені, серце, травний канал, нирки), в основі якого лежать порушення мікроциркуляції, запалення і генералізований фіброз. ССД відносять до групи склеродермічних хвороб, яка також включає обмежену (осередкову) склеродермію, дифузний еозинофільний фасцит, склеродерму Бушке, мультифокальний фіброз, індуковані форми склеродермії і псевдосклеродермічні синдроми.
Код за МКХ-10
М34 Системний склероз.
М34.0 Прогресуючий системний склероз.
М34.1 Синдром CREST.
М34.2 Системний склероз, спричинений лікарськими засобами і хімічними сполуками.
М34.8 Інші форми системного склерозу.
М34.9 Системний склероз неуточнений.
Епідеміологія
Випадки ССД реєструють у всіх частинах світу, проте поширеність захворювання в різних географічних зонах і етнічних групах неоднакова. Первинна захворюваність коливається від 3,7 до 20,0 на 1 млн населення в рік. Поширеність в середньому становить 240–290 на 1 млн населення; найвищу частоту ССД (в 20 разів вищу, ніж у популяції в цілому) виявлено в племені чокто (Choctaw) (Оклахома, США). Представники негроїдної раси схильні до захворювання більше, ніж європеоїдної. ССД вражає в основному жінок (співвідношення жінок і чоловіків — 7:1). Хворіють переважно особи у віці 30–50 років, хоча початок захворювання можливий в будь-якому віці (за даними літератури, від 10 міс до 80 років) (Гусева Н.Г., 2001; Сигидин Я.А. и соавт., 2004; Clements Ph.J., Furst D.E., 2004). Трапляються, хоч і не часто, сімейні випадки ССД, що включають близнюків, родичів першого та другого ступеня спорідненості. Про спадкову схильність до захворювання свідчить і більш висока поширеність інших захворювань сполучної тканини, а також імунних і хромосомних аномалій серед родичів хворих на ССД.
Етіологія і патогенез. Етіологія ССД недостатньо вивчена і на сьогодні є багатокомпонентною проблемою, основу якої становить взаємодія факторів зовнішнього середовища з генетичною схильністю до захворювання. Участь генетичних факторів доведено наявністю сімейних випадків ССД, близьких захворювань і синдромів, виявленням імунологічних порушень у родичів, частотою хромосомних аномалій, у тому числі й у родичів, виявленої, хоча і не чіткої, асоціації ССД з антигенами НLА (А9, В8, В35, ДR1, ДRЗ, ДR5), більш чітко — з гаплотипом В8/ДR3 і німими алелями в С4А-локусі. Виявлено підвищення частоти накопичення антигену В8 при ранньому початку захворювання, антигену А10 — при ураженні легенів, а ДRЗ — при СRESТ-синдромі. Виявлено асоціації ССД із певними антигенами і алелями системи гістосумісності (НLА), особливо алелями класу II, які варіюють у різних популяціях (НLА-DR5 і НLА-ВКЗ — у європейців і білих американців, НLА-DR2 — у японців і представників племені Choctaw). Принципово важлива кореляція імуногенетичних маркерів з імунологічними (специфічні аутоантитіла) і клінічними проявами ССД (табл. 15.1).
Таблиця 15.1
Аутоантитіла, генетичні маркери і клінічна картина ССД
|
Аутоантитіла до антигенів |
HLA |
Клінічні особливості |
|
Антитопоізомеразні Scl-70 (топоізомераза-1) |
DR5 (DR11), DR3/DRw52, DQ7 |
Дифузна ССД, швидко прогресуючий перебіг, фіброз легень |
|
Антицентромерні (центромери) |
DR1, DR11, DR4, DQB1 |
Лімітована ССД, повільно прогресуючий перебіг |
|
Анти-РМ-Scl |
DR3/DRw52 |
Легенева гіпертензія, оverlар ССД-поліміозит/ДМ, підгострий перебіг, ураження м’язів |
Передбачуваний мультифакторний тип успадкування сприяє реалізації патологічного процесу лише при взаємодії генетичних і факторів зовнішнього середовища. До останніх належать охолодження, вібрація, інфекція, вплив кремнієвого пилу, хлорвінілу та ін.
Важлива роль вірусної інфекції (групи ретровірусів і герпес-вірусів), якій властива схильність до персистенції у вигляді латентних форм, а потім активація під впливом різних хімічних, біологічних та інших факторів. Так, активація латентної інфекції, викликаної вірусом Епштейна — Барр призводить до вираженого дефекту Т-супресорів, а отже до активації В-лімфоцитів.
Патогенез ССД надзвичайно складний. У його основі — порушення імунітету, мікроциркуляції і фіброзоутворення, які взаємодіють на рівні основних клітинних (імунокомпетентні клітини → ендотелій → фібробласти → клітини крові) і рецепторно-лігандних систем (молекули клітинної адгезії, фактори росту, цитокіни).
Для ССД характерний широкий спектр різноманітних порушень клітинного та гуморального імунітету: наявність Т-клітинної активації та дерегуляції в системі Тh1- і Тh2-клітин, підвищення рівня окремих імунорегуляторних цитокінів, наявність специфічних антинуклеарних і антинуклеолярних антитіл антицентромерних, антитопоізомеразних, або анти-Scl-70 і РНК-антитіл, а також антинейтрофільних цитоплазматичних антитіл, антитіл до ендотелію, різних компонентів сполучної тканини та ін.
Участь імунних механізмів у розвитку ССД є незаперечною. Підтвердження тому — такі порушення клітинного та гуморального імунітету, як зменшення кількості циркулюючих Т-лімфоцитів при нормальній кількості В-лімфоцитів, дисбаланс хелперної та супресорної активності Т-клітин за рахунок зниження супресорної, гіпергаммаглобулінемії, висока частота антинуклеарних антитіл (антицентромерних, антицентріольних, анти-Scl-70 та ін.), ЦІК, РФ, тканинна лімфоцитарна і плазмоцитарна інфільтрація, відкладення імуноглобулінів, комплементу та ін.
Доведено патогенетичну роль Т-клітинних порушень, їх участь у розвитку судинної патології та фіброзу при ССД. У ранній стадії захворювання виявляють периваскулярну інфільтрацію дерми з переважанням СD4+ Т-лімфоцитов, мукоїдне набухання стінки судин, скупчення фібробластів і активування раніше тучних клітин у периваскулярному просторі, експресію IСАМ-1 на ендотеліальних клітинах.
На ранніх стадіях розвитку ураження дерми (клінічно щільний набряк, початкова індурація) клітинний інфільтрат містить активні Т-лімфоцити, чим пояснюється зменшення їх кількості в циркуляції. Відзначають паралелізм між виразністю інфільтрації та наступним фіброзом шкіри, що можна пояснити тісним зв’язком імунокомпетентних клітин з фібробластами через лімфокіни і монокіни (фібронектин, ІЛ-1). Процеси посиленого колагено- і фіброзоутворення займають центральне місце в патогенезі ССД.
Характерне для ССД підвищене колагено- і фіброзоутворення займає центральне місце в патогенезі та визначає нозологічну специфіку захворювання. Фенотипічно стійка гіперактивність фібробластів, можливо генетично детермінована, веде до надлишкової продукції компонентів міжклітинного матриксу, підвищення неофібриногенезу і генералізованого фіброзу, властивого ССД. Сьогодні активно досліджують регуляторні механізми експресії матриксних генів, зокрема стимулюючу роль фактора росту бета, промоторні функції різноманітних генів колагену.
Підвищений біосинтез колагену (I і III типу), глікозаміногліканів, протеогліканів, фібронектину, глікопротеїну фібробластами при ССД підтверджується електронно-мікроскопічними дослідженнями.
Показана аномальна функція і дефектність мембранної рецепції фібробластів шкіри, що проявляється збільшенням швидкості транспорту Са2; через клітинну мембрану порушенням внутрішньоклітинного вмісту циклічних нуклеотидів — зниження рівня циклічного аденозинмонофосфату і підвищення — циклічного гуанозинмонофосфату, зниженням чутливості клітин до естрадіолу, зниженою або парадоксальною реакцією на катехоламіни. Виявлені порушення функціональної активності склеродермічних фібробластів зберігаються при перенесенні клітин в інше середовище, що може свідчити про їх природжений дефект.
Важливим фактом є і те, що колагенові білки і глікозаміноглікани при ССД мають антигенні властивості, що викликає аутоімунні реакції, які призводять до колагенової стимуляції. Антитіла, виявлені при ССД, специфічні до компонентів базальних мембран, ядерних антигенів ендотелію, що зумовлює ураження мікроциркуляторного русла.
Важливою ланкою патогенезу ССД є порушення мікроциркуляції, зумовлені ураженням судин, з одного боку, і внутрішньосудинними змінами, з іншого. У механізмі розвитку цих змін, очевидно, важлива роль належить підвищеній активності міофібробластів судинної стінки. У результаті виникають: деструкція ендотелію мікросудин, проліферація гладком’язових клітин із гіперпродукцією колагену (III тип) і підвищення схильності до вазоконстрикції; внутрішньосудинні зміни у вигляді агрегації клітинних елементів крові (тромбоцитів, лейкоцитів й еритроцитів), підвищеної в’язкості і коагуляційних властивостей крові, мікротромбозів, потовщення судинних стінок зі звуженням просвіту, що клінічно проявляється генералізованим синдромом Рейно і зумовлює характерну для ССД редукцію мікроциркуляторного русла. Останнім часом виявлено, що сироватка хворих на ССД має шкідливу дію на ендотеліальні клітини і цитотоксичну активність щодо фібробластів, проте генез цієї цитотоксичності вимагає уточнення.
Про ступінь ураження ендотелію можна судити за серологічними ендотеліальними маркерами: характерне підвищення рівня антигену фактора фон Віллебранда і розчинних клітинних молекул адгезії (pVCAM-1, Е-селектин тощо), зменшення вмісту АПФ, що корелює з активністю патологічного процесу.
Таким чином, у питанні етіології ССД залишаються багато нез’ясованих моментів. В основі патогенезу головними є процеси посиленого колагено- і фіброзоутворення, а також судинні ураження.
Залежно від поширеності ураження шкіри (індурація) і основного симптомокомплексу виділяють клінічні форми ССД (табл. 15.2). Крім того, у вітчизняну класифікацію включено варіанти перебігу та стадії розвитку ССД.
Таблиця 15.2
Класифікація ССД
|
Клінічна форма |
Характерні ознаки |
|
Пресклеродермія |
Синдром Рейно; капіляроскопічні зміни; специфічні аутоантитіла |
|
Дифузна форма |
Розвиток шкірних змін протягом 1 року після появи синдрому Рейно. Залучення шкіри кінцівок і тулуба. Наявність симптому тертя сухожиль. Ранній розвиток інтерстиціального захворювання легень, олігоуричного ураження нирок, дифузного ураження ШКТ і залучення міокарда. Розширення і редукція капілярів нігтьового ложа. Антитіла до топоізомерази-1 (Scl-70) |
|
Лімітована форма |
Синдром Рейно протягом багатьох років передує іншим симптомам хвороби. Залучення шкіри обмежується дистальними відділами кінцівок (дистальних ліктьових і колінних суглобів) і обличчям. Пізніше розвиток легеневої гіпертензії з або без інтерстиціального захворювання легень, кальцинати і ураження ШКТ. Висока частота антицентромерних антитіл. Розширення капілярів нігтьового ложа, зазвичай без редукції |
|
Склеродермія без склеродермії (Scleroderma sine scleroderma) |
Синдром Рейно є/немає. Немає ущільнення шкіри. Початок хвороби — з легеневого фіброзу, склеродермічного ниркового кризу, ураження серця і ШКТ. Можуть виявлятися антинуклеарні антитіла |
Клінічні форми
- Дифузна форма:
- генералізоване ураження шкіри кінцівок, обличчя та тулуба протягом 1 року; синдром Рейно (одночасно або після ураження шкіри);
- ранній розвиток вісцеральної патології (інтерстиціальне ураження легень, ураження ШКТ, міокарда, нирок);
- значна редукція капілярів нігтьового ложа з формуванням аваскулярних ділянок (за даними капіляроскопії нігтьового ложа);
- виявлення антитіл до топоізомерази-1 (Scl-70).
- Лімітована форма:
- тривалий період ізольованого синдрому Рейно;
- ураження шкіри обмежене ділянкою обличчя, кистей і стоп;
- пізній розвиток легеневої гіпертензії, ураження травного тракту, телеангіектазій, кальцинозу (СREST-синдром);
- розширення капілярів нігтьового ложа без виражених аваскулярних ділянок;
- виявлення антицентромерних антитіл.
- Перехресна форма (оverlap-syndrome). Характерне поєднання клінічних ознак ССД і ще одного або декількох системних захворювань сполучної тканини.
- Вісцеральна форма:
- відсутність ущільнення шкіри;
- синдром Рейно;
- ознаки легеневого фіброзу, гострої склеродермічної нирки, ураження серця і ШКТ;
- виявлення антинуклеарних антитіл (Scl-70, антицентромерні антитіла).
- Ювенільна склеродермія:
- початок хвороби до 16 років;
- ураження шкіри нерідко за типом вогнищевим або лінійним (геміформа);
- склеродермії;
- схильність до утворення контрактур, можливі аномалії розвитку кінцівок.
- Індукована склеродермія: поширене, частіше дифузне, ураження шкіри (індурація), іноді в поєднанні з судинною патологією, що розвивається після впливу хімічних та інших факторів зовнішнього середовища.
- Пресклеродермія: клінічно ізольований синдром Рейно в поєднанні з капіляроскопічними та/або імунологічними порушеннями, характерними для ССД.
Варіанти перебігу
- Гострий швидко прогресуючий перебіг. Характерний розвиток генералізованого фіброзу шкіри (дифузна форма) і внутрішніх органів (серце, легені, нирки) у перші 2 роки від початку захворювання. Раніше нерідко закінчувалося смертю; сучасна адекватна терапія поліпшила прогноз у цієї категорії хворих.
- Підгострий помірно прогресуючий перебіг. Характерне переважання клінічних та лабораторних ознак імунного запалення (щільний набряк шкіри, артрит, міозит), нерідкісні overlар-синдроми.
- Хронічний повільно прогресуючий перебіг. Характерне переважання судинної патології: на початку захворювання — багаторічний синдром Рейно, поступово розвиваються помірні шкірні зміни (лімітована форма), збільшується вираженість судинних ішемічних розладів, вісцеральної патології (ураження травного тракту, легенева гіпертензія).
Прогностичні відмінності варіантів перебігу ілюструє 5- і 10-річна виживаність, яка при гострому перебігу становила раніше 4 і 0%, при підгострому — 75 і 61%, а при хронічному — 88 і 84% відповідно. На сьогодні за більш ранньої діагностики та сучасної терапії прогноз ССД покращився, але відмінності в дебюті, основних клінічних проявах й еволюції зберігаються.
Стадії ССД:
- I — початкова — виявляють 1–3 локалізації хвороби;
- II — стадія генералізації, що відображає системний, полісиндромний характер процесу;
- II — пізня (термінальна), коли є недостатність одного або декількох органів (серце, легені, нирки).
Бажано при формулюванні діагнозу, визначенні прогнозу і виборі адекватної терапії використовувати всі 3 параметри класифікації ССД (табл. 15.3).
Таблиця 15.3
Клінічні та лабораторні характеристики ССД
|
Ураження |
Основні прояви |
|
Шкіри |
Щільний набряк, індурація, атрофія; гіперпігментація, ділянки гіпопігментації, осередкове ураження |
|
Судин |
Синдром Рейно, судинно-трофічні зміни, дигітальні виразки/рубчики; некрози; телеангіектазії |
|
Опорно-рухового апарату |
Артралгія, артрит, фіброзні контрактури; міалгія, міозит, атрофія м’язів; кальциноз; остеоліз |
|
Травного тракту |
Дисфагія, дилатація стравоходу, звуження у нижній третині, ослаблення перистальтики, рефлюкс-езофагіт, виразки — іноді, стриктури стравоходу; дуоденіт, часткова непрохідність кишечнику, синдром порушення усмоктування |
|
Органів дихання |
Фіброзуючий альвеоліт; базальний пневмофіброз (компактний, кістозний); функціональні порушення за рестриктивним типом, легенева гіпертензія; плеврит (частіше адгезивний) |
|
Серця |
Інтерстиціальний міокардит; кардіофіброз (вогнищевий, дифузний); ішемія міокарда, порушення ритму та провідності; склероз ендокарда, вади серця (рідко); перикардит (частіше адгезивний) |
|
Нирок |
Гостра склеродермічна нефропатія (склеродермічний нирковий криз); хронічна нефропатія від прогресуючого гломерулонефриту до субклінічних форм |
|
Ендокринної та нервової системи |
Порушення функцій щитоподібної залози (частіше — гіпотиреоїдизм), рідше — статевих залоз, імпотенція, тригемініт, поліневропатія |
|
Загальні |
Втрата маси тіла (10 кг і більше); лихоманка (частіше субфебрилітет) |
|
Лабораторні |
Аутоантитіла анти-Scl-70 або антитопоізомеразні; антицентромерні Дилатація, зміна форми капілярів, аваскулярні поля |
Клінічна картина
Клінічна картина ССД характеризується полісиндромним поєднанням ознак фіброзу та генералізованого ендартеріїту.
Однією з найбільш ранніх ознак ССД є синдром Рейно. Цей синдром майже постійно супроводжує ССД і може на місяці і навіть роки випереджати виникнення інших ознак хвороби. Синдром Рейно характеризується онімінням і циклічною зміною кольору шкіри (збліднення, ціаноз, гіперемія) після охолодження, перевтоми, хвилювань. Вазоспастичні порушення при синдромі Рейно не обмежуються пальцями рук, а поширюються на кисті, стопи, нерідко хворі відчувають оніміння в ділянці губ, носа, вух, кінчика язика. При огляді, поряд з мінливим забарвленням шкіри, похолодінням кінцівок, відзначають появу рубчиків, а також ранок, які тривалий час не загоюються (рис. 15.1).
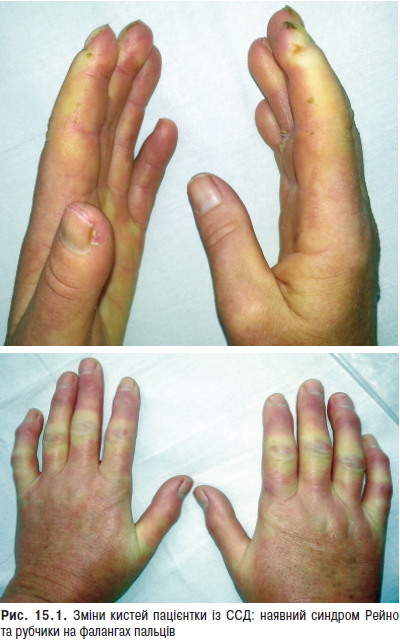
Склеродермічне ураження шкіри доповнюється синдромом Рейно, проявляється спочатку стадією щільного набряку, а пізніше індурацією й атрофією. Зміни найчастіше починаються на II–IV пальці рук. Ущільнення і потовщення шкіри супроводжується втратою придатків шкіри, набряком та порушенням пігментації (чергування гіперпігментації і депігментації). З прогресуванням процесу пальці стають стрункими, щільними, витонченими («пальці мадонни») (рис. 15.2), розвивається згинальна контрактура. Шкіра пальців стає тонкою, часто покривається виразками з повільним загоєнням і утворенням рубців, розвиваються склеродактилія й акросклероз, деформація нігтів, можлива гангрена пальців, самоампутація, остеоліз дистальних фаланг (виявляють рентгенологічно), у результаті чого — вкорочення пальців. У ряді випадків у ділянці кистей, на розгинальній поверхні ліктьових суглобів, головним чином періартикулярно, в м’яких тканинах відзначають відкладення солей кальцію (синдром Тіб’єржа-Вейссенбаха) (рис. 15.3).
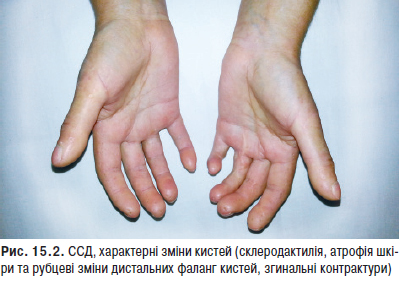
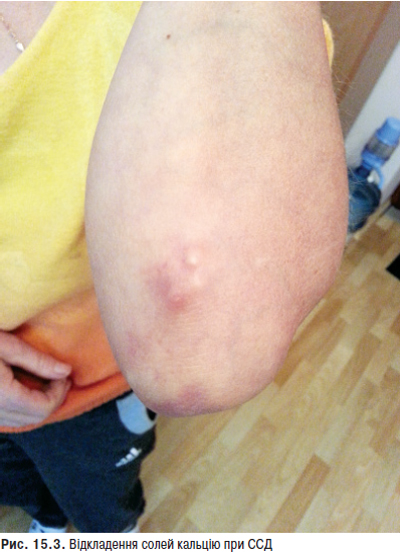
Характерною ознакою змін шкірних покривів при ССД є утворення телеангіектазій з переважною локалізацією в ділянці обличчя та грудей. Часто уражається обличчя, набуває нерухомого, маскоподібного вигляду, шкіра на ньому воскоподібна, натягнута, блискуча, не береться в складку через спаяність з прилеглими тканинами, носогубні складки згладжені, при цьому ніс загострюється, розвивається мікростомія та внаслідок посилення радіальних складок рот виглядає, як кисет (рис. 15.4).
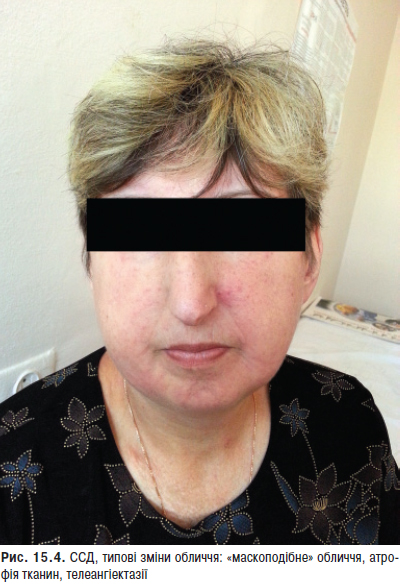
Нерідко має місце випадіння волосся. Значно змінюються повіки. Спочатку утворюються щільні набряки повік із псевдоптозом, пізніше розвиваються рубцеві зміни, випадають вії. У тяжких випадках шкірні покриви можуть підлягати ураженню на всьому протязі. Склерозування шкіри грудної клітки може викликати у хворого відчуття панцира. Для ранньої діагностики необхідна орієнтація на початкові зміни за типом щільного набряку (особливо зміна пальців кисті, які набувають сосископодібного вигляду). При дифузній швидкопрогресуючій ССД розвивається практично тотальне ураження шкірних покривів, у тому числі шкіри тулуба і кінцівок; характерне переважання індуративних змін.
Обмежені склеротичні перетворення відбуваються також і в ділянці слизової оболонки рота. Раннім симптомом є вкорочення та потовщення вуздечки язика (що призводить до порушення мови та ковтання), а пізніше може розвиватися атрофія язика, зморщування м’якого піднебіння та язичка. Відзначають також ураження зубів, сухість і згладженість слизової оболонки порожнини рота, які можуть проходити за типом синдрому Шегрена.
Ураження опорно-рухового апарату
Суглобовий синдром є одним із частих і ранніх ознак захворювання (поступаючись лише синдрому Рейно). Виділяють 3 основні його варіанти:
1. Поліартралгії.
2. Склеродермічний поліартрит з переважанням ексудативно-проліферативних або фіброзно-індуративних змін.
3. Псевдоартрит або періартрит з деформацією суглобів та розвитком контрактур, головним чином за рахунок ураження періартикулярних тканин і сухожильно-м’язового синдрому.
Переважно на ранніх стадіях хвороби розвивається поліартрит з ураженням дрібних суглобів кисті. Іноді в патологічний процес залучаються інші суглоби. З’являється припухлість м’яких тканин, зумовлена підгострим синовітом, яка в поєднанні з набряком шкіри призводить до виникнення типової картини «пальців-сосисок». При русі в уражених суглобах і сухожиллях може проявлятися характерна крепітація. Такі зміни суглобів можуть бути помилково розцінені як ранній прояв РА, особливо при розвитку контрактур суглобів кистей, зумовлених змінами м’яких тканин. Однак прогресуючого перебігу суглобового синдрому з формуванням ерозивних змін, таких характерних для РА, не відзначають. При ССД буває складно визначити, результатом чого є припухлість пальців і дискомфорт — наслідком ураження шкіри, суглобів чи сухожиль.
Можливе ураження кісток. Найбільш характерним є остеоліз дистальних фаланг, який клінічно проявляється характерним укороченням і деформацією пальців. Рідше підлягають ураженню відростки променевих кісток та нижньої щелепи. Зумовлений остеоліз судинно-трофічними порушеннями, можливі зміни й у власне колагеновій матриці (рис. 15.5).
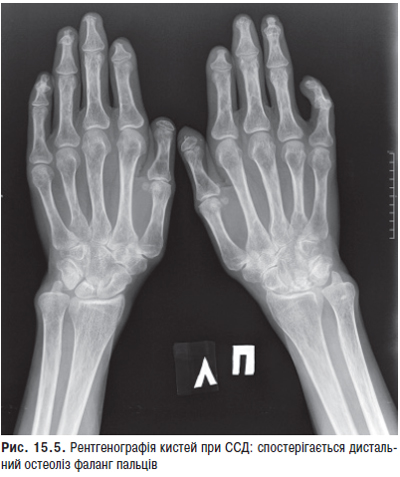
Ураження м’язів при ССД проявляється у вигляді фіброзного інтерстиціального міозиту з розростанням сполучної тканини і атрофією м’язових волокон, а також істинним міозитом з дистрофічними і некротичними змінами, з міастенічним синдромом та порушенням рухів.
Ураження травного тракту
При ССД можливе ураження всіх відділів травного тракту, але найбільш поширена і характерна зміна стравоходу і кишечнику (50–80%).
Склеродермічний езофагіт проявляється дисфагією, відчуттям клубка за грудиною, стійкою печією, яка посилюється в горизонтальному положенні. Рентгенологічно виявляють гіпотонію (зникнення перистальтичних хвиль з уповільненням проходження барію), розширення верхніх двох третин стравоходу і звуження в нижньому відділі, іноді грижі стравохідного отвору діафрагми, ерозії та виразки — наслідок гастроезофагеального рефлюксу. Ерозії та виразки, як і рідкісні випадки пухлинного ураження стравоходу, виявляють і при ендоскопічному дослідженні з біопсією. Хронаксиметрія дозволяє об’єктивізувати ознаки ураження стравоходу за часом уповільнення проходження барію (у нормі в горизонтальному положенні хворого становить 4–10 с). Морфологічну основу ураження стравоходу становлять поширений фіброз й атрофія гладком’язового шару. В окремих хворих виявляють так званий стравохід Барретта (метаплазія епітелію дистального відділу стравоходу за кишковим типом), що підвищує ризик розвитку аденокарциноми.
Шлунок і дванадцятипала кишка також можуть бути атонічні та розширені, можливий біль й важкість в епігастральній ділянці, швидко настає відчуття насичення внаслідок порушення евакуації.
Ураження кишечнику діагностують рідше, але воно іноді виходить на перший план у картині захворювання. При переважному ураженні тонкої кишки характерні абдомінальний біль, діарея, синдром порушення всмоктування, при ураженні товстої кишки — виражений запор аж до розвитку кишкової непрохідності (псевдообструкція). Рентгенологічними методами виявляють уповільнення перистальтики, зміну рельєфу слизової оболонки, асиметричне розташування петель кишечнику з дивертикулоподібними випинаннями, а також ригідність стінки товстої кишки (при іригоскопії). Нерідко має місце поєднання ССД (лімітована форма хронічного перебігу) з первинним біліарним цирозом печінки.
Ураження дихальної системи
Ураження легень виявляють більше ніж у 70% хворих. В останні десятиліття воно виходить на перший план серед причин смерті від ССД. Характерні 2 типи ураження легень.
- Інтерстиціальне захворювання легень (фіброзуючий альвеоліт і дифузний пневмосклероз) переважно в базальних відділах легень.
- Легенева гіпертензія — ізольована або в поєднанні з базальним пневмофіброзом.
Перший тип ураження з переважанням фіброзу властивий дифузній ССД швидкопрогресуючого перебігу (розвивається в перші роки хвороби), другий тип частіше виявляють при лімітованій формі з тривалим хронічним перебігом і переважанням судинної патології. Клінічні прояви інтерстиціального захворювання легень неспецифічні та включають задишку, сухий кашель, швидку втомлюваність. При аускультації чутно двобічну крепітацію в задньобазальних відділах легень. Характерне порушення функції зовнішнього дихання за рестриктивним типом.
Рентгенологічно при інтерстиціальному захворюванні легень виявляють характерне посилення і деформацію легеневого малюнка в нижніх відділах (дифузний пневмосклероз). Більш детальну характеристику ураження легень дає КТ високої роздільної здатності: поряд із картиною «стільникової легені» (кінцевий етап патологічного процесу) можливе виявлення більш раннього симптому «матового скла» — нерівномірне «склоподібне» затемнення легеневої тканини внаслідок активного альвеоліту.
Бронхоальвеолярний лаваж і відкрита біопсія легень дозволяють уточнити характер і активність патологічного процесу. На прогресування легеневого фіброзу вказують посилення задишки, зменшення форсованої життєвої ємності легень і дифузійної здатності легень для С02 протягом наступних 6–12 міс, розповсюдження змін типу «матового скла» і «стільникової легені» (при КТ високої роздільної здатності). При тривалості захворювання від 2 до 10 років поступово розвивається легенева недостатність, у частини хворих — вторинна легенева гіпертензія. Інтерстиціальне захворювання легень нерідко поєднується з ознаками облітеруючого бронхіоліту. Можливий розвиток бронхоектазів, емфіземи, перифокальної пневмонії, рідше — розрив субплевральних кіст з пневмотораксом, абсцедування, рак легені на тлі склеродермічного пневмосклерозу. Фактори ризику інтерстиціального захворювання легень — дифузна форма ССД, наявність антитіл до топоізомерази-1 (Scl-70). Ризик розвитку раку легені у хворих на ССД вищий у 3–5 разів порівняно з аналогічними віковими групами в популяції.
Часто виявляють ознаки адгезивного плевриту (спайки), рідко — невеликий випіт у плевральній порожнині.
Легеневу гіпертензію виявляють у 10–40% хворих, вона розвивається частіше через 10 і більше років від початку захворювання. Характерне підвищення тиску в легеневій артерії вище 25 мм рт ст. у спокої або 30 мм рт. ст. при фізичних навантаженнях. Основна клінічна ознака — прогресуюча задишка; аускультативна ознака — акцент і роздвоєння ІІ тону на легеневій артерії та тристулковому клапані, більш виразні на висоті вдиху. Провісник легеневої гіпертензії — ізольоване зменшення дифузійної здатності легень для CO2. Діагностують захворювання за допомогою ехоКГ і катетеризації правих відділів серця. В основі легеневої гіпертензії лежать порушення газообміну, вазоспазм і потовщення стінки легеневих артеріол. Ускладнення тяжкої легеневої гіпертензії: розвиток легеневого серця і правошлуночкової недостатності, тромбоз легеневих судин (нерідка причина летального результату).
Ураження серця
Серце — одна з частих (16–90%) і найбільш несприятливих локалізацій патологічного процесу, його ураження займає перше місце серед причин раптової смерті хворих на ССД. В основі кардіологічної патології лежать властиві захворюванню фіброз, ураження дрібних судин і порушення мікроциркуляції, які призводять до ішемії міокарда при інтактності основних коронарних артерій.
Симптоми — відчуття дискомфорту або тривалий тупий біль в ділянці серця, серцебиття й аритмії, задишка в спокої або при навантаженнях. Іноді ураження серця при ССД проходить безсимптомно і виявляється тільки при інструментальному обстеженні (збільшення розмірів серця, наявність зон гіпокінезії за ехоКГ, інфарктоподібні зміни на ЕКГ). Добове моніторування ЕКГ, ехоКГ, сцинтиграфія міокарда та інші дослідження сприяють ранній діагностиці ураження серця, виявленню несприятливих форм електричної нестабільності міокарда, прихованої серцевої недостатності, дефектів перфузії.
Фіброз міокарда шлуночків — характерна ознака склеродермічного ураження серця, причина систолічної та діастолічної дисфункції лівого шлуночка.
Аритмії та порушення провідності, які діагностують у 70% хворих, можуть бути причиною раптової смерті. Найчастіші порушення ритму — суправентрикулярна тахікардія, політопні та групові екстрасистоли. Виразність аритмій корелює з тяжкістю ураження серця і суттєво погіршує прогноз. Порушення провідності проявляються подовженням інтервалу Р–Q, дефектами внутрішньошлуночкової провідності і блокадою передньої гілки лівої ніжки пучка Гіса.
Можливе ураження ендокарда і клапанів серця з формуванням відносно доброякісного склеродермічного пороку (найчастіше мітрального); декомпенсація розвивається рідко.
Перебіг перикардиту (адгезивного, рідше ексудативного) зазвичай безсимптомний, патологію виявляють переважно при ехоКГ. В окремих випадках відзначають значний випіт у перикард, що може призвести до тампонади серця.
Серцева недостатність розвивається рідко, але в разі появи вирізняється резистентністю до терапії та несприятливим прогнозом.
Ураження нирок
Ураження нирок, що діагностують у 10–20% хворих, варіює від гострих фатальних до хронічних латентних форм. Функціональні та морфологічні дослідження підвищують частоту виявлення патології нирок. Для гострої нефропатії (склеродермічний нирковий криз) характерні швидкопрогресуюча ниркова недостатність, злоякісна (гіперренінова) артеріальна гіпертензія (проте у 10% хворих артеріальний тиск залишається нормальним), наростаюча протеїнурія, оліго- і анурія, мікроангіопатична гемолітична анемія, тромбоцитопенія (менше 100 тис. на 1 мкл), енцефало- і ретинопатія.
Особливість склеродермічного ниркового кризу — раптовий початок і відсутність проявів. Ризик розвитку склеродермічного ниркового кризу найбільший при дифузійній ССД в перші 2–5 років захворювання. В основі патології — ураження інтерлобулярних і малих кортикальних артеріол з розвитком ішемічного некрозу. В окремих хворих можливий розвиток прогресуючого гломерулонефриту, асоційованого з антинейтрофільними цитоплазматичними антитілами. Нерідко спостерігають ураження нирок за типом хронічної латентної нефропатії, перебіг якої субклінічний (переважно функціональні порушення) або з помірною лабораторною та клінічною симптоматикою. Морфологічно, крім судинної патології та ураження клубочків, виявляють помірні зміни канальців і строми. 15-річна виживаність хворих на ССД без ураження нирок становить 72%, за наявності ураження нирок — 13%, причому при гострій нефропатії лише 23% хворих живуть більше 5 років. На сьогодні гостра склеродермічна нефропатія розвивається рідше (можливо, завдяки протективному ефекту сучасної фармакотерапії).
Ураження нервової та ендокринної системи
Неврологічну симптоматику рідше реєструють при ССД. Неврологічна та ендокринна симптоматика також значною мірою пов’язана із судинними і фіброзними змінами відповідних структур. Найбільш характерні периферична невропатія та вегетативні розлади. Зрідка раннім симптомом хвороби буває порушення чутливості в зоні іннервації V пари черепно-мозкових нервів, тобто оніміння щік. В основі ураження лежать судинні, сполучнотканинні (фіброзні) і дегенеративні зміни нервової системи. Можливі тригемінальна сенсорна невропатія, частіше в рамках поліневритичного синдрому, тиреоїдит із розвитком гіпотиреозу, у окремих хворих — імпотенція.
Загальні прояви
Найбільш характерною є значна втрата маси тіла, яку відзначають у період генералізації або швидкого прогресування хвороби. Гарячкова реакція зазвичай мало виражена (субфебрилітет).
Лабораторні дослідження
Приблизно у половини хворих виявляють гіпохромну анемію, помірне підвищення ШОЕ, зниження гематокриту. Для ураження нирок характерний сечовий синдром, вираженість якого варіює залежно від клінічної форми. Біохімічне дослідження крові дозволяє оцінити стан і функцію внутрішніх органів, схильних до ураження при ССД.
Імунологічні дослідження включають визначення АНФ (виявляють у 95% хворих на ССД, зазвичай в середньому та помірному титрі). Важливу роль відіграє визначення так званих склеродермоспецифічних аутоантитіл: до топоізомерази-1 (анти-Scl-70), характерних для дифузної ССД; їх наявність у поєднанні з носійством HLA-DR3/DRw52 в 17 разів підвищує ризик розвитку легеневого фіброзу. Титр антитіл корелює з поширеністю ураження шкіри та активністю хвороби. Виявлення антитіл Scl-70 у хворих з ізольованим синдромом Рейно асоціюють з подальшим розвитком розгорнутої клінічної картини ССД. Антицентромерні антитіла виявляють у 30% осіб із ССД, частіше при лімітованій формі (60–70%). Крім того, їх реєструють у 12% хворих на первинний біліарний цироз (половина з яких мають ознаки ССД), дуже рідко — при хронічному активному гепатиті та первинній легеневій гіпертензії. Антицентромерні антитіла розглядають як попередник розвитку ССД при ізольованому феномені Рейно. Антитіла до РНК-полімерази III можна виявити у 20–25% хворих, переважно з дифузною формою ССД і ураженням нирок, вони асоційовані з несприятливим прогнозом. Крім зазначених аутоантитіл, при ССД з нижчою частотою виявляють інші антинуклеарні антитіла: антитіла до Рm-Scl у 3–5% пацієнтів із ССД у поєднанні з поліміозитом (ССД-поліміозит, перехресний синдром); антитіла до U3-рибонуклеопротеїну у 5% хворих, асоційовані з дифузною формою хвороби, первинною легеневою гіпертензією, ураженням скелетних м’язів; антитіла до U1-рибонуклеопротеїну у 6% хворих ССД, асоційовані з ССД-СЧВ перехресним синдромом, артритами, ізольованою легеневою гіпертензією і раннім дебютом хвороби. РФ виявляють у 45% хворих, частіше при поєднанні з синдромом Шегрена і РА.
Інструментальні дослідження
Капіляроскопія нігтьового ложа виявляє характерні для ССД зміни (дилатація і редукція капілярів), володіє високою чутливістю і специфічністю.
Щоб своєчасно виявляти і оцінювати ступінь ураження внутрішніх органів, необхідно проводити відповідні інструментальні дослідження (табл. 15.4).
Таблиця 15.4
Інструментальні дослідження
|
Досліджуваний орган |
Вид ураження |
Діагностичні дослідження |
|
Стравохід |
Гіпотонія |
Манометрія/рентгенографія |
|
Рефлюкс-езофагіт |
Ендоскопія/рН-метрія |
|
|
Стриктура |
Рентгенографія/ендоскопія |
|
|
Шлунок |
Парез |
Сцинтиграфія |
|
НПЗП-індукована виразка |
Ендоскопія |
|
|
Тонка кишка |
Гіпотонія |
Рентгеноконтрастне дослідження |
|
Надмірний ріст мікрофлори |
Дихальний вуглецевий тест |
|
|
Псевдообструкція, НПЗП-індукована виразка, пневматоз |
Оглядова рентгенографія |
|
|
Товста кишка |
Гіпотонія |
Барієва клізма |
|
Псевдодивертикули, псевдообструкція |
Оглядова рентгенографія |
|
|
Аноректальний відділ |
Ураження сфінктера |
Манометрія |
|
Легені |
Інтерстиціальний фіброз |
Рентгенографія, КТ високого розрізнення, ФЗД, сцинтиграфія, ДЗсо, ТБЛ |
|
Легенева гіпертензія |
Допплер-ехоКГ, ЕКГ, ДЗсо, рентгенографія, КПШ |
|
|
Серце |
Аритмії, вогнищевий фіброз міокарда, перикардит |
ЕКГ, моніторування ЕКГ за Холтером, ехоКГ, рентгенографія, сцинтиграфія |
|
Нирки |
Склеродермічний нирковий криз |
Добове моніторування артеріального тиску, аналіз сечі, визначення креатиніну, електролітів, реніну в крові, проба Реберга, ЗАК (гемоглобін, лейкоцити, тромбоцити), офтальмоскопія, біопсія нирки |
Примітки: ДЗсо — дифузійна здатність легень для синовіальної оболонки; КПШ — катетеризація правого шлуночка; ЗАК — загальний аналіз крові; ТБЛ — торакоскопічна біопсія легень; ФЗД — функція зовнішнього дихання.
Морфологічні дослідження: біопсія різних тканин (шкіра, м’язи, синовіт, фасції) та органів (стравохід, легені, нирки та ін.) дозволяють уточнити характер патології та надають істотну допомогу в діагностиці та диференційній діагностиці ССД. Проте вирішальною у встановленні діагнозу залишається клінічна симптоматика захворювання.
При аналізі результатів клініко-лабораторного обстеження хворого слід враховувати основні (характерні тільки для ССД) діагностичні та додаткові (властиві як ССД, так і ряду інших захворювань) ознаки.
Діагностичні ознаки системної склеродермії
- Основні:
- склеродермічне ураження шкіри;
- синдром Рейно, дигітальні виразки/рубчики;
- суглобово-м’язові прояви;
- остеоліз;
- кальциноз;
- базальний пневмофіброз;
- кардіосклероз із порушенням ритму і провідності;
- склеродермічне ураження травного тракту;
- гостра склеродермічна нефропатія;
- наявність специфічних антинуклеарних антитіл (анти-Scl-70 і антицентромерні антитіла); капіляроскопічні ознаки (за даними широкопольної капіляроскопії).
- Додаткові:
- гіперпігментація шкіри, телеангіектазії, трофічні порушення;
- артралгії, міалгії, поліміозит;
- полісерозит (частіше адгезивний);
- поліневрит, тригемініт;
- втрата маси тіла (>10 кг);
- збільшення ШОЕ (>20 мм/год);
- гіпергаммаглобулінемія (>23%);
- наявність антитіл до ДНК або АНФ, РФ.
Американською колегією ревматологів запропоновано наступні класифікаційні критерії ССД:
- «Великий» критерій.
- Проксимальна склеродермія: симетричне потовщення та індурація шкіри пальців, що поширюються проксимально від п’ястково-фалангових і плесно-фалангових суглобів. Зміни можуть торкатися обличчя, шиї, грудної клітки, живота.
- «Малі» критерії:
- склеродактилія: вищеперелічені зміни шкірних покривів, обмежені пальцями;
- дигітальні рубчики ділянки западання шкіри на кінчиках пальців або втрата речовини подушечками пальців;
- двобічний базальний пневмофіброз: сітчасті або лінійно-вузлові тіні, найбільш виражені в базальних ділянках легень при стандартному рентгенологічному обстеженні; можуть бути прояви за типом «стільникової легені».
Для встановлення діагнозу ССД потрібен «великий» критерій або 2 «малих». Чутливість — 97%, специфічність — 98%. Критерії придатні для виявлення вираженої ССД, але не охоплюють всіх клінічних форм захворювання, у тому числі ранньої лімітованої, перехресної та вісцеральної ССД.
Диференційна діагностика
Диференційну діагностику ССД проводять з іншими захворюваннями склеродермічної групи: при більшості з них відсутні синдром Рейно і ураження внутрішніх органів.
- Обмежена форма склеродермії (морфеа) може спостерігатись в будь-який період життя, але найчастіше виявляється у дітей. Ця форма захворювання характеризується ураженням виключно шкіри, переважно із залученням прилеглих м’язів. Шкірні прояви поділяються на 5 підтипів: бляшкова, генералізована, лінійна, пансклеротична та змішана форми. Синдром Рейно та ураження внутрішніх органів практично не відзначаються. Пальці кистей в патологічний процес не залучаються, хоча у хворих можуть бути наявні антинуклеарні антитіла до хроматину, гістонів чи нуклеосом (рис. 15.6).
- Дифузний еозинофільний фасцит — індурація тканин починається з передпліч та/або гомілок з можливим поширенням на проксимальні відділи кінцівок та тулуба, пальці кистей і обличчя залишаються інтактними. Характерні ураження шкіри за типом апельсинової шкірки, згинальні контрактури, еозинофілія, гіпергаммаглобулінемія і підвищення ШОЕ. Приблизно в ⅓ випадків простежується зв’язок із попереднім надмірним фізичним навантаженням або травмою. Можливий розвиток апластичної анемії.
- Склеродерма Бушке — виражена індурація в ділянках обличчя, шиї, плечового пояса. Часто пов’язана з попередньою інфекцією верхніх дихальних шляхів.
- Мультифокальний фіброз. Основні локалізації: ретроперитонеальний, інтраперитонеальний та медіастинальний фіброз; рідше виявляють осередки фіброзу в легенях, очній ямці (псевдопухлина очної ямки), щитоподібній залозі (тиреоїдит Ріделя) та ін. До малих форм відносять також контрактури Дюп’юїтрена і келоїд. Нерідко відзначають поєднання двох і більше локалізацій процесу.
- Пухлиноасоційована (паранеопластична) склеродермія — варіант паранеопластичного синдрому. Характерні переважний розвиток фіброзу в періартикулярних тканинах, контрактури. Можливі переважання периферичної симптоматики і резистентність до лікування.
- Псевдосклеродермія — зміни шкіри, які виявляють при вроджених або набутих порушеннях метаболізму: порфірія, фенілкетонурія, амілоїдоз, синдроми Вернера і Ротмунда; діабетична псевдосклеродермія, склеромікседема та ін.
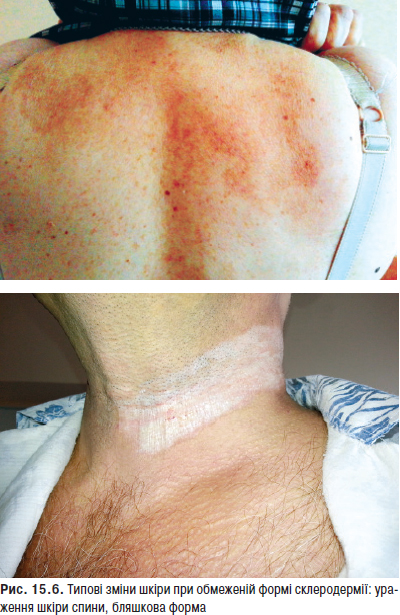
Порфірія, фенілкетонурія, глікогенози і мукополісахаридоз можуть супроводжуватися розвитком склеродермоподібного ураження шкіри, прилеглих тканин, м’язів, стійкими контрактурами, проте на відміну від ССД відсутні синдром Рейно, характерна вісцеральна патологія, імунні порушення, а при біопсії виявляють морфологічні особливості, властиві кожній із нозології.
Синдром Вернера (прогерія дорослих, дефект гена ламіна) проявляється склеродермоподібними змінами шкіри (особливо кінцівок) і скелетних м’язів, розвитком катаракти, гіпогеніталізму, передчасного артеріосклерозу, інсулярної недостатності, підвищеним ризиком розвитку остеосаркоми. Діагностують частіше у чоловіків у віці 20–30 років.
Синдром Ротмунда — Томсона (атрофічна пойкілодермія). Характерною є пойкілодермія обличчя та кінцівок, двостороння катаракта, дистрофія волосся, нігтів і зубів, гіпогонадизм, порушення ендохондрального окостеніння, артеріосклероз і карликовість, гіперпігментація шкіри, телеангіектазії, атрофічний дерматоз, анемія, підвищення ризику розвитку остеогенної саркоми. Синонім — дистрофія Ротмунда. Діабетична псевдосклеродермія проявляється індуративними змінами шкіри і прилеглих тканин, розвивається частіше при тривалому і важкому інсулінзалежному діабеті. Ілюструє роль метаболічних ендокринних порушень у розвитку склеродермоподібної патології.
Склеромікседема характеризується ущільненням шкіри і папульозними висипами в ділянках голови, шиї, рук і верхніх відділів тулуба; поєднується з моноклональною гаммапатією (IgG), іноді з поліміозитом, множинною мієломою, амілоїдозом.
Хронічна реакція «трансплантат проти господаря» при алогенній трансплантації стовбурових клітин може проявлятися склеродермоподібним ураженням шкіри (осередковим або генералізованим) і наявністю аутоантитіл, частіше антинуклеарних і антимітохондріальних. Описано випадки загострення ССД після гемотрансфузії.
Індукована склеродермія може виникнути під впливом хімічних агентів, окремих ліків, харчових добавок та ін. Можливі шкірна, судинна і вісцеральна патологія, які неможливо відрізнити від ССД (у зв’язку з цим індуковану склеродермію включено до класифікації).
Ізольований синдром Рейно визначає необхідність диференційної діагностики ССД з іншими системними захворюваннями сполучної тканини: ЗЗСТ, антисинтетазним синдромом в рамках поліміозиту і ДМ. При диференційній діагностиці вторинного і первинного синдрому Рейно слід враховувати, що останній частіше розвивається в молодому віці, проявляється переважно поширеними ціанозом і гіперемією, нерідко поєднується з гіпергідрозом (вегетативна дисфункція) і зазвичай не супроводжується розвитком ішемічних ускладнень.
Приклади формулювання діагнозів
ССД: хронічний перебіг, II стадія розвитку (генералізована), активність I ступеня, з ураженням шкіри — набряк, індурація, гіперпігментація; судин — синдром Рейно; суглобів — склеродактилія; легень — базальний пневмосклероз, дихальна недостатність I ступеня; стравоходу — езофагіт.
ССД: гострий перебіг, II стадія розвитку (генералізована), активність III ступеня, з ураженням шкіри — індурація шкіри тулуба, плечового пояса, передпліч; серця — дифузний кардіофіброз, серцева недостатність IIA, функціональний клас II; нирок — склеродермічна нефропатія, хронічна ниркова ниркова недостатність II ступеня.
Лікування
Мета лікування
- Корекція судинних порушень і профілактика їх ускладнень.
- Уповільнення прогресування фіброзу.
- Профілактика та лікування уражень внутрішніх органів.
- Покращання якості життя.
- Збільшення тривалості життя.
Показання до госпіталізації
- Уточнення діагнозу та вибір лікування.
- Вперше виявлена ССД, особливо рання стадія дифузної форми.
- Прогресуючий синдром Рейно, рецидивуючі виразкові ураження шкіри і гангрена пальців кистей і стоп.
- Прогресуюче ураження легень (фіброзуючий альвеоліт, легенева гіпертензія), серця (неконтрольована аритмія, серцева недостатність, ексудативний перикардит), ШКТ (атонія шлунка, синдром мальабсорбції, псевдоілеус, кровотеча).
- Поява ознак склеродермічного ниркового кризу (злоякісна гіпертонія, порушення функції нирок).
- Виражена анемія.
Немедикаментозне лікування
Хворим на ССД необхідно дотримуватися певного режиму, уникати психоемоційних навантажень, тривалого впливу холоду і вібрації, зменшити перебування на сонці. Для зниження частоти та інтенсивності нападів вазоспазму рекомендовано носити теплий одяг, у тому числі нижню білизну, яка зберігає тепло, головні убори, вовняні шкарпетки і рукавиці замість рукавичок. З цією ж метою необхідно рекомендувати хворому кинути палити, відмовитися від вживання кави і напоїв, які містять кофеїн, уникати прийому лікарських засобів, що викликають вазоспазм: симпатоміметиків (ефедрин, амфетамін, ерготамін), блокаторів бета-адренорецепторів. Хворим з виявленою гіпотонією стравоходу рекомендують харчуватися часто, але в малих кількостях, не лягати протягом 2 год після прийому їжі, спати на ліжку з піднятим узголів’ям.
Медикаментозне лікування
Лікування хворих на ССД має бути раннім, патогенетично обґрунтованим, комплексним (з урахуванням складного патогенезу захворювання) і диференційованим залежно від перебігу, клінічної форми та характеру органної патології. Оскільки захворювання має хронічний прогресуючий характер, лікування проводять довгостроково, іноді довічно.
Основні напрямки медикаментозного впливу при ССД — судинна, протизапальна і антифіброзна терапія.
Судинна терапія
Існує великий арсенал судинних препаратів, з яких на перший план виходять блокатори кальцієвих каналів (ніфедипін та ін.), інгібітори АПФ (каптоприл та ін.) і простагландин Е1, синтетичні аналоги простацикліну.
Показання до їх призначення:
- синдром Рейно і його ускладнення (ішемія, некрози);
- легенева гіпертензія;
- ниркова артеріальна гіпертензія.
Блокатори кальцієвих каналів призначають уже на початку захворювання в адекватній дозі з урахуванням вираженості судинних порушень і переносимості лікування. Маючи певні переваги ніфедипін призначають у дозі 30–60 мг/добу окремими курсами або протягом тривалого часу (1 рік). Ніфедипін знижує частоту та інтенсивність епізодів вазоспазму, прискорює загоєння дигітальних виразок, підвищує швидкість кровотоку. Побічні явища (рефлекторна тахікардія, головний біль, гіперемія обличчя, набряки щиколоток та ін.) виявляють у ⅓ хворих. Застосування ніфедипіну пролонгованої дії знижує частоту побічних ефектів при збереженні високої клінічної ефективності. При непереносимості ніфедипіну можливе призначення інших блокаторів кальцієвих каналів: амлодипіну (5–20 мг/добу), фелодипіну (5–10 мг/добу), верапамілу (120–360 мг/добу), дилтіазему (90–240 мг/добу). За наявності побічних явищ рекомендують зниження дози, перерву в лікуванні, індивідуальний підбір препарату.
Важливе місце в лікуванні хворих на ССД займають інгібітори АПФ (каптоприл та ін.), які істотно змінили прогноз у хворих з істинною склеродермічною ниркою, що супроводжується вираженою артеріальною гіпертензією і вазоконстрикцією. Інгібітори АПФ призначають за вітальними показаннями в дозах, які контролюють артеріальний тиск (каптоприл по 50–150 мг/добу, максимально — до 300 мг/добу). Доцільне їх поєднання з блокаторами кальцієвих каналів, дезагрегантами; при наростанні ниркової недостатності — з гемодіалізом, гемодіафільтрацією. Ефект інгібіторів АПФ виражається в зниженні та нормалізації артеріального тиску, зменшенні або зникненні головного болю, стабілізації функції нирок, загальному поліпшенні стану. Позитивну дію інгібітори АПФ чинять при легеневій гіпертензії, явищах серцевої недостатності, порушеннях мікроциркуляції, що зумовлює широке застосування препаратів цієї групи у хворих на ССД.
Алпростадил (простагландин Е1) призначають при прогресуючому синдромі Рейно як потужний вазодилататор і антиагрегант. Рекомендовано повільне (протягом 120–360 хв) в/в введення препарату в дозі, еквівалентній 10–20 мкг алпростадилу (вміст 1 ампули розводять в 100–200 мл 0,9% розчину натрію хлориду), 20 перфузій на курс, 2 курси на рік.
У лікуванні легеневої гіпертензії паралельно з патогенетичною терапією важливе місце займають: бозентан — інгібітор ендотеліну (в Україні не зареєстрований), сильденафіл, синтетичний аналог простацикліну I2 ілопрост, які включені в міжнародні клінічні рекомендації з лікування легеневої гіпертензії.
Ілопрост — синтетичний аналог простацикліну. Простациклін утворюється безпосередньо в ендотелії судин і є найбільш потужним із вазодилататорів, відкритих на сьогодні. Крім того, простациклін одночасно має цитопротекторну дію та інгібує агрегацію тромбоцитів (рис. 15.7).
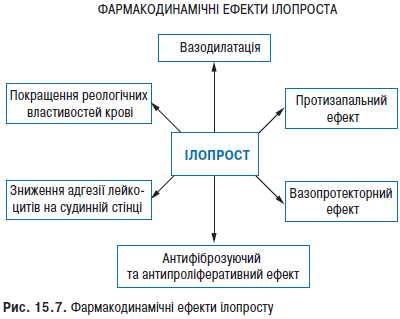
Ілопрост як синтетичний аналог простацикліну I2 в 1000 разів підвищує концентрацію внутрішньоклітинного циклічного аденозинмонофосфату порівняно з іншими простациклінами і тим самим пригнічує синтез трансформуючого фактора росту (TGF)-β, що пояснює його виражений антифіброзний ефект і клінічну дію при ЗЗСТ та ССД. Також ілопрост істотно знижує вміст адгезивних молекул, сироваткових концентрацій фактора росту судинного ендотелію (VEGF) й ендотеліну-1, що пояснює розвиток клінічної ремісії при синдромі Рейно (на 5–7-му добу).
Крім того, ілопрост викликає зниження експресії гена фактора росту сполучної тканини, яке призводить до гальмування синтезу колагену фібробластами та знижує проліферативну відповідь, інгібуючи трансформуючий фактор росту β.
Судинорозширювальна і вазопротекторна дія ілопросту полягає в його безпосередньому впливі на зниження судинного тонусу та протидії вазоспастичному ефекту лейкотрієнів, тромбоксану А2, ендотеліну-1.
Також ілопрост чинить протизапальну дію, оскільки він є антагоністом біологічних ефектів ендотеліну і тромбоксану та інгібітором ФНП-α.
Механізм дії ілопросту, його висока клінічна ефективність і профіль безпеки, доведені в багатоцентрових клінічних дослідженнях з найвищим рівнем доказовості А, дозволяють рекомендувати його застосування в терапії судинних уражень, у тому числі легеневої гіпертензії, викликаних ревматичними хворобами. Його доза становить 20 мкг для повільного введення протягом 6 год, на курс — 5 ін’єкцій кожні 4 міс.
Існує інгаляційний ілопрост, який є препаратом вибору при легеневій гіпертензії (рис. 15.8).
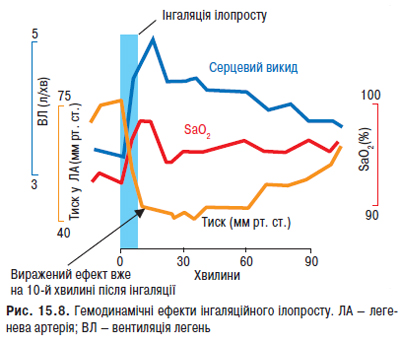
Вазодилататори доцільно поєднувати з антиагрегантами: пентоксифіліном (перорально 400–800 мг/добу, при необхідності в/в, або по 600–1200 мг/добу), дипіридамолом (150–200 мг/добу), парентеральним введенням декстрану (крапельно по 400 мл через день, 8–12 перфузій на курс), іншими ангіопротекторами. Доцільно проведення 2–3 курсів в/в введення на рік, в інтервалах — пероральний прийом антиагрегантів.
За наявності ознак гіперкоагуляції, мікротромбозів рекомендовано включення в терапевтичний комплекс антикоагулянтів: гепарину по 5000 ОД підшкірно 2–3 рази на добу або надропарину кальцію з подальшим переходом на варфарин або феніндіон. Можливе поєднання антикоагулянтної терапії з прийомом антиагрегантів (низькі дози ацетилсаліцилової кислоти).
Протизапальна терапія
Протизапальну терапію застосовують в ранній стадії ССД при швидкопрогресуючому перебігу й за високої активності патологічного процесу.
- НПЗП в стандартних терапевтичних дозах показані для лікування м’язово-суглобових проявів ССД, стійкої субфебрильної лихоманки (висока температура тіла для ССД нехарактерна).
- ГК показані при прогресуючому дифузному ураженні шкіри та клініко-лабораторних ознаках запальної активності (артрит, теносиновіт, міозит, серозит) в невисоких (15–20 мг/добу, або бетаметазон 0,5–2 мл залежно від патології ін’єкції в уражені м’які тканини 1 раз на тиждень) дозах. При фіброзуючому альвеоліті доза ГК може бути підвищена до 30–40 мг/добу, при поліміозиті (ССД-поліміозит) — до 40–50 мг/добу. Прийом високих доз може збільшити ризик розвитку склеродермічного ниркового кризу.
- Циклофосфамід застосовують при інтерстиціальному захворюванні легень (фіброзуючий альвеоліт), ранній ССД, швидкопрогресуючому перебігу, зазвичай в комбінації з ГК та пеніциламіном. Призначають в/в в дозі 800–1000 мг 1 раз на місяць або всередину 2 мг/кг/добу. Рекомендовано в/в введення, оскільки відзначають нижчу частоту побічних ефектів (у тому числі геморагічного циститу) порівняно з прийомом всередину. Пульс-терапію циклофосфамідом продовжують щонайменше протягом 6 міс (за відсутності побічних ефектів). При позитивній динаміці легеневих функціональних тестів рентгенологічних змін інтервал між сеансами пульс-терапії циклофосфамідом збільшують до 2 міс, а при збереженні позитивної динаміки — до 5 міс. Пульс терапію циклофосфамідом необхідно проводити щонайменше протягом 2 років.
- Метотрексат показаний при вираженому ураженні суглобів і м’язів, перехреcній формі ССД; здатний зменшувати поширеність і вираженість ущільнення шкіри, але не впливає на вісцеральну патологію.
- Можливе застосування азатіоприну, хлорамбуцилу. Є позитивний досвід застосування циклоспорину при ССД, проте його застосування утруднене у зв’язку з нефротоксичністю.
- В/в імуноглобулін (IVIg) використовується порівняно рідко при ССД, але може бути рекомендований у випадках активного, торпідного та/або асоційованого з інфекцією захворювання. Призначають за загальною схемою в дозі від 0,4 до 2 г/кг на добу в/в протягом 2–5 днів щомісяця до настання ефекту. Переноситься добре.
Антифіброзна терапія
Антифіброзна терапія показана вже в ранній стадії дифузної ССД.
Із групи антифіброзних засобів найбільшим ефектом володіє пеніциламін, має багатосторонню дію на метаболізм сполучної тканини і активно пригнічує надмірне фіброзоутворення. Це засіб вибору при швидкопрогресуючій склеродермії, дифузній індурації шкіри та фіброзі внутрішніх органів. Антифіброзна дія препарату реалізується при тривалому (не менше 6–12 міс) застосуванні в дозі 450–900 мг/добу з наступним зниженням і використанням підтримувальних доз (250–400 мг/добу) протягом 2–5 років. Клінічний ефект проявляється позитивною динамікою шкірного синдрому (зменшення індурації та ін.), суглобово-м’язових (збільшення обсягу рухів) і судинних порушень (зменшення проявів синдрому Рейно, поліпшення трофіки тканин). У частини хворих відзначено позитивну динаміку з боку серця, легень і травного тракту, уповільнення прогресування і навіть регресію (часткову) патологічного процесу. Підвищується виживаність хворих. У разі виникнення побічної дії препарату (дерматит, диспептичні порушення, нефропатія та ін., які мають місце у ⅓ хворих) необхідні жорсткий лікарський контроль, припинення лікування або зниження дози пеніциламіну при ускладненнях. Найбільш небезпечними та такими, що вимагають скасування препарату, є пригнічення кровотворення і нефротоксична дія.
В окремих дослідженнях показаний сприятливий ефект рекомбінантного гамма-інтерферону, переважно стосовно фіброзу шкіри. Однак відзначено небажані побічні дії препарату, включаючи розвиток ниркового кризу. Помірним антифіброзним ефектом володіє також колхіцин, але його застосування обмежене великою кількістю побічних явищ.
При ураженні травного тракту, крім основної терапії, хворі отримують додатково антисекреторні та прокінетичні препарати (омепразол, ранітидин, метоклопрамід та ін.). Омепразол — інгібітор протонної помпи, ефективний у лікуванні ерозивного рефлюкс-езофагіту. Прийом 1–2 капсул (20–40 мг) протягом 2 тиж дає чіткий ефект (зникнення болісної печії, загоєння ерозій). Ранітидин та інші блокатори Н2-рецепторів гістаміну (циметидин, фамотидин) також зменшують прояви рефлюкс-езофагіту, за необхідності можливе поєднання з омепразолом.
Ураження кишечнику з синдромом порушення всмоктування супроводжується, як правило, надмірним ростом мікрофлори, що зумовлює застосування антибактеріальних препаратів широкого спектра дії (тетрациклін, амоксицилін, ципрофлоксацин). При інтестинальній псевдообструкції для поліпшення перистальтики рекомендовано призначення довгостроково діючого соматостатину — октреотиду по 50 мг/добу підшкірно.
Екстракорпоральні методи лікування (плазмаферез) використовують за показаннями, головним чином при активній швидкопрогресуючій ССД, ураженні нирок, легень (у поєднанні з патогенетичною медикаментозною терапією).
У систему комплексного лікування та реабілітації хворих на ССД входять також лікувальна гімнастика, масаж, локальна терапія (мазі, аплікації тощо), фізіотерапія та санаторно-курортне лікування (бальнео- і грязелікування) — при неактивному процесі та хронічному перебігу ССД.
Особливості лікування вісцеральних ускладнень при ССД представлено в табл. 15.5.
Таблиця 15.5
Лікування вісцеральних ускладнень при ССД
|
Локалізація |
Вид ураження |
Вид лікування |
Препарати |
|
Легені |
Фіброзуючий альвеоліт |
Імуносупресія Програмний плазмаферез |
Циклофосфамід ГК |
|
Легенева гіпертензія |
Блокада рецепторів ендотеліну-1 Простагландин I2 Простагландин Е1 Інгібування фосфодіестерази-5 |
Бозентан Епопростенол Алпростадил Сильденафіл |
|
|
Серце |
Аритмія |
Антиаритмічні препарати |
Аміодарон Верапаміл |
|
Ексудативний перикардит |
Протизапальне |
ГК НПЗП |
|
|
Серцева недостатність |
Кардіопротекція |
Триметазидин Фосфокреатинін |
|
|
Нирки |
Склеродермічна нефропатія |
Інгібітори АПФ |
Каптоприл Еналаприл |
|
ШКТ |
Рефлюкс-езофагіт |
Інгібітори протонної помпи |
Омепразол Лансопразол |
|
Мальабсорбція |
Ротаційні антибіотики Аналог соматостатину |
Доксициклін Еритроміцин Октреотид |
Лікування склеродермічної нирки
Поточний протокол лікування
1. Інгібітори АПФ
Особливо при порушенні функції нирок
2. Ілопрост — тривала інфузія при контролі артеріального тиску
3. Поступове зниження артеріального тиску (щодня на 20 мм рт. ст.)
-
-
- Додаткові антигіпертензивні засоби
- Інфузія нітратів/штучна вентиляція легень
- Біопсія нирки при нормальних артеріальному тиску і показниках згортання крові
-
4. Гемодіаліз
5. Плазмаферез при вираженій тромботичній мікроангіопатії.
ССД і вагітність
Протипоказання до вагітності — дифузна форма ССД, виражені порушення функцій внутрішніх органів (серце, легені та нирки). При лімітованій формі та хронічному перебігу ССД вагітність не протипоказана. Однак під час вагітності може розвинутися органна патологія, у зв’язку з чим важливі спостереження та регулярне обстеження, спільне ведення пацієнток ревматологом і акушером-гінекологом. У випадках виявлення ССД під час вагітності необхідний ретельний моніторинг функцій нирок і серця.
Приблизні терміни непрацездатності
Хворі з гострою швидкопрогресуючою ССД непрацездатні та повинні бути переведені на інвалідність. При хронічному перебігу лімітованої ССД хворі обмежено працездатні та мають бути звільнені від важкої фізичної роботи, уникати охолодження, впливу хімічних агентів (необхідна медико-соціальна експертиза). Приблизні терміни тимчасової непрацездатності — 45–90 днів.
Диспансерне спостереження
Усі хворі на ССД підлягають динамічному спостереженню, що дозволяє проводити оцінку активності хвороби, своєчасно виявляти органну патологію та за показаннями коригувати терапію. Лікарський огляд, залежно від перебігу хвороби, наявності та вираженості вісцеральних уражень, проводять кожні 3–6 міс. Одночасно роблять загальні та біохімічні аналізи крові та сечі. При повторних візитах хворого лікар зобов’язаний провести активне опитування, щоб оцінити динаміку синдрому Рейно, проявів стравохідного рефлюксу, задишки, аритмії тощо.
При огляді слід звертати увагу на поширеність і вираженість ущільнення шкіри, базальної крепітації в легенях, на підвищення артеріального тиску, наявність дигітальних виразок та набряків. Необхідне дослідження функції зовнішнього дихання та проведення ехоКГ. У хворих, які приймають варфарин, слід контролювати протромбіновий індекс і МНВ, а при лікуванні циклофосфамідом — кількість лейкоцитів у периферичній крові та рівень трансаміназ.
Глава 16. Ідіопатичні запальні міопатії
Запальні міопатії — набуті захворювання, що характеризуються запаленням м’язів. У цю категорію входить велика кількість м’язових захворювань, у тому числі інфекційні міопатії (бактеріальні, вірусні, грибкові та ін.), токсичні міопатії, міозити, асоційовані з хворобою «трансплантат проти господаря», вогнищевий, гранулематозний, еозинофільний міозит, а також ІЗМ, не пов’язані з конкретним етіологічним фактором.
ІЗМ — група хронічних захворювань, основним проявом яких є м’язова слабкість, пов’язана із запаленням поперечносмугастих м’язів.
Код за МКХ-10
М33 Дерматополіміозит (ДМ).
М33.2 Поліміозит.
М33.6 Паранеопластичний міозит.
МЗЗ.9 Неуточнений дерматоміозит/поліміозит.
Ознаки міопатії можуть превалювати в клінічній картині ССД, СЧВ, РА, синдрому Шегрена, системних васкулітів.
Сучасна клінічна класифікація ІЗМ наведена в табл. 16.1.
Таблиця 16.1
Клінічна класифікація ІЗМ
|
Форма |
Поліміозит ідіопатичний ДМ ідіопатичний Поліміозит (ДМ), пов’язаний із пухлинами Поліміозит (ДМ), пов’язаний з васкулітами (дитячий) Overlap-синдром (перехресний) Антисинтетазний синдром Міозит із включеннями Інші форми запальних міопатій:
Міопатії, пов’язані з інфекцією Міопатії, пов’язані з дією лікарських препаратів |
|
|
Перебіг |
Гострий Підгострий Хронічний |
|
|
Ступінь активності |
0 (відсутня) I (мінімальна) II (помірна) III (висока) |
|
|
Клініко-морфологічна характеристика уражень |
М’язи |
Міозит, міопатія |
|
Шкіра |
Кальциноз, телеангіектазія Специфічні — еритема шиї («декольте»), синдром Готтрона, геліотропний параорбітальний набряк тощо |
|
|
Серце |
Міокардит, кардіоміопатія |
|
|
Легені |
Фіброзуючий альвеоліт, пульмоніт, пневмоніт |
|
|
Система травлення |
Езофагіт, порушення ковтання, дисфагії, псевдобульбарний синдром, гастрит тощо |
|
|
Суглоби |
Артралгії, поліартрит дрібних і великих суглобів |
|
|
Нервова система |
Поліневропатія |
|
|
Нирки |
Гломерулонефрит |
|
Епідеміологія
Частота ДМ в популяції становить від 2 до 10 випадків на 1 млн населення на рік. ДМ діагностують приблизно у 20% від усіх випадків запальних міопатій. Частота злоякісних новоутворень при ДМ в 12 разів вища, ніж у популяції. На тлі злоякісних новоутворень частіше виникає ДМ, ніж поліміозит. Співвідношення чоловіків і жінок становить 1:1.
Частота міозиту з «включеннями» серед хворих із запальними міопатіями становить від 15 до 28%. Він виникає у хворих похилого віку (середній вік — 60 років); частіше у чоловіків, ніж у жінок (співвідношення 2:1).
Етіологія
Незважаючи на те що причина ДМ невідома, існує ряд доказів впливу чинників зовнішнього середовища на розвиток імунних порушень у схильних до них осіб з подальшим виникненням аутоімунного міозиту. На роль генетичної схильності вказує можливість розвитку ДМ у монозиготних близнюків та кровних родичів хворих. Носійство деяких генетичних маркерів (НLА В8/DRЗ, НLА В14, НLА В40) тісно пов’язано з певними імунними порушеннями при ДМ (зокрема, з гіперпродукцією міозитспецифічних аутоантитіл, хоча їх значення залишається нез’ясованим).
«Виявлена» експресія антигенів НLА класу 1 в скелетних м’язах, залучених в патологічний процес при ДМ, може бути ключем до з’ясування механізму, за допомогою якого м’язові волокна стають чутливими до дії цитотоксичних Т-клітин. Однак це не пояснює, який чинник викликає і підтримує імунологічні порушення, а також питання про можливу участь інфекції. На користь ролі спадкової схильності свідчить підвищений вміст НLА В8 і Drwз, між якими є нестійка рівновага. Відомо, що з антигеном НLА В8 асоціюються патологічні стани, при яких існують імунні порушення. Цей факт поєднує ДМ з іншими аутоімунними хворобами.
Про можливу етіологічну роль інфекційних чинників непрямо свідчить початок захворювання, який частіше припадає на зиму та ранню весну (особливо у хворих з ювенільним ДМ), що за часом збігається з епідеміями інфекцій.
Вивчають роль вірусних інфекцій, зокрема міксовірусів, пікорнавірусів, вірусів Коксакі В, А9. На користь цієї гіпотези свідчать випадки ДМ, виявленого після перенесених грипу, краснухи, оперізуючого лишаю та інших вірусних інфекцій, а також виділення з ураженої м’язової тканини вірусу Коксакі А9.
Висловлювалося також припущення про зв’язок захворювання з туберкульозом, скарлатиною і токсоплазмозом. Однак переконливих даних на користь цього на сьогодні ще немає.
Більш очевидна асоціація ДМ зі злоякісними новоутвореннями, яку можна пояснити за допомогою «аутоімунної гіпотези», якщо припустити, що пухлина виділяє антигени, які перехресно реагують (ймовірно, як стрептокок при ревматизмі) та здатні порушувати толерантність до нормальних тканин організму.
В основі патогенезу ДМ найбільш визнаною є імунопатологічна теорія, виникнення якої зумовлено розвитком запальних процесів у результаті клітинних і гуморальних імунних реакцій.
При ДМ та поліміозиті розвивається запальна клітинна інфільтрація скелетних м’язів, а також гуморальні імунні реакції з підвищеним синтезом аутоантитіл до м’язової тканини.
При імуногістологічному дослідженні уражених м’язів виявляють мононуклеарну інфільтрацію Т- і В-лімфоцитами і макрофагами.
Однак між поліміозитом і ДМ існують принципові імунопатологічні відмінності. При ДМ найістотніше значення мають гуморальні імунні порушення, пов’язані з відкладенням у дрібних судинах імунних комплексів, з активацією комплементу і розвитком васкулопатії, що супроводжується запальною інфільтрацією скелетних м’язів. У складі клітинного інфільтрату переважають CD4-лімфоцити, макрофаги і В-лімфоцити. При поліміозиті в інфільтраті домінують CD8, Т-лімфоцити, при цьому відсутні ознаки васкулопатії або імунокомплексного ураження судин.
У хворих на ДМ більш поліклональний характер Т-клітинних рецепторів. Водночас у пацієнтів з поліміозитом і міозитом із «включеннями» відзначають більш олігоклональний характер Т-клітинних рецепторів.
Крім того, у периферичній крові хворих на ДМ нерідко містяться РФ, антинуклеарні антитіла, LЕ-клітини, антитіла до міоглобіну, а також низка антитіл до екстрагуючих компонентів ядра (Jo-1, РМ-1-антигенів). Усе це свідчить про участь у запальній міопатії як клітинних, так і гуморальних факторів імунітету.
Патоморфологія
Біопсія клінічно зміненого м’яза є цінним методом для підтвердження діагнозу ДМ. Проте дані морфологічного дослідження не завжди корелюють із клінічною картиною захворювання. До характерних змін належать дегенерація м’язових волокон із ознаками фагоцитозу (або без них) і регенерація. Часто відзначають ознаки круглоклітинної інфільтрації. Характерні також набухання м’язових волокон із втратою поперечної смугастості та їх некрозом. При тривалому перебігу захворювання можуть проявлятися фіброз і кальциноз, а також атрофія м’язових волокон. При морфологічному дослідженні уражених ділянок шкіри відзначають лише неспецифічні запальні зміни. При аутопсії у дітей, хворих на ДМ, часто виявляють ознаки поширеного васкуліту з периваскулярними запальними клітинними інфільтратами, гіперплазією інтими і тромбозом.
Первинний поліміозит розвивається переважно у жінок у віці 30–50 років і спостерігається у 34% хворих. Початок, як правило, поступовий, перебіг хронічний прогресуючий. Для цієї форми поліміозиту характерні часті нетипові шкірні висипи, синдром Рейно, артралгії, різні системні прояви.
Первинний ДМ виявляють у 29% хворих, виникає на 2–7-му десятилітті життя, його перебіг відзначається характерними змінами шкірних покривів. Початок захворювання гострий чи підгострий. Жінки хворіють частіше за чоловіків.
Частота злоякісних новоутворень при ІЗМ у 12 разів вища, ніж у популяції. На тлі злоякісних новоутворень частіше розвивається ДМ, ніж поліміозит. Співвідношення чоловіків і жінок — 1:1.
ДМ або поліміозит зі злоякісними новоутвореннями переважає у чоловіків (3:1) у віці старше 40 років і зустрічається приблизно у 10–20% хворих. Перебіг цієї форми ДМ може бути гострим і підгострим з ураженням шкіри, м’язів, з поліартралгією або поліартритом та іншими проявами захворювання. У більшості випадків пухлини локалізуються в легені, яєчнику, передміхуровій залозі, молочній залозі та кишечнику. У деяких випадках симптоми онкологічних захворювань з’являються одночасно з виникненням поліміозиту і навіть раніше. Однак частіше їх не вдається виявити протягом перших 3 років після початку захворювання. ДМ, асоційований із пухлинами, характеризується особливо тяжким перебігом. Патологічні зміни прогресують, незважаючи на проведену терапію, і хворі помирають зазвичай внаслідок порушення функції дихальних і глоткових м’язів, а не від прямого впливу самої пухлини. Необхідно пам’ятати про те, що всі хворі (особливо старше 40 років) на ДМ (поліміозит) повинні бути обстежені з метою виключення первинного пухлинного захворювання.
ДМ дитячого віку (ДМ з васкулітами) діагностують приблизно у 7% хворих. Перебіг захворювання гострий інтермітуючий або частіше хронічно прогресуючий. Як правило, у хворих цієї групи розвиваються тяжкі контрактури, виразки шкіри, артралгії, при тривалому перебігу — кальциноз у м’язах, шкірі та підшкірній клітковині. Ураження судин за типом васкулітів (від інфільтрації до проліферації інтими і некротичних змін) зумовлює досить часте залучення в процес внутрішніх органів, зокрема ШКТ, що проявляється болем у животі, шлунково-кишковою кровотечею або навіть перфорацією.
Поліміозит (частіше, ніж ДМ) асоціюється з симптомами оverlap-синдрому приблизно в 21% хворих. Характерно те, що в такому поєднанні перебіг хвороби менш тяжкий, а ГК ефективні в нижчих дозах. За наявності м’язової слабкості у всіх хворих біль в м’язах відзначають лише у ½ випадків, рідше залучаються в патологічний процес м’язи глотки і гортані. Кальциноз розвивається часто, але більш обмежений. Характерними є ознаки СЧВ, ССД, синдрому Шегрена, а також виявлення LЕ-клітин, високих титрів РФ, антитіл до нативної ДНК, нуклеопротеїдів.
Частота міозиту з «включеннями» в популяції хворих із запальними міопатіями коливається від 15 до 28%. Захворювання розвивається у хворих похилого віку (середній вік — 61 рік), частіше у чоловіків, ніж жінок (співвідношення 2:1).
Крім клінічних форм ДМ, виділяються варіанти його перебігу (гострий, підгострий і хронічний), що допомагає у виборі тактики лікування і визначенні прогнозу.
Приблизно у 30% хворих ДМ починається гостро, для нього характерні загальні симптоми — підвищення температури до 38–39 °С, тахікардія, підвищене потовиділення, головний біль і нездужання, а також еритема, слабкість і біль у м’язах з подальшим прогресуючим наростанням симптоматики ДМ.
При гострому перебігу хвороби через 3–6 тиж від початку відзначають катастрофічно наростаюче генералізоване ураження поперечносмугастих м’язів, аж до повного знерухомлення, розвиток дисфагії та дизартрії. Констатують загальний тяжкий лихоманково-токсичний стан із різноманітними шкірними висипами. Причиною смертельного результату зазвичай є аспіраційні пневмонії або легенево-серцева недостатність, зумовлена ураженням легень і серця. При лікуванні хворих великими дозами ГК прогноз хвороби кращий.
Підгострий перебіг процесу вирізняється циклічністю, але все ж неухильно наростає адинамія, відзначають ураження шкіри і внутрішніх органів (протягом 1–2 років). До терапії ГК хвороба також закінчувалася смертю, проте на сьогодні можливе одужання хворих із розвитком виражених аміотрофій, контрактур, кальцинозу, які значно погіршують їх здатність пересуватися.
Хронічний перебіг — найбільш сприятливий, уражаються лише окремі групи м’язів. Тому, незважаючи на значну кількість загострень, загальний стан хворих залишається задовільним, вони довго зберігають працездатність. Виняток становлять молоді люди, в яких розвиваються великі кальцинози в шкірі, підшкірній клітковині, м’язах з виникненням стійких контрактур та майже повної нерухомості.
Лабораторні показники і вимірювання температури тіла допомагають визначити вираженість активності процесу. ДМ властивий хвилеподібний перебіг з періодами загострень і ремісій різної тривалості. Чітко сформульована стадійність протягом ДМ. Перший період — це початковий від декількох днів до місяця і більше, проявляється нерідко лише м’язовими або шкірними ознаками, гіперемією або набряком верхніх повік, іноді в поєднанні з нездужанням, підвищенням температури тіла або іншими невизначеними симптомами. Цей період може бути відсутній. Другий період — маніфестний, з вираженими основними синдромами — шкірним, м’язовим, загальним. Третій період — пізній, дистрофічний, кахексичний, термінальний, період ускладнень. Ускладнення можуть бути бактеріальні, грибкові, стероїдні, з можливим розвитком виразок, іноді з катастрофічним закінченням хвороби внаслідок кровотеч, перитоніту та ін.
Клінічна картина
Клінічні варіанти дебюту (табл. 16.2):
- у більшості хворих: нездужання, загальна слабкість, ураження шкіри з наступним поступово прогресуючим (протягом кількох тижнів) наростанням слабкості в проксимальних групах м’язів;
- у дітей та осіб молодого віку: гострий початок, часто поєднується з вираженими конституціональними проявами (лихоманка, схуднення, тощо) і міалгією;
- у літніх хворих з міозитом з «включеннями»: повільне (протягом декількох років) наростання м’язової слабкості;
- у хворих з аміопатичним ДМ: протягом тривалого часу типове для ДМ ураження шкіри за відсутності м’язової слабкості;
- у хворих з «антисинтетазним» синдромом: феномен Рейно, поліартралгії або поліартрит і задишка, зумовлена інтерстиціальним легеневим фіброзом.
Таблиця 16.2
Симптоматика ДМ (за: Pearson C.M.)
|
Симптоми |
Частота, % |
|
Слабкість м’язів |
|
|
100 |
|
80 |
|
63 |
|
62 |
|
10 |
|
Болі в м’язах |
49 |
|
Контрактури |
32 |
|
Атрофія м’язів |
53 |
|
Зміни шкіри |
|
|
40 |
|
22 |
|
21 |
|
Зміни суглобів |
32 |
Ураження м’язів — провідний клінічний симптом захворювання. Слабкість поперечносмугастих м’язів є постійним і провідним симптомом ДМ. За його відсутності діагноз ІЗМ сумнівний. М’язові ураження частіше бувають симетричними. Характерні припухлість, тістоподібна консистенція і болісність при пальпації уражених м’язів, а також нерідко набряклість шкіри над ними. М’язи тазового пояса і стегон уражаються раніше, ніж плечового. У результаті слабкості в м’язах у хворих виникають труднощі при підйомі по сходах, причісуванні, стає важко встати з ванни. Іноді виникає спонтанний біль в уражених м’язах. У міру прогресування слабкості — хода хворого нагадує качину, йому важко підняти голову або повернутися у ліжку. При ураженні м’язів глотки і гортані можуть виникати ослаблення голосу і гугнявість, а також дисфагії з поперхуванням і потраплянням їжі в носоглотку аж до картини бульбарного паралічу, явищ аспірації.
При тяжкому перебігу захворювання можливе залучення в патологічний процес дихальних м’язів (у тому числі діафрагми). Одночасне ураження глоткових і дихальних м’язів може призвести до загибелі хворого внаслідок аспірації слини та їжі й дихальної недостатності.
Найчастіше стан хворого стабілізується через кілька тижнів або місяців. Відбувається поступове відновлення сили уражених м’язів. Репаративні процеси супроводжуються заміщенням частини м’язових волокон фіброзної тканиною, яке може прогресувати, супроводжуючись кальцинозом, а також атрофією уражених м’язів, формуванням контрактур і вкороченням м’язів. При рентгенологічному дослідженні виявляють кальциноз цих м’язів.
Перебіг ІЗМ надзвичайно варіабельний — від тяжких форм, які закінчуються протягом декількох місяців летально, до легких, прояви яких протягом багатьох років хворі не вважають ознаками захворювання. Так, в дуже легких випадках захворювання слабкість м’язів може проявлятися лише нездатністю хворого утримувати відведену підняту випрямлену ногу або руку при наявності опори або сісти з положення лежачи з руками, схрещеними на грудях. Через залучення до процесу мімічних м’язів з’являється характерний плоский вираз обличчя, хворий не може заплющити очі, надути щоки; може розвинутися диплопія, птоз.
Ураження дистальних м’язів відзначають рідко, головним чином при міозиті з «включеннями», які виражені меншою мірою, ніж ураження проксимальної.
У половини хворих міалгії або хворобливість м’язів виявляють при пальпації, а також відзначають набряк м’язів.
М’язові атрофії розвиваються тільки в осіб, які тривалий час хворіють на ІЗМ, особливо за відсутності адекватної терапії.
Ураження шкіри різноманітні, їх локалізація, час появи не завжди пов’язані з міозитом. Характерна ознака ДМ — це еритематозний (геліотропний) висип, що локалізується на верхніх повіках, вилицях, крилах носа, в ділянці носогубної складки, зоні «декольте» і на верхній частині спини, над ліктьовими і колінними суглобами, п’ястково-фаланговими і проксимальними міжфаланговими суглобами, на волосистій частині голови (рис. 16.1, 16.2).
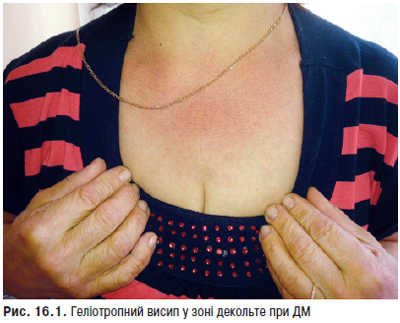
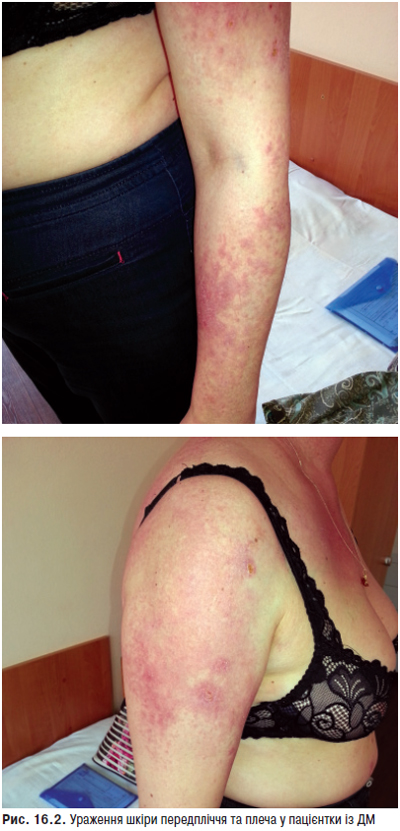
Другою характерною ознакою ДМ є ознака Готтрона — це злегка припідняті або плоскі еритематозні полущені висипи, які локалізуються над п’ястково-фаланговими і проксимальними міжфаланговими суглобами або над розгинальними поверхнями ліктьових і колінних суглобів (рис. 16.3).
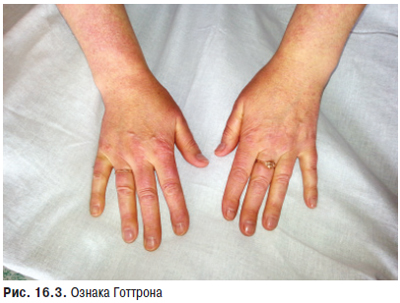
Крім того, характерною ознакою є почервоніння, лущення і тріщини на шкірі долонь («рука механіка»).
До інших проявів ураження шкіри належать гіпертрофія кутикули нігтя, навколонігтьова еритема, інфаркти навколонігтьового ложа, петехії, сітчасте ліведо, папульозні, бульозні висипи, телеангіектазії, гіперкератоз, гіпер- і депігментація, фотодерматит, свербіж шкіри, панікуліт (рідко).
У деяких пацієнтів ураження шкіри передує розвитку м’язової слабкості за кілька місяців або років (так званий аміопатичний міозит).
Кальциноз. Кальцинати локалізовані підшкірно або в сполучній тканині, навколо м’язових волокон, у зонах мікротравматизації над ліктьовими суглобами, на згинальних поверхнях пальців і сідницях. Кальциноз розвивається в пізніх стадіях ІЗМ, частіше при ювенільному ДМ.
Ураження суглобів діагностують приблизно у ⅓ випадків у вигляді артралгій. Двобічне симетричне ураження — частіше дрібних суглобів кистей і променезап’ясткових суглобів, рідше — ліктьових і колінних: іноді передує м’язовій слабкості, нагадує ураження при РА, зазвичай має тимчасовий характер, швидко купірується при призначенні ГК. Описано розвиток хронічного артриту з підвивихами суглобів кистей, але без ерозивних змін, за рентгенологічними даними (артропатія Жакку). Однак артрити для ІЗМ нетипові. Деструкції відзначають вкрай рідко.
Ураження органів дихання при ІЗМ виявляють у 80% випадків найчастіше у вигляді 3 типів:
- залучення в процес дихальних м’язів;
- розвиток вторинних бактеріальних пневмоній;
- розвиток фіброзуючого альвеоліту.
Ураження міжреберних м’язів і діафрагми виникає у переважної більшості пацієнтів, що призводить до експіраторної задишки і розвитку дихальної недостатності за рестриктивним типом (40% випадків). Відбувається зниження кашльового кліренсу легень, легеневого кровотоку, що створює «сприятливі» умови для розвитку гіповентиляційної пневмонії та легеневої гіпертензії. Крім того, порушення ковтання, пов’язане з ураженням глоткових м’язів, може призводити до аспірації їжі та слини з подальшим розвитком аспіраційної пневмонії.
Пневмонія — найбільш часта форма ураження легенів (при ІЗМ виникає в 30–55% випадків). Головну роль в її розвитку відіграє аспірація їжі, пов’язана з ураженням м’язів глотки і верхньої третини стравоходу. Частота аспіраційного синдрому у хворих на ІЗМ досягає 30%. З іншого боку, важливе етіологічне значення в розвитку пневмонії мають інфекційні збудники, які виявляють у 30% хворих на ІЗМ.
Ризик розвитку пневмоній і труднощі їх лікування зростають при тривалому прийомі високих доз ГК (близько 1 мг/кг/добу). Водночас створює додатковий негативний вплив на стан хворого гіповентиляційний синдром, що призводить до додаткового ризику розвитку пневмонії.
Фіброзуючий альвеоліт — найбільш тяжкий варіант ураження легень при ІЗМ, його частота, за зарубіжними даними, становить 10–60%. Для фіброзуючого альвеоліту при ІЗМ характерний широкий спектр клінічних проявів, який має схожість з ідіопатичним фіброзуючим альвеолітом, а також виявлення антисинтетазних антитіл (анти-Jo-1).
Форми фіброзуючого альвеоліту: швидкопрогресуючий фіброзуючий альвеоліт, більш повільний і субклінічний.
Швидкопрогресуючий фіброзуючий альвеоліт (синдром Хаммена — Річа). Для нього характерні гостро виникаючий непродуктивний кашель, задишка в спокої, лихоманка. При цьому симптоми ураження мускулатури можуть відійти на другий план. На рентгенограмах виявляють множинні дрібновогнищеві затемнення й сітчасту деформацію легеневого малюнка. Прогноз несприятливий, такий варіант перебігу дуже швидко може закінчуватися фатально.
Більш повільний розвиток альвеоліту. Захворювання дебютує задишкою при фізичному навантаженні, у ряді випадків — непродуктивним кашлем. Ці симптоми можуть передувати м’язовому ураженню або виникати на фоні наявного тяжкого міозиту. У хворих обох груп аускультативно можна виявляти крепітацію у нижніх відділах легень. У рідкісних випадках відзначають розвиток легеневого серця. Симптом «барабанних паличок» виникає рідко.
Субклінічне ураження. Яскрава клінічна легенева симптоматика відсутня. Інтерстиціальні зміни в легеневій тканині виявляють за допомогою спеціальних методів дослідження (рентгенографія, КТ). Синдром фіброзуючого альвеоліту може випереджати розвиток м’язового синдрому в 10% випадків або розвиватися одночасно з ним. Однак найбільш часто клінічні ознаки фіброзуючого альвеоліту виникають на тлі розгорнутої клінічної картини ІЗМ, як правило, на ранніх етапах захворювання.
Ураження серця при ІЗМ у більшості випадків протікає безсимптомно. Іноді при спеціальному обстеженні виявляється порушення ритму і провідності (тахікардія, аритмія); застійна серцева недостатність розвивається рідко. Однак, за даними різних авторів, у 20–50% хворих можливе ураження всіх трьох оболонок серця, але найбільш часто в процес залучається міокард (з розвитком міокардиту або дистрофічних змін). Особливо виразно ураження серця проявляється в період загострення захворювання.
Найчастіше картина міокардиту малосимптомна — у вигляді тахікардії, лабільного пульсу, короткочасного болю в ділянці серця, властивого для вогнищевого ураження міокарда. Для дифузного міокардиту характерними є розширення меж серця, переважно вліво, послаблення 1 тону, порушення ритму, тахікардія, ритм галопу, поява систолічного шуму на верхівці, гіпотонія. На ЕКГ відзначають неспецифічні зміни, які відповідають тяжкості ураження міокарда. Частіше за все — зміна зубця Т, який може бути згладженим, двофазним або негативним, депресія або елевація сегмента SТ, різні порушення ритму і провідності, зниження вольтажу. Іноді спостерігається гостра ішемія міокарда з інфарктоподібною ЕКГ.
Ураження ендокарда при ІЗМ — досить рідкісне явище. Процес в основному локалізується на клапанному апараті і, як правило, має «німий» перебіг або ж маскується ознаками ураження міокарда. У діагностиці ендокардиту важливе місце займає ехокардіографія і фотокардіографія.
Перикардит при ІЗМ розвивається рідко, діагностика його зазвичай утруднена, оскільки шум тертя перикарда (важлива діагностична ознака) звичайно короткочасний. Для перикардиту характерні постійні болі за грудиною, задишка, тахікардія, а також зміни на ЕКГ, властиві перикардиту.
При ІЗМ можливий також розвиток хронічного легеневого серця в результаті фіброзуючого альвеоліту та гіпертензії малого кола.
Ураження коронарних судин нетипове для ДМ, як і підвищення артеріального тиску, яке в деяких випадках може бути зумовлено дифузним ураженням нирок.
Можливе ураження серця (більш ніж в 22% випадків), викликане супутнім атеросклерозом чи гіпертонічною хворобою.
Ураження судин при ДМ рідкісні, проявляються продуктивним васкулітом дрібних артерій, артеріол і дрібних вен, іноді розвиваються запустіння капілярної сітки і поширені склеротичні зміни дрібних артерій і артеріол в шкірі, скелетних м’язах, ШКТ. Можливі також зміни в артеріолах пальців за типом синдрому Рейно, однак вони нетипові для ІЗМ і можуть бути використані в якості диференційного тесту зі склеродермією.
Ураження ШКТ відзначають майже у половини хворих, що зумовлено м’язової патологією, рідше васкулітом дрібних судин. У патологічний процес можуть залучатися всі відділи травного тракту. Одним з найбільш частих і важливих симптомів ураження ШКТ є порушення ковтання. Дисфагія зазвичай розвивається поступово і має прогресуючий характер. Повна неможливість проковтнути їжу, воду, слину створює особливо гнітюче становище для хворого, може сприяти розвитку аліментарної кахексії та прирікає людину на голодну смерть, це викликає необхідність годування через зонд.
Водночас у таких хворих можуть виникати порушення жування і ковтання внаслідок ураження жувальних м’язів і м’язів органів мовлення, стравоходу, слизової оболонки порожнини рота та глотки, нерідко з набряком, інфільтрацією та їх виразкуванням, а також підвищена салівація. Виразки також можуть утворюватися в шлунку, кишечнику, що може стати причиною кровотечі. У літературі описано випадки перфорації шлунка і кишечнику, кишкової непрохідності, які можуть бути також і наслідком ГК-терапії. Рентгенологічно виявляють відсутність перистальтики стравоходу (верхня частина), уповільнений пасаж барієвої суспензії по стравоходу, гіпотонію шлунка і кишечнику.
Клінічні симптоми ураження печінки, за винятком деякого її збільшення (⅓ випадків), як правило, відсутні. Описано лише поодинокі випадки хронічного гепатиту у дорослих і дітей.
Збільшення селезінки у дорослих зустрічається порівняно нечасто (близько 10%) і зазвичай у поєднанні з лімфаденопатією, що відображає активність процесу за участю ретикулоендотеліальної системи.
Ураження нирок при ІЗМ розвивається порівняно рідко. Про характер ураження нирок можна зробити наступний висновок: серйозні ураження нирок діагностують у невеликої кількості хворих на ІЗМ, вони досить різноманітні, тому не можна виділити типової для цього захворювання ниркової патології, хоча швидше можна говорити про дифузний гломерулонефрит. У ряді випадків нерідкі дистрофічні ураження та інші прояви малої патології нирок, які зазвичай відображають загальну тяжкість, активність, гостроту процесу ІЗМ і самі не впливають на результат захворювання. Однак ураження нирок частіше може виникати при перехресних синдромах, в основному з ССД. Можлива протеїнурія, зумовлена мембранозною нефропатією. Відзначено поодинокі випадки гострої ниркової недостатності внаслідок масивної міоглобінурії.
Ураження нервової системи найчастіше проявляється астеновегетативним синдромом — порушенням сну, підвищеною дратівливістю, плаксивістю, головним болем, запамороченням. У поодиноких випадках можливе ураження периферичної нервової системи за типом поліневриту з порушенням чутливості (за типом «шкарпеток» і «рукавичок»), а також центральної — енцефаліти, менінгоенцефаліти, вогнищеві ураження мозку з гіперкінезами, епілепсією, парезами, зумовлені, як правило, судинними процесами. Нерідко виявляють ураження черепно-мозкових нервів. При ІЗМ відзначають зниження або відсутність сухожильних рефлексів, що найбільш ймовірно спричинено важким ураженням м’язів і сухожиль, проте диференціювати його з ураженням нервової системи досить важко, оскільки набряклі м’язи можуть викликати компресійний неврит.
Зміни з боку ендокринної системи трапляються рідко і проявляються головним чином порушенням функції статевих залоз (аменорея, дисменорея, імпотенція), надниркової залози, щитоподібної залози переважно в період високої активності захворювання.
Із загальних симптомів ІЗМ найбільш характерними є тривала субфебрильна температура, прогресуюча втрата маси тіла, трофічні зміни шкіри (сухість, тріщини, ламкість нігтів, випадіння волосся), при тривалому перебігу карієс зубів, ураження кісткової системи (ОП).
При ДМ можливий розвиток синдрому Шегрена.
«Антисинтетазний» синдром — один з найбільш тяжких варіантів ІЗМ. Він пов’язаний з утворенням антитіл до аміноацил-тРНК-синтетази і має такий симптомокомплекс, що характеризується наступними ознаками:
- важким ураженням легень за типом фіброзуючого альвеоліту;
- гострим початком;
- лихоманкою;
- симетричним неерозивним артритом або артралгією;
- феноменом Рейно;
- ураженням шкіри долонь за типом «руки механіка»;
- дисфагією;
- кальцинозом м’яких тканин;
- склеродактилією;
- антитілами Jo-1, рідше іншими антисинтетазними антитілами.
Міозит із «включеннями» (inclusion body myositis) характеризується такими особливостями:
- дуже повільним розвитком слабкості та атрофії не тільки в проксимальних, а й дистальних групах м’язів;
- асиметричними ураженнями;
- нормальною активністю креатинфосфокінази (КФК) або помірним підвищенням її активності;
- рідкісним поєднанням зі ЗЗСТ і злоякісними новоутвореннями;
- резистентністю до ГК та інших методів фармакотерапії ідіопатичного запального міозиту.
Діагностика
Діагностика ІЗМ заснована на даних анамнезу, результатах клінічного обстеження, морфологічного дослідження біоптатів скелетних м’язів і електроміографії (ЕМГ).
Анамнез
Основні принципи збору анамнезу у хворого з підозрою на ІЗМ включають з’ясування обставин захворювання та відповіді на запитання:
Що обмежує обсяг рухів: біль в м’язах чи слабкість певних груп м’язів? Яких (проксимальних або дистальних)?
Які фактори підсилюють, а які зменшують м’язову слабкість?
Чи приносить відпочинок полегшення? Чи відбувається збільшення м’язової сили на тлі відпочинку?
За який проміжок часу відбулося посилення м’язової слабкості та як швидко вона прогресує?
Обставини захворювання
Уточнюють розвиток інших (не м’язових) симптомів (стомлюваність, висипи, задишка, порушення ковтання, артралгії).
Визначають зв’язок розвитку захворювання з впливом міотоксичних речовин (прийом препаратів), попередньою інсоляцією, інфекцією або подорожжю.
Визначають симптоми, які свідчать про ураження щитоподібної залози.
Оцінюють залучення інших м’язів (серце, м’язи глотки, ШКТ, очні й дихальні м’язи).
Якщо хворий вже отримує ГК, з’ясовують, скільки часу пройшло від початку їх прийому до збільшення м’язової сили (для ІЗМ нехарактерний швидкий клінічний ефект).
При зборі анамнезу дуже важливо відрізняти м’язову слабкість від загальної стомлюваності або міалгій. Сімейний анамнез свідчить на користь спадкової міодистрофії.
З діагностичною метою виконують загальний і біохімічний аналізи крові, імунологічні дослідження.
Лабораторні дослідження
Загальний аналіз крові
Зміни неспецифічні: збільшення ШОЕ відзначають рідко (переважно при розвитку системних проявів).
Біохімічне дослідження
- Можливе збільшення концентрації так званих м’язових ферментів — загальної КФК, МВ-фракції КФК, альдолази, а також АлАТ, АсАТ, ЛДГ, у більшості хворих підвищення рівня міоглобіну.
- Активність ферментів слід визначати до проведення голкової ЕМГ (неспецифічне підвищення концентрації ферментів).
Характерне збільшення концентрації хоча б одного ферменту в різні терміни хвороби практично у всіх пацієнтів. КФК — найбільш чутливий і специфічний маркер м’язового запалення, рівень КФК певною мірою корелює з вираженістю м’язової слабкості. Збільшення МВ-фракції КФК свідчить про ураження м’язів, а не міокарда. Збільшення вмісту тропоніну-1 — більш специфічний маркер ураження міокарда при ІЗМ, ніж МВ-КФК.
Функція щитоподібної залози
Визначення Т3, Т4 і тиреотропного гормону рекомендується всім пацієнтам з м’язовою слабкістю за відсутності характерної шкірної висипки для виключення ендокринної патології.
Імунологічні дослідження
АНФ проявляється у 50–60% випадків, але не має значення для встановлення діагнозу. За наявності дуже високих титрів вірогідний «перехресний» синдром з іншими ЗЗСТ.
Визначення антитіл до аміноацил-тРНК-синтетази (антисинтетазне антитіло), в першу чергу до гістидинсинтетази (Jo-1), який є діагностичним критерієм антисинтетазного синдрому. Проводиться також визначення простатспецифічного антигену з метою виключення раку передміхурової залози і карциноембріонального антигену (СА-125, СА-15.3) для виключення раку яєчника і молочної залози.
Інструментальні дослідження
ЕМГ — чутливий, але не специфічний метод діагностики, корисний для моніторингу ефективності лікування, особливо при сумнівних результатах лабораторних і клінічних досліджень. Більш ніж у 90% хворих на ІЗМ при дослідженні проксимальних і параспінальних м’язів виявляють ознаки патологічної спонтанної активності м’язових волокон (потенціали фібриляції, складні повторювані розряди та ін.) при подразненні та в спокої, короткі малоамплітудні поліфазні потенціали при скороченні. Нормальна електрична активність при ЕМГ у більшості випадків дозволяє виключити діагноз ІЗМ, проте дані ЕМГ погано корелюють з клінічними проявами м’язової слабкості. При міозиті з «включеннями» проявляються змішані міопатичні і невропатичні зміни.
Рентгенологічна денситометрія: ГК-ОП: визначення вихідної мінеральної щільності тканини, потім 1 раз на рік.
Мамографія: виключення раку молочної залози.
Капіляроскопія судин навколонігтьового ложа: дилатація капілярних петель (частіше при перехресному синдромі, рідше при ДМ).
Рентгенологічне дослідження легенів показано всім хворим для оцінки ураження легень. До більш чутливих методів відносять рентгенологічну КТ високого розрізнення.
МРТ.В останні роки значного поширення набула МРТ м’язової тканини, яка дозволяє провести ранню діагностику захворювання завдяки виявленню набряку м’язової тканини. Це особливо актуально у випадку ДМ, коли ураження шкірних покривів передує появі клінічних ознак міозиту, а також при аміопатичному варіанті ДМ. Набряк м’язової тканини — індикатор активності хвороби.
МРТ має більшу чутливість у діагностиці м’язового запалення порівняно з УЗД і КТ. Крім того, МРТ дозволяє вибрати більш точне місце для виконання діагностичної біопсії, а також виявити ознаки м’язового запалення у хворих з нормальною активністю КФК з нормальними результатами голкової ЕМГ і м’язової біопсії. МРТ допомагає в диференційній діагностиці між запальним набряком м’язів і м’язовою атрофією з жировим заміщенням, її можна використовувати для динамічного контролю проведеного лікування значно частіше, ніж виконувати біопсію і/або проводити голкову ЕМГ.
Зменшення або відсутність набряку при повторних МРТ — ознака позитивної відповіді на лікування. Дані МРТ-дослідження м’язової тканини включені в алгоритм оцінки індексу пошкодження, запропонований Міжнародною групою з клінічного вивчення й оцінки активності міозиту.
Холтерівське моніторування ЕКГ. Дослідження показано всім хворим для раннього виявлення прогностично несприятливих порушень ритму і провідності.
М’язова біопсія — золотий стандарт діагностики ІЗМ. Її необхідно проводити навіть за наявності характерних клінічних, лабораторних та інструментальних ознак захворювання. При поліміозиті виявляють інфільтрацію мононуклеарними клітинами (в основному лімфоцитами), що локалізуються в ендомізію, некроз і регенерацію м’язових волокон. При ДМ мононуклеарну інфільтрацію визначають в основному навколо фасцій і кровоносних судин, виявляють ознаки васкулопатії (тромбоз капілярів, набряк, гіперплазія, вакуолізація і дегенерація клітин ендотелію). У пізніх стадіях ІЗМ виявляють атрофію м’язових фібрил, їх заміщення жировою тканиною, фіброз. При міозиті з «включеннями» поряд з ознаками міозиту виявляють внутрішньоклітинні лінійні вакуолі (при світловій мікроскопії), цитоплазматичні або внутрішньоядерні тубулярні включення з періодичною поперечною і поздовжньою смугастістю (при електронній мікроскопії).
Біопсія легень. При гістологічному дослідженні біоптатів легень пацієнтів із фіброзуючим альвеолітом при ІЗМ ділянки ураження нерівномірно розподілені між ділянками здорової тканини. Виявляють гіперплазію пневмоцитів 2-го типу, потовщення альвеолярних стінок і перегородок, розростання перибронхіальної і периваскулярної сполучної тканини, гіперпластичний склероз малих артерій. Запальний інфільтрат складається переважно з лімфоцитів, плазматичних клітин, у той час як еозинофіли і лейкоцити трапляються рідко.
Діагностичні критерії
Для діагностики ІЗМ слід використовувати такі діагностичні критерії:
1. Ураження шкіри:
а) геліотропний висип (пурпурно-червоні еритематозні висипи на повіках);
б) ознака Готтрона (пурпурно-червона, лущиться, атрофічний еритема або плями на розгинальній поверхні кистей над суглобами);
в) еритема на розгинальній поверхні кінцівок над ліктьовими і колінними суглобами.
2. Проксимальна м’язова слабкість (верхні та нижні кінцівки і тулуб).
3. Підвищення рівня КФК та/або альдолази в сироватці крові.
4. Біль в м’язах при пальпації або міалгії.
5. Міогенні зміни при ЕМГ (короткі, поліфазні потенціали моторних одиниць зі спонтанними потенціалами фібриляції).
6. Виявлення антитіл Jo-1 (антитіла до гістидил-тРНК-синтетази).
7. Недеструктивний артрит або артралгії.
8. Ознаки системного запалення (температура тіла вище 37 °С, підвищення концентрації СРБ або збільшення ШОЕ >20 мм/год).
9. Морфологічні зміни, які відповідають запальному міозиту (запальні інфільтрати в скелетних м’язах з дегенерацією або некрозом м’язових волокон; активний фагоцитоз або ознаки активної регенерації).
Діагноз ДМ (чутливість — 94,1%, специфічність — 90,3%) встановлюють за наявності принаймні одного типу ураження шкіри і не менше чотирьох інших ознак (пункти 2–9). Діагноз поліміозит (чутливість — 98,9%, специфічність — 95,2%) встановлюють, якщо наявні не менше чотирьох ознак (пункти 2–9).
Диференційний діагноз
ДМ. У ранній стадії захворювання в клінічній практиці як при аміопатичній формі ДМ, так і при класичному ДМ переважають ураження шкіри і загальна слабкість, а ознаки міопатії (проксимальна м’язова слабкість, підвищення КФК і характерні зміни при морфологічному дослідженні м’язових біоптатів) можуть бути мінімальні або взагалі відсутні. У багатьох хворих (особливо літнього віку) необхідно проводити додаткове обстеження для виключення пухлинної природи ДМ.
Слід проводити диференційну діагностику поліміозиту та:
- спадкових м’язових захворювань (м’язова дистрофія Дюшена, Беккера та ін.) — слід враховувати сімейний анамнез;
- вроджених м’язових захворювань (немалінова, мітохондріальна міопатії та ін.), які зазвичай розвиваються в дитячому віці;
- міастенії, синдрому Ламберта — Ітона і метаболічних міопатій: характерною ознакою є м’язова слабкість, яка виникає епізодично та різко посилюється після фізичного навантаження;
- медикаментозних міопатій (ГК, антималярійні препарати, статини, пеніциламін та ін.); для стероїдної міопатії характерні нормальний рівень КФК, збільшення м’язової сили на тлі зниження дози ГК, відсутність ознак м’язового запалення в м’язових біоптатах;
- метаболічних міопатій (порушення метаболізму глікогену, ліпідів, пуринів). Характерна ознака — зниження толерантності до фізичного навантаження і відновлення м’язової сили на тлі відпочинку;
- мітохондріальних міопатій;
- ендокринних міопатій, які виникають при гіпотиреозі, тиреотоксикозі, хворобі Аддісона, ендогенному гіперкортицизмі, акромегалії, гіперпаратиреозі;
- міопатій при порушеннях електролітного обміну;
- прогресуючих м’язових дистрофій (м’язова дистрофія Дюшена, міодистрофія Беккера, міодистрофія Роттауфа — Мортьє — Бейєра, попереково-кінцівкова міодистрофія Ерба — Рота, плечолопатково-лицьова міопатія Ландузі — Дежерина та ін.);
- непрогресуючих м’язових дистрофій (міотубулярна, немалінова, хвороба центрального стрижня);
- амілоїдозу;
- рабдоміолізу;
- інших захворювань (РПМ: виражена болісність м’язів плечового і тазового пояса без об’єктивного зменшення м’язової сили у поєднанні з підвищенням ШОЕ і лихоманкою в осіб старше 50 років).
Приклади формулювання діагнозів
Ідіопатичний дерматополіміозит, гострий перебіг, активна фаза, III ступінь активності, з ураженням м’язів грудної клітки, ковтальних м’язів з явищами дисфагії, м’язів нижніх та верхніх кінцівок, параорбітальний набряк, синдром Готтрона в ділянках колінних і п’ястково-фалангових суглобів, поліартрит суглобів кистей, суглобова функціональна недостатність ІІ ст., фіброзуючий альвеоліт, дихальна недостатність (ДН) ІІІ, кардіоміопатія, серцева недостатність ІІА, ФК III, поліневропатія за типом синдрому «шкарпеток».
Поліміозит ідіопатичний, підгострий перебіг з дифузним ураженням м’язів нижніх кінцівок; серця — міокардит з порушенням ритму і провідності за типом синусової тахікардії, блокади лівої ніжки пучка Гіса, серцева недостатність ІІА, ФК III.
Вторинний ДМ на тлі центрального раку легені з дифузним ураженням м’язів верхніх і нижніх кінцівок, плечового пояса, діафрагми, грудних м’язів, ДН III.
Лікування
Нефармакологічні методи
- Адаптація рівня фізичної активності до стану пацієнта.
- Уникнення втрати рівноваги (ризик остеопорозних переломів).
- Дотримання низькокалорійної дієти з достатнім вмістом кальцію і вітаміну D для:
- зниження ризику розвитку цукрового діабету та ожиріння на тлі ГК-терапії;
- зниження ризику ГК-ОП.
Реабілітаційні заходи слід проводити диференційовано (залежно від стадії захворювання).
У гострій фазі показані пасивні вправи і напруження м’язів, у стадії одужання — ізометричні, а потім ізотонічні вправи.
У хронічній стадії — анаеробні вправи.
Медикаментозне лікування
Ранній початок лікування (протягом перших 3 міс від появи симптомів) асоціюється з більш сприятливим прогнозом, ніж пізній.
Глюкокортикоїди
Принципи лікування ГК:
- якомога більш ранній початок терапії;
- адекватна початкова доза (1,0–2,0 мг/кг/добу);
- тривалість прийому початкової дози не менше 2–3 міс;
- повільний темп зниження дози;
- адекватна підтримувальна доза.
Залежно від тяжкості захворювання початкова доза коливається від 1 до 2 мг/кг/добу. Протягом перших тижнів добову дозу слід ділити на 3 прийоми, потім — приймати всю дозу одноразово вранці.
Поліпшення стану хворих на ІЗМ відбувається повільніше, ніж пацієнтів з іншими ревматичними захворюваннями (в середньому через 1–3 міс). За відсутності позитивної динаміки протягом 4 тиж слід підвищити дозу ГК.
Після досягнення ефекту (нормалізація м’язової сили і КФК) дозу ГК поступово знижують до підтримувальної, щомісяця приблизно на ¼ від сумарної. Зниження дози має проводитися під суворим клінічним і лабораторним контролем. У разі загострення захворювання необхідно припинити зниження або навіть підвищити дозу препарату до нормалізації клінічних та лабораторних показників.
Пульс-терапія ГК рідко ефективна, застосовується головним чином при ювенільному міозиті. У цих хворих вона може запобігти швидкому прогресуванню міопатії і розвитку кальцинозу. При ІЗМ у дорослих пульс-терапію ГК слід використовувати у випадку швидкого прогресування дисфагії (ризик аспіраційної пневмонії) і розвитку системних проявів (міокардит, альвеоліт).
За відсутності позитивної динаміки на фоні тривалого прийому високих доз ГК слід виключити стероїдную міопатію, міозит з «включеннями», інші захворювання м’язів.
На сьогодні рекомендують раннє призначення метотрексату або азатіоприну, а саме за наявності маркерів несприятливого прогнозу (пізніше призначення ГК-терапії, тяжка м’язова слабкість, наявність дисфагії), неможливості призначити адекватну дозу ГК через побічні ефекти, при недостатній ефективності ГК.
Метотрексат дозволяє швидше перевести хворих на підтримувальну дозу ГК. Доза метотрексату варіює від 7,5 до 25 мг/тиж. При поганій переносимості пероральної форми (особливо високих доз) препарат доцільно вводити парентерально.
Азатіоприн в дозі 2–3 мг/кг/добу (100–200 мг/добу) поступається метотрексату щодо ефективності та швидкості настання ефекту (в середньому через 6–9 міс), особливо у пацієнтів з антисинтетазним синдромом.
Хлорамбуцил. Іноді резистентність до ГК, метотрексату й азатіоприну можна подолати, якщо призначити хлорамбуцил (2–4 мг/добу) або метотрексат у комбінації з азатіоприном.
Циклофосфамід рідко ефективний при ІЗМ, але його відносять до препаратів вибору при розвитку фіброзуючого альвеоліту. У хворих із резистентним міозитом доза циклофосфаміду становить у середньому 1–2 мг/кг/добу.
Циклоспорин у дозі 2,0–3,5 мг/кг/добу призначають пацієнтам при резистентності до ГК. Препарат чинить позитивний ефект при прогресуванні інтерстиціальних захворювань легень.
Антималярійні препарати (наприклад гідроксихлорохін в дозі 200–400 мг/добу) іноді дозволяють контролювати шкірні прояви ДМ. Їх також призначають для підтримувальної терапії в поєднанні з низькими дозами ГК.
Мікофенолова кислота — селективний імунодепресант антиметаболічного типу дії. Початкова доза становить 500 мг 2 рази на добу з подальшим її підвищенням до 1000 мг 2 рази на добу. Мікофенолову кислоту рекомендують застосовувати також при резистентному шкірному синдромі.
В/в імуноглобулін. Отримано дані про доцільність в/в застосування імуноглобуліну в низьких дозах (1 г/кг) 2 рази на місяць протягом 4–6 міс у хворих на ІЗМ, резистентних до стандартної терапії ГК і цитотоксичними препаратами. У половини пацієнтів клінічне поліпшення зберігається протягом 3 і більше років після завершення лікування. Іншим можливим показанням до в/в введення імуноглобуліну вважають тяжке ураження стравоходу.
Плазмаферез слід проводити головним чином у хворих з тяжкою, резистентною до інших методів лікування ІЗМ у поєднанні з ГК і цитотоксичними препаратами.
Нові аспекти фармакотерапії
В останні роки для лікування ІВМ застосовують інгібітори ФНП-α, зокрема інфліксимаб. Існують поодинокі спостереження успішного прийому при ДМ препаратів, що блокують проліферацію В-лімфоцитів (ритуксимаб).
Хірургічне лікування
Включає оперативне видалення пухлини у хворих із паранеопластичною ІЗМ.
Критерії якості лікування:
- зменшення або відсутність м’язової слабкості або болю у м’язах;
- нормалізація активності ферментів КФК, альдолази, АсАТ, АлАТ;
- нормалізація показників гострофазового запалення (фібриноген, серомукоїд, дифеніламінова проба, СРБ, ШОЕ, глобуліни);
- нормалізація або поліпшення даних біопсії м’язів, а також даних ЕМГ.
Періодичність обстеження:
- загальний огляд: при кожному візиті (але не рідше 1 раз на 2–3 міс);
- визначення КФК: кожні 2–3 міс;
- лабораторне дослідження для моніторингу токсичності терапії: залежно від характеру терапії;
- диспансеризація для виявлення онкологічної патології: але не рідше 1 разу на рік.
Профілактика
Профілактика ІЗМ головним чином вторинна, спрямована на попередження загострення. Слід уникати травм, оперативних втручань, інфекцій, охолодження, щеплень, лікарських препаратів, на які відзначено побічні ефекти, контакту з хімічними речовинами, лікувальних процедур, таких як радонові, сульфідні ванни та ін. Жінкам рекомендовано не допускати вагітності.
При гострому та підгострому перебігу хворих на ІЗМ переводять на інвалідність I і II групи, і не раніше ніж через рік може обговорюватися питання про повернення до роботи або навчання.
При хронічному перебігу захворювання в період ремісії хворі можуть зберігати працездатність, якщо робота не пов’язана з фізичною працею, охолодженням, контактом з хімічними речовинами, які подразнюють шкіру, слизові.
Прогноз
До відкриття ГК смертність становила 30–50%. Упровадження в клінічну практику ГК значно підвищило виживаність хворих на ІЗМ, яка в цілому в групі (за винятком хворих на міозит на тлі злоякісних новоутворень) становить 90% через 5 років після встановлення діагнозу.
До факторів ризику несприятливого прогнозу при ІЗМ відносять похилий вік, пізно встановлений діагноз, неадекватну терапію на початку хвороби, важкий перебіг міозиту, антисинтетазний синдром. Прогноз паранеопластичного ДМ залежить від своєчасності виявлення пухлини та ефективності її лікування.
Глава 17. Хвороба Шегрена
Хвороба Шегрена (первинний синдром Шегрена) (М35.0).
Хвороба Шегрена — системне захворювання сполучної тканини з переважним ураженням секреторних епітеліальних залоз, основними проявами якого є ксеростомія та ксерофтальмія.
Код за МКХ-10
М35.0 Сухий синдром [Шегрена].
Поряд з хворобою Шегрена (у зарубіжній літературі зустрічається наступна термінологія — первинний синдром Шегрена, сухий синдром, синдром Гужеро — Шегрена, атрофічна дакріосіалопатія, сухий кератокон’юнктивіт Шегрена та ін.) виділяють синдром Шегрена, що поєднується з РА, ССД, СЧВ, ІЗМ, хронічним активним гепатитом, біліарним цирозом печінки та іншими аутоімунними захворюваннями.
Найбільш докладно симптомокомплекс, який включає сухий кератокон’юнктивіт, ксеростомію зі зниженням функції інших секретуючих епітеліальних залоз (потових, залоз бронхів, трахеї, шлунка та ін.), описаний шведським офтальмологом Шегреном (Sjogren Н., 1933). При цьому Шегрен зазначав часте виявлення цього симптомокомплексу в осіб з РА, дифузними захворюваннями сполучної тканини та іншими аутоімунними захворюваннями.
За даними К.Г. Барнсу (1990), приблизно у третини хворих сухий синдром протікає самостійно, без ознак інших захворювань сполучної тканини. Найбільш часто синдром Шегрена поєднується з РА, що виявляють у 50% хворих з сухим кератокон’юнктивітом і/або ксеростомією. Серед хворих на РА частота синдрому Шегрена не перевищує 10%. Поєднання синдрому Шегрена і РА має місце лише при значній давності суглобової патології та за наявності РФ. Однак чіткої кореляції з активністю синовіту не простежується, оскільки в багатьох випадках синдром Шегрена розвивається у хворих при зменшенні вираженості проявів суглобового синдрому. У решти 20% випадків синдром Шегрена поєднується з іншими захворюваннями, включаючи склеродермію (при якій синдром Шегрена виявляють у 50% випадків), ІЗМ, СЧВ, тиреоїдит Хашимото, аутоімунні захворювання печінки і такі рідкісні хвороби, як гемохроматоз і гіперліпопротеїнемію V типу.
Хвороба Шегрена в класифікації ВООЗ (1975, IX перегляд) включена в групу системних захворювань сполучної тканини. Причому це захворювання є найбільш частим серед ЗЗСТ і займає третє місце серед ревматичних захворювань, поступаючись ХРХС і РА.
Розповсюдженість хвороби Шегрена варіює від 0,1 до 3,3% в загальній популяції та від 2,8 до 4,8 — серед осіб віком старше 50 років. Жінки хворіють частіше, ніж чоловіки (співвідношення приблизно 9:1), зазвичай у віці 20–50 років, хворобу виявляють переважно в осіб віком старше 30 років, значно рідше діагностують у дітей.
Хворим з рецидивуючим сіаладенітом, збільшенням слинних/слізних залоз, сухістю рота, офтальмологічними порушеннями, ураженням суглобів, синдромом Рейно, рецидивуючою пурпурою, підвищенням ШОЕ, гіпергаммаглобулінемією необхідно проводити тест Ширмера, сіалометрію, досліджувати РФ і антиядерні антитіла (апти-Rо/SS-А, анти-Lа/SS-В). При отриманні позитивних результатів необхідно спеціальне обстеження для виключення хвороби та синдрому Шегрена.
Етіологія і патогенез хвороби Шегрена вивчені недостатньо. Існує вірусна гіпотеза її виникнення, заснована на виявленні при електронній мікроскопії вірусоподібних тубулоретикулярних структур, НВs-антигену в епітелії слинних залоз, клубочках нирок і визначенні в крові хворих підвищеного рівня антитіл до цитомегаловірусу, вірусу Епштейна — Барр, вірусу герпесу 6-го типу, ВІЛ, Т-лімфотропного вірусу людини 1-го типу. Однак прямих доказів вірусної етіології хвороби Шегрена не отримано.
На користь генетичної детермінованості хвороби Шегрена свідчать сімейні випадки захворювання, а також висока частота виявлення антигенів гістосумісності НLА DrЗ (69% випадків) і НLА В8 (59% асоціація), що перебувають у стані неврівноваженого зчеплення, а при синдромі Шегрена, асоційованому з РА, — НLА Drw4.
Існує також думка про вплив статевих гормонів на імунну регуляцію, засновану на експериментальних дослідженнях з новозеландськими чорними мишами лінії NZB та їх гібридами — білими лінії NZB/w, у результаті яких виявлено, що андрогени пригнічують захворювання і пролонгують виживаність, а естрогени викликають протилежний ефект. Цей факт, ймовірно, може пояснити таку високу захворюваність на хворобу Шегрена серед жінок.
Враховуючи вищесказане, патогенез хвороби Шегрена можна представити таким чином. Передбачається, що для початку процесу потрібний первинний вплив вірусу (цитомегаловірус, вірус Епштейна — Барр) на тлі генетично зумовленої схильності, пов’язаної з наявністю антигенів НLА (Drw3 і В8), зниженням природної кілерної активності та функціональної активності Т-супресорів. Первинне ураження клітин органів-мішеней (наприклад слинних залоз) призводить до вивільнення аутоантигенів, які через макрофаги активують Т-хелпери і В-лімфоцити. Помірна локальна активація (у слинних залозах) В-клітин призводить до продукування аутоантитіл, чому сприяє функціональна недостатність Т-супресорів. Активація Т-хелперів спричиняє їх надмірну проліферацію та інфільтрацію слинних залоз (вторинне ураження органів-мішеней — клітинне). Ці зміни можуть зумовити локальне ураження (перша ланка патогенезу) з накопиченням органоспецифічних антитіл.
У тих випадках, коли виникає поліклональна активація В-лімфоцитів, процес набуває системного характеру з утворенням органонеспецифічних антитіл. Існує також думка, що хвороба Шегрена являє собою слабовиражене лімфопроліферативне захворювання, яке може, розвиваючись від локального поліклонального до дифузного поліклонального, трансформуватися в злоякісне моноклональне В-лімфоцитарне захворювання, причому цей процес починається на дуже ранніх стадіях хвороби.
Аутоімунний генез захворювання підтверджується виявленням великої кількості органоспецифічних (до епітелію проток підшлункової та слинних залоз, парієтальних клітин шлунка, клітинних мембран печінки, тиреоглобуліну тощо) і органонеспецифічних (АНФ, антитіла до апарата Гольджі, анти-Rо і анти-Lа, LЕ-клітин та ін.) аутоантитіл, а також ЦІК, що складаються з JgG-РФ-анти-JgG, гіперпротеїнемії, гіпергаммаглобулінемії, кріоглобулінемії та ін.
Патоморфологія. Основною морфологічною патогномонічною ознакою хвороби Шегрена, яка веде до ураження секреторних залоз, є лімфоплазмоклітинна інфільтрація, яка заміщає паренхіму. У багатьох випадках інфільтрація набуває генералізованого характеру з залученням в процес м’язів, ШКТ, легень, нирок, ретикулоендотеліальної системи. У ранніх стадіях захворювання залози можуть збільшуватися. Це зумовлено запальними змінами з вираженою інфільтрацією лімфоцитами і плазматичними клітинами, які концентруються головним чином навколо проток. Спостерігається також набрякання паренхіми. Надалі залози зменшуються в розмірах внаслідок атрофії ацинусів і втрати нормальної архітектоніки. Розвивається фіброз, який супроводжується звуженням просвіту проток. Відбувається проліферація клітин проток, а лімфоцити, які інфільтрують залози, агрегуються в фолікули з зародковими центрами.
У фінальній стадії патологічного процесу утворюється зменшена в розмірах, дезорганізована, фіброзно змінена залоза з лімфоїдними фолікулами й острівцями проліферуючого епітелію проток, оточеними лімфоцитами (епіміоепітеліальні острівці).
Нерідко розвивається генералізована лімфоїдна гіперплазія, відбувається підвищення частоти злоякісних В-лімфоцитарних захворювань і лімфобластної саркоми. Описано гістологічні зміни, характерні для злоякісної лімфоми; при застосуванні кортикостероїдів вдавалося домогтися ремісії. Подібні зміни відзначають у хворих з вираженою гіпергаммаглобулінемією, імунологічними порушеннями та слабовираженим артритом.
Класифікація
Загальноприйнятої класифікації хвороби Шегрена не існує. У розробленій Інститутом ревматології РАМН класифікації за характером початку і подальшого перебігу хвороби Шегрена розрізняють хронічну (переважно залозисту) і підгостру (із позазалозистими проявами) форму захворювання. Початкову, виражену і пізню стадію діагностують на підставі сіалографічного методу дослідження привушних слинних залоз (табл. 17.1).
Таблиця 17.1
Клінічна класифікація хвороби Шегрена
|
Перебіг |
Підгострий Хронічний |
|
|
Стадія розвитку |
I (початкова) II (генералізована) III (термінальна) |
|
|
Ступінь активності |
Відсутня (0) Мінімальна (I) Помірна (II) Висока (III) |
|
|
Клініко-морфологічна характеристика уражень |
Слинні залози та порожнина рота |
Паренхіматозний паротит (рецидивуючий), збільшення слинних залоз I, II, III ступеня, стоматит |
|
Сльозні залози та очі |
Сухий кон’юнктивіт/блефарокон’юнктивіт, сухий кератокон’юнктивіт (дистрофія епітелію кон’юнктиви I, II ступеня та рогівки I–III ступеня, нитчастий кератит, ксероз рогової оболонки), гіполакримія I–III ступеня |
|
|
Слизові оболонки |
Сухий атрофічний ринофаринголарингіт, сухий трахеобронхіт, сухий кольпіт |
|
|
Шкіра |
Сухість шкіри, хейліт, уртикарна висипка, фотодерматоз, рецидивуюча гіпергамма-/кріоглобулінемічна та змішана пурпура, сітчасте ліведо |
|
|
Ретикулоендотеліальна система |
Регіонарна (генералізована) лімфаденопатія, гепатомегалія, спленомегалія, псевдолімфома, лімфома |
|
|
Суглоби |
Артралгії, рецидивуючий неерозивний артрит |
|
|
М’язи |
Міалгії, міозит |
|
|
Серозні оболонки |
Полісерозит (плеврит, перикардит) сухий, випітний |
|
|
Судини |
Синдром Рейно, рецидивуюча гіпергамма- та кріоглобулінемічна пурпура |
|
|
Легені |
Інтерстиціальна пневмонія, альвеолярний легеневий фіброз, рецидивуюча пневмонія |
|
|
Нирки |
Канальцевий ацидоз, імунокомпетентний гломерулонефрит, дифузний гломерулонефрит |
|
|
ШКТ |
Гіпотонія стравоходу, атрофічний гастрит із секреторною недостатністю, панкреатит |
|
|
Нервова система |
Поліневропатія, поліневрит, неврити трійчастого та лицьового нерва, цереброваскуліт |
|
|
Щитоподібна залоза |
Аутоімунний тиреоїдит |
|
Хронічний варіант перебігу, який відзначають у 30–40% хворих більш похилого віку, характеризується поступовим збільшенням вираженості симптомів (хворі можуть вказати лише рік появи того чи іншого симптому). Першими клінічними ознаками хвороби при цьому варіанті перебігу є сухість слизової порожнини рота і утворення пришийкового карієсу, різі в очах, поступове збільшення привушних і підщелепних слинних залоз, рідше артралгії та синдром Рейно. Лабораторні показники змінюються мало, за винятком періоду загострення, коли відбувається помірне підвищення ШОЕ (20–35 мм/год), незначна гіпергаммаглобулінемія (23–26%), а також РФ в низьких титрах, рідше в середніх і високих, лейкопенія. При цьому перебіг хвороби Шегрена характеризується практичною відсутністю вісцеральних проявів, у зв’язку з чим захворювання залишається моносимптомним протягом багатьох років, проте відзначають значні функціональні порушення з боку слинних, слізних залоз і залоз шлунка. За відсутності патогенетичної адекватної терапії можлива трансформація в підгострий варіант хвороби Шегрена. Морфологічними особливостями хронічного варіанта даного захворювання є переважання дистрофічних процесів в слинних і потових залозах, а також в епітелії кон’юнктиви і рогової оболонки. Пізня діагностика і відсутність лікування може призводити до тяжкого ураження рогівки і майже повної втрати зору.
Підгострий варіант перебігу діагностують у 60–70% хворих більш молодого віку. Захворювання починається раптово (хворі можуть назвати місяць або день початку хвороби). Першими клінічними симптомами частіше є паротит з високою температурою, кон’юнктивіт, неерозивний артрит (частіше дрібних суглобів кистей) або інтенсивні артралгії, рідше міалгії та синдром Рейно. При лабораторному обстеженні в перші дні і місяці захворювання виявляють стійке підвищення ШОЕ, анемію, лейкопенію, гіпергаммаглобулінемію, високі титри РФ. Надалі у цих хворих паротит, кератокон’юнктивіт, артрит мають рецидивуючий характер, протягом усього захворювання зберігаються ознаки високої лабораторної активності з розвитком системних проявів уже в перші роки.
Розрізняють 3 ступені активності хвороби Шегрена. Активність визначають залежно від клініко-морфологічного синдрому, даних інструментального та лабораторного обстеження, а також динаміки хвороби в процесі спостереження.
Високий (III) ступінь активності характеризується рецидивуючими паротитами, стоматитами, кератокон’юнктивітами, неерозивними артритами та іншими загальними ознаками запалення з переважанням гострих, підгострих інтерстиціальних і судинних проявів у вигляді рецидивуючої гіпергамма- і кріоглобулінемічної пурпури, міозиту, виразково-некротичного васкуліту, гломерулонефриту, інтерстиціальної пневмонії з залученням до процесу ретикулоендотеліальної системи.
При помірній (II ступінь) активності поряд зі зменшенням вираженості яскравих запальних проявів захворювання (зниження частоти рецидивів паротиту, пурпури та ін.) відзначають тенденцію до розвитку деструктивних процесів у секретуючих епітеліальних залозах, що супроводжується менш вираженими тестами, переважно запальної та імунологічної активності.
Мінімальна активність (I ступінь) характеризується переважанням в клінічній картині хвороби функціональних, дистрофічних і склеротичних змін різної локалізації, що призводить до значних функціональних порушень з боку слинних, слізних залоз і залоз шлунка з розвитком тяжкого сухого кератокон’юнктивіту, ксеростомії й атрофічного гастриту.
Як правило, існує паралелізм між офтальмологічними, стоматичними і системними проявами захворювання. Найбільш тяжкі системні прояви бувають у вираженій і пізній стадії хвороби Шегрена.
Клінічна картина
Клінічні прояви хвороби Шегрена можна розділити на 2 категорії: симптоми, пов’язані з гіпофункцією секретуючих епітеліальних залоз, і позазалозисті — системні прояви захворювання.
Клінічні ознаки ураження екзокринних залоз при хворобі Шегрена:
- сухий кератокон’юнктивіт;
- ксеростомія; збільшення слинних і слізних залоз;
- рецидивуючий паротит;
- сухість слизових оболонок носа, глотки, гортані, трахеї, бронхів, зовнішніх жіночих статевих органів та піхви;
- сухість шкіри;
- хронічний атрофічний гастрит із секреторною недостатністю, хронічний панкреатит.
Практично у всіх пацієнтів із хворобою Шегрена виявляють ксерофтальмію, що є клінічною ознакою ураження слізних залоз. У результаті зниження секреції слізної рідини розвивається сухий кератокон’юнктивіт, ступінь вираженості якого знаходиться в прямій залежності від інтенсивності ураження слізних залоз і порушення їх функції. Так, у початковій стадії захворювання скарги або відсутні, або зводяться до відчуття свербежу, незначного печіння в очах, легкої світлобоязні, дискомфорту. При огляді відзначають невиражену ін’єкцію і набряк слизової оболонки ока, а також невелике прозоре виділення в кон’юнктивальній порожнині. Спершу проявляються лише ознаки рецидивуючого кон’юнктивіту.
Подальше зниження слізної секреції призводить до вираженої стадії захворювання, розвитку ерозії рогівки або нитчастого кератиту з порушенням зору. Хворі скаржаться на різі в очах, відчуття присутності піску і чужорідного тіла, почервоніння і свербіж повік, виражену світлобоязнь, стомлюваність і біль в очних яблуках при напруженні, зниження гостроти зору, що веде до втрати працездатності. У рідкісних випадках результатом таких змін може бути тяжке порушення зору або навіть сліпота внаслідок васкуляризації рогівки. Ускладненнями сухого кератокон’юнктивіту можуть бути виразки або перфорація рогівки, що вимагають хірургічного втручання.
Для діагностики сухого кератокон’юнктивіту, ступеня його вираженості обов’язково проводять офтальмологічне обстеження з використанням спеціальних методів (тест Ширмера, тест з бенгальським рожевим, а також тест з флуоресцеїном).
Другою найбільш частою ознакою хвороби Шегрена є ураження слинних залоз з такими клінічними проявами, як ксеростомія, збільшення слинних залоз, рецидивуючий паротит, сухість червоної облямівки губ, хейліт, стоматит, гінгівіт, атрофія сосочків язика, множинний карієс пришийковий, випадіння зубів, збільшення регіонарних лімфатичних вузлів.
Ступінь вираженості даної симптоматики відповідає стадії хвороби Шегрена. Так, у початковій стадії хворі частіше скарг не пред’являють, але при фіксованому опитуванні реєструють періодичну сухість слизової оболонки рота при тривалій розмові, емоційному та фізичному навантаженні, печіння слизової оболонки рота при прийомі їжі, яка викликає подразнення, а пальпаторно — невелике збільшення слинних залоз. Клінічні прояви паротиту в цю стадію відзначаються не у всіх хворих і залежать від варіанта перебігу хвороби Шегрена. При огляді порожнини рота: слизова оболонка, як правило, звичайного кольору, досить зволожена, проте вільної слини мало, крім того, вона піниста; вже у початковій стадії хвороби Шегрена проявляється гіперестезія емалі зубів і пришийковий карієс.
У виражену стадію хвороби Шегрена реєструють скарги на періодичну сухість в роті, потребу запивати суху їжу, утруднення ковтання, зумовлене труднощами формування харчової грудки, оскільки їжа прилипає до піднебіння, язика і зубів через недостатнє її змочування, необхідність під час розмови перерозподіляти слину в роті язиком. Збільшення привушних та підщелепних слинних залоз відзначають досить часто. Збільшення привушних залоз може бути одностороннім, що викликає асиметрію обличчя і навіть його деформацію, нагадуючи мордочку хом’ячка. Дуже часто, особливо при підгострому перебігу хвороби Шегрена, виявляють значне збільшення привушних слинних залоз. При рецидивуючих паротитах, які супроводжуються збільшенням, припухлістю залоз, утрудненням при відкритті рота, з’являється гнійне виділення зі слинних проток, підвищення температури тіла до 39–40 °C. Процес при цьому, як правило, двосторонній з частими загостреннями та абсцедуванням. Рідше збільшення привушних слинних залоз розвивається безсимптомно за типом пухлиноподібного утворення в цій ділянці. При огляді порожнини рота відзначають гіперемію слизової оболонки (легко травмується), значне зменшення вільної слини (в’язка, піниста), явища глоситу, приєднання грибкової інфекції, швидкопрогресуючий карієс зубів, який призводить до часткової їх втрати.
Пізня стадія хвороби Шегрена характеризується ознаками тяжкого ураження слинних залоз. У багатьох хворих виникають труднощі при ковтанні навіть рідкої їжі, крім того вони не можуть вживати гарячу та гостру їжу. Слинні залози в пізню стадію можуть бути збільшені (значно або незначно) або атрофовані. При цьому частота загострень паротиту знижується. Слина з проток слинних залоз, як правило, не виділяється. При огляді порожнини рота губи сухі з ділянками лущення і тріщинами, слизова оболонка різко гіперемована з ознаками ороговіння, легко травмується, поверхня язика гладка, яскраво-червона, слина відсутня. Швидкопрогресуючий карієс зубів призводить за короткий час до їх повної втрати.
У діагностиці ураження слинних залоз важливе і вирішальне місце займають спеціальні методи дослідження (сіалометрія, сіалографія, сцинтиграфія), які проводять стоматологи, а також морфологічне дослідження біоптату малих слинних (губних) залоз, розташованих у слизовій оболонці порожнини рота.
Поряд з іншими залозами зовнішньої секреції схильними до ураження є залози слизової оболонки носа, глотки, гортані, трахеї, бронхів. Так, сухість носоглотки з утворенням сухих кірок у носі, просвіті євстахієвої труби може призводити до тимчасової глухоти і розвитку отиту. Сухість глотки, а також голосових зв’язок викликає утруднення ковтання сухої їжі й осиплість голосу. Сухість носоглотки і гортані призводить до розвитку субатрофічного, атрофічного риніту, фарингіту, ларингіту. Частими ускладненнями сухості носоглотки, трахеї та бронхів є синусити, рецидивуючі трахеїти і пневмонії (у результаті підвищеної схильності до інфекції).
Досить часто при хворобі Шегрена відзначають ураження залоз зовнішніх статевих органів, внаслідок чого розвивається сухість шкіри та слизової оболонки, клінічні ознаки атрофічного вагініту.
Ураження шкіри при хворобі Шегрена часто проявляється вираженою сухістю, особливо верхніх і нижніх кінцівок, лущенням і гіперкератозом, зниженням потовиділення (навіть при високій температурі навколишнього середовища), випадінням волосся, трофічними змінами нігтів.
Ураження ШКТ зумовлено патогенетичними механізмами захворювання — лімфоїдною інфільтрацією зі зниженням секреторної функції і загибеллю екзокринних залоз ШКТ. Часто при хворобі Шегрена відзначають дисфагію, зумовлену ксеростомією, а також нерідко — гіпокінезією стравоходу, що підтверджено результатами рентгенологічного дослідження. У переважної більшості хворих (близько 80%) розвивається атрофічний гастрит з вираженою секреторною недостатністю. Як правило, простежується пряма залежність між ступенем ксеростомії і зниженням секреторної функції шлунка. Клінічна симптоматика при цьому зазвичай мізерна, часто — втрата апетиту, зміни смаку. Відзначають також ознаки ураження дванадцятипалої кишки (частіше за типом гіпомоторної дискінезії та бульбостазу) при проведенні фіброгастроскопії і рентгенологічного дослідження. Ураження інших відділів кишечнику відзначають у 85–95% хворих і, як правило, поєднується з ураженням шлунка, підшлункової залози, печінки і жовчовивідних шляхів. У таких хворих відзначають запори, нестійкі випорожнення, біль на протяжності товстої кишки (пальпаторно), а у 5–10% хворих — поліпоз товстої кишки, наявність дивертикулів, геморою, тріщин в ділянці ануса, а також дисбактеріозу переважно за рахунок збільшення кількості кишкових паличок.
Ураження підшлункової залози, що супроводжується зниженням зовнішньосекреторної функції, клінічно проявляється найчастіше больовим і диспептичним синдромом.
Нерідко при хворобі Шегрена (50–65%) виявляють ураження печінки і жовчовивідних шляхів. Клінічно і морфологічно патологія печінки проявляється у вигляді хронічного персистуючого гепатиту, а у ½ хворих гістологічно виявляють лімфогістіоцитарні інфільтрації та дистрофічні зміни гепатоцитів. Однак завжди необхідно пам’ятати про те, що хронічний активний гепатит і первинний біліарний цироз печінки можуть проходити в поєднанні з синдромом Шегрена, а при хворобі Шегрена може бути ураження печінки, тому необхідно проводити диференційну діагностику між ними. Частіше при хворобі Шегрена відзначають хронічний холецистит, нерідко в поєднанні з дискінезією жовчовивідних шляхів за гіпокінетичним типом.
Позазалозисті системні прояви при хворобі Шегрена: рецидивуючий, переважно неерозивний артрит; міозит; генералізована лімфаденопатія, гепатомегалія, спленомегалія, псевдолімфома, лімфома; інтерстиціальна пневмонія, альвеолярний легеневий фіброз; канальцевий ацидоз, гломерулонефрит; синдром Рейно, рецидивуючі гіпергамма- і кріоглобулінемічний пурпури; виразково-некротичний васкуліт: периферична поліневропатія, поліневрит, неврити лицьового і трійчастого нерва; цереброваскуліт.
Серед позазалозистих системних проявів хвороби Шегрена найбільш часто (75–85%) виявляють суглобовий синдром. Найчастіше діагностують рецидивуючі артралгії, рідше артрити (4–16%) дрібних (міжфалангових) суглобів (іноді великих), які поєднуються з больовими згинальними контрактурами кистей. Характерним для хвороби Шегрена є повна клінічна ремісія суглобового синдрому при призначенні ГК вже на 2–3-й день лікування та відсутність протягом багатьох років ознак ерозивного процесу в суглобах. При хворобі Шегрена, як правило, не відзначають розвитку деформації, анкілозування і підвивихів суглобів, навіть при тривалому перебігу артритів.
Ураження м’язів при хворобі Шегрена, яке реєструють приблизно у 10–15% хворих, проявляється міалгіями, помірно вираженим міозитом, підвищенням активності сироваткових ферментів (КФК, трансаміназа), у біоптатах м’язів — змінами, характерними для поліміозиту.
Досить часто (39–70%) при хворобі Шегрена діагностують лімфоаденопатію. Регіонарну лімфаденопатію (збільшення підщелепних, шийних, потиличних, надключичних лімфатичних вузлів) відзначають, як правило, у переважної більшості хворих (70%), у той час як генералізовану — приблизно у ⅓ хворих, нерідко в поєднанні зі збільшенням печінки і селезінки. При тривалому перебігу хвороби Шегрена (10–15 років) у 4–5% хворих розвиваються лімфопроліферативні захворювання — псевдолімфома, макроглобулінемія Вальденстрема. Псевдолімфома може трансформуватися в злоякісну лімфому. У таких випадках у хворих виявляли значне збільшення слинних залоз, генералізовану лімфаденопатію, гепатомегалію, спленомегалію, лихоманку та ін., а також продукцію моноклонального імуноглобуліну М. Крім того, розвиток злоякісної лімфоми супроводжується зниженням ШОЕ, зменшенням гаммаглобулінів, титру РФ на відміну від псевдолімфоми, для якої характерні ознаки високої лабораторної активності. Лімфоми при хворобі Шегрена розвиваються в 44 рази частіше, ніж у загальній популяції.
Ураження легень при хворобі Шегрена розрізняють двох видів. При першому відзначають переважно дифузне ураження легеневої тканини у вигляді дифузної деформації легеневого малюнка сітчастого характеру за типом фіброзуючого альвеоліту з вираженими функціональними порушеннями дихання та легеневого кровотоку, нерідко з формуванням легеневого серця, а також з кріоглобулінемією і зниженням комплементу. Другий тип характеризується посиленням легеневого малюнка за рахунок деформуючого бронхіту і повнокров’я легень при помірних функціональних порушеннях дихання. Крім того, у цих хворих частіше діагностують рецидиви бронхіту, пневмоній, високий рівень гаммаглобулінів, ЦІК і АНФ. Плеврити при хворобі Шегрена розвиваються порівняно рідко, однак при рентгенологічному дослідженні досить часто виявляють потовщення міжчасточкової плеври, плевродіафрагмальні та плевроперикардіальні спайки.
Приблизно в 10–12% пацієнтів із хворобою Шегрена виявляли ознаки сухого перикардиту, міокардиту. Ця патологія найчастіше має місце при підгострому перебігу і високій активності процесу.
При хворобі Шегрена реєструють різні ураження нирок, зумовлені лімфоцитарними інфільтратами, васкулітами, гіпергаммаглобулінемією, кріоглобулінемією або підвищеною фільтрацією і реабсорбцією легких ланцюгів та інших фрагментів імуноглобулінів. Найчастіше відзначають ураження канальцевого апарата — канальцевий ацидоз, генералізовану аміноацидурію і фосфатурію з розвитком нефрогенного діабету. Значно рідше виникає дифузний гломерулонефрит мембранозного або імунокомплексного характеру. Важкі форми гломерулонефриту з нирковою недостатністю розвиваються у хворих з кріоглобулінемією і наявністю пурпури. Часто діагностують хронічний пієлонефрит.
Ураження судин мікроциркуляторного русла при хворобі Шегрена виникає за типом синдрому Рейно, рецидивуючих гіпергамма- і кріоглобулінемічного висипів. Синдром Рейно при хворобі Шегрена реєструють приблизно у половини хворих. Проте переважають стерті форми, рідше проявляється класичний двофазний синдром Рейно.
Гістологічно виділяють три типи васкулітів при хворобі Шегрена: некротизуючий васкуліт, лімфоцитарний васкуліт, облітеруючий ендартеріїт. Усі гістологічні типи васкулітів виявляють і при гіпергамма- і кріоглобулінемічній пурпурі, однак некротизуючий васкуліт і облітеруючий ендартеріїт частіше діагностують у хворих на цю патологію з кріоглобулінемією, а лімфоцитарний васкуліт — у хворих з гіпергаммаглобулінемією.
Найбільш тяжкі системні прояви хвороби Шегрена розвиваються у хворих із некротизуючим васкулітом. Геморагічні висипи частіше з’являються на шкірі гомілок, однак можливі також на шкірі стегон, живота і тулуба. У хворих з довгостроково рецидивуючою гіпергаммаглобулінемічною пурпурою зазвичай розвивається гіперпігментація шкіри внаслідок відкладення гемосидерину. Обсяг геморагічних висипів збільшується до кінця робочого дня, при тривалому перебуванні в ортостатичному положенні та зменшується після відпочинку. Поява висипів супроводжується свербінням, болісним печінням і підвищенням температури шкіри в ділянці висипу. Важкі васкуліти з розвитком множинних виразково-некротичних висипів на шкірі обличчя, тулуба, кінцівок, утворенням виразок, які довго не загоюються, нижньої третини гомілки частіше розвиваються на тлі рецидивуючої кріоглобулінемічної пурпури. Для неї характерною є поява висипів за типом пухирців і пустул з геморагічним умістом всередині. Нерідко висипи супроводжуються різкими больовими відчуттями, похолодінням кінцівок, вираженим синдромом Рейно, артралгією, періартикулярним набряком суглобів, парестезіями і зниженням чутливості за типом шкарпеток і рукавичок.
При хворобі Шегрена можуть розвиватися клінічні ознаки периферичної поліневропатії (у тому числі неврити лицьового і трійчастого нервів). Важкі поліневрити і цереброваскуліти при хворобі Шегрена трапляються порівняно рідко і, як правило, на тлі кріоглобулінемії.
Нерідко у пацієнтів із хворобою Шегрена діагностують ураження щитоподібної залози з порушенням її функції, особливо з гіпофункцією (у тому числі тиреоїдит Хашимото).
При хворобі Шегрена простежується підвищена схильність до алергічних реакцій на лікарські препарати, особливо на антибактеріальні, препарати золота, а також харчові продукти, побутові хімічні засоби.
В осіб із хворобою Шегрена нерідко (⅓ випадків) розвиваються ознаки синдрому гіперв’язкості, зумовлені очевидно підвищенням концентрації імуноглобулінів G та М, можливо кріоглобулінемією. У діагностиці синдрому гіперв’язкості виділяють 9 клінічних симптомів: головний біль, запаморочення, сонливість, дзвін у вухах (слухові ілюзії), зниження слуху, порушення зору, кровотечі (носові, шлункові, гемороїдальні, маткові та ін.), порушення апетиту, втомлюваність.
Найбільш частою ознакою синдрому гіперв’язкості є порушення зору не тільки суб’єктивно, але й об’єктивно — диплопія, ретинопатія, зниження гостроти зору, набряк сосочка зорового нерва. На другому місці головний біль; втрата апетиту, втомлюваність, ортостатичне запаморочення — на третьому місці, інші зустрічаються рідше. Симптоми синдрому гіперв’язкості з’являються при в’язкості сироватки крові 2–3,5 відносних одиниць (визначеною у віскозиметрі Оствальда).
Лабораторні дані
Найбільш інформативними в діагностиці хвороби Шегрена є гіперпротеїнемія, гіпергаммаглобулінемія, високі рівні ЦІК, кріоглобулінів, високі титри РФ, антитіл до розчинних ядерних антигенів (антитіла до RO SS-А і Lа/ S-В), а також висока ШОЕ, лейкопенія, помірна нормохромна анемія, позитивна тимолова проба. LЕ-клітини і антитіла до нативної ДНК виявляють виключно рідко. Для хвороби Шегрена характерні високі або нормальні цифри комплементу; гіпокомплементемія розвивається порівняно рідко і є прогностично несприятливою ознакою, особливо в поєднанні з кріоглобулінемією. Один з найбільш частих лабораторних проявів хвороби Шегрена — високі титри РФ — частіше зустрічається при підгострому перебігу, наявності артриту і гіпергаммаглобулінемічної пурпури. З іншого боку, у пацієнтів із хворобою Шегрена та СЧВ і синдромом Шегрена ураження нирок асоціювалося з низькими титрами РФ. У хворих з кріоглобулінемічною пурпурою наявність низьких титрів РФ одночасно з низькими рівнями гаммаглобулінів, JgG і комплементу пов’язана з наявністю незагоюваних протягом тривалого часу трофічних виразок. ЦІК виявляють у осіб із хворобою Шегрена в 36% випадків, який, як правило, асоціюється з гіпергаммаглобулінемічною пурпурою, пізньою стадією сухого кератокон’юнктивіту і неефективним лікуванням. При хворобі Шегрена часто підвищена в’язкість крові, визначення якої важливе для діагностики синдрому гіперв’язкості. В ході лабораторного дослідження виявляють помірну анемію, яка розвивається при хронічних захворюваннях (20–40%), рідше аутоімунну гемолітичну анемію та тромбоцитопенію. Вкрай рідко розвивається апластична анемія. Лейкопенію діагностують у більшості хворих, лейкопоез при цьому непригнічений. У частини хворих (5–10%) відзначають еозинофілію, лімфоцитоз, моноцитоз. Частота лімфо- і моноцитозу значно підвищується при розвитку хронічного лімфолейкозу і В-клітинних неходжкінських лімфомах маргінальної зони лімфатичних вузлів. У третини хворих в крові виявляють кріоглобуліни, в 40% випадках — це змішані моноклональні кріоглобуліни II типу. Моноклональні імуноглобуліни, переважно IgMk, рідше IgGк, IgАк в сироватці крові, а легкі ланцюги імуноглобулінів в сечі (білок Бенс-Джонса) виявляють у 15–20% хворих. Це вимагає диференційної діагностики хвороби Шегрена з моноклональною секрецією і вже розвиненими різними варіантами лімфопроліферативних захворювань.
Як правило, при хворобі Шегрена виявляють слідову моноклональну секрецію; визначення високих рівнів моноклональних імуноглобулінів і розвиток синдрому гіперв’язкості вимагає виключення макроглобулінемії Вальденстрема і плазмоцитоми. Гіпокомплементемія, зниження СЗ-, С4-фракцій комплементу з підвищенням показників гострофазової відповіді — прогностично несприятливі ознаки, їх виявляють переважно у хворих з деструктивними формами васкуліту, лімфомами і при розвитку вторинної інфекції. Гіпопротеїнемія з дефіцитом поліклональних імуноглобулінів, зникнення моноклональної секреції в крові та виявлення білка Бенс-Джонса в сечі хворих з клінічними ознаками деструктивного васкуліту і симптомами лімфопроліферативних захворювань (лихоманка з профузним потовиділенням, значне зменшення маси тіла за короткий проміжок часу) характеризують розвиток великоклітинних неходжкінських лімфом з високим ступенем агресивності при хворобі Шегрена. Поява моноклональних імуноглобулінів, змішаних моноклональних кріоглобулінів в крові та білка Бенс-Джонса в сечі — лабораторні попередники розвитку неходжкінських лімфом при хворобі Шегрена. У 50–60% хворих з моноклональною секрецією після 10–15 років перебігу хвороби Шегрена формуються лімфоми; нерідко діагностують лімфому одночасно з виявленням моноклональної секреції.
Гіпертрансаміназемія, підвищення рівня лужної фосфатази, позитивні антимітохондріальні антитіла в дебюті захворювання — прояви первинного біліарного цирозу печінки, викликаного вторинним синдромом Шегрена. Поява цих ознак в пізніх стадіях розвитку хвороби Шегрена може бути наслідком ураження печінки (хронічний холангіт, холестатичний холангіт, початкові ознаки біліарного цирозу печінки). Антитіла до тиреоглобуліну, мікросомальної фракції щитоподібної залози і тиреоцитів виявляють у 60% хворих, функціональні порушення щитоподібної залози — у 20–25% випадків.
Ознаки гіперкоагуляції, за даними коагулограми, виявляють у значної кількості пацієнтів з хворобою Шегрена, проте тяжкі прояви спостерігають тільки у хворих з деструктивним васкулітом і II типом кріоглобулінемії. Рецидивуючі тромбози глибоких вен гомілок, ілеофеморальні тромбози, тромбоемболії судин легенів, нирок та головного мозку за відсутності АКЛ нерідко ускладнюють перебіг захворювання у пацієнтів із тромбоцитопенією. У разі відсутності антитіл до тромбоцитів необхідно проводити обстеження для виключення лімфопроліферативних захворювань і мієлодиспластичного синдрому (стернальна пункція, імуногістоцитохімічне дослідження клітин периферичної крові і трепанобіоптатів).
Інструментальні дослідження
Для виявлення сухого кератокон’юнктивіту використовують тест Ширмера: зменшується сльозовиділення після стимуляції нашатирним спиртом (<10 мм/5 хв).
Крім тесту Ширмера, для виявлення сухого кератокон’юнктивіту використовують забарвлення епітелію кон’юнктиви і рогівки бенгальським рожевим і флуоресцеїном: це дозволяє виявити поверхневі ерозії, вогнища дистрофії епітелію, зменшення часу розриву слізної плівки на рогівці <10 с при біомікроскопії.
При діагностиці паренхіматозного сіаладеніту використовують сіалографію — рентгеноконтрастне дослідження слинних залоз для виявлення анатомічних змін. При хворобі Шегрена реєструють локальні розширення вивідних проток. Важливий метод діагностики сіаладеніту в рамках хвороби/синдрому Шегрена — біопсія малих слинних залоз: виявлення 100 і більше клітин в 4 мм при перегляді двох полів зору і не менше чотирьох малих слинних залоз. Про наявність сіаладеніту свідчить зменшення стимульованої секреції слини <2,5 мл/5 хв при виконанні сіалометрії (тест необов’язковий, його можна не використовувати як критерій, але необхідно застосовувати при диференційній діагностиці, контролі за проведеною терапією).
Рентгенологічні та морфологічні зміни можуть демонструвати трахеобронхіт (з рецидивуючою інфекцією або без неї), бронхопневмонію, лімфоцитарну інтерстиціальну пневмонію, дифузний інтерстиціальний фіброз, дископодібні ателектази, бронхоектази, адгезивні і випітні серозити і фокуси лімфоїдної інфільтрації в паренхімі легень. КТ органів грудної клітки порівняно з бронхоальвеолярним лаважем, рентгенологічними та функціональними дослідженнями легень — найінформативніший метод для уточнення характеру патологічного процесу і легеневої тканини при хворобі Шегрена. При функціональному дослідженні легень можна виявити рестриктивні й обструктивні зміни, пов’язані з ураженням верхнього та нижнього відділів дихального тракту.
Для діагностики аутоімунного захворювання визначають ревматоїдний/антинуклеарний фактор і антитіла до Rо/Lа-ядерних антигенів.
Діагностичні критерії (Європейська група з вивчення хвороби Шегрена, 1993)
1. Суб’єктивні очні симптоми: позитивна відповідь не менш ніж на 1 з нижчеперелічених питань:
- Чи турбує вас протягом останніх 3 міс постійна болісна сухість очей?
- Чи турбує вас періодично відчуття піску в очах?
- Чи використовуєте ви штучні сльози частіше ніж 3 рази на день?
2. Суб’єктивні симптоми з боку ротової порожнини: позитивна відповідь не менш ніж на 1 з нижчеперелічених питань:
- Чи турбує вас постійне відчуття сухості в роті протягом останніх 3 міс і більше?
- Чи турбує вас постійно або періодично припухлість слинних залоз?
- Ви часто п’єте воду, щоб проковтнути суху їжу?
3. Об’єктивні ознаки ураження ока: виявлення об’єктивних даних ураження ока на підставі позитивного результату не менше одного з наступних тестів:
- Тест Ширмера (≤5 мм за 5 хв)
- Тест із бенгальським рожевим (≥4 за бальною системою ван Бийстервельда)
4. Дані біопсії: середній бал при біопсії слинної залози ≥1 (вогнище-скупчення не менше 50 лімфоцитів, середній бал визначається як кількість вогнищ у 4 мм2 залозистої тканини).
5. Ураження слинних залоз: виявлення об’єктивних даних ураження слинних залоз на підставі наступних тестів (не менше одного):
- Сцинтиграфія слинних залоз
- Сіалографія привушних залоз
- Нестимульована слинотеча (≤1,5 мл за 15 хв)
6. Антитіла: присутність у сироватці крові анти-Ro/SS-A і/або анти-La/SS-B-антитіл:
- Позитивний РФ
- Позитивний АНФ
Необхідно виключити: лімфома, СНІД, саркоїдоз, реакція «трансплантат проти господаря», сіаладеноз, застосування антидепресантів, нейролептиків, гіпотензивних, ваголітичних засобів.
За наявності 4 або більше із 6 вищеперелічених критеріїв можна встановити діагноз ймовірного первинного синдрому Шегрена. Наявність першого або другого критерію і двох критеріїв із групи № 3–5 дозволяє діагностувати ймовірний первинний синдром Шегрена.
Чутливість становить 97,3–97,5%, специфічність — 91,8–94,2%.
Диференційний діагноз хвороби Шегрена необхідно проводити насамперед із захворюваннями, при яких найбільш часто розвивається синдром Шегрена, а саме: РА, ССД, СЧВ. Для полегшення цього завдання пропонуємо розглянути табл. 17.2, в якій узагальнено дані літератури.
Таблиця 17.2
Порівняльна характеристика клінічних та лабораторних показників при хворобі/синдромі Шегрена
|
Показник |
ХШ, % |
СШ + РА, % |
СШ + ССД, % |
СШ + СЧВ, % |
|
Ксерофтальмія |
89–91 |
84 |
68–80 |
60 |
|
Ксеростомія |
86–96 |
42–84 |
32–80 |
30–60 |
|
Збільшення привушних слинних залоз |
80–82 |
23–45 |
20–64 |
10–35 |
|
Рецидивуючі паротити |
65–66 |
15–32 |
20–36 |
10–40 |
|
Сухий ринофарингіт |
58–74 |
23–35 |
10–12 |
0–12 |
|
Субфебрилітет |
41–62 |
30–43 |
44–80 |
55–80 |
|
Артралгії |
67–74 |
100 |
90–100 |
80–89 |
|
Артрит |
4–16 |
100 |
20–22 |
44–60 |
|
Лімфаденопатія |
39–70 |
23–25 |
30–32 |
60–80 |
|
Синдром Рейно |
43–44 |
5–15 |
100 |
20–30 |
|
Гіпергаммаглобулінемічна пурпура |
13–17 |
0 |
0 |
0–5 |
|
Ураження: легень, серця, нирок |
13–14 |
5–23 |
40–68 |
25–40 |
|
Дисфагія |
10–15 |
15–23 |
30–64 |
20–60 |
|
Медикаментозна алергія |
35–40 |
23–25 |
20–28 |
20–45 |
|
Гемоглобін <110 г/л |
66–85 |
50–54 |
50–60 |
0–10 |
|
Лейкоцити <4×109/л |
8–66 |
7–45 |
20–28 |
10–30 |
|
ШОЕ >15 мм/год |
18 |
25 |
17 |
44 |
|
Загальний білок >85 г/л |
38 |
45 |
8 |
22 |
|
Глобуліни
|
92 |
95 |
50 |
89 |
|
62 |
30 |
17 |
28 |
|
|
Фібриноген >4 г/л |
48 |
70 |
42 |
72 |
|
Серомукоїд >0,30 ОД |
74 |
70 |
42 |
72 |
|
Церулоплазмін >0,300 г/л |
36 |
75 |
50 |
56 |
|
СРБ >3,0 мг% |
46 |
95 |
50 |
83 |
|
Тимолова проба >6,0 ОД |
46 |
70 |
42 |
33 |
|
РФ ≥ 1/80 |
38 |
65 |
17 |
61 |
|
АНФ ≥1/16 |
90 |
55 |
42 |
56 |
|
Комплемент СН50
|
90 |
95 |
67 |
56 |
|
40 |
45 |
25 |
78 |
|
|
ЦІК
|
48 |
55 |
17 |
6 |
|
16 |
5 |
25 |
61 |
|
|
Кріоглобуліни від +++ до ++++ |
86 |
75 |
50 |
56 |
|
Антитіла до нативної ДНК радіоімунологічним методом, помірне/значне підвищення |
46 |
20 |
17 |
22 |
|
Анти-Ro |
16 |
30 |
8 |
11 |
|
Анти-Sm |
58–74 |
23–35 |
10–12 |
0–12 |
|
Анти-РНП |
41–62 |
30–43 |
44–80 |
55–80 |
ХШ — хвороба Шегрена; СШ — синдром Шегрена.
При односторонньому ураженні слинних залоз хворобу Шегрена слід диференціювати з новоутвореннями (частіше первинні пухлини слинних залоз). Офтальмологічні та імунологічні зміни, а також наявність або відсутність ЗЗСТ допомагає уточнити діагноз. Однак у таких випадках вирішальним є морфологічне дослідження.
Дифузне збільшення слинних і слізних залоз зі зниженням секреції може бути зумовлено лейкозом, лімфомою або амілоїдозом (синдром Микулича). Аналогічні зміни можуть бути і при саркоїдозі. Диференційну діагностику в цих випадках проводять з урахуванням ознак, властивих цим захворюванням, а також характерних лабораторних і морфологічних проявів.
Лікування хвороби Шегрена досі становить невирішену проблему. Незважаючи на застосування ГК і цитостатичних препаратів, у багатьох хворих не вдається істотно зменшити прояв сухості, особливо при хронічному варіанті хвороби Шегрена і високій стадії захворювання.
Приклади формулювання діагнозів
Хвороба Шегрена (первинний синдром Шегрена), підгострий перебіг, виражена стадія, ІII ступінь активності, двосторонній паротит із гіпофункцією слинних залоз ІI ступеня, стоматит, гіполакримія ІI ступеня, сухий кератокон’юнктивіт; ураження суглобів — рецидивуючий неерозивний артрит; судин — рецидивуюча гіпергаммаглобулінемічна і кріоглобулінемічна пурпура; гіпотонія стравоходу, атрофічний гастрит із секреторною недостатністю, панкреатит.
Хвороба Шегрена, хронічний перебіг, пізня стадія, активність І ступеня з ураженням слинних залоз і гіпофункцією ІII ступеня, стоматит, пришийковий карієс з випадінням зубів, ураження слізних залоз і очей з гіполакримією ІII ступеня, дистрофія рогівки ІI ступеня, гастрит із вираженою секреторною недостатністю.
Хвороба Шегрена, підгострий перебіг, активна фаза, III ступінь активності з ураженням слинних залоз (двобічний паротит, синдром ксеростомії II ступеня зі стоматитом, парадонтозом і пришийковим карієсом), слізних залоз (ксерофтальмія I ступеня, кон’юнктивіт); поліартрит з переважним ураженням суглобів кистей, променезап’ясткового суглоба, суглобова функціональна недостатність II, з ураженням судин (гіпергаммаглобулінемічна пурпура), травної системи (гастрит, значне зниження секреторної функції).
Лікування
Терапія хвороби Шегрена залежить від варіанта перебігу хвороби, стадії розвитку і ступеня активності процесу, а також вираженості системних проявів, включаючи симптоматичне лікування (яке, як правило, є основним).
Наявність у більшості хворих різних клінічних проявів системності процесу — підвищення температури тіла, стійких поліартралгій, поліартриту, серозиту, вісцеральної патології, створює передумови для призначення протизапальної та цитостатичної імуносупресивної терапії.
Немедикаментозне лікування
Хворобу Шегрена діагностують переважно у жінок, схильних до розвитку різних форм психопатологічних розладів (тривожно-фобічні, іпохондричні, неврастенічні), тому їм слід обмежувати психоемоційне навантаження. Необхідно виключати інсоляцію внаслідок поганої переносимості та частого розвитку фотодерматозів. Небажане тривале перебування в районах із сухим, жарким кліматом.
У приміщеннях доцільно використовувати зволожувачі повітря. При значній функціональній недостатності слинних і слізних залоз необхідно виключити великі голосові та зорові навантаження, роботу в запилених приміщеннях і з хімічними речовинами.
Пацієнтки повинні постійно застосовувати фторовмісні та протизапальні зубні пасти, проводити санацію вогнищ інфекції, профілактику грибкової інфекції порожнини рота і піхви.
У хворих зі значним збільшенням привушних/піднижньощелепних слинних і слізних залоз з паренхіматозним сіаладенітом рецидивуючого характеру не можна проводити рентгенотерапію в ділянці привушних/піднижньощелепних слинних і слізних залоз.
Для профілактики ОП особам, які довго отримують низькі дози преднізолону і особливо інтенсивну терапію (пульс-терапія ГК і цитостатиками, поєднання екстракорпоральних методів лікування з пульс-терапією), рекомендують приймати їжу з високим вмістом кальцію.
При шлунково-кишковій патології необхідно дотримуватися відповідної дієти.
Медикаментозне лікування
Лікування кожного хворого проводять індивідуально залежно від характеру перебігу, залозистих, позазалозистих проявів та активності захворювання. Найважливіші групи лікарських препаратів — ГК, імуносупресивні препарати з антилімфопроліферативною дією (хлорамбуцил, циклофосфамід), НПЗП (диклофенак натрію, мелоксикам, німесулід та ін.) у стандартних терапевтичних дозах можна застосовувати короткими курсами при загостренні хронічного сіаладеніту і суглобових проявів.
Антималярійні препарати (гідроксихлорохін) не слід використовувати в лікуванні хвороби Шегрена, оскільки вони виявилися неефективними в контрольованих дослідженнях.
ГК (5 мг/добу або 5 мг через добу) призначають хворим з рецидивуючими сіаладенітами і мінімальними системними проявами — суглобовим синдромом і нирковим канальцевим ацидозом. Низькі дози ГК знижують частоту рецидивів сіаладеніту, згладжують системні прояви і покращують якість життя, але не збільшують салівацію. Є дані, що терапія ГК, впливаючи на загальні системні прояви хвороби Шегрена, сприяє кращому перебігу паротиту та «сухого синдрому», тобто вже в 1-шу добу нормалізується температура тіла, протягом тижня зменшуються розміри привушних залоз, хворі відзначають зменшення сухості в роті та очах. Найчастіше призначають преднізолон у дозі 10–40 мг/добу або інші препарати в еквівалентних дозах (метилпреднізолон, тріамцинолон) залежно від вираженості процесу протягом 3–4 тиж з наступним зниженням на 2,5–5 мг/тиж до підтримувальних доз (5–15 мг/добу).
Розроблено показання до цитостатичної терапії. Вона небезпечна не тільки у зв’язку з частотою алергічних реакцій на лікарські препарати, а й через потенційний ризик розвитку злоякісних лімфом. Однак клінічний досвід Інституту ревматології РАМН показує, що цитостатики в поєднанні з ГК показані за високої імунної активності (високі титри РФ, ЦІК, гіпергаммаглобулінемія, кріоглобулінемія та ін.), яка супроводжує міозит, рецидивуючий артрит, поліневрити, цереброваскуліт, ураження нирок, розвиток дифузної лімфаденопатії, ознаки васкуліту. В якості препаратів вибору рекомендують азатіоприн (100–150 мг/добу), хлорамбуцил (4–8 мг/добу), в окремих випадках циклофосфамід (зокрема, при проведенні пульс-терапії).
В окремих випадках за показаннями призначають антикоагулянти (гепарин 5000–10 000 ОД 4 рази на добу підшкірно (під контролем часу згортання крові), еноксипарин натрію 20 мг 1 раз на добу підшкірно, надропарин кальцію 0,3 мл 1 раз на добу підшкірно), ейкозаноїди (простагландин Е1 20 мкг у 250 мл фізіологічного розчину в/в крапельно протягом 3 год через день або щодня № 5–20; аналог простацикліну — ілопрост 20 мкг в/в протягом 6 год № 5 на курс).
Лікування основного захворювання при вторинному синдромі Шегрена
Інститутом ревматології РАМН пропонуються схеми використання ГК, цитостатиків і пульс-терапії в лікуванні системних проявів хвороби Шегрена (табл. 17.3).
Таблиця 17.3
Схеми застосування гормональних, цитостатичних препаратів і пульс-терапії в лікуванні системних проявів хвороби Шегрена
|
Клінічні симптоми |
Преднізолон (мг) |
Хлорамбуцил (мг) |
Пульс-терапія |
|
Рецидивуючий неерозивний артрит, інтенсивні артралгії |
5–10 |
— |
— |
|
Помірний міозит |
20–30 |
4–6 |
— |
|
Тяжкий міозит |
30–80 |
6–8 |
+ |
|
Дифузна лімфаденопатія, |
30–60 |
6–8 |
+ |
|
Плеврит, перикардит випітний |
30–60 |
6–8 |
+ |
|
Інтерстиціальна пневмонія, альвеолярний легеневий фіброз |
30–60 |
— |
— |
|
Імунокомплексний гломерулонефрит |
30–40 |
4–6 |
— |
|
Гломерулонефрит |
40–60 |
6–8 |
— |
|
Гломерулонефрит при кріоглобулінемії |
40–60 |
Циклофосфамід 200 мг — в/м 1 раз на тиждень |
+ |
|
Кріоглобулінемічна пурпура |
20–30 |
Циклофосфамід 200 мг — в/м 1 раз на тиждень |
+ |
|
Виразково-некротичний васкуліт |
20–30 |
Циклофосфамід 200 мг — в/м 1 раз на тиждень |
+ |
|
Поліневрит з руховими чутливими порушеннями |
60–100 |
Циклофосфамід 200 мг — в/м 1 раз на тиждень |
+ |
|
Цереброваскуліт |
60–100 |
Циклофосфамід 200 мг — в/м 1 раз на тиждень |
+ |
|
Гіпергаммаглобулінемічна пурпура |
60–100 |
6–8 |
— |
|
Аутоімунна гемолітична анемія |
5–10 |
Циклофосфамід 200 мг — в/м 1 раз на тиждень |
+ |
в/м — внутрішньом’язово.
При тяжких системних проявах хвороби Шегрена застосовують також пульс-терапію метилпреднізолоном у великих дозах (1000 мг в/в протягом 3 днів) і циклофосфамідом (одноразово 1000 мг) з подальшим призначенням преднізолону в помірних дозах (30–40 мг) і цитостатиків (хлорамбуцил 6 мг/добу або циклофосфамід 200 мг внутрішньом’язово 2 рази на місяць).
Показання до проведення екстракорпоральної терапії
Абсолютні:
- виразково-некротичний васкуліт на тлі кріоглобулінемії з моноклональними антитілами до РФ в кріопреципітаті;
- кріоглобулінемічний гломерулонефрит;
- енцефаломієлополірадикулоневрит, демієлінізуюча мієлопатія, поліневрит, ішемія верхніх і нижніх кінцівок внаслідок кріоглобулінемічного васкуліту.
Відносні:
- гіпергаммаглобулінемічна пурпура;
- мононеврит;
- медикаментозний дерматит, набряк Квінке, феномен Артюса;
- альвеолярний легеневий фіброз.
Екстракорпоральну терапію проводять у комбінації з пульс-терапією ГК або комбінованою пульс-терапією.
При алергічних реакціях, інтерстиціальному нефриті з ознаками хронічної ниркової недостатності, тяжких офтальмологічних проявах (бульозно-нитчастий кератит, виразки рогівки) переважно застосовують гемосорбцію. При інших системних проявах ефективніше призначати плазмаферез, кріоаферез і подвійну фільтрацію плазми. Останні два методи ефективні при змішаній моноклональній кріоглобулінемії. Зазвичай процедури проводять з інтервалом у 2–5 днів з введенням після закінчення процедури 250–1000 мг преднізолону і 200–1000 мг циклофосфаміду залежно від тяжкості системних проявів та імунозапальної активності захворювання. Для хворих з нормальним або низьким загальним білком переважно застосовують подвійну фільтрацію плазми, кріоаферез із кріофракціонуванням плазмових білків. Усього проводять 3–5 процедур за наявності гіпергаммаглобулінемічної пурпури і 5–8 процедур при кріоглобулінемічній пурпурі. При васкуліті, зумовленому змішаною моноклональною кріоглобулінемією, доцільно використовувати програмний плазмаферез протягом року до досягнення стійкої клініко-лабораторної ремісії.
Зважаючи на наявність при хворобі Шегрена В-клітинної гіперреактивності та підвищеного ризику розвитку В-клітинних неходжкінських лімфом може бути виправданим використання моноклональних анти-СВ20-антитіл (ритуксимаб). На сьогодні результати призначення при хворобі Шегрена ритуксимабу для лікування екстранодальних МАLТ-лімфом і генералізованого кріоглобулінемічного васкуліту свідчать про перспективність цього методу. Наскільки ефективним є застосування даного препарату при залозистих формах хвороби Шегрена і у хворих із системними проявами захворювання, уточнюють.
З інших методів загального лікування хвороби Шегрена слід виділити застосування інгібіторів протеаз для поліпшення слізної та слинної секреції. Рекомендується в/в введення апротиніну в дозі 10 000–30 000 ОД або 25 000–50 000 ОД на фізіологічному розчині через день. Курс лікування — 4–5 введень. При першому введенні рекомендується доза апротиніну 10 000 ОД або 25 000 ОД. При гарній переносимості препарату дозу підвищують в 2–3 рази залежно від ступеня активності процесу.
При сухості трахеї і бронхів рекомендується тривалий прийом бромгексину по 8–16 мг 3 рази на добу і протягом 2–3 міс. Для поліпшення обмінно-трофічних процесів (особливо при атрофічному гастриті) застосовують метаболічні препарати по 2–4,0 мл на добу внутрішньом’язово протягом 2 тиж. Оскільки при хворобі Шегрена відзначають активацію перекисного окиснення ліпідів і накопичення малонового діальдегіду (що є його найбільш токсичним продуктом), некерованого в результаті недостатності антиоксидантної системи, таким хворим показано прийом антиоксидантів.
При патології травної системи залежно від наявності секреторної недостатності в якості замісної терапії застосовуються ферментні препарати (підшлункової залози та ін.).
Для регуляції тонусу і перистальтики сприятливу дію має метоклопрамід. Враховуючи часту наявність дискінезій жовчного міхура та жовчовивідних шляхів за гіпотонічним типом при хворобі Шегрена, ефективним є призначення холеретиків, а також дуоденального зондування та беззондового тюбажу. У випадках розвитку кишкового дисбактеріозу показані біфідобактерії.
Місцеве лікування включає застосування різних препаратів, які зменшують сухість слизових оболонок. При ксерофтальмії з метою профілактики сухого кератокон’юнктивіту та його ускладнень (перфорації рогівки і розвитку сліпоти) необхідно раннє застосування замісної терапії. Найбільшого поширення набуло використання 5–10% розчину ацетилцистеїну, 0,5% розчину метилцелюлози, фізіологічного розчину, полімерних розчинів та інших, які потрібно закрапувати кожні 1–2 год. Олійних крапель слід уникати через небезпеку розвитку аспіраційної пневмонії. З метою впливу на регенерацію епітелію рогівки і кон’юнктиви використовують вітамінні краплі 1–2 рази на добу. Для посилення функції слізних залоз призначають електрофорез безпосередньо на ділянку слізних залоз із дексаметазоном. Лікування сухого кератокон’юнктивіту при хворобі Шегрена здійснюється обов’язково спільно з лікарем-окулістом.
Ксеростомія важко піддається лікуванню. Через нестачу слини хворі змушені з метою її заміщення постійно змочувати порожнину рота рідиною, запивати водою грудочки їжі. Деякі засоби, що застосовуються для змащування слизової оболонки рота, такі як 1% розчин метилцелюлози, мають тенденцію викликати відчуття ще більшої сухості, коли їх дія закінчується. Місцеве застосування 5–10% метилурацилової та мазей метаболічних препаратів дає нетривалий та непостійний ефект. Для лікування паротиту (сіаладеніту) ефективним є застосування диметилсульфоксиду у вигляді місцевих аплікацій 30% розчину. Найчастіше диметилсульфоксид (30% розчин 150 мл) призначають у поєднанні з гідрокортизоном (125 мг), гепарином (25 000т ОД) і 5% розчином аскорбінової кислоти у вигляді аплікацій на ділянку привушних залоз тривалістю 20–30 хв щоденно, на курс лікування — 10–15 процедур (кількість таких курсів протягом року — 3–4). Застосовують також у вигляді аплікацій на слизову оболонку порожнини рота (комплекс рибонуклеотидів) протягом 2 тиж. Препарат має протизапальну, імуномодулюючу дію, сприяє нормалізації десквамативних і регенераторних процесів слизової оболонки рота. Застосування у вигляді аплікацій сприяє гідролізуванню слизової оболонки рота, має протимікробну дію, полегшує прийом їжі та знижує можливість травматизації слизової оболонки, не викликаючи при цьому подразнюючої дії. Для лікування сіаладеніту застосовують внутрішньопротокове введення метилпреднізолону від 20 до 50 мг, розчиненого в 0,5–1,0% прокаїну, в кожну залозу (курс лікування — 6 введень), за необхідності — антибіотики, а також місцеві новокаїнові блокади. Лікування проводять спільно зі стоматологом.
При сухості слизової оболонки носа застосовують часті аплікації турундами, змоченими у фізіологічному розчині. Сухість слизової оболонки піхви зменшується при використанні калію йодиду.
Критерії якості лікування:
- Позитивна динаміка або нормалізація клінічних проявів захворювання.
- Нормалізація лабораторних показників, включаючи титри в сироватці крові анти-Ro/SS-A і/або анти-La/SS-B антитіл.
- Поліпшення гістологічної картини біопсії слинної залози.
- Негативні результати об’єктивних тестів ураження очей.
Профілактика при хворобі Шегрена спрямована на попередження загострень хвороби, правильне працевлаштування, своєчасне призначення симптоматичної терапії, лікування інтеркурентних інфекцій, за показаннями — санація вогнищ інфекції. Дані хворі підлягають комплексному диспансерному спостереженню у ревматолога, офтальмолога і стоматолога.
Прогноз
При тривалому перебігу хвороби Шегрена відзначають прогресування стоматологічних ознак захворювання, розвиток лімфом у слинних залозах у 14,8% хворих. Паралельно зі збільшенням вираженості ксеростомії прогресують ураження ЖКТ, інтерстиціальні зміни в легенях, тубулоінтерстиціальне ураження нирок з розвитком хронічної ниркової недостатності, підвищується частота ураження периферичної нервової системи. Для хвороби Шегрена характерне повільне прогресування залозистих і позазалозистих проявів, за винятком хворих з деструктивними формами васкуліту. Змішана моноклональна кріоглобулінемія служить прогностично несприятливим проявом хвороби Шегрена: 20-річна виживаність при хворобі Шегрена зі змішаною моноклональною кріоглобулінемією становить 41,9% порівняно з 81,3% у хворих, які не мають змішаної моноклональної кріоглобулінемії. Змішана моноклональна кріоглобулінемія асоційована з виразково-некротичною формою деструктивного васкуліту та розвитком лімфом. Незалежні фактори ризику летального результату при даному захворюванні: генералізована лімфаденопатія, спленомегалія, кріоглобулінемічна пурпура, поліневропатія, значне збільшення привушних залоз, анемія, кріоглобулінемія, тромбоцитопенія, лейкопенія і зниження СЗ-/С4-фракцій комплементу.
Середній вік на момент летального результату становить близько 57 років. Генералізований васкуліт (37,7%), неходжкінські лімфоми (34,5%), неоплазії (переважно рак шлунка, 16,4%) та аутоімунні панцитопенії (6,6%) — основні причини випадків смерті при даному захворюванні.
Глава 18. Змішане захворювання сполучної тканини
Синдром Шарпа (М35.1).
ЗЗСТ — своєрідний клініко-імунологічний синдром системного запального ураження сполучної тканини, який проявляється поєднанням окремих ознак ССД, ДМ, СЧВ, появою антитіл до розчинного ядерного рибонуклеопротеїду у високих титрах. Прогноз результату ЗЗСТ більш сприятливий, ніж при тих захворюваннях, з ознак яких складається синдром. Крім того, у 25–50% хворих розпізнають синдром Шегрена. Більшість ревматичних хвороб мають певну нозологічну окресленість, яка відображена в діагностичних критеріях для кожної з них. Однак у порівняно невеликої кількості пацієнтів діагностика цього виду патології значно ускладнена, оскільки захворювання не відповідає лише одній нозологічній формі.
Термін «змішане захворювання сполучної тканини», запропонований G. Sharp і співавторами (1972), об’єднав різні клінічні прояви за спільною ознакою — наявністю антитіл до рибонуклеопротеїду — в одну клінічну форму, яку іноді називають синдромом Шарпа. Для ЗЗСТ, крім одночасного або послідовного виникнення симптомів двох або трьох захворювань (ССД, СЧВ, ДМ), високих титрів сироваткових антитіл до U1 рибонуклеопротеїду, характерні відносна рідкість ураження нирок, висока ефективність ГК і досить сприятливий прогноз. Незважаючи на численні дослідження, проведені в різних країнах, у тому числі й Росії (Інститут ревматології РАМН), сутність ЗЗСТ все ще залишається нерозкритою. Так, при тривалому спостереженні за хворими відзначено зміну клінічних симптомів й імунологічних порушень. Антитіла до U1 рибонуклеопротеїду — характерний імунологічний маркер ЗЗСТ, присутність якого дозволило включити ЗЗСТ у підклас «системні ураження сполучної тканини» — хвороба Шегрена за класом МКХ-10.
Поширеність ЗЗСТ не вивчено. За даними дослідження, проведеного в Японії, ЗЗСТ виявляють лише в 2,7% випадків при системних ураженнях сполучної тканини, тоді як СЧВ — в 20,9%, а ССД та поліміозит — в 5,7 і 4,9% відповідно. Хворіють переважно жінки у віці 20–30 років, їх кількість відносно чоловіків становить від 9:1 до 16:1. Відмінностей за расовими або етнічними ознаками не виявлено.
Етіологія і патогенез
На сьогодні обговорюється роль генетичних факторів при ЗЗСТ, оскільки описано сімейні випадки захворювання з ідентичним НLА-фенотипом. Характерно, що у 66% хворих антитіла до U1 рибонуклеопротеїду асоціюються з НLА-DR4-фенотипом. У розвитку хвороби основне значення мають імунні порушення, які проявляються постійно високим титром антитіл до U1 рибонуклеопротеїду (антинуклеарних антитіл крапчастого типу світіння за непрямої імунофлуоресценції, що належать до класу IgG і спрямовані до ядерного рибонуклеопротеїду), гіпергаммаглобулінемією, зниженням вмісту комплементу, наявністю ЦІК, а також зміною імунорегуляторних функцій Т-лімфоцитів (зниження супресорної функції).
Припускають, що розвиток ЗЗСТ зумовлений молекулярною мімікрією між 33 кД поліпептиду U1 рибонуклеопротеїду і подібним компонентом у ретровірусах тварин.
Припускають також, що антитіла до U1 рибонуклеопротеїду проникають у клітини через Fс-рецептори і руйнують їх. У зв’язку з тим, що Fс-рецептори локалізуються переважно на Т-супресорах, відбувається руйнування останніх, у результаті чого відзначають проліферацію аутореактивних Т-хелперів, що і пояснює аутоімунний характер патогенезу ЗЗСТ.
У стінках судин м’язів, клубочків нирок і дермоепідермальному стику дерми виявлено депозити IgG, IgМ і комплементу, а в уражених тканинах — лімфоїдні та плазмоклітинні інфільтрати. Для ЗЗСТ характерний також розвиток проліферативних процесів у внутрішній і середній оболонках великих судин з клінічними ознаками легеневої гіпертензії та іншими проявами патології судин.
Клінічний перебіг
Клінічний перебіг ЗЗСТ характеризується значним поліморфізмом і динамізмом.
У ранній період ЗЗСТ переважають «периферичні» ознаки, які зазвичай асоціюються з початком ССД, — артралгія та артрит (65–42% випадків), синдром Рейно (65%), набряк пальців рук і всієї кисті, рідше міозит (23%), лімфаденопатія, лихоманка; типові шкірні симптоми СЧВ і ДМ виникають рідше, приблизно в 8% випадків. Вісцерити у ранній період ЗЗСТ у вигляді серозиту, міокардиту відзначають в поодиноких випадках. У середині перебігу захворювання разом зі збільшенням кількості хворих з «периферичними» симптомами значне місце займає вісцеральна патологія. Відзначають симптоми, властиві ССД (гіпотензія стравоходу, базальний пневмофіброз, міокардоз тощо), а також СЧВ (серозит, пневмоніт, ураження нирок, ЦНС та ін.); приблизно у 65% хворих — міозит, слабкість проксимальних м’язових груп; у 23% — синдром Шегрена. У цей період нерідко (близько у 27% випадків) виявляють окремі шкірні симптоми СЧВ (еритематозні висипи у вигляді метелика, капілярити), а також (в 23%) — ССД (ділянки ущільнення шкіри на кистях, амімія, гіперпігментація, телеангіектазії).
У термінальний період значно знижується частота виникнення артралгії, набряку кистей, міозиту, лімфаденопатії, лихоманки, еритематозних висипів у вигляді метелика, капіляритів, серозити відзначають у вигляді плевральних і/або перикардіальних спайок. Крім того, помітно регресують симптоми запальної активності. Водночас частіше проявляються склеродермоподібні шкірні синдроми, у 50% хворих зберігаються ураження суглобів, у ⅔ — синдром Рейно. G.G. Sharр і співавтори (1975), узагальнивши дані багаторічних спостережень 200 хворих, виділили клінічні симптоми ЗЗСТ, які найчастіше виникають (табл. 18.1).
Таблиця 18.1
Клінічна характеристика анти-U1-рибонуклеопротеїд-позитивних хворих на ЗЗСТ
|
Клінічні прояви |
Частота, % |
|
Артрит/артралгії |
95 |
|
Синдром Рейно |
85 |
|
Гіпотонія стравоходу |
67 |
|
Зниження дифузної здатності легень |
67 |
|
Набряк кистей |
66 |
|
Міозит |
63 |
|
Лімфаденопатія |
39 |
|
Шкірні висипи |
38 |
|
Акросклероз |
33 |
|
Лихоманка |
33 |
|
Серозит |
27 |
|
Спленомегалія |
19 |
|
Гепатомегалія |
15 |
|
Гломерулонефрит |
10 |
|
Неврологічні розлади |
10 |
В останні роки відзначають підвищення частоти розвитку більш важкої легеневої патології — фіброзуючого альвеоліту і легеневої гіпертензії як найбільш важливих ознак хвороби, які є особливо несприятливими прогностично.
Наведені дані збігаються з даними В.А. Насонової та співавторів (1997), за винятком частоти ураження нирок і виникнення синдрому Шегрена, які у 26 спостережуваних автором хворих виникали в 10 разів частіше.
Ураження опорнорухового апарату при ЗЗСТ супроводжується ознаками, властивими трьом нозологічними формами — РА, СЧВ, ССД. Таке поєднання часто виявляють у одного хворого. При цьому процес відрізняється доброякісністю, що не властиво класичним варіантам кожного з цих захворювань (особливо РА і ССД).
Ураження суглобів характеризується нестійким мігруючим поліартритом з переважним ураженням періартикулярних тканин, а також мігруючими поліартралгіями.
Клінічні ознаки поліартриту, як правило, відзначають у ⅔ хворих, при цьому відсутні істинно прогресуючі ураження суглобового хряща і кістки, що властиво РА. Однак зрідка при ЗЗСТ розвивається ульнарна девіація, відзначаються підвивихи в суглобах деяких пальців кисті, рентгенологічно в одиничних випадках визначаються поверхневі ерозії в деяких дрібних суглобах кистей і/або стоп. Характерне втягнення в процес великих суглобів поряд з ураженням дрібних суглобів кистей, як при СЧВ.
Крім того, при ЗЗСТ можливе виникнення аваскулярного некрозу кісток суглобів, раніше не уражених запальним процесом (двосторонній некроз головок стегнових кісток, некроз головки плечової кістки тощо), характерне для СЧВ, а також рентгенологічними методами виявляють кальциноз м’яких тканин кінцівок, остеоліз кінцевих фаланг пальців верхніх кінцівок (характерні для ССД).
Синдром Рейно — одна з найбільш частих і ранніх ознак ЗЗСТ. Тільки в 50% випадків цей синдром характеризується типовою дво- і трифазною зміною кольору акральних відділів кінцівок і на відміну від ССД — вкрай рідко ішемічним некрозом або виразками.
Набряк пальців («сосископодібні пальці») або всієї кисті, як правило, супроводжується синдромом Рейно, характеризується м’яким, подушкоподібними набряком, рідко щільним, практично не завершується індурацією й атрофією шкіри зі стійкими згинальними контрактурами (як при ССД).
Клінічна картина при ураженні м’язів у пацієнтів із ЗЗСТ різна — від минущих локальних міозитів і міалгій до вираженої картини поліміозиту з типовим ураженням проксимальних м’язових груп і значною м’язовою слабкістю. Вміст сироваткових ферментів (КФК, альдолази, АсАТ) підвищується помірно і швидко нормалізується (так само, як і клінічна картина міозиту) під впливом терапії ГК у середніх дозах.
Результати тривалого спостереження пацієнтів із ЗЗСТ свідчать, що поліміозит може бути причиною смерті майже 40% з них. Ураження стравоходу, зокрема його гіпокінезія, — досить частий синдром при ЗЗСТ, однак скарги при ньому вкрай рідкісні (в основному дисфагія, печія). У діагностиці ураження стравоходу важливе місце займають рентгенологічні дослідження, за допомогою яких виявляють уповільнення пасажу барію, ослаблення і зникнення перистальтичних хвиль, недостатність кардії, а також манометричні — для визначення порушень рухливості стінок стравоходу (на рівні нижніх двох третин), які украй рідко досягають таких значень, як при ССД.
Ураження серозних оболонок і легень виявляють більш ніж у половини хворих на ЗЗСТ. Серозит, як правило, проходить за типом адгезивного плевриту і/або перикардиту, діагностується переважно рентгенологічно. В активну фазу хвороби відзначено також випадки випітного плевриту і перикардиту. Ураження легень проявляється задишкою, легеневою гіпертензією (але частіше симптоми відсутні), порушенням дифузії. Зниження життєвої ємності легень описують у більш пізні стадії захворювання. Рентгенологічно при ЗЗСТ виявляють посилення і деформацію легеневого малюнка за типом базального пневмосклерозу, рідше дифузного з утворенням у легенях множинних кіст. Іноді в активний період хвороби відзначають симптоми вовчакового пневмоніту, фіброзуючого альвеоліта з легеневою гіпертензією, останнім часом — все частіше.
Часто діагностують перикардит, однак у дітей можливий розвиток міокардиту.
Ураження шкіри при ЗЗСТ характеризується наявністю специфічних і неспецифічних ознак різних системних і шкірних захворювань. Так, окрім так званих ознак склеродермії (м’якого або щільного набряку пальців рук або всієї кисті, амімії, натягнутості та блиску шкіри обличчя, телеангіектазій, порушення пігментації та ін.), у різні періоди хвороби виявляють специфічні для СЧВ симптоми: еритематозні висипи у вигляді метелика, капілярити, дискоїдний вовчак, алопецію, ураження слизових оболонок, особливо порожнини рота, фотосенсибілізацію, а також ознаки, характерні для ДМ (періорбітальний набряк з геліотропним забарвленням шкіри повік, симптом Готтрона). В ⅓ випадків при ЗЗСТ описано неспецифічні шкірні прояви — кропив’янку, еритематозні плями, мультиформну еритему, вітиліго та ін. Важливим критерієм ЗЗСТ є рідкість ураження нирок (за даними літератури, від 5 до 10%), а за його наявності — доброякісний перебіг. Як правило, відзначають ізольовану помірну протеїнурію та/або гематурію. При морфологічному дослідженні виявляють зазвичай сегментарний, вогнищевий або мезангіальний гломерулонефрит. Тяжкий нефротичний синдром і «склеродермічна нирка» з нирковою недостатністю розвиваються рідко.
Ураження ЦНС (цереброваскуліт, асептичний менінгіт, мігрень) виявляють рідко, набагато частіше при ЗЗСТ діагностують поліневропатії.
При ЗЗСТ може розвиватися синдром Шегрена (його перебіг переважно більш доброякісний, ніж первинного), а також тиреоїдит Хашимото.
Із загальних клінічних проявів хвороби відзначають лихоманку різного ступеня вираженості, лімфаденопатію, рідко — спленомегалію та гепатомегалію.
Показники лабораторних досліджень
Дані загальноклінічних лабораторних досліджень при ЗЗСТ неспецифічні. Більш ніж у 50% хворих виявляють помірну анемію (Hb <110 г/л), лейкопенію (рівень лейкоцитів <4–109/л), підвищення ШОЕ (>25 мм/год), рідко — тромбоцитопенію. Крім того, при ЗЗСТ найбільш часто відзначають гіпергаммаглобулінемію (>22%), причому в ⅔ випадків — при першому обстеженні.
Імунологічним маркером ЗЗСТ вважають антитіла до ядерного U1 рибонуклеопротеїду у високих титрах (за даними G.G. Shагр і співавторів — у 100% випадків, а за даними В.А. Насонової та співавторів — у 78,2%). Досить часто (76,8–94%) при ЗЗСТ імунофлуоресцентним методом визначають АНФ крапчастого типу світіння, також можлива поява РФ, рідше LЕ-клітин, гіпокомплементемії, ЦІК (частіше в активну фазу хвороби).
На тлі симптомів поліміозиту характерне підвищення активності КФК, альдолази, АсАТ, ЛДГ.
Діагностичні критерії, запропоновані D. Alarcon-Segovia і співавторами (1987), підтверджені J.M. Amigues і співавторами (1996)
Серологічні критерії:
1. Високі титри анти-U1-рибонуклеопротеїдних антитіл
2. Титр гемаглютинації (1:1600)
Клінічні критерії:
1. Набряк кистей
2. Синовіт
3. Міозит (підтверджений при біопсії або лабораторними даними)
4. Синдром Рейно
5. Склеродактилія
За наявності 3 або більше з 5 вищеперелічених критеріїв можна встановити діагноз ЗЗСТ.
Чутливість становить 62,5%, специфічність — 86,2%.
Проте для встановлення остаточного діагнозу ЗЗСТ часто необхідне багаторічне спостереження і повторне ретельне обстеження хворого, оскільки іноді ЗЗСТ — лише етап у розвитку іншого ревматичного захворювання.
Диференційна діагностика ЗЗСТ та інших ревматичних захворювань, окремі симптоми якого і становлять його клінічну картину, представлена в табл. 18.2.
Таблиця 18.2
Диференційна діагностика ЗЗСТ та інших ревматичних захворювань
|
Симптом, показник |
ЗЗСТ |
СЧВ |
ССД |
ДМ |
РА |
|
Синдром Рейно |
++++ |
++ |
++++ |
Рідко |
+ |
|
Набряк кистей |
+++ |
Рідко |
+++ |
+ |
+ |
|
Артрит (в тому числі ерозивний) |
+++/+ |
++/0 |
++/+ |
Рідко |
+++/ ++++ |
|
Міозит |
+++ |
+ |
+ |
++++ |
Редко |
|
Гіпотензія стравоходу |
++ |
+ |
++++ |
++ |
+ – |
|
Пневмофіброз |
++ |
++ |
+++ |
+ |
+ |
|
Лімфаденопатія |
++ |
+++ |
Рідко |
Рідко |
+ |
|
Серозит |
+++ |
+++ |
+ |
Рідко |
+ |
|
Гломерулонефрит з нефротичним синдромом |
+/+ – |
+++/ ++++ |
+/0 |
0 |
+/+ |
|
Цереброваскуліт |
+ |
+++ |
Рідко |
0 |
Рідко |
|
Лихоманка (температура тіла вище 38 °С) |
++ |
+++ |
Рідко |
Рідко |
+ |
|
Симптом метелика, капілярити |
++ |
+++ |
0 |
0 |
0 |
|
Щільний набряк, склеродактилія |
++ |
+/– |
+++ |
– |
– |
|
Періорбітальний набряк, симптом Готтрона |
+ |
Рідко |
Рідко |
+++ |
0 |
|
Синдром Шегрена |
+ |
+ |
++ |
+- |
++ |
|
Гіпергаммаглобулінемія >22% |
++++ |
++ |
+++ |
+ |
+ |
|
Анти-U1-рибонуклеопротеїд |
++++ |
+ |
+ |
0 |
+ |
|
Антитіла до ДНК |
+ |
++++ |
+ |
Рідко |
+ |
|
LЕ-клітини |
+ |
++++ |
Рідко |
0 |
+ |
|
Підвищення ШОЕ >25 мм/год |
+++ |
++++ |
+ |
Рідко |
+++ |
|
РФ |
++ |
+ |
++ |
+/+ – |
+++ |
|
Зниження ризику серцевої недостатності на 50% |
+ |
+++ |
Рідко |
Рідко |
Рідко |
|
Анемія, Нb <110 г/л |
+ |
+++ |
Рідко |
0 |
Рідко |
|
Лейкопенія, рівень лейкоцитів <4–109/л |
+ |
+++ |
Рідко |
0 |
Рідко |
Приклад формулювання діагнозів
ЗЗСТ, початковий період, активна фаза, активність ІII ступеня з ураженням судин (повний синдром Рейно), ураженням шкіри (набряк), езофагіт, міопатія, ексудативний плеврит, перикардит.
Лікування
При ЗЗСТ високоефективні ГК (преднізолон, метилпреднізолон та ін.). Доза ГК і тривалість лікування залежать від активності процесу та характеру органних уражень. Як правило, ГК застосовують в середніх і низьких дозах курсом тривалістю 2–3 міс. ГК у високих дозах призначають при вираженому синдромі поліміозиту, а також при активному процесі і прогресуючих ознаках СЧВ (ураження нирок і ЦНС). Тривалість лікування може становити від кількох місяців до декількох років.
У разі недостатньої ефективності ГК застосовують цитостатичні препарати, частіше азатіоприн (протягом 2–24 міс).
Широкого застосування набули НПЗП, на сьогодні переважно селективні інгібітори ЦОГ-2, зокрема німесулід в дозі 100 мг 2 рази на добу, мелоксикам у дозі 7,5 мг/добу, особливо при суглобовому синдромі. За показаннями призначають судинорозширювальні засоби, антиагреганти, антикоагулянти, проводять аплікації диметилсульфоксиду, а також симптоматичну терапію.
Критерії якості лікування:
1. Позитивна динаміка клінічних проявів.
2. Зниження титрів антирибонуклеопротеїдних антитіл у сироватці крові.
3. Половина хворих, як правило, залишаються працездатними.
Прогноз при ЗЗСТ в цілому задовільний. Смертність серед хворих, за даними А.L. Claudy (1981), становить 7%. Найбільш часто причиною смерті є ниркова недостатність або легенева гіпертензія (легенева недостатність).
Глава 19. Ревматична поліміалгія
Код за МКХ-10
М35.3 Ревматична поліміалгія.
РПМ — запальне захворювання опорно-рухового апарата, розвивається в осіб у віці не молодше 50 років, характеризується сильним болем стереотипної локалізації (ділянки шиї, плечовий і тазовий пояс), порушенням рухів, значним підвищенням лабораторних показників запалення, а також настанням ремісії при призначенні ГК в невеликих дозах. РПМ часто поєднується з гігантоклітинним (темпоральним) артеріїтом (хвороба Хортона).
Поширеність. Захворювання реєструють практично на всіх континентах, у представників різних рас. За даними епідеміологічних досліджень, частота виявлення нових випадків РПМ за рік у різних країнах коливається від 4,9 до 11,1 на 100 тис. усіх жителів (12,7–68,3 на 100 тис. жителів у віці 50 років і старше). Відзначено тенденцію до меншої поширеності захворювання в країнах, розташованих ближче до екватора. У віці молодше 50 років РПМ не розвивається (відомі лише поодинокі випадки виникнення хвороби у віці 49 років). Пік захворюваності припадає на 7-й десяток життя. Приблизно в 2 рази частіше хворіють жінки.
Етіологія і патогенез
Етіологія невідома. Причина розвитку РПМ тільки в другій половині життя, переважно в осіб літнього віку, нез’ясована. Зв’язку з відомими інфекційними агентами не встановлено. Специфічних аутоімунних порушень не виявлено. Частота злоякісних пухлин при цьому захворюванні не підвищена. Існують дві гіпотези патогенезу РПМ. Відповідно до першої з них РПМ розглядають як ідіопатичне захворювання опорно-рухового апарата, симптоми якого зумовлені запаленням синовіальної оболонки (переважно плечових і кульшових суглобів) і/або періартикулярних тканин. На підтвердження цього наводяться наступні аргументи. Больовий синдром схожий на такий при артритах плечових або кульшових суглобів різної етіології (наприклад, у хворих з анкілозуючим спондилітом) і особливо при захворюваннях навколосуглобових м’яких тканин цих анатомічних ділянок. При морфологічному дослідженні синовіальної оболонки і періартикулярних тканин плечових суглобів у хворих з РПМ відзначено запальні зміни. У певної частини хворих з РПМ розвивається синовіт різних (не тільки плечових і кульшових) суглобів. При РПМ, так само як при РА, підвищена частота виявлення НLА DR4.
Часте (до 50%) поєднання захворювання з гігантоклітинним (темпоральним) артеріїтом дозволило припустити, що РПМ завжди є лише одним із синдромів цього системного васкуліту, в одних випадках клінічно явного, а в інших — прихованого. Дійсно, у частини хворих з ревматичною поліміалгією за відсутності клінічних ознак запального ураження судин у біоптатах скроневих артерій виявляють типову морфологічну картину артеріїту. Це припущення поки не вдалося повністю ні підтвердити, ні відкинути через обмеження діагностичних можливостей існуючих методів прижиттєвого дослідження судин; секційні дані при РПМ невідомі. Сумніви в обов’язковості зв’язку РПМ з артеріїтом полягають в тому, що її постійний прояв — своєрідний за локалізацією больовий синдром — не вдається пояснити запаленням відповідних судин. У синовіальній оболонці плечових суглобів і періартикулярних тканин у хворих на РПМ запальних змін судин не виявлено. При гігантоклітинному артеріїті у разі явного ураження артерій шиї, верхніх або нижніх кінцівок больовий синдром, характерний для РПМ, відзначають далеко не завжди. Подібний больовий синдром може проявлятися при таких захворюваннях, як серонегативний спондилоартрит, плечолопатковий періартрит, парапротеїнемічні гемобластози, яким васкуліт невластивий.
Патоморфологічна картина
Патоморфологічна картина РПМ вивчена недостатньо. Відомо про лише невелику кількість досліджень біоптатів скелетних м’язів і тканин плечових суглобів. Проте можна вважати доведеним, що скелетні м’язи при РПМ незмінені (ні тільки морфологічно, за даними світлової та електронної мікроскопії, але й функціонально, наприклад при ЕМГ дослідженні). При артроскопії плечових суглобів з подальшою біопсією синовіальної оболонки виявлено ознаки помірно вираженого неспецифічного синовіту. Морфологічне вивчення біоптатів періартикулярних тканин плечових суглобів показало наявність неспецифічного запалення у капсулі, міжм’язових сполучнотканинних прошарках, піддельтоподібній сумці (набряк, невелика периваскулярна лімфоїдна інфільтрація без змін стінок судин). Картина вогнищевого синовіту описана і при дослідженні синовіальної оболонки колінних суглобів (у тих випадках, коли відзначали клінічні симптоми артриту цих суглобів). Зміни синовіальної рідини, проаналізовані в тих самих випадках, класифіковані переважно як незапальні. Проте у окремих хворих кількість клітин у синовіальній рідині досягає 10 000–20 000 в 1 мм3, що свідчить про запальний характер ураження суглобів.
Клінічна картина
Клінічні прояви захворювання в переважній більшості випадків розвиваються гостро, повна картина хвороби формується на 2–4-му тижні. Першим завжди виникає сильний, стереотипний за локалізацією біль. Він охоплює ділянку шиї, плечові суглоби і плечі, кульшові суглоби і стегна. Больових відчуттів поза цими локалізаціями (нижче ліктьових і колінних суглобів, в грудному та поперековому відділі хребта, грудній клітці), як правило, немає. Біль в ділянках плечового і тазового пояса двосторонній й симетричний, постійний, посилюється при рухах і зменшується у спокої. Характер больових відчуттів ниючий, деякі пацієнти описують їх як хворобливу втому, яку вони відчували раніше після значного фізичного навантаження. Характерна також скутість, найбільш виражена вранці після сну і після будь-якого тривалого періоду нерухомості. Постійною ознакою РПМ є обмеження рухів у плечових, кульшових суглобах, а також у ділянці шиї. Активні рухи порушені зазвичай більшою мірою, ніж пасивні, але в ряді випадків і обсяг пасивних рухів, особливо в плечових суглобах, може бути суттєво зменшений. Через біль суттєво порушується можливість самообслуговування (важко зачісуватися, вмиватися, вдягатися, піднімати і утримувати що-небудь руками, сідати на низьке сидіння і вставати з нього), а також здатність пересуватися. У ряді випадків хворі змушені більшу частину часу проводити в ліжку.
При огляді та пальпації ділянок плечового і тазового пояса, а також шиї змін не виявляють. Може проявлятися тільки помірна болісність при пальпації горбків плечових кісток або великих вертлюгів стегнових кісток, яка за інтенсивністю значно нижча, ніж спонтанні больові відчуття. Хворі часто скаржаться на слабкість у м’язах, здійснюють рухи обережно і повільно, що особливо помітно при вставанні з ліжка, це нагадує картину, властиву запальним міопатіям. Але справжньої слабкості м’язів немає, утруднення рухів зумовлене болем.
Приблизно у ¼ хворих розвивається артрит. Найчастіше схильні до ураження променезап’ясткові, колінні, акроміально-ключичні суглоби і дуже рідко дрібні суглоби кистей або стоп. Як правило, кількість запалених суглобів не перевищує 1–3, симетричність ураження відсутня. Ступінь вираженості артриту переважно невисокий. Біль в уражених суглобах зазвичай несильний, він набагато менший, ніж в плечовому і тазовому поясі. Нерідко припухлість суглобів і болісність при рухах в них можна помітити тільки при цілеспрямованому огляді. У деяких хворих вдається отримати синовіальну рідину, зміни якої в більшості випадків мають незапальний або слабкозапальний характер. Артрит рідко виникає на самому початку хвороби, зазвичай він приєднується через декілька тижнів після розвитку типової клінічної картини. На тлі лікування ГК артрит швидко проходить і зазвичай надалі не рецидивує.
У 10–15% хворих розвивається слабовиражений синдром карпального каналу з типовими проявами у вигляді оніміння в кінчиках I–IV пальців кистей. Синдром карпального каналу часто поєднується з синовітом променезап’ясткових суглобів, в більшості випадків проходить після призначення ГК усередину. У невеликої частини хворих на РПМ розвивається картина долонного фасциту: помірний набряк кисті, формування невеликих згинальних контрактур пальців, ущільнення і болісність долонної фасції та сухожиль м’язів згиначів пальців. Вираженість долонного фасциту поступово (зазвичай трохи повільніше, ніж інші прояви РПМ) зменшується після початку ГК-терапії.
Нерідко відзначають лихоманку, зазвичай субфебрильну, але іноді до 38 °С і вище. Вона ніколи не передує типовим больовим відчуттям, а зазвичай приєднується в їх розпал, обтяжуючи стан хворих. У багатьох випадках досить швидко відбувається зменшення маси тіла, іноді значне, що зазвичай супроводжується втратою апетиту. Характерні також загальна слабкість, знижений настрій.
У всіх хворих з перших днів захворювання різко підвищується ШОЕ, значення якої досягає 40–50 мм/год і більше. Ступінь підвищення ШОЕ краще за інші лабораторні показники відповідає вираженості больового синдрому і порушенню рухів. Інші лабораторні показники запалення (рівень СРБ, фібриногену та ін.) також підвищуються, але на відміну від ШОЕ не мають настільки чіткого зв’язку з вираженістю клінічних проявів й ефектом терапії ГК. У багатьох хворих виникає гіпохромна анемія. Приблизно у 25% хворих виявляють підвищення рівня трансаміназ і лужної фосфатази в крові. Причина цих змін не з’ясована, але швидше за все вони відображають функціональні зміни печінки, що виникають внаслідок циркуляції в крові великої кількості медіаторів запалення. Уже після декількох днів лікування ГК активність трансаміназ і лужної фосфатази повертається до норми.
У перші 1–2 тиж хвороби біль може проявлятися тільки в якійсь одній анатомічній ділянці, наприклад тільки плечі або тазовому поясі. Але в термін до 1 міс перебігу хвороби у переважної більшості розгортається повна клінічна картина, настає пік больових відчуттів, який супроводжується значним знерухомленням, пацієнт припиняє виходити зі свого будинку і змушений проводити основний час у ліжку. Аналіз больових явищ саме в цей період захворювання особливо важливий для діагностики РПМ, до і після нього клінічна картина може відхилятися від класичної. Настання піку хвороби може бути попереджено прийомом НПЗП, найбільш сильних у відношенні протизапальної дії. Ці лікарські препарати, проте, ніколи не усувають повністю больових відчуттів і обмеженості рухів.
Призначення преднізолону всередину в невеликий дозі (15 мг/добу) практично у всіх хворих призводить до швидкого (протягом декількох днів) і значного поліпшення. Через 2–3 тиж лікування всі прояви захворювання зазвичай повністю купіруються, нормалізуються ШОЕ та інші лабораторні показники запалення.
Ще до того, як при РПМ стали застосовуватися ГК, констатовано можливість розвитку спонтанної ремісії і повного одужання. Зазвичай такий сприятливий результат розвивався через кілька років від початку захворювання.
Діагностика
Відомі такі діагностичні ознаки захворювання:
- вік пацієнта на початку хвороби не менше 50 років;
- біль принаймні у 2 з наступних 3 ділянок: плечовий, тазовий пояс і шия;
- двобічна локалізація больових відчуттів у плечовому і тазовому поясі;
- переважання зазначеної локалізації болю під час піку хвороби;
- підвищення ШОЕ >35 мм/год;
- швидкий і виражений ефект при лікуванні преднізолоном у добовій дозі не більше 15 мг;
- відсутність ознак РА.
Для діагностування РПМ необхідна наявність всіх зазначених ознак.
Велике значення має ретельне опитування пацієнтів, метою якого є виявлення типих рис РПМ або, навпаки, пересвідчення у невідповідності скарг та перебігу захворювання класичним уявленням про це захворювання. Остаточне рішення про діагноз виносять тільки після оцінки ефекту преднізолону, застосовуваного всередину в дозі 15 мг/добу (в окремих випадках 20 мг, але не більше) на термін не менше 2–3 тиж. За відсутності через цей строк повної клініко-лабораторної ремісії можна з достатньою впевненістю стверджувати, що діагноз РПМ малоймовірний.
Кожен хворий на РПМ повинен бути цілеспрямовано обстежений на предмет виявлення гігантоклітинного (скроневого) артеріїту, оскільки його наявність вимагає призначення істотно вищої дози ГК. З цією метою проводять опитування (виявлення скарг, характерних для хвороби Хортона), пальпаторне дослідження скроневих артерій, дослідження пульсу на дистальних артеріях верхніх і нижніх кінцівок, аускультацію великих артерій в ділянці шиї і в надключичних ямках, а також аорти. У тих випадках, коли фізичні локальні дані не дозволяють повністю виключити васкуліт, необхідне застосування інструментальних методів оцінки стану артерій і кровотоку в них; при підозрі на скроневий артеріїт показана біопсія цієї артерії.
До типових помилок у діагностиці РПМ належать переоцінка клінічної значущості змін, які не виходять за межі інволюційних (правильний діагноз заміняють формулюванням «поширений остеоартроз і остеохондроз»); недостатня увага до нетипових для захворювання клініко-лабораторних проявів, особливо зберігається після призначення ГК; застосування преднізолону в явно недостатній дозі (<15 мг/добу) або призначення всієї дози препарату одноразово протягом доби, або на недостатній термін.
Диференційна діагностика
Диференційну діагностику проводять з парапротеїнемічними гемобластозами (мієломна хвороба тощо), РА, серонегативними спондилоартритами (головним чином із псоріатичним спондилоартритом, який може початися в літньому віці), поліміозитом, системними васкулітами, захворюваннями м’яких тканин опорно-рухового апарату, остеомаляціями, гіперпаратиреозом, гострими інфекціями, що супроводжуються міалгіями. При підозрі на РПМ доцільно оцінити загальний аналіз крові і сечі, рівень лужної фосфатази, креатинкінази, кальцію і фосфору в крові, РФ, загального білка і його фракції. Іноді у пацієнтів на фоні довгострокового помірного підвищення ШОЕ неясного походження розвиваються регіонарні больові синдроми, подібні за локалізацією до РПМ. Зазвичай вони зумовлені різними м’якотканинними захворюваннями опорно-рухового апарату і/або остеохондрозом шийного відділу хребта та ОА кульшових суглобів, які часто розвиваються в літньому віці. Відмінності з РПМ в цих випадках стають очевидними при уважному аналізі джерела больових відчуттів кожної локалізації, часу їх розвитку і перебігу; в ряді випадків при обстеженні хворого з’являється можливість пояснити і підвищення ШОЕ (наприклад виявляють хронічні вогнищеві інфекції).
У багатьох важких для діагностування випадках вирішальне значення має оцінка результатів пробного застосування невеликої дози ГК.
Лікування
Відповідно до найбільш поширеної думки, єдиним ефективним засобом лікування РПМ є ГК. Їх застосування вважають обов’язковим, тому що вони усувають (точніше, значно знижуть) ризик приєднання хвороби Хортона. Початкова доза преднізолону (цьому препарату зазвичай надають перевагу) становить 15 мг/добу, її розподіляють на 2–3 прийоми. Якщо позитивний ефект очевидний, але на 2–3-му тижні лікування не відзначається повної клініко-лабораторної ремісії захворювання, доза преднізолону може бути підвищена до 20 мг. Після розвитку ремісії ту саму дозу преднізолону зберігають ще протягом 1 міс, а потім починають поступово знижувати до повної відміни препарату. Зниження проводять на 1,25 мг преднізолону через кожні 7–10 днів за умови відсутності ознак загострення захворювання. Якщо ж загострення (відновлення типового для РПМ больового синдрому, що супроводжується підвищенням ШОЕ) розвивається, необхідне тимчасове підвищення дози преднізолону на величину, достатню для досягнення ремісії захворювання. Загострення, які частіше виникають під час прийому невеликих підтримувальних доз преднізолону, можуть неодноразово повторюватися.
Повного одужання (зі скасуванням преднізолону) вдається досягти у всіх хворих, але час, необхідний для цього, буває різним: від 6 міс до 2–3 років. Відомі поодинокі випадки рецидивів хвороби.
Незважаючи на похилий вік хворих, преднізолон у невеликих дозах переноситься в переважній більшості випадків добре. Серйозні ускладнення, що вимагають безумовного припинення терапії, виникають рідко, особливо, якщо своєчасно і адекватно контролюються супутні захворювання. На практиці у хворих на РПМ побічні дії ГК найчастіше проявляються несиметричним болем у ділянках плечових або кульшових суглобів. Цей біль виникає на фоні попередньої ремісії через кілька місяців після початку застосування ГК та імітує загострення захворювання. Він зумовлений негативним впливом препаратів на скелетні м’язи та сухожилля, у тому числі стероїдною міопатією, не супроводжується підвищенням ШОЕ, не усувається при підвищенні дози преднізолону і добре піддається локальній терапії. Ці побічні дії ГК зменшуються у міру подальшого зниження дози преднізолону і після призначення лікарських засобів, що виявляють анаболічні властивості, та препаратів калію.
Інші лікарські засоби для лікування РПМ зазвичай не застосовують.
Глава 20. Рецидивуючий поліхондрит
М94.1 Рецидивуючий поліхондрит.
РПХ (панхондрит, системна хондромаляція, хронічний атрофічний поліхондрит) — генералізоване прогресуюче запальне захворювання хрящової тканини із залученням до процесу органів чуття (очі, вуха, вестибулярний апарат), що призводить до структурних змін хряща аж до його повного зникнення. Перший випадок РПХ описаний в 1923 р. R. Jackson-Wartenhorst; саму назву запропонували C. Pearson і співавтори в 1960 р. РПХ є рідкісним захворюванням, на яке найчастіше хворіють особи у віці 30–40 років незалежно від статі, переважно європеоїдної раси. Це захворювання може розвиватися також у дітей, підлітків, людей похилого віку.
Етіологія захворювання невідома. Вважають, що ураження хряща може бути опосередковане імунопатологічними механізмами. У хворих виявляють антитіла до колагену II типу і хряща, а також ЦІК. Крім гуморальних, є ознаки клітинних імунних реакцій — лімфоцити периферичної крові хворих на РПХ на відміну від лімфоцитів здорових активно реагують на екстракт здорового хряща, що також свідчить на користь аутоімунних механізмів розвитку РПХ.
Вважають, що деградація матриксу хряща є наслідком дисбалансу в системі протеїназно-антипротеїназної активності, що, в свою чергу, веде до розвитку аутоімунних реакцій на колаген, протеоглікани і еластин. Кисневі метаболіти, що утворюються при активації поліморфно-нуклеарних клітин, моноцитів, макрофагів, також беруть участь у пошкодженні хряща. При РПХ виявляють підвищений рівень антиколагенових антитіл, переважно до колагену IX і XI типу (Herman J.H., Dennis T.W., 1973).
Патоморфологія
При гістологічному дослідженні ураженого хряща реєструють ділянки метахромазії, дегенерації хондроцитів, входження в товщу хряща активних фібробластів, розволокнення, лізиссеквестрацію хрящового матриксу. У перихондральних тканинах виявляють вогнищеву або дифузну інфільтрацію лімфоїдними і плазматичними клітинами. У деяких відділах (склери, дуга аорти) відзначають зміни еластичних волокон (дегенерація, некроз), розпад колагенових фібрил, наступні фіброзуючі зміни, запальний процес в стінках судин (Реаrson О., 1979).
При електронно-мікроскопічному дослідженні (Наshimoto К. et al., 1977) в хрящової тканини виявлено велику кількість гранул, схожих на лізосоми. Автори висловили припущення, що ці лізосоми вивільняються з пошкоджених хондроцитів; наявність їх може зумовити деградацію протеїнополісахаридного комплексу основної речовини хряща і клітинних елементів із подальшим руйнуванням і фіброзом хрящової тканини.
Клінічна картина
РПХ має хвилеподібний перебіг. Захворювання, як правило, починається гостро, потім може набувати хронічного характеру з періодичними загостреннями тривалістю від декількох днів до місяців. Епізоди активного запалення зазвичай поєднуються із загальним нездужанням, лихоманкою і підвищенням ШОЕ. Найбільш типовими ознаками РПХ (Агабабова Е.Р., 1989) є наступні:
- запальні зміни вушних раковин, які виявляють у 58% хворих; у класичних випадках вушні раковини виглядають почервонілими, значно припухлими, вони різко болісні при доторканні, слух в цей період може бути значно знижений через запальний набряк зовнішнього слухового проходу; надалі через розвиток фіброзних змін вушні раковини деформуються, у 48% хворих відбувається обтурація слухових ходів і слух стійко знижується;
- залучення в патологічний процес хрящової перегородки носа (відзначають у 82% хворих), що проявляється його сідлоподібною деформацією; ця ознака може з’явитися навіть при першому виникненні захворювання, але частіше при рецидивах процесу; больовий синдром не завжди супроводжує розвиток деформації носа;
- лихоманка, яку частіше відзначають при перших атаках поліхондриту (у 81% хворих);
- суглобовий синдром (у 71–78% пацієнтів) може бути у вигляді артралгій; поліартриту (оскільки в патологічний процес може залучатися суглобовий хрящ), який супроводжується симптомами, що імітують запальне захворювання суглобів (хоча синовіт відсутній); ураженню підлягають багато з’єднань —як синхондрози (лобкове, грудиноключичне з’єднання), так і синовіальні суглоби; відзначають припухлість, ексудативні зміни, болісність при пальпації і обмеження рухів; рентгенологічними методами виявляють звуження суглобових щілин і ерозії кісток, що нагадують РА (проте РФ у сироватці хворих не визначається);
- ураження хрящів гортані та трахеї (у 70% хворих) проявляється хворобливістю при їх пальпації; у тяжких випадках через набряк у цій ділянці, спадання стінок може розвинутися утруднене дихання, що іноді вимагає проведення трахеотомії; можуть залучатися до процесу і бронхи; розвивається колапс дрібних бронхів, що веде до активації інфекції дихальних шляхів, дихальної недостатності, а іноді асфіксії;
- ураження очей (у 60% хворих); у процес залучені практично всі відділи ока, описано періорбітальний набряк, параліч очних м’язів, кон’юнктивіт, кератит, епісклерит і увеїт. Васкуліт сітківки та оклюзії вен і артерій зорового нерва можуть призвести до сліпоти;
- ураження нервової системи діагностують рідко. Є свідчення про залучення до процесу деяких черепних нервів. Органічне ураження ЦНС з подальшим розвитком деменції є наслідком васкуліту.
У 10% хворих відзначають васкуліти різних типів. При ураженні дрібних судин розвивається лейкопластичний васкуліт, середніх — періартеріїт, великих — картина, яка нагадує хворобу Такаясу. Розширення аорти в висхідному її відділі може супроводжуватися формуванням відносної недостатності аортального клапана. Вторинний тромбоз може мати місце в низхідному і черевному відділах аорти. Зрідка розвивається проліферативний гломерулонефрит.
Лабораторні показники неспецифічні. Відзначають збільшення ШОЕ, підвищення рівня α2— і β-глобулінів, іноді помірну анемію і РФ у невисоких титрах.
Діагностика
Відповідно до пропозицій L.P. McAdam та співавторів (1976) для достовірного діагнозу РПХ необхідна наявність у хворого 3 та більше перерахованих критеріїв:
- двобічне ураження хрящів слухового апарата;
- неерозивний серонегативний запальний поліартрит;
- хондрит носової перегородки;
- запалення ока (кон’юнктивіт, кератит, склерит, епісклерит, увеїт);
- хондрит респіраторного тракту: хрящів гортані та/або трахеї;
- кохлеарна та/або вестибулярна дисфункція.
Більш ніж в 30% випадків РПХ асоціюється з іншими аутоімунними або гематологічними захворюваннями.
Диференційна діагностика
Диференційний діагноз проводять з такими захворюваннями, як сифіліс, лепра, при яких ураження перегородки носа може вести до деформації його за типом сідлоподібного носа. У коло захворювань, з якими слід диференціювати РПХ, мають бути включені гранулематоз Вегенера і пухлинний процес. Вирішальне значення в цих випадках має дослідження біопсійного матеріалу. Зрідка локальна травма, грибкова інфекція можуть бути причиною запальних змін, схожих на хондрит.
Лікування
ГК пригнічують активну фазу хвороби і знижують тяжкість чергового рецидиву. Доза преднізолону коливається від 30 до 60 мг/добу, потім поступово знижується до підтримувальної (5–15 мг/добу). Більш високі дози преднізолону призначають у разі дуже тяжких проявів ураження трахеї, бронхів і очей. Імуномодулювальний ефект виявляє сульфонилбісбензоламін, який застосовують у дозі 50–200 мг/добу для зменшення вираженості клінічних ознак хвороби, після чого його призначають у підтримувальних дозах: 50 мг/добу або через день.
В останні роки перевагу віддають цитотоксичним засобам: хлорамбуцил призначають по 4–6 мг/добу, азатіоприн — по 150–200 мг/добу, циклофосфамід — із розрахунку 1,0–1,5 мг на 1 кг маси тіла хворого. Усі ці препарати застосовують для зменшення активності процесу з наступним переходом на підтримувальні дози протягом тривалого періоду.
Є поодинокі роботи, в яких відображено вплив циклоспорину А і моноклональних антитіл до СD4 в особливо рефрактерних випадках РПХ.
Перебіг і прогноз
Перебіг РПХ прогресуючий. З кожним загостренням в процес залучаються все більше хрящових структур з ураженням органів чуття. Прогноз щодо видужання несприятливий. Тривалість варіює від декількох місяців до 20 років. Смерть найчастіше настає від дихальної недостатності, рідше — від недостатності кровообігу.
Глава 21. Системні васкуліти. Васкулопатії. Синдром Гудпасчера
Васкулітом (ангіїтом) називають запалення стінок кровоносних судин, внаслідок якого розвивається ішемія і некроз їх та периваскулярних тканин, погіршується кровоток. Ці процеси призводять до руйнування стінки судини, розривів, крововиливів у оточуючі тканини. Пошкодження ендотелію супроводжуються утворенням тромбів, ішемією тканин. Васкуліти можуть бути первинними (ідіопатичними) і вторинними. При первинних васкулітах ураження судин є основним або єдиним проявом хвороби, вторинні васкуліти розвиваються на тлі інших хвороб. Клінічний перебіг васкулітів залежить від активності системного запалення, типу судин, їх розміру і локалізації.
Епідеміологія
Поширеність системних васкулітів коливається від 0,4 до 14 і більше випадків на 100 тис. населення (Насонов Е.Л., Насонова В.А., 2010 (ред.)). Зараз спостерігається тенденція до збільшення їх поширеності. Системні васкуліти переважно діагностують у чоловіків. Вони можуть розвиватися в будь-якому віці, але частіше виникають на 4–5 десятку життя. У дитячому і юнацькому віці найбільш часто виявляють геморагічний васкуліт і хворобу Кавасакі.
Етіопатогенез
Етіологія більшості первинних системних васкулітів невідома. Спостерігається зв’язок ВП з вірусом гепатиту В, а ЕКВ — з вірусом гепатиту С. В основі розвитку (патогенезу) васкулітів лежать клітинні або гуморальні імунні реакції (табл. 21.1).
Таблиця 21.1
Патогенез васкулітів
|
Характер імунних механізмів |
Хвороба |
Зміни в судинах |
|
Клітинні реакції з утворенням гранульом (гранулематозне запалення) |
|
У відповідь на невідомий антиген у стінках судин і навколо них внаслідок взаємодії CD4+-Т-клітин і макрофагів формуються гранульоми. Включенню в патологічний процес якого-небудь тканинного антигену можуть сприяти вікові зміни стінок судин (приклад ГКА) і призводити до порушення імунного нагляду, розвитку клітинної імунної реакції |
|
Імунокомплексні реакції |
|
При ВП, лейкоцитокластичному васкуліті ЦІК відкладаються в судинній стінці, стимулюють активацію комплементу, міграцію фагоцитів і призводять до ушкодження стінки |
При ряді васкулітів (гранулематоз Вегенера, МПА, синдром Черджа — Стросс, ВП (класичний), деякі медикаментозні васкуліти) відзначають продукцію антитіл до лізосомальних ферментів нейтрофілів (АНЦА). Хвороба Кавасакі супроводжується продукцією органонеспецифічних антитіл. Гістологічне дослідження при системних васкулітах виявляє інфільтрацію судинної стінки нейтрофілами, мононуклеарами та/або гігантськими багатоядерними клітинами, а також руйнування всіх шарів судини (фібриноїдний некроз). Периваскулярна інфільтрація є неспецифічною ознакою васкулітів. Механізми пошкодження судинної стінки при васкулітах ділять на 2 групи (табл. 21.2).
Таблиця 21.2
Механізми пошкодження судинної стінки при васкулітах
|
I. Імунологічні |
|
|
1. |
Відкладення в судинній стінці імунних комплексів (надходження їх з крові або утворення в стінці) |
|
2. |
Синтез антитіл до цитоплазми нейтрофілів |
|
3 |
Вплив на ендотеліальні клітини АНЦА |
|
4. |
Синтез антитіл до ендотелію та інших елементів судинної стінки |
|
5. |
Розвиток цитотоксичних алергічних реакцій |
|
6. |
Дія цитотоксичних Т-лімфоцитів |
|
7. |
Розвиток реакцій уповільненого типу з утворенням гранульом у стінках судин і периваскулярних тканинах |
|
8. |
Дія цитокінів (ІЛ-1, ФНП-α та ін.) |
|
II. Неімунологічні |
|
|
1. |
Інвазія мікроорганізмів у судинну стінку і навколишні тканини |
|
2. |
Безпосередній вплив хімічного агента на судинну стінку |
|
3. |
Проникнення пухлинних клітин у судинну стінку |
|
4. |
Інші |
Відкладенню імунних комплексів у судинній стінці сприяють (підвищують проникність) гістамін, брадикінін, лейкотрієни, які продукуються гладкими клітинами і тромбоцитами під дією IgЕ. Відкладення імунних комплексів супроводжується активацією комплементу (утворенням компонента С5а), який залучає нейтрофіли. Нейтрофіли фагоцитують імунні комплекси, вивільняють ферменти, які пошкоджують судинну стінку. Надалі в місця ураження надходять лімфоцити і макрофаги. Гранулематозне запалення опосередковують реакції уповільненого типу. Вважається, що утворення гранульом можуть спровокувати самі імунні комплекси.
Ендотеліальні клітини під дією цитокінів (гамма-інтерферону) експресують HLA класу II. Вони секретують ІЛ-1, який активує Т-лімфоцити та ініціює запалення в судинній стінці. Цитотоксичні Т-лімфоцити, антитіла до ендотелію, цитотоксичні алергічні реакції беруть участь у пошкодженні судин.
Важливим серологічним маркером та патогенетичним медіатором васкулітів дрібних судин є АНЦА — гетерогенна популяція антитіл, які реагують з різними ферментами нейтрофілів і, у першу чергу, з протеїназою-3 і мієлопероксидазою. АНЦА найбільш часто (у 80–90% випадків) зустрічаються при гранулематозі Вегенера, МПА і синдромі Черджа — Стросс, тобто при АНЦА-асоційованих васкулітах. Слід зазначити, що васкуліти виникають і за відсутності антитіл, їх титр не відповідає тяжкості хвороби й зберігається роками. Значення має спадкова схильність, особливості імунної відповіді на окремі антигени.
Фактори, які визначають величину і ступінь пошкодження судинної стінки при васкулітах, наведено в табл. 21.3.
Таблиця 21.3
Фактори, які визначають величину і ступінь пошкодження судинної стінки при васкулітах
|
1. |
Тривалість циркуляції антигенів |
|
2. |
Концентрація імунних комплексів у крові |
|
3. |
Фізико-хімічні характеристики імунних комплексів, їх розміри |
|
4. |
Здатність ретикулоендотеліальної системи видаляти імунні комплекси |
|
5. |
Індивідуальні відмінності імунної відповіді |
|
6. |
Адгезивні можливості ендотеліальних клітин, наявність пошкоджень ендотелію |
|
7. |
Зміни артеріального тиску (гідростатичної сили) |
|
9. |
Ступінь турбулентності потоку крові в судинах |
|
9. |
Зміни діаметра і проникності судин |
Ці фактори пояснюють ураження при васкулітах певних судин (наприклад судин нижніх кінцівок), а також той факт, що тільки певні типи імунних комплексів викликають розвиток васкуліту. Механізми розвитку аутоімунних васкулітів представлено в табл. 21.4.
Таблиця 21.4
Механізми розвитку аутоімунних васкулітів
|
Утворення імунних комплексів у стінці судин |
Антигени надходять у судинну стінку, де зв’язуються з антитілами, що надійшли з крові. Імунні комплекси, що утворилися, активують систему комплементу, залучають лейкоцити і запускають запалення в стінці. Подібні зміни відбуваються при ВП |
|
Відкладення ЦІК у стінці судин |
Комплекси формуються в крові й осідають у стінках дрібних судин, де активують систему комплементу, залучають лейкоцити й ініціюють запалення |
|
Відкладення кріоглобулінів |
Кріоглобуліни — білкові комплекси, які випадають в осад при зниженні температури. Кріоглобулінові комплекси часто складаються з часток вірусу гепатиту С і антитіл класу IgG. При їхньому відкладенні відбувається пошкодження дрібних судин, з’являється пурпура |
Класифікація
Ознаки, які враховуються при класифікації васкулітів, наведено в табл. 21.5.
Таблиця 21.5
Ознаки, які враховуються при класифікації васкулітів
|
Етіологія і патогенез |
Аутоімунні, інфекційні, променеві |
|
Типи судин, які уражуються |
Аортит, артеріїт, капілярит, венуліт (флебіт) |
|
Поширеність уражень |
Локальні та системні |
|
Тривалість перебігу |
Гострі і хронічні васкуліти |
Клінічні ознаки, які дозволяють вважати васкуліт основною хворобою, включають:
- наявність лихоманки нез’ясованої етіології;
- полісистемні ураження;
- патологічні зміни шкіри;
- наявність симптомів ішемії;
- множинний мононеврит.
Труднощі, які виникають при створенні класифікацій васкулітів, полягають у тому, що різні васкуліти мають багато спільних рис, відзначають взаємне нашарування синдромів. У табл. 21.6 подано класифікацію васкулітів за Е. Фаучі (2005).
Таблиця 21.6
Класифікація васкулітів (за: Фаучі Е., 2005)
|
I |
Cистемні некротичні васкуліти |
ВП МПА Синдром Черджа — Стросс Змішаний васкуліт |
||
|
II |
Гранулематоз Вегенера |
|||
|
III |
ГКА |
|||
|
IV |
Аортоартеріїт |
|||
|
V |
Геморагічний васкуліт |
|||
|
VI |
Васкуліти з переважним ураженням шкіри |
А. За участю екзогенного антигену |
Медикаментозний васкуліт Сироваткова хвороба Васкуліт при інфекційних хворобах |
|
|
Б. За участю ендогенного антигену |
Паранеопластичний васкуліт Васкуліт при ревматичних хворобах Інші вторинні васкуліти Васкуліт при вродженій недостатності комплементу |
|||
|
VII |
Інші васкуліти |
А. Хвороба Кавасакі Б. Первинний нейроваскуліт В. ОТА Г. Хвороба Бехчета Д. Інші |
||
Вторинні васкуліти виникають при: інфекціях; паразитарних хворобах; медикаментозній хворобі; злоякісних пухлинах; системних захворюваннях сполучної тканини; професійних хворобах. Групу інших васкулітів за Е. Фаучі (2005) представлено в табл. 21.7.
Таблиця 21.7
Група інших васкулітів (за: Фаучі Е., 2005)
|
Синдром Когана |
Для нього характерна наявність паренхіматозного кератиту, вестибулярних порушень, зниження слуху. Можуть уражуватися судини різного діаметра, аортальний клапан |
|
Стійка припіднята еритема |
Хронічне рідкісне захворювання шкіри нез’ясованої етіології, яке характеризується симетричними шкірними висипаннями (бляшки, вузлики, червоні, фіолетові папули) на розгинальних поверхнях кінцівок. У біоптатах виявляють запальну інфільтрацію дерми, запалення венул з лейкоклазією |
|
Васкуліти, безпосередньо зумовлені інфекцією |
Запалення судин викликають рикетсії (проникають в ендотелій і в ньому розмножуються) |
Типи судин, які уражуються при різних васкулітах, описано в табл. 21.8.
Таблиця 21.8
Типи судин, які уражуються при різних васкулітах
|
Вид васкуліту |
Типи уражуваних судин |
|
Алергічні васкуліти шкіри |
Капіляри, венули |
|
Хвороба Шенлейна — Геноха |
Артеріоли, капіляри, венули |
|
Кріоглобулінемічний васкуліт |
Артеріоли, капіляри, венули |
|
МПА |
Артерії малого калібру, артеріоли, капіляри, венули |
|
Гранулематоз Вегенера |
Артерії середнього і малого калібру, артеріоли, капіляри, венули |
|
Синдром Черджа — Стросс |
Артерії середнього і малого калібру, артеріоли, капіляри, венули |
|
ВП |
Артерії середнього і малого калібру |
|
Хвороба Кавасакі |
Артерії великого, середнього і малого калібру |
|
Скроневий артеріїт |
Аорта, артерії великого, середнього і малого калібру |
|
Хвороба Такаясу |
Аорта, артерії великого і середнього калібру |
Виділяють васкуліти судин великого, середнього і малого калібру (табл. 21.9).
Таблиця 21.9
Судини різного калібру
|
Артерії великого калібру |
Аорта і її найбільші розгалуження, що прямують до великих частин тіла (голова, шия, кінцівки та ін.) |
|
Артерії середнього калібру |
Головні вісцеральні артерії (ниркові, печінкові, коронарні, мезентеріальні) |
|
Судини малого калібру |
Венули, капіляри, артеріоли |
Номенклатуру системних васкулітів (за: Jennette J.C. et al., 1994) представлено в табл. 21.10.
Таблиця 21.10
Номенклатура системних васкулітів
|
Форма |
Коротка характеристика |
|
|
Васкуліти судин великого калібру |
||
|
Гігантоклітинний (скроневий) артеріїт |
Розвивається у осіб у віці старше 50 років, часто поєднується з РПМ. Характеризується гранулематозним запаленням аорти та її великих відгалужень з ураженням екстракраніальних гілок сонної артерії, особливо скроневої |
|
|
Артеріїт Такаясу |
Хвороба починається у віці до 50 років. Розвивається гранулематозне запалення аорти та її основних гілок |
|
|
Васкуліти судин середнього калібру |
||
|
ВП |
Розвивається некротизуюче запалення середніх і дрібних артерій (без розвитку гломерулонефриту), васкуліт артеріол, капілярів і венул |
|
|
Хвороба Кавасакі |
Артеріїт зустрічається у дітей, уражує великі, середні і дрібні артерії. Переважно уражуються коронарні артерії, можливе залучення до патологічного процесу вен. Часто спостерігається поєднання зі слизово-шкірно-лімфонодулярним синдромом |
|
|
Васкуліти судин малого калібру |
||
|
Гранулематоз Вегенера |
Гранулематозне запалення уражує дихальні шляхи. Наслідком некротизуючого васкуліту, який уражує дрібні і середні судини (капіляри, венули, артеріоли, артерії), є некротизуючий гломерулонефрит |
|
|
Синдром Черджа — Стросс |
Розвивається гранулематозне запалення верхніх дихальних шляхів, бронхіальна астма, еозинофілія. Некротизуючий васкуліт уражує дрібні й середні судини |
|
|
МПА (поліартеріїт) |
Некротизуючий васкуліт уражує дрібні судини (капіляри, венули, артеріоли) і середні артерії. Розвивається некротизуючий гломерулонефрит, легеневий капілярит |
|
|
Пурпура Шенлейна — Геноха |
Розвивається васкуліт з переважними IgА-депозитами, уражуються артеріоли, капіляри, венули. До розвитку патологічного процесу залучається шкіра, кишечник, клубочки нирок, суглоби |
|
|
ЕКВ |
Для васкуліту характерне відкладення кріоглобулінемічних імунних депозитів у дрібних судинах (артеріолах, капілярах, венулах). Переважно уражуються судини шкіри, клубочків нирок. У сироватці крові визначаються кріоглобуліни |
|
|
Шкірний лейкоцитокластичний (гіперсенситивний) васкуліт |
Відзначають ізольований лейкоцитокластичний ангіїт (системний васкуліт і гломерулонефрит відсутні) |
|
ОТА проявляється хронічним запаленням артерій середнього і дрібного калібру, вен і нервових стовбурів. Переважно залучаються дистальні відділи судин верхніх і нижніх кінцівок (зрідка — церебральні та вісцеральні). При первинному нейроваскуліті частіше уражуються артеріоли, але можуть бути залучені судини будь-якого калібру. При цьому утворюються лімфоцитарні інфільтрати, нерідко — гранульоми.
Клінічні особливості
При системних васкулітах уражуються різні органи і системи. Характер цих уражень наведено в табл. 21.11.
Таблиця 21.11
Характер уражень різних органів і тканин при васкулітах
|
Орган, система |
Характер ураження |
Назва васкуліту |
|
Конституціональні синдроми |
Лихоманка, міалгії, артралгії, зменшення маси тіла |
Усі васкуліти |
|
Ураження шкіри |
Сітчасте ліведо, дигітальні інфаркти, виразки, вузлики |
ВП Синдром Черджа — Стросс Гранулематоз Вегенера |
|
Пурпура, яку можна пропальпувати |
Усі васкуліти, за винятком артеріїту Такаясу і ГКА |
|
|
Ураження ЛОР-органів |
Риніт, синусит, середній отит |
Гранулематоз Вегенера МПА Синдром Черджа — Стросс |
|
Алергічний риніт |
Синдром Черджа — Стросс |
|
|
Ураження легенів |
Інфільтрація, розпад, некротизуючий альвеоліт, легеневі геморагії, плеврит |
Гранулематоз Вегенера, МПА Синдром Черджа — Стросс |
|
Бронхіальна астма |
Синдром Черджа — Стросс |
|
|
Ураження нирок |
Ішемічне ураження |
ВП Артеріїт Такаясу |
|
Гломерулонефрит |
МПА Гранулематоз Вегенера Кріоглобулінемічний васкуліт Синдром Черджа — Стросс Пурпура Шенлейна — Геноха |
|
|
Ураження нервової системи |
Множинний мононеврит |
ВП Кріоглобулінемічний васкуліт Гранулематоз Вегенера Синдром Черджа — Стросс |
|
Ураження суглобів |
Олігоартрит (недеструктивний) |
ВП Гранулематоз Вегенера Синдром Черджа— Стросс Пурпура Шенлейна — Геноха |
Легеневі прояви, які спостерігаються при системних васкулітах, наведено в табл. 21.12.
Таблиця 21.12
Легеневі прояви при системних васкулітах
|
Симптоми |
Назва системного васкуліту |
|
Кровохаркання |
Гранулематоз Вегенера МПА |
|
Бронхіальна обструкція |
Синдром Черджа — Стросс МПА ВП |
|
Бронхіальна астма |
Синдром Черджа — Стросс |
|
Альвеоліт |
МПА |
|
Кашель, утруднення носового дихання |
Гранулематоз Вегенера |
При усіх системних васкулітах на рентгенограмах легень визначається посилення судинного малюнка, периваскулярна і вогнищева інфільтрація. У цілому клінічні особливості васкулітів представлено в табл. 21.13.
Таблиця 21.13
Клінічні особливості різних васкулітів
|
ВП |
Початок хвороби |
Переважно поступовий, але може бути гострим з лихоманкою, міальгією, болем у суглобах, шкірними висипаннями, зменшенням маси тіла |
|
М’язи |
Типовим є біль в литкових м’язах, втрата здатності ходити. Часто біль передує нейроміопатії |
|
|
Суглоби |
У ¼ хворих розвивається транзиторний, мігруючий, недеформуючий артрит. Уражуються гомілковостопні, колінні, плечові, ліктьові, променезап’ясткові суглоби, меншою мірою — дрібні суглоби кистей та стоп |
|
|
Шкіра |
Її ураження відзначають у 25–60% хворих, що може бути першим проявом хвороби. Виявляють судинну папулопетехіальна пурпуру, бульозні та везикулярні висипання, підшкірні вузлики (аневризми артерій діаметром 0,5–2 см), сітчасте ліведо. Розвиваються інфаркти шкіри, ішемічні зміни дистальних фаланг пальців |
|
|
Нервова система |
Найбільш частим проявом (у 50% пацієнтів) є периферична невропатія у вигляді множинного мононевриту, мононевропатії. Характерні моторні й сенсорні порушення в нижніх кінцівках в результаті залучення гомілкових нервів, їх гілок та інших нервів. Відзначають біль, парестезії, рухові розлади, які можуть передувати порушенням чутливості. Іноді уражується ЦНС з головним болем, гіперкінетичним синдромом, судомами, інфарктом головного мозку, інсультом (ішемічним, геморагічним) |
|
|
Нирки |
У період від 3 до 6 міс від початку хвороби у 60–87% хворих уражуються ниркові артерії, клубочки. При цьому переважає судинний тип ураження (а не гломерулонефрит, як при МПА). Множинні безбольові інфаркти нирок призводять до швидкого розвитку ниркової недостатності. Часто відзначають помірну протеїнурію до 3 г/добу), мікрогематурію (ознака активності хвороби), лейкоцитурію (без сечової інфекції). Артеріальна гіпертензія розвивається у ⅓ хворих та пов’язана з нирковим васкулітом, інфарктом нирок, вторинним ураженням клубочків. У цілому ураження нирок — прогностично несприятлива ознака |
|
|
Серце |
У 40% хворих розвивається кардіомегалія, тахікардія, порушення ритму, стенокардія, дрібновогнищевий інфаркт міокарда. Перебіг стенокардії характеризується з нетиповим больовим синдромом, інфаркту міокарда — латентний, без ангінозного болю |
|
|
Периферичні артерії кінцівок |
Виникає ішемія та гангрена пальців |
|
|
Кишечник |
Наслідком ішемії кишечнику і тромбозу мезентеріальних судин є абдомінальний біль (у ⅓ хворих), нудота, блювання, мелена |
|
|
Печінка |
Гепатомегалія, підвищення рівня лужної фосфатази внаслідок виникнення інфарктів печінки, розривів внутрішньопечінкових судин |
|
|
Підшлункова залоза |
Утворення кіст |
|
|
Ендокринні органи |
Типовим ураженням є орхіт, епідидиміт |
|
|
Лабораторні зміни |
Підвищення ШОЕ, рівня СРБ, лужної фосфатази, АсАТ, АлАТ, лейкоцитоз, нормохромна анемія, зрідка — еозинофілія. Зниження рівня С3 та С5 комплементу, збільшення — ЦІК, титрів РФ (іноді), АНФ. Виявляють антитіла до кардіоліпіну, у 7–63% хворих — HbsAg |
|
|
Основа діагнозу |
Відповідність діагностичним критеріям (Lightfoot R.W. et al., 1990), характерні клінічні прояви із залученням будь-яких органів (найбільш часто — шкіри, суглобів, периферичних нервів, кишечнику, нирок), морфологічне підтвердження васкуліту (біопсія шкіри, мязів, нирок), ангіографія. На відміну від МПА, виникає незначне порушення функції нирок, переважає ураження суглобів, периферичної нервової системи та ЦНС, інфаркти внутрішніх органів. Для ВП не характерно утворення перинуклеарних АНЦА |
|
|
МПА |
Суглоби |
У більшості хворих відзначають поліартралгії, у половині випадків — стійкий неерозивний артрит великих суглобів. Може бути позитивним РФ |
|
Шкіра |
Розвиваються виразки, іноді — некрози (у тому числі м’яких прилеглих тканин) |
|
|
Верхні дихальні шляхи |
Відзначають атрофію слизової оболонки носа, некротичний риніт (деструкція та деформація носа не виникають), гранульом не виявляють. У 30–40% хворих змінюються пазухи носа і середнього вуха |
|
|
Легені |
Некротизуючий альвеоліт з кашлем, болем у грудній клітці, кровохарканням, легеневою кровотечею. При рентгенографії виявляють інфільтрати без розпаду, з реакцією плеври, ознаки альвеоліту |
|
|
Орган зору |
Епісклерит |
|
|
ШКТ |
Біль в животі |
|
|
Нервова система |
Периферична поліневропатія на фоні тяжкої патології нирок |
|
|
Нирки |
Ураження нирок виявляють у 100% хворих у вигляді швидкогресуючого фокального сегментарного некротизуючого екстракапілярного гломерулонефриту з «півмісяцями». Специфічних симптомів не виявлено. На відміну від ВП, швидко розвивається ниркова недостатність без симптоматичної артеріальної гіпертензії |
|
|
Основа діагнозу |
Діагностичні критерії відсутні. Діагноз встановлюють з урахуванням клінічної картини, результатів імунологічних та морфологічних досліджень.У 80% хворих виявляють перинуклеарні АНЦА (які реагують з мієлопероксидазою), у 100% — мікрогематурію, у 80–100% — підвищення рівня креатиніну, у 90% — протеїнурію. У клінічній картині домінують явища некротизуючого гломерулонефриту (характерне прогресуюче ураження нирок), іноді — легеневого капіляриту |
|
|
Гранулематоз Вегенера |
ЛОР-органи |
У 70% хворих відзначають постійний нежить з гнійно-геморагічними виділеннями, виразкування слизової оболонки з розвитком перфорації носової перегородки, сідлоподібної деформації носа; гранульоми вух, пазух носа, отит (серозний, гнійний). |
|
Верхні дихальні шляхи |
Гранулематозне ураження трахеї, стеноз гортані. |
|
|
Легені |
Ураження легень (відзначають у дебюті у 45% хворих) може перебігати безсимптомно. Множинні білатеральні інфільтрати з тенденцією до розпаду, формування порожнин. Рідше — плеврит, легеневі дифузні геморагії, збільшення лімфатичних вузлів середостіння. Супутня інфекція (бактеріальні пневмонії) |
|
|
Нирки |
Гломерулонефрит у дебюті хвороби у 11–18% випадків, в цілому — у 77–85%. Прогресуюче ураження: протеїнурія, гематурія, розвиток ниркової недостатності, смерть через 6 міс за відсутності лікування. На фоні терапії хронічної ниркової недостатності розвивається у 40% хворих та вимагає гемодіалізу, пересадки нирок |
|
|
Орган зору |
Ураження (у 50% хворих) оболонок ока, судин сітківки, псевдопухлини ока |
|
|
Шкіра |
Пурпура (що пальпується), підшкірні вузлики, папули, везикули, виразки, гангренозна піодермія, феномен Рейно |
|
|
М’язи |
Біль у м’язах |
|
|
Суглоби |
Моноартрит, мігруючий олігоартрит або асиметричний поліартрит дрібних і великих суглобів. У 50–60% хворих визначається РФ |
|
|
Нервова система |
Нервові порушення рідко спостерігаються в дебюті хвороби. У подальшому вони мають місце у ⅓ хворих у вигляді множинного мононевриту, дистальної симетричної поліневропатії |
|
|
ШКТ |
Біль в животі, діарея, кровотечі внаслідок ентероколіту, виразкування тонкого або товстого кишечнику |
|
|
Сечостатева система |
Геморагічний цистит, який може бути наслідком лікування циклофосфамідом, некротизуючого васкуліту судин сечового міхура |
|
|
Серцево-судинна система |
Уражується рідко |
|
|
Лабораторні зміни |
Визначаються АНЦА |
|
|
Основа діагнозу |
Відповідність діагностичним критеріям (Levitt R.V. et al., 1990), присутність АНЦА (які реагують з протеїназою-3) в крові. Вирішальне значення в діагностиці гранулематозу Вегенера мають результати біопсії |
|
|
Синдром Черджа — Стросс |
Органи дихання |
Розвитку васкуліту передує бронхіальна астма або вона є основним проявом хвороби, перебігає важко, у 79% випадків вимагає застосування ГК. На ранніх стадіях спостерігається еозинофілія і гранульоми. У хворих з плевритами (у ⅓ хворих) в плевральній рідині містяться еозинофіли |
|
Верхні дихальні шляхи |
Алергічний риніт супроводжується порушенням носового дихання, рецидивуючим синуситом, поліпозом носа |
|
|
ШКТ |
Біль в животі, діарея, кровотечі, перфорація, перитоніт, кишкова обструкція, які пов’язані з еозинофільним гастроентеритом, васкулітом судин кишечнику, іноді — з виразковим колітом |
|
|
Серцево-судинна система |
У ⅓ хворих розвивається гострий, у тому числі конструктивний, перикардит, серцева недостатність. Зрідка розвивається інфаркт міокарда, артеріальна гіпертензія, ендокардит Леффлера |
|
|
Шкіра та нервова система |
Ураження подібні до таких при ВП |
|
|
Нирки |
Ураження у вигляді некротизуючого гломерулонефриту, менш злоякісного, ніж при гранулематозі Вегенера, ВП, поєднується з визначенням перинуклеарних АНЦА |
|
|
Суглоби |
Поліартрит або поліартралгія у 50% хворих |
|
|
Лабораторні зміни |
Еозинофілія більше 109/л у 97% хворих у будь-якій стадії хвороби. Може спостерігатися еозинофільна інфільтрація тканин без периферичної еозинофілії. Підвищується рівень IgE, більше ніж у 50% хворих визначаються перинуклеарні АНЦА |
|
|
Основа діагнозу |
Враховують діагностичні критерії (Masi A.T. et al., 1990), вік хворих (середній), бронхіальну астму в анамнезі або алергічний риніт, еозинофілію, множинний мононеврит, легеневі інфільтрати, кардіоміопатію |
|
|
Геморагічний васкуліт |
Шкіра |
Її ураження відзначають в 50% випадків: з’являються петехіальні висипи та/або пурпура, яку можна пропальпувати, еритематозні плями, папули, везикули, які супроводжуються свербінням. Вираженість висипань посилюється у вертикальному положенні. Вони переважно локалізуються в дистальних відділах ніг, на стегнах та сідницях, рідше — на руках, животі, спині. При хронічному рецидивуючому перебігу зберігаються ділянки гіперпігментації |
|
Суглоби |
Артралгії та артрити відзначають у 59–100% хворих. Мігруючі артралгії виникають одночасно з ураженням шкіри. У ¼ хворих артралгії, артрити можуть передувати ураженню шкіри. Часто уражуються суглоби ніг, ліктьові, променезап’ясткові. Суглобовий синдром триває 1 тиж або більше |
|
|
ШКТ |
Біль у животі, нудота, блювання, зрідка — шлунково-кишкові кровотечі. Виникає набряк стінки тонкого кишечнику |
|
|
Сечостатеві органи |
Їх ураження відзначають у 10–60% випадків і може бути першим проявом хвороби. Може розвинутися нефротичний синдром, хронічна ниркова недостатність, артеріальна гіпертензія. Реєструють безсимптомну ізольовану мікро- або макрогематурію, помірну протеїнурію (у дітей виникає запалення і набряк мошонки, геморагії) |
|
|
Лабораторні зміни |
Підвищення в сироватці крові концентрації IgA, титру АСЛ-О (у 30%), ШОЕ, СРБ, РФ(+) (у 30–40% хворих) |
|
|
Основа діагнозу |
Враховують діагностичні критерії (Miles I.A. et al., 1990), типові зміни шкіри, кишечнику та нирок у поєднанні з артралгіями або артритом; зв’язок розвитку хвороби у дітей з інфекцією верхніх дихальних шляхів |
|
|
ГКА |
Неспецифічні симптоми |
Лихоманка, загальна слабкість, анорексія, зменшення маси тіла, депресія |
|
Ураження скроневих артерій |
Головний біль з локалізацією в скроневій, лобній, потиличній ділянках, болісність при дотику до шкіри черепа, набухання і набряк скроневих артерій, послаблення пульсації |
|
|
Ураження потиличної артерії |
Головний біль у ділянці потилиці |
|
|
Ураження верхньощелепної артерії |
«Переміжна кульгавість» язика та при жуванні, зубний біль без причини |
|
|
Ураження зовнішньої сонної й артерій, які забезпечують кров’ю очі |
Набряк обличчя, порушення ковтання, слуху, зору, ішемічний хоріоретиніт, набряк рогівки, ірит, кон’юнктивіт, епісклерит, диплопія |
|
|
Ураження аорти |
Синдром дуги аорти, аневризма її та великих артерій |
|
|
Лабораторні дані |
Збільшення ШОЕ більше 50 мм/год, вмісту в крові СРБ, ІЛ-6, підвищення активності лужної фосфатази (у 30–62% хворих), трансаміназ (у 15%) |
|
|
Основа діагнозу |
Враховують діагностичні критерії ГКА (Hunder G.G. et al., 1990), вік хворих (старше 50 років), клінічну картину з вираженим головним болем, порушенням зору, симптомами РПМ, збільшенням ШОЕ, анемією, відсутність АНЦА |
|
|
РПМ |
М’язово-скелетна система |
Симетричний біль та скутість в м’язах плечового та тазового пояса, в ділянці шиї, ранкова скутість. Біль та скутість посилюються при рухах, зменшуються у спокої. Ураження суглобів у вигляді моно-, олігоартриту або симетричного серонегативного поліартриту, який нагадує РА у людей похилого віку (уражуються переважно колінні, променезап’ясткові, гомілковостопні, зрідка — проксимальні міжфалангові, плеснофалангові суглоби) |
|
Лабораторні зміни |
Різке збільшення ШОЕ |
|
|
Основа діагнозу |
Враховують діагностичні критерії РПМ (Bird H.A. et al., 1979), вік хворих (>60–65 років), характерну клінічну картину хвороби, значення ШОЕ >40 мм/год |
|
|
Артеріїт Такаясу |
Неспецифічні симптоми |
Тривале підвищення температури тіла, лихоманка, зменшення маси тіла, слабкість, сонливість, артралгії, симетричний поліартрит, міалгії |
|
Судини шиї та верхніх кінцівок |
Переміжна кульгавість верхніх кінцівок, слабкість, втомлюваність, біль в проксимальних відділах рук, переважно з одного боку, посилення вираженості цих симптомів при фізичному навантаженні |
|
|
Орган зору |
Звуження полів зору, швидка втомлюваність очей, поступове зниження гостроти зору, диплопія. Можлива гостра оклюзія центральної артерії сітківки з раптовою втратою зору на одне око |
|
|
Серцево-судинна система |
Ущільнення, дилатація, формування аневризми висхідного відділу аорти з виникненням аортальної недостатності. Біль в грудній клітці, задишка, серцебиття, зрідка — напади стенокардії. Розвивається міокардит (некроз міофібрил з мононуклеарною інфільтрацією інтерстиціальної тканини, утворенням фокальних гранульом) зі збільшенням розмірів серця, гіпертрофією лівого шлуночка, міжшлуночкової перегородки. У 33–76% хворих розвивається артеріальна гіпертензія внаслідок реноваскулярних механізмів, стенозу ниркових артерій |
|
|
ШКТ |
Розвивається ішемія вісцеральних органів внаслідок запалення черевного відділу аорти, з’являється біль в епігастрії та попереку |
|
|
Нервова система |
З причини запальних змін лівої загальної сонної артерії, внутрішньої гілки правої загальної сонної артерії та хребетних артерій у хворих розвиваються дисциркуляторна енцефалопатія, головний біль, запаморочення, порушення пам’яті, уваги, зниження працездатності. Виникають епізоди гострого порушення мозкового кровообігу, втрати свідомості. При ураженні периферичної нервової системи виникає симетрична поліневропатія, яка найбільше виражена в руках |
|
|
Легені |
У 20% хворих формується легенева гіпертензія |
|
|
Шкіра |
Виникає вузлувата еритема, гангренозна піодермія, феномен Рейно, шкірний васкуліт |
|
|
Лабораторні зміни |
Анемія, тромбоцитоз, гіпергаммаглобулінемія, збільшення ШОЕ (у ⅓ хворих може бути нормальною), концентрації СРБ (у 60–70% пацієнтів) |
|
|
Основа діагнозу |
Діагноз базується на врахуванні діагностичних критеріїв (Arend W.P. et al., 1990), анамнезу хвороби, скарг, результатів дослідження пульсу, рівня артеріального тиску на руках та ногах, ангіографії, УЗД судин |
|
|
ЕКВ |
Шкіра |
Пурпура, яку можна пропальпувати, локалізується на нижніх кінцівках, у ділянці живота, сідниць. Ортостатична пурпура посилюється на холоді та тривалому перебуванні в одній позі |
|
Суглоби |
Симетричні мігруючі поліартралгії, асоційовані з ранковою скутістю, залученням проксимальних міжфалангових суглобів, променезап’ясткових, колінних, гомілковостопних та ліктьових суглобів |
|
|
Нервова система |
Першою ознакою хвороби може бути периферична невропатія з парестезіями та онімінням нижніх кінцівок |
|
|
Нирки |
На пізніх стадіях хвороби виникають гематурія, протеїнурія, нефротичний синдром, артеріальна гіпертензія |
|
|
ШКТ |
Часто відмічають абдомінальний біль |
|
|
Печінка |
Часто в крові визначаються маркери вірусного гепатиту С або В |
|
|
Легені |
Можливі легеневі кровотечі |
|
|
Серцево-судинна система |
Іноді розвивається інфаркт міокарда |
|
|
Синдром Шегрена |
Виникає у 14–40% хворих |
|
|
Синдром Рейно |
Є раннім проявом хвороби, розвивається у ⅓ хворих |
|
|
Лабораторні зміни |
Підвищення в сироватці крові концентрації кріоглобулінів, високі титри РФ класу IgM; зниження концентрації комплементу C1q, С4, С2, СН50 (при нормальній концентрації С3); поява АНФ; часто визначаються маркери вірусного гепатиту С, зрідка — В |
|
|
Основа діагнозу |
Враховують факт виявлення в сироватці крові кріоглобулінемії, клінічні прояви — в першу чергу, ураження шкіри, нирок, інфікування гепатотропними вірусами |
|
|
ОТА (хвороба Вінівартера — Бюргера) |
Серцево-судинна система |
Відзначають біль в ногах, який посилюється вночі та в спокої, відчуття печіння, оніміння пальців, мерзлякуватість. Стійкий ціаноз дистальних фаланг, набряки пальців, виразки та некрози, нерідко — гангрена. Переміжна кульгавість спостерігається не завжди так, як уражуються проксимальні відділи артерій у 50–80% пацієнтів. Наслідком ураження коронарних артерій може бути розвиток стенокардії та інфаркт міокарда. У 10% хворих виявляють феномен Рейно |
|
ШКТ |
Іноді уражуються мезентеріальні артерії. Виникає біль у животі, розвиваються виразки, некрози кишечнику та шлунково-кишкові кровотечі. Можливий розвиток кишкової непрохідності |
|
|
Нирки |
При тромбозі ниркової артерії виникає інфаркт нирки, який проявляється болем, гіпертермією, гематурією, підвищенням артеріального тиску |
|
|
ЦНС |
Виникають порушення мозкового кровообігу (у тому числі повторні), ішемічні неврити зорових нервів, тромбоз судин сітківки, крововиливи на очному дні |
|
|
Лабораторні зміни |
Характерними є нейтрофільний лейкоцитоз, збільшення ШОЕ, вмісту серомукоїду, сіалових кислот, гаптоглобіну, фібриногену, γ-глобуліну. Підвищується агрегація тромбоцитів, згортання крові. Виявляють антигени до HLA-A9, -B5, -DR4 |
|
|
Основа діагнозу |
Враховують відповідність діагностичним критеріям ОТА (Moses M. et al., 1970), характерну клінічну картину (зокрема клінічну тріаду — оклюзію дистальних артерій, синдром Рейно, мігруючий тромбофлебіт поверхневих вен), результати радіоізотопного дослідження (порушення мікроциркуляції в ділянці нижніх кінцівок), ангіографічні ознаки, наявність венуліту у біоптатах |
Діагностика
Установити точний діагноз васкуліту дозволяє поєднання клінічних, лабораторних, гістологічних і ангіографічних даних. Перелік досліджень, необхідних при системних васкулітах, наведено в табл. 21.14.
Таблиця 21.14
Дослідження, проведення яких необхідне при системних васкулітах
|
Характер дослідження |
Мета дослідження |
|
|
Загальний аналіз крові, визначення печінкових ферментів, креатиніну, клінічний аналіз сечі |
Проводять у усіх випадках |
|
|
КФК |
Визначають при підозрі на міопатію |
|
|
Бактеріальне дослідження крові |
Виключення інфекції, сепсису |
|
|
Серологічне обстеження |
АНФ РФ |
Для виключення системного ревматичного захворювання |
|
АНЦА |
Для підтвердження діагнозу гранулематозу Вегенера, МПА, синдрому Черджа — Стросс |
|
|
Кріоглобулінемія |
Підтвердження кріоглобулінемічного васкуліту |
|
|
Антитіла до фосфоліпідів |
Виключення (підтвердження) АФС |
|
|
Антитіла до базальної мембрани капілярів |
Виключення синдрому Гудпасчера |
|
|
Визначення маркерів вірусів |
Гепатит В і С, ВІЛ |
За наявності підозри на ВП і кріоглобулінемічний васкуліт |
|
Цитомегаловірус, вірус Епштейна — Барр, парвовірус В19 |
Геморагічний васкуліт, ВП |
|
|
Біопсія тканин |
Виконується при гранулематозі Вегенера, МПА, ВП, синдромі Черджа — Стросс, кріоглобулінемічному васкуліті, ГКА |
|
При системних васкулітах у крові відзначають нейтрофільний лейкоцитоз, тромбоцитоз, нормохромну анемію, підвищення ШОЕ. Рівень СРБ корелює з активністю васкуліту. Дослідження, необхідні для визначення типу васкуліту:
- бактеріологічний посів крові;
- дослідження ліквору;
- антитіла до інфекційних збудників;
- антитіла до цитоплазми нейтрофілів (АНЦА);
- антинуклеарні антитіла;
- РФ;
- АФЛ;
- кріоглобуліни;
- КФК;
- компоненти комплементу С3, С4, СН50.
Тканини, з яких беруть біоптати при васкулітах, а також характер очікуваних змін, наведено в табл. 21.15.
Таблиця 21.15
Тканини, з яких беруть біоптати при васкулітах, і характер очікуваних змін
|
Васкуліт |
Місце взяття біоптату, |
Характер клітинної інфільтрації у біоптатах |
|
ВП |
Біопсія уражених органів. Вісцеральна ангіографія |
Некротизуючий артеріїт. Ураження судин дрібного і середнього калібру виникають одночасно на різних стадіях хвороби |
|
Синдром Черджа — Стросс |
Біопсія уражених органів (як правило, легенів) |
Гранулематозні еозинофільні інфільтрати |
|
Гранулематоз Вегенера |
Біопсія легенів; АНЦА |
Некротизуючі гранульоми у верхніх і нижніх дихальних шляхах. Некротизуючий вогнищевий артеріїт у легенях, некротизуючий гломерулонефрит |
|
ГКА |
Біопсія скроневих артерій |
Гігантоклітинні інфільтрати в стінках судин |
|
Хвороба Такаясу |
Ангіографія; МРТ грудної клітки |
Гігантоклітинні інфільтрати в стінках судин |
|
Ізольований васкуліт ЦНС |
Біопсія мозкових оболонок або уражених ділянок мозку |
Гранулематозне запалення дрібних і середніх артерій мозку |
|
Лейкоцитокластичні васкуліти (медикаментозний, харчовий, гіпокомплементарний, хвороба Шенлейна — Геноха, есенціальна змішана кріоглобулінемія) |
Біопсія шкіри |
Лейкоцитокластичний васкуліт з ураженням дрібних судин (капілярів, венул) на тій самій стадії розвитку |
У клінічній практиці найчастіше зустрічаються гіперсенситивні (лейкоцитокластичні) васкуліти, включаючи геморагічний васкуліт. При прийомі лікарських препаратів можливе зв’язування їх з білками організму, у результаті чого вони розцінюються імунною системою як чужорідні. Продукуються антитіла, виникають імунні комплекси, які відкладаються в стінках венул шкіри. Розрив судин супроводжується появою пурпури. У біоптатах виявляють нейтрофільну інфільтрацію стінок судин, вихід еритроцитів у оточуючі тканини.
Щодо ролі у патогенезі васкулітів антитіл до цитоплазми нейтрофілів, то вважається, що ці антитіла (АНЦА) спрямовані проти специфічних антигенів, які є в цитоплазмі нейтрофілів. Вони зв’язуються з вмістом цитоплазми, і викид активованими нейтрофілами протеолітичних ферментів призводить до пошкодження судин. Ці антитіла виявляються при ряді аутоімунних захворювань, зокрема МПА і гранулематозі Вегенера. Відмінності між цитоплазматичними і перинуклеарними АНЦА виявляють за допомогою методу непрямої імунофлуоресценції. Залежно від характеру світіння виділяють 2 типи АНЦА (табл. 21.16).
Таблиця 21.16
Типи АНЦА
|
Цитоплазматичні АНЦА |
У реакції з фіксованими етанолом нейтрофілами з’являється дифузне зернисте світіння цитоплазми. Антитіла зв’язуються із сериновою протеїназою-3, що міститься в азурофільних гранулах нейтрофілів. Їх виявляють при гранулематозі Вегенера |
|
Перинуклеарні АНЦА |
У реакції з нейтрофілами з’являється перинуклеарне зернисте світіння. Ці аутоантитіла зв’язуються з мієлопероксидазою, а також еластазою і лактоферином. Їх виявляють у хворих з МПА і синдромом Черджа — Стросс |
У цілому АНЦА виявляють при різних захворюваннях, але найчастіше — при васкулітах (табл. 21.17).
Таблиця 21.17
Визначення перинуклеарних (П) та цитоплазматичних (Ц) АНЦА при різних патологіях
|
Патологія |
П АНЦА |
Ц АНЦА |
|
МПА |
+ |
+ |
|
Синдром Черджа — Стросс |
+ (рідко) |
+ |
|
Гранулематоз Вегенера |
– |
+ |
|
Аутоімунні захворювання печінки |
+ |
– |
|
Гломерулонефрит |
+ |
– |
|
Виразковий коліт (МПО-негативний) |
+ (МПО-негативний) |
– |
|
ВІЛ-інфекція |
+ |
– |
|
Інші інфекції |
+ (рідко) |
– |
|
Новоутворення |
+ (рідко) |
– |
При гранулематозі Вегенера титр цитоплазматичних АНЦА перебуває в прямій залежності від активності хвороби, і його визначення може використовуватися для прогнозування загострень. АНЦА необхідно визначати при підозрі на МПА, поліміозит, а також первинний біліарний цироз печінки. Частоту виявлення АНЦА при різних аутоімунних хворобах подано в табл. 21.18 (Emlen W., 1999).
Таблиця 21.18
Частота виявлення АНЦА при різних аутоімунних захворюваннях
|
Захворювання |
Цитоплазматичні антигени |
Частота виявлення,% |
|
Поліміозит |
Синтетаза тРНК (анти- Jo-1 та ін.) |
20–30 |
|
СЧВ |
Рибосомальний протеїн |
5–10 |
|
Гранулематоз Вегенера |
Серинова протеїназа-3 |
90 |
|
МПА і деякі інші васкуліти |
Мієлопероксидаза та ін. |
70 |
|
Первинний біліарний цироз печінки |
Мітохондрії |
80 |
Оцінку активності васкулітів здійснюють за допомогою шкали BVAS — Birmingham Vasculitis Activity Score (табл. 21.19).
Таблиця 21.19
Оцінка активності васкулітів у балах за шкалою BVAS (за: Lugmani R.A. et al., 1994)
|
Прояви |
Бал |
Прояви |
Бал |
|
1. Системні прояви |
Інфільтрат |
4 |
|
|
Змін немає |
0 |
Вузлики або фіброз |
2 |
|
Слабкість |
1 |
Кровохаркання |
4 |
|
Лихоманка <38,5 °С |
1 |
Легенева кровотеча |
6 |
|
Лихоманка >38,5 °С |
2 |
Плевральний випіт/плеврит |
4 |
|
Біль у м’язах |
1 |
Максимально можлива сума балів |
6 |
|
Артрит/артралгії |
1 |
6. Серцево-судинна система |
6 |
|
Зменшення маси тіла (<2 кг) |
2 |
Змін немає |
0 |
|
Зменшення маси тіла (>2 кг) |
3 |
Відсутність пульсу, яка виникла знову |
4 |
|
Максимально можлива сума балів |
3 |
Шуми |
2 |
|
2. Шкірні покриви |
Аортальна недостатність |
4 |
|
|
Змін немає |
0 |
Перикардит |
4 |
|
Пурпура |
2 |
Інфаркт міокарда, який виник знову |
6 |
|
Інший васкуліт шкіри |
2 |
Кардіоміопатія, серцева недостатність |
6 |
|
Інфаркт |
2 |
Максимально можлива сума балів |
6 |
|
Виразки |
4 |
7. ШКТ |
|
|
Гангрена |
6 |
Змін немає |
0 |
|
Множинна гангрена пальців |
6 |
Біль у животі |
3 |
|
Максимально можлива сума балів |
6 |
Кривава діарея |
6 |
|
3. Слизові оболонки/очі |
Перфорація жовчного міхура |
9 |
|
|
Змін немає |
0 |
Інфаркт кишечнику |
9 |
|
Кон’юнктивіт |
1 |
Панкреатит |
9 |
|
Епісклерит/склерит |
2 |
Максимально можлива сума балів |
9 |
|
Увеїт |
6 |
8. Нирки |
|
|
Набряк сітківки |
6 |
Змін немає |
0 |
|
Крововилив у сітківку |
6 |
Артеріальна гіпертензія (діастолічний артеріальний тиск >90 мм рт. ст.) |
4 |
|
Виразки порожнини рота |
1 |
Протеїнурія (>1 г/добу) |
4 |
|
Виразки геніталій |
1 |
Гематурія (>1 ер. у полі зору або >1000 ер./мл) |
8 |
|
Максимально можлива сума балів |
6 |
Креатинін 125–249 мкмоль/л |
8 |
|
4. ЛОР-органи |
Креатинін 250–499 мкмоль/л |
10 |
|
|
Змін немає |
0 |
Креатинін >500 мкмоль/л |
12 |
|
Виділення з носа або утруднення носового дихання |
2 |
Максимально можлива сума балів |
12 |
|
Скоринки |
4 |
9. Нервова система |
|
|
Синусит |
2 |
Змін немає |
0 |
|
Носова кровотеча |
4 |
Органічні порушення, деменція |
3 |
|
Виділення з вух |
4 |
Судоми |
9 |
|
Середній отит |
4 |
Інсульт |
9 |
|
Глухота, яка виникла знову |
6 |
Ураження спинного мозку |
9 |
|
Ларингіт/захриплість голосу |
2 |
Периферична невропатія |
6 |
|
Стеноз гортані |
6 |
Множинний руховий мононеврит |
9 |
|
Максимально можлива сума балів |
6 |
Максимально можлива сума балів |
9 |
|
5. Легені |
Загальна максимально можлива сума балів |
63 |
|
|
Змін немає |
0 |
||
|
Задишка/утруднене дихання |
2 |
Клініко-лабораторні дані, які дозволяють підозрювати наявність у пацієнта васкуліту, наведені у табл. 21.20.
Таблиця 21.20
Клініко-лабораторні дані, які дозволяють підозрювати наявність у пацієнта васкуліту
|
Анамнестичні дані |
|
|
Скарги |
Слабкість, анорексія, втрата маси тіла, лихоманка, порушення зору, стенокардія, утруднені рухи кінцівками, нижньою щелепою та ін. |
|
Наявність різних захворювань |
Гепатит В, С, ВІЛ-інфекція, ЗЗСТ, онкопатологія та ін. |
|
Прийом лікарських засобів |
Будь-якої групи |
|
Дані фізикального обстеження |
|
|
Шкіра |
Геморагічна пурпура, виразки, некрози |
|
Судини |
Болісність судин при пальпації, вислуховування шуму над ними, вузлики; дефіцит пульсу, артеріальна гіпертензія |
|
М’язи |
Скутість, болісність при скороченні і пальпації |
|
Нервова система |
Вогнищеві та дифузні порушення функції ЦНС, сенсорні і моторні невропатії |
|
Статева система |
Болісність яєчок, еритема (почервоніння мошонки) |
|
Лабораторні дані |
|
|
Клінічний аналіз крові |
Анемія, тромбоцитоз, збільшення ШОЕ |
|
Біохімічні дослідження |
Підвищення рівня КФК, амінотрансфераз, креатиніну, сечовини |
|
Імунологічні дослідження |
Виявлення HbsAg, антинуклеарні антитіла, АНЦА, кріоглобулінів, збільшення вмісту γ-глобулінів |
Активність васкулітів контролюють за допомогою лікарських засобів. Для ушкодження органів або тканин при васкулітах не характерний зворотний розвиток, тому оцінка його ступеня дозволяє прогнозувати подальший перебіг хвороби. Оцінку фази хвороби за шкалою активності патологічного процесу представлено в табл. 21.21.
Таблиця 21.21
Оцінка фази хвороби за шкалою активності патологічного процесу
|
Повна ремісія |
Ознаки активності відсутні, загальна сума балів 0–5. При нормальному рівні СРБ необхідності в терапії немає |
|
Часткова ремісія |
Зменшення загальної суми балів на тлі лікування на 50% від початкової |
|
Неактивна фаза |
Спостерігається повна ремісія, підтримувальна терапія не потрібна |
|
«Велике» загострення |
Реєструють залучення в запальний процес життєво важливих органів — ЦНС, серцево-судинної системи, легенів, нирок. Відзначається збільшення загальної суми на 6 і більше балів. Потрібне призначення ГК, цитостатиків, в/в імуноглобуліну, плазмаферезу |
|
«Мале» загострення |
Відзначають збільшення загальної суми балів від 1 до 5 |
Вузликовий поліартеріїт
Вузликовий поліартеріїт (ВП) — це некротизуюче запалення середніх і дрібних артерій без гломерулонефриту або васкуліту артеріол, капілярів і венул.
Код за МКХ-10
М30 Вузликовий поліартеріїт.
Клініцист A. Kussmaul і патологоанатом R. Maier в 1866 р. виділили ВП в окрему нозологічну форму і вперше дали детальну клініко-морфологічну характеристику захворюванню в публикації «про своєрідне, не описане раніше захворювання артерій — periarteriitis nodosa, яке проходить з брайтовою хворобою нирок і швидкопрогресуючим загальним паралічем». Синонімом ВП є також назва хвороба Куссмауля — Мейєра, яка відображає заслуги цих вчених. Пізніше із ВП виділили окремі нозологічні форми, такі як гранулематоз Вегенера, синдром Черджа — Стросс, МПА. Поширеність ВП становить 0,46–7,7 випадка на 100 тис. населення, щорічно виявляють 0,2–1 новий випадок на 100 тис.
Хворіють на ВП однаково часто і чоловіки, і жінки. Захворювання може розвинутися в будь-якому віці, навіть у дітей грудного віку, але частіше хворіють особи у віці 40–50 років. Діти становлять від 10 до 21,4% усіх хворих на ВП.
Етіологія
Етіологія ВП невідома. До середини XIX ст. будь-яку судинну патологію вважали наслідком сифілітичної інфекції. Після виділення ВП в окрему нозологічну форму ймовірною стала бактеріальна (стрептококова, туберкульозна), токсична, вірусна етіологія захворювання. Першу експериментальну модель ВП отримано в 1943 р. A. Rich и J. Gregory при введенні кроликам в/в кінської сироватки і сульфадимезину. Пізніше встановлено зв’язок між ВП і вірусом гепатиту B (частота випадків інфікування становить 5–40%), виділено вірус-індуковану форму захворювання. Вважається, що має значення також інфекція, асоційована з HCV, цитомегаловірусом, парвовірусом В-19, Т-лімфотропним вірусом 1-го типу, ВІЛ. Зв’язку з HLA-системою не виявлено. Провокуючими факторами є інсоляція, переохолодження, пологи, тривалі емоційні навантаження, вакцинація, прийом деяких лікарських препаратів (алопуринол, сульфаніламідні препарати та ін.). ВП може поєднуватися з аутоімунними захворюваннями (СЧВ, РА, ДМ, синдромом Когана), а також з волосистоклітинним лейкозом.
Патогенез
В стінці судин відкладаються імунні комплекси, які містять імуноглобуліни A, M, що призводить до активації системи комплементу і хемотаксису нейтрофілів, які за рахунок викиду біологічно активних речовин призводять до деструкції судинної стінки. В останні роки встановлено важливу роль в патогенезі ВП клітинних імунних реакцій. За даними C. Lid і співавторів (1994), в запальному інфільтраті судинної стінки переважають макрофаги, активовані СD4+ Т-лімфоцити, що несуть HLA-DR, ІЛ-2Р і трансферинові рецептори.
Патоморфологія
У патологічний процес при ВП можуть бути залучені судини будь-яких органів, але найбільш часто страждають шкіра, суглоби, периферичні нерви, кишечник, нирки. Ушкоджуються переважно артерії м’язового типу середнього, рідше дрібного калібру, частіше запалення локалізується в ділянці їх біфуркації, рідко до процесу залучаються артеріоли і вкрай рідко — венули. Типовим є фокальний і сегментарний некротизуючий панваскуліт, що поширюється тільки на частину окружності судинної стінки. У гостру фазу розвивається запалення стінки артерії з фібриноїдним некрозом її середньої оболонки. Запальний інфільтрат представлений різними клітинами, переважно нейтрофілами, меншою мірою лімфоцитами і еозинофілами. Пізніше відбувається повне руйнування нормальної архітектоніки судинної стінки, включаючи еластичну мембрану з заміщенням її аморфним матеріалом. У місці пошкодження артерії виявляють формування аневризми з пристінковим тромбозом. При організації затромбованих аневризм з’являються щільні вузлики за ходом судин, що є патогномонічною, але не обов’язковою ознакою захворювання. Частіше вузлики розташовані за ходом судин серця, брижі, нирок, печінки, кишечнику, у підшкірній клітковині. Наслідками ураження судин можуть бути розриви стінки судини, стенози, оклюзія просвіту, розвиток склеротичних змін. Типовим є хвилеподібний розвиток артеріїту, тому на різних ділянках ураженої судини відзначають різні фази запалення. Характерною ознакою ВП є поліморфізм морфологічних змін в органах и тканинах. У різних органах діагностують ішемію, крововиливи, некрози, кровотечі.
Клінічна картина
Клінічні прояви ВП надзвичайно різноманітні. Ще у 1907 р. М.А. Трахтенберг стверджував, що неможливо дати єдину характеристику клінічної картини ВП, оскільки жоден випадок захворювання не схожий на інший. Багато клініцистів підкреслювали хамелеоноподібні, маскуючі прояви цього захворювання.
Г.А. Даштаянц (1966 г.) виділив 3 стадії розвитку ВП: перша — початкова або лихоманково-больова; друга — розгорнута, виражені ураження органів; третя — кінцева з розвитком недостатності функцій різних органів і систем.
Частіше захворювання розпочинається поступово, рідше — гостро з швидким розвитком симптомів. Найбільш характерними симптомами дебюту захворювання є: лихоманка, схуднення, міалгії, артралгії, шкірні висипи. Іноді захворювання розпочинається з ураження внутрішніх органів, симптоми цих уражень можуть з’явитися за декілька місяців і навіть років до системних проявів.
Лихоманку відзначають у 75–88% хворих на ВП. Характерною є постійна фебрильна лихоманка, яка не реагує на антибіотики, а температура тіла знижується лише під дією ГК. Іноді захворювання розпочинається як лихоманка невідомого генезу, але тривалий ізольований лихоманковий синдром для ВП не характерний. Обмінно-дистрофічні зміни в організмі при запаленні зумовлюють прогресуюче схуднення хворого на ВП. При значній активності захворювання схуднення продовжується аж до кахексії.
Шкірні прояви при ВП виявляють в 25–60% випадків. Найбільш інформативними є сітчасте ліведо (livedo reticularis) і підшкірні вузлики, типові для цього захворювання, але тепер трапляються рідко. Часто у хворих наявна геморагічна пурпура, яку виявляють пальпаторно, рідше — везикулярні та бульозні висипи, некрози шкіри і м’яких тканин дистальних фаланг пальців.
Ураження кістково-м’язової системи при ВП, пов’язані з ураженням судин, які кровопостачають ці тканини, реєструють у 30–70% випадків. Типовим є інтенсивний біль у гомілкових м’язах, іноді настільки значний, що хворий не може пересуватися. Ураження суглобів у дебюті хвороби відзначають більш ніж у 50% випадків. Суглобовий синдром варіює від мігруючих артралгій до транзиторного недеформуючого артриту з ураженням одного або декількох суглобів (гомілковостопного, колінного, плечового, ліктьового, променезап’ясткового). Іноді залучаються суглоби кистей і стоп, що може нагадувати РА.
Особливістю клінічного перебігу ВП розгорнутої стадії є ураження різних органів, які виникають іноді одночасно, але частіше послідовно, в найбільш неочікуваному поєднанні. Для клінічної картини цієї стадії характерні 5 клінічних синдромів: неврологічний, нирковий, абдомінальний, кардіальний, легеневий.
Ураження нервової системи, пов’язане з ураженням vasa nervorum, яке призводить до ішемії нервових стовбурів або волокон, є однією із найбільш частих і ранніх ознак захворювання, яку реєструють у 50–75% випадків. Характерним проявом є периферична поліневропатія з множинними асиметричними моно- або поліневритами та руховими і чутливими розладами. Частіше ці зміни пов’язані з ураженням гомілкових нервів і їх гілок, рідше — з ураженням променевого, ліктьового, серединного нервів. Найбільш типовими є чутливі та рухові розлади за типом шкарпеток і рукавичок. Ураження нервової системи проявляється інтенсивним болем, парестезіями з розвитком у подальшому рухових розладів. Можливий розвиток поліневропатії за типом mononeuritis multiplex. При цьому варіанті уражаються чутливі та рухові компоненти багатьох окремих периферичних нервів. У більшості хворих ці два типи ураження периферичної нервової системи поєднуються. Рідко відзначають розвиток синдрому Гійєна — Барре, при якому ушкоджується переважно руховий компонент периферичного нерва. У цих хворих основним проявом є не біль, а м’язова слабкість, яка виникає раптово. При ВП неврологічні порушення різноманітні — від мононевриту до важкого енцефалополімієлорадикулоневриту з тетрапарезом. У процес залучаються і черепні нерви, частіше лицьовий. Рідко можливі ураження ЦНС у вигляді гіперкінетичного синдрому, інфарктів мозку, порушення психіки.
Нирки при ВП уражаються в 45–50% випадків. Морфологічна основа клінічних проявів — васкуліт дугових, інтерлобарних, інтерлобулярних артерій нирок з формуванням множинних аневризм, інфаркти нирок. Рідше клінічні прояви зумовлені ураженням артеріол, а у пізній період хвороби — вторинним ураженням клубочків, стенозом ниркових артерій. Розвиток гломерулонефриту не характерний для ВП. Зазвичай симптоми, які свідчать про ураження нирок, з’являються через 3–6 міс від початку хвороби і належать до несприятливих ознак. Ураження нирок частіше проявляється сечовим синдромом із помірною протеїнурією (втрата білка <3 г/добу), мікрогематурією, яку розцінюють як ознаку активності васкуліту, незначною лейкоцитурією. Артеріальну гіпертензію реєструють у 30–35% хворих. Швидке прогресування ниркової недостатності у хворих на ВП пов’язане з множинними інфарктами нирок. Ниркова недостатність розвивається більш ніж у 20% хворих. Рідше фіксують такі ускладнення, як розрив аневризми ниркових судин з утворенням навколониркової гематоми.
Ураження серцево-судинної системи при ВП відзначають у 40–75% випадків. Найчастіше виявляють коронарит, клінічними проявами якого можуть бути: вторинна стенокардія, гострий коронарний синдром з розвитком інфаркту міокарда, який частіше має атиповий перебіг, у безбольовій формі. Характерним проявом є ураження серця за типом швидкопрогресуючого кардіосклерозу з розвитком порушення серцевого ритму і провідності, хронічної серцевої недостатності.
Ураження ШКТ відзначається у 50% хворих і є типовим проявом та найбільш тяжкою формою органної патології при ВП. Частіше ушкоджуються судини тонкого, товстого кишечнику, шлунка, можуть залучатися судини селезінки, підшлункової залози та інших органів. Ішемія, що розвивається в органах ШКТ, проявляється абдомінальним болем, нудотою, блюванням, діареєю, появою виразок, ускладнених кровотечею, перфорацією, інфарктами органів. Також можна відзначати клінічну картину холециститу, панкреатиту, апендициту. Ішемічний біль у животі, що супроводжується меленою, реєструють при тромбозі мезентеріальних артерій. У літературі описано випадки інфаркту печінки, утворення гематоми в результаті розриву дрібних печінкових судин. Іноді утворюються кісти в підшлунковій залозі.
Легені ушкоджуються у 15% хворих. Частіше реєструють легеневий васкуліт або пневмоніт, рідше діагностують інфаркт легень і плеврит.
Типовим для ВП є розвиток орхіту, епідидиміту, з характерною болісністю при пальпації або болем в яєчках, який є одним із критеріїв діагнозу ВП. Симптоми нецукрового діабету з’являються при артеріїті судин гіпофіза. Ураження очей виявляють у 10% випадків, частіше це васкуліт з оклюзією центральної артерії сітківки, рідко розвиваються кон’юнктивіт, ірит, негранулематозний увеїт.
У частини хворих ушкоджуються периферичні артерії нижніх або верхніх кінцівок, що призводить до ішемії тканин, гангрени пальців, розриву аневризм. Можливе ураження скроневої артерії, що нагадує клінічну картину скроневого артеріїту.
У випадку ВП, асоційованого з вірусом гепатиту В, значних відмінностей у клінічному перебігу не відзначають, але частіше відмічають ураження яєчка, розвиток нефропатії, злоякісної артеріальної гіпертензії, інфаркту нирок. При асоціації ВП з вірусом гепатиту С частіше розвиваються патологія шкіри, печінки, гіпокомплементемія.
Виділяють обмежену або моноорганну форму ВП, для якої характерне ураження судин одного органа: апендикулярного відростка, жовчного міхура, матки, яєчок, серця, периферичних судин. Іноді захворювання проявляється лише поліневропатією. У 10% випадків ВП діагностують шкірну форму, ознаками якої є зміни шкіри у вигляді пурпури, що пальпується, болісних вузликів, сітчастого ліведо, некротичних виразок.
Лабораторні дослідження
Специфічних лабораторних показників для ВП немає. В активний період відзначають лейкоцитоз, тромбоцитоз, підвищення ШОЕ, рідше — незначну еозинофілію, помірну нормохромну анемію. Рівень підвищення СРБ корелює з активністю хвороби. У 25% випадків в активній фазі ВП знижується рівень С3, С4 компонентів комплементу, підвищується рівень ЦІК, може бути незначне підвищення титру РФ, антинуклеарних антитіл, рідко визначаються антинейтрофільні цитоплазматичні антитіла (АНЦА). При ураженні нирок розвивається помірна протеїнурія, еритроцитурія. Усім хворим на ВП необхідні також серологічні дослідження крові для визначення маркерів вірусу гепатиту B, C, ВІЛ-інфекції.
Інструментальні дослідження
Для підтвердження діагнозу ВП доцільно проводити ангіографію судин нирок і черевної порожнини. На ангіограмах відзначають аневризми і сегментарні стенози артерій середнього калібру й вкорочення периферичних судинних гілок (судин брижі, нирок, печінки). Розміри аневризм становлять 1–1,5 мм, здебільшого судин брижі, нирок, печінки. Потрібно зазначити, що на фоні терапії ці зміни можуть зникати, а можуть розвиватися клиноподібні інфаркти. При проведенні ультразвукової допплерографії зміни у судинах нирок реєструють у 60% хворих. У половині випадків вони представлені стенозами.
Морфологічні зміни
Морфологічне підтвердження діагнозу ВП необхідне, оскільки це один із критеріїв встановлення діагнозу. Переважно застосовують біопсію органів, залучених у патологічний процес. Більш доступним та інформативним є гістологічне дослідження шкіри і м’язів, результати позитивні у 30–50% хворих. Рідше проводять біопсію стегнового нерва, печінки, прямої кишки, яєчок. За неможливості виконання біопсії показана ангіографія, яка також необхідна у випадку, якщо передбачено біопсію печінки і нирок з метою виявлення аневризм, травма яких може зумовити кровотечу. Біопсія нирок доцільна лише у тому випадку, якщо відсутні ураження інших органів, оскільки морфологічні зміни нирок при ВП подібні до таких при всіх васкулітах з ураженням судин середнього калібру. Біопсія нирок також необхідна для диференційної діагностики ВП і МПА.
Діагностика
Діагноз ВП ґрунтується на детальному клінічному обстеженні хворого, вивченні історії хвороби, даних лабораторного, інструментального, морфологічного обстеження хворого.
Класифікаційні критерії ВП розроблено у 1990 р. R. Lightfoot і співавторами (табл. 21.22).
Таблиця 21.22
Класифікаційні критерії ВП
|
Критерій |
Визначення |
|
Зменшення маси тіла >4 кг |
На початку захворювання, не пов’язана з особливостями харчування |
|
Сітчасте ліведо |
Сітчасті зміни судинного малюнка шкіри на кінцівках і тулубі |
|
Біль чи болісність в яєчках при пальпації |
Не пов’язані з інфекцією, травмою |
|
Міалгії, слабкість або болісність у м’язах нижніх кінцівок при пальпації |
Дифузні міалгії (виключно плечового пояса і поперекової ділянки), слабкість або болісність у м’язах ніг |
|
Мононеврит, множинна моно- або поліневропатія |
Відповідні неврологічні прояви |
|
Діастолічний артеріальний тиск >90 мм рт. ст. |
Підвищення артеріального тиску |
|
Підвищення рівня сечовини або креатиніну в крові |
Сечовина >14,4 ммоль/л (40 мг%), креатинін >0,133 ммоль/л (1,5 мг%), не пов’язані з дегідратацією або порушенням уродинаміки |
|
Інфікування вірусом гепатиту В |
Наявність HBsAg або антитіл до вірусу гепатиту В у сироватці крові |
|
Ангіографічні зміни |
Аневризми або оклюзії вісцеральних артерій, не пов’язані з атеросклерозом та іншими запальними захворюваннями |
|
Біопсія артерій |
Гранулоцитарна і мононуклеарно-клітинна інфільтрація стінки судин |
Діагноз ВП можна встановити за наявності 3 і більше будь-яких критеріїв (чутливість — 82,2%, специфічність — 86,6%).
Диференційний діагноз
Симптоми, які нагадують ВП, реєструють при багатьох інших захворюваннях, тому ВП часто є діагнозом виключення. Потрібно зазначити, що ВП має бути виключеним у всіх хворих з лихоманкою, схудненням, ознаками поліорганного ураження (судинна пурпура, множинний мононеврит, сечовий синдром). Виключати необхідно ряд інфекцій, особливо гепатити В, С, стрептококову, ВІЛ-інфекції. Подібні симптоми можуть відзначати при багатьох ревматичних захворюваннях — СЧВ, РА, синдромі Шегрена, ДМ, ССД, есенціальній кріоглобулінемії, рецидивуючому поліхондриті, ГКА, МПА, хворобі Кавасакі, а також гігантоклітинному лейкозі, запальних захворюваннях кишечнику, амфетаміновій наркоманії.
Основні диференційно-діагностичні ознаки ВП і МПА, а також ВП, СЧВ і РА з системними проявами наведено у табл. 21.23, 21.24.
Таблиця 21.23
Диференційно-діагностичні ознаки ВП і МПА
|
Ознака |
ВП |
МПА |
|
Патологія нирок |
Судинний тип нефропатії |
Гломерулонефрит |
|
Реноваскулярна гіпертензія |
+++ |
– |
|
Інфаркт нирок |
++ |
– |
|
Мікроаневризми |
++ |
– |
|
Швидкопрогресуючий гломерулонефрит |
– |
+++ |
|
Хронічна ниркова недостатність |
+ |
+++ |
|
Патологія легень |
Легеневий васкуліт, пневмоніт 10–15% |
Некротизуючий альвеоліт 12–30% |
|
Легеневі кровотечі |
– |
++ |
|
Периферична поліневропатія |
50–80% |
10–20% |
|
Рецидиви захворювання |
+ |
++ |
|
АНЦА до мієлопероксидази |
– |
50–80% |
|
Ознаки інфікування гепатитом В |
++ |
– |
|
Ангіографія: мікроаневризми, стенози |
++ |
– |
|
Гістологія |
||
|
Некротичний васкуліт |
+++ |
+++ |
|
Змішано-клітинна інфільтрація |
+++ |
+++ |
|
Гранульома |
+ |
– |
|
Калібр уражених судин |
||
|
Середні і дрібні артерії |
+++ |
+ |
|
Артеріоли |
+ |
++ |
|
Капіляри |
– |
+++ |
|
Венули |
– |
+++ |
Таблиця 21.24
Диференційно-діагностичні ознаки ВП, СЧВ і РА із системними проявами
|
Ознака |
ВП |
СЧВ |
РА |
|
Частіше хворіють |
Чоловіки й жінки |
Жінки |
Жінки |
|
Лихоманка |
+++ |
+++ |
+ |
|
Артралгії, міалгії |
+++ |
+++ |
+++ |
|
Зв’язок із вірусною інфекцією; психологічною травмою |
+++ ++ |
+ ++ |
+++ ++ |
|
Ураження шкіри |
Папулопетехіальна пурпура, бульозні, везикульозні висипи |
«Метелик», еритема, капілярити, фотодерматоз |
Множинні екхімози, поліморфний висип геморагічного характеру |
|
Лімфаденопатія |
– |
+++ |
+++ |
|
Ранкова скутість |
– |
++ |
+++ |
|
Артрит |
– |
++ |
+++ |
|
Рентгенологічні зміни: узури, ОП |
– |
+ |
+++ |
|
Ураження нервової системи: судомний синдром; моно-, поліневрити |
+ +++ |
+++ + |
+ ++ |
|
Ураження серця: ендокардит; перикардит |
Коронарит – + |
Люпус-кардит + +++ |
Міокардіодистрофія + ++ |
|
Ураження легень: інтерстиціальні пневмонії; плеврит |
++ + |
+ +++ |
+++ +++ |
|
Ураження ШКТ |
+++ |
+ |
+ |
|
Ураження очей |
+ |
– |
++ |
|
Ураження нирок |
Інфаркти |
Люпус-нефрит |
Гломерулонефрит, амілоїдоз |
|
Лейкоцити Тромбоцити |
Підвищені |
Знижені |
Підвищені |
|
Антинуклеарні антитіла |
+(+) |
+++ |
+(+) |
|
Судинна аневризма |
+++ |
+ |
– |
|
РФ |
+(+) |
+(+) |
+++ |
|
Калібр уражених судин |
Середні |
Середні, дрібні |
Великі, середні, дрібні |
У проведенні диференційної діагностики допоможе той факт, що у хворих з АНЦА-асоційованими васкулітами аневризми судин розвиваються рідко, але останні також можуть відзначатися при СЧВ, передсердній міксомі, хворобі Кавасакі, тромбоцитопенічній пурпурі, ендокардиті.
Лікування
Лікування хворого на ВП має відповідати сучасним вимогам, бути комплексним та індивідуальним. Метою лікування є досягнення ремісії, запобігання ураженню життєво важливих органів, підтримка ремісії, зниження ризику загострень, запобігання розвитку побічних ефектів медикаментозної терапії.
Для індукції ремісії необхідне призначення ГК разом із цитостатиками. У якості пригнічувальної терапії призначають преднізолон у дозі 1 мг/кг/добу (per os) у кілька прийомів (максимально 60 мг/добу), при позитивній клініко-лабораторній динаміці через 7–10 днів дають повну дозу в один прийом вранці. Після досягнення ефекту, частіше через 3–4 тиж, дозу ГК знижують по 5 мг кожні 2 тиж з поступовим уповільненням швидкості зниження протягом 6 міс. Підтримувальна доза становить 0,15–0,2 мг/кг/добу (5–10 мг/добу).
Циклофосфамід призначають (per os) у дозі 1–2 мг/кг/добу (максимально 200 мг/добу) або у вигляді пульс-терапії в дозі 15 мг/кг/добу. Перші 3 курси пульс-терапії проводять з інтервалом 2 тиж, а потім через 3 тиж. Після досягнення ремісії (через 3–6 міс) дозу циклофосфаміду поступово знижують на 1,5 мг/кг/добу. При призначенні циклофосфаміду необхідно контролювати рівень лейкоцитів у периферичній крові, який має бути не нижчим 3–3,5•109/л, нейтрофілів — 1–1,5•109/л. На початку лікування контроль має проводитися через день, після стабілізації рівня лейкоцитів — не рідше ніж 1 раз на 2 тиж. При нирковій недостатності (креатинін >0,17 ммоль/л) та в осіб літнього віку доза циклофосфаміду має бути знижена на 25–50%. Застосування циклофосфаміду більш ніж 6 міс поспіль часто викликає побічні реакції (респіраторну патологію з морфологічними симптомами ураження легень).
Для підтримувальної терапії використовують азатіоприн у дозі 1,5–2 мг/кг/добу або циклофосфамід у дозі 1 мг/кг/добу. Терапію ГК і цитостатиками доцільно проводити протягом не менш ніж 24 міс після досягнення ремісії.
При швидкопрогресуючому тяжкому перебігу, з порушенням функції життєво важливих органів використовують ескалаційну терапію. Призначають 7–10 процедур плазмаферезу протягом 14 днів (видалення плазми в обсязі 60 мл/кг маси тіла з заміщенням її рівнозначним обсягом 4,5–5% людського альбуміну) у комбінації з пульс-терапією метилпреднізолоном у дозі 15 мг/кг/добу й циклофосфамідом у дозі 10 мг/кг/добу, в/м ін’єкції бетаметазону 1 мл, за необхідності повторне введення через тиждень або внутрішньошкірно 0,2 мл/см2 бетаметазону (рівномірне обколювання місця ураження, не більше 1 мл/тиж).
При обмеженому ураженні судин і відсутності ознак прогресування захворювання призначають середні дози ГК (преднізолон 40–60 мг/добу) протягом 30–45 днів, при досягненні клінічно-лабораторного ефекту дозу поступово знижують до підтримувальної.
У хворих із ушкодженням лише судин шкіри ефективним є метотрексат (17,5–20 мг/тиж).
У разі виявлення у хворого маркерів реплікації вірусу гепатиту B призначають противірусні препарати. Інтерферон-альфа зараз застосовують рідко, що пов’язано з великою кількістю побічних проявів. Частіше призначають ламівудин в дозі 100 мг/добу (упродовж 6 міс) та середні дози ГК з плазмаферезом.
Також при лікуванні ВП використовують лімфоцитаферез, імуносорбцію, проводять лікування артеріальної гіпертензії та іншу симптоматичну терапію.
Прогноз
Із введенням у терапію хворих на ВП комбінації ГК і цитостатиків 5-річна виживаність становить 60–80%, у той час як до цього відзначали лише 15%. Несприятливий прогноз зумовлений ураженням серцево-судинної системи, ЦНС, ШКТ, нирок, а також початком захворювання у віці старше 50 років.
Мікроскопічний поліангіїт
МПА (поліартеріїт) — це некротизуючий васкуліт з мінімальною кількістю або відсутністю імунних депозитів, при якому ушкоджуються переважно дрібні судини (капіляри, венули або артеріоли), рідко — артерії малого й середнього калібру, у клінічній картині якого домінують явища некротизуючого гломерулонефриту і легеневі капілярити.
Ще в середині минулого сторіччя відзначено, що в ряді випадків при ВП спостерігається переважно васкуліт дрібних судин і домінує патологія нирок. З відкриттям АНЦА інтерес до цієї проблеми посилився, оскільки в 70–90% хворих на МПА виявляють саме ці антитіла. Клінічні спостереження й досягнення в галузі імунології дозволили в 1985 р. виділити з ВП ще одну нозологічну форму — МПА. Разом з гранулематозом Вегенера та синдромом Черджа — Стросс МПА належить до групи АНЦА-асоційованих захворювань.
Захворюваність на МПА становить 0,36 на 100 тис. населення на рік. МПА діагностують у 10 разів частіше, ніж ВП, і у 2 рази частіше, ніж гранулематоз Вегенера. Хворіють переважно чоловіки середнього віку (співвідношення чоловіків і жінок 1,4:1).
Етіологія
Причина захворювання невідома. Чіткої генетичної схильності не встановлено, але в останні роки відзначено підвищення частоти Hla-dqw7 при АНЦА-васкулітах, включаючи МПА. На сьогодні домінує думка щодо важливої ролі АНЦА в патогенезі МПА. До 30% хворих на МПА є АНЦА-негативними.
Морфологія
За морфологічними і клінічними ознаками МПА займає проміжне становище між ВП і гранулематозом Вегенера.
Васкуліт має наступні особливості:
- до патологічного процесу залучаються переважно дрібні судини мікроциркуляторного русла (капіляри, венули, артеріоли);
- некротизуючий васкуліт має розповсюджений характер;
- мікроскопічно в запальному інфільтраті переважають нейтрофіли, немає імунних комплексів або антитіл до базальної мембрани;
- морфологічною особливістю є відсутність гранульом, тому при МПА, на відміну від гранулематозу Вегенера, не відбувається розпаду тканин, на відміну від ВП — рідко пошкоджуються судини середнього калібру та не формуються аневризми;
- ушкоджуються судини різних органів, але найбільш характерні зміни виявляють у нирках, шкірі, легенях;
- в нирках формується сегментарний некротизуючий гломерулонефрит, який у 80–90% випадків поєднується з екстракапілярною проліферацією епітеліальних клітин і формуванням півмісяців.
Клінічна картина
Продромальний період короткий. Звичайно МПА починається із загальних, неспецифічних скарг, так званого грипоподібного синдрому, який супроводжує більшість запальних захворювань. Хворого турбують слабкість, міалгії, артралгії, схуднення. Але, по-перше, ці симптоми менш яскраво виражені, ніж при ВП, а по-друге, — для МПА не характерний тривалий ізольований продромальний період, а типовим є швидке залучення до патологічного процесу інших органів. Найбільш значні патологічні зміни виникають у шкірі, нирках і легенях.
При МПА у ⅓ хворих в дебюті відзначають зворотне ураження верхніх дихальних шляхів у вигляді атрофії слизової оболонки носа, некротичного риніту. У 30–40% випадків реєструють патологію придаткових пазух носа й середнього вуха. На відміну від гранулематозу Вегенера, деструкцій тканин верхніх дихальних шляхів, деформацій носа не виявляють, гранульоми в біоптатах не визначаються. У незначної частини хворих реєструють стійкі неерозивні артрити великих суглобів, які на початку хвороби помилково розцінюють як ознаки РА.
У початковій стадії МПА типовим є ураження шкіри, які відзначають у 80% хворих, у вигляді пурпури, що пальпується, папул, везикуло-бульозних висипів, некрозів шкіри й підлеглих тканин, виразок, крововиливів під нігті (табл. 21.25).
Таблиця 21.25
Клінічна класифікація МПА (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічний |
|
|
Ступінь активності |
0 (відсутній), I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика ураження |
Нирки |
Фокальний, сегментарний, некротизуючий екстракапілярний гломерулонефрит |
|
Кістково-м’язова система |
Артралгії, артрит, міалгії |
|
|
Шкіра |
Пурпура, виразково-некротичні зміни |
|
|
Дихальна система |
Атрофічний некротизуючий риніт, синусит, некротизуючий альвеоліт з геморагіями, плеврит |
|
|
Периферична нервова система |
Поліневропатія |
|
Ураження нирок при МПА, яке найчастіше клінічно проявляється швидкопрогресуючим гломерулонефритом, діагностують у всіх хворих. Типовим є нефротичний синдром, рідше відзначають сечовий синдром з відповідними змінами сечі та крові. Особливістю ураження нирок є, як правило, відсутність артеріальної гіпертензії або наявність м’якої артеріальної гіпертензії, швидкий розвиток ниркової недостатності, про що свідчать зниження швидкості клубочкової фільтрації і підвищення в сироватці крові рівня креатиніну.
Легені уражаються у 12–29% хворих. Частіше виявляють легеневий капілярит, але трапляються випадки інтерстиціального фіброзу легень. Ознаки останнього можуть з’явитися за декілька років до інших симптомів. Найбільш часті клінічні прояви: кашель, біль у грудній клітці. Характерні кровохаркання й легенева кровотеча, в 10% випадків — смертельні.
У цій стадії хвороби в 70% хворих відзначають ураження кістково-м’язової системи у вигляді артралгій, неерозивних артритів великих суглобів, міалгій. У 25–32% пацієнтів ушкоджуються судини ШКТ, що клінічно проявляється болем в животі, шлунково-кишковими кровотечами. У результаті ураження нервової системи у 14–58% хворих розвивається периферична поліневропатія або множинний мононеврит. Ураження очей проявляється склеритом, епісклеритом, увеїтом. Ураження серця для МПА не характерне.
Виділяють гострий, підгострий і хронічний варіанти перебігу, а також ступені активності: 0 (відсутній), I (мінімальний), II (помірний), III (високий).
Характерною рисою перебігу МПА є часті рецидиви хвороби із залученням у патологічний процес органів, незалучених у дебюті. Важливою ознакою початку рецидиву є виникнення слабкості, симптомів з боку м’язів і суглобів.
При так званих абортивних формах можуть відзначати або осередковий гломерулонефрит, який проявляється мікрогематурією, або легеневий капілярит, який призводить надалі до кровохаркання.
Лабораторні дослідження
У хворих реєструють помірну гіпохромну анемію, нейтрофільний лейкоцитоз, підвищення ШОЕ, підвищення концентрації в крові СРБ. Майже в половини хворих виявляють РФ у низьких титрах, рідше визначається АНФ. Маркери вірусу гепатиту B, гіпокомплементемія, еозинофілія відсутні. При дослідженні сечі в 100% випадків захворювання виявляють мікрогематурію, трохи рідше — протеїнурію. При порушенні функції нирок визначається креатинемія і зниження швидкості клубочкової фільтрації. За допомогою імунофлуоресценції виявляють перинуклеарні АНЦА, імуноферментним методом — АНЦА, що реагують із мієлопероксидазою. Позитивний результат імунофлуоресцентного дослідження бажано підтвердити імуноферментним методом.
Морфологічні зміни
Для діагностики використовують біоптати шкіри, нирок й легень. У шкірі знаходять лейкоцитокластичний васкуліт. У нирках при МПА розвивається капілярит клубочків і виникає сегментарний некротизуючий гломерулонефрит, який у 80–90% випадків поєднується з екстракапілярною проліферацією епітеліальних клітин і формуванням півмісяців. До ранніх гістологічних проявів належать мікротромбози у капілярах клубочків. Також виявляють гострий тубулярний некроз та тубулоінтерстиціальний нефрит. Відсутність імуноглобулінів і комплементу при імуногістологічному дослідженні, а також електронно-щільних депозитів при електронному мікроскопічному дослідженні в капілярах клубочків не дає можливості говорити про імунокомплексний генез ураження при МПА. У легенях відзначають септальні капілярити з нейтрофільною інфільтрацією, рідше — некротизуючий альвеоліт з геморагічним синдромом.
Інструментальні дослідження
При рентгенологічному дослідженні виявляють транзиторні легеневі інфільтрати без розпаду, ознаки альвеоліту.
Діагноз
Діагноз МПА ґрунтується на клінічній картині, даних морфологічного та імунологічного дослідження. Гістопатологічні зміни на фоні типової клінічної картини відповідають достовірному діагнозу. Якщо дані біопсії негативні, основне значення для діагностики має результат аналізу на АНЦА. Сьогодні не існує класифікаційних критеріїв захворювання. Zashin S. та співавтори у 1990 р. запропонували наступні діагностичні критерії МПА:
- лихоманка, нездужання, схуднення;
- артрит, міалгія;
- легеневі інфільтрати, часто з фатальною кровотечею;
- швидкопрогресуючий гломерулонефрит;
- шкірні прояви (некротизуючий васкуліт дрібних судин);
- HNO-cимптом;
- моно- або поліневропатія;
- серологія: АНЦА, що реагують із мієлопероксидазою.
Диференційний діагноз
Найчастіше доводиться проводити диференціювання із ВП, МПА, гранулематозом Вегенера та синдромом Черджа — Стросс. При МПА, на відміну від ВП, практично не пошкоджуються судини середнього калібру і не формуються аневризми. У нирках при ВП уражаються міжчасточкові судини, виникають мікроаневризми, стенози, інфаркти нирок, реноваскулярна гіпертензія, а при МПА відмічають некротичний гломерулонефрит, який поєднується з екстракапілярною проліферацією епітеліальних клітин і формуванням півмісяців, а клінічно формується швидкопрогресуючий гломерулонефрит. Для МПА не характерний тривалий ізольований продромальний період — грипоподібний синдром, симптоми його виражені меншою мірою, ніж при ВП, і швидко приєднуються зміни інших органів. При ВП частіше розвиваються й переважають ураження суглобів, периферичної і центральної нервової системи, інфаркти внутрішніх органів, функція нирок порушується незначно. Для ВП не характерні рецидиви захворювання, для МПА вони типові. У сироватці крові хворих на МПА присутні АНЦА до мієлопероксидази, але відкладення імунних комплексів у стінках уражених судин відсутні. Відсутні при МПА й маркери хронічного вірусного гепатиту В. Зниження концентрації С3, С4 компонентів комплементу не характерне для МПА.
Диференціація МПА із гранулематозом Вегенера та синдромом Черджа — Стросс представлено у табл. 21.26.
Таблиця 21.26
Диференціація МПА із гранулематозом Вегенера та синдромом Черджа — Стросс
|
Клінічна ознака |
Гранулематоз Вегенера |
МПА |
Синдром Черджа — Стросс |
|
Ураження вуха, горла, носа |
Перфорація носової перегородки, деформація носа, кондуктивна або нейросенсорна туговухість, підзв’язковий стеноз |
Ураження відсутні або легкі |
Поліпи носа, алергічний риніт, кондуктивна туговухість |
|
Ураження очей |
Орбітальна псевдопухлина, склерит, епісклерит, увеїт |
Іноді склерит, епісклерит, увеїт |
Іноді склерит, епісклерит, увеїт |
|
Ураження легень |
Вузлики, інфільтрати або порожнини, альвеолярні кровотечі |
Альвеолярні кровотечі |
Астма, легкі інфільтрати, альвеолярні кровотечі |
|
Ураження нирок |
Cегментарний некротизуючий гломерулонефрит, іноді гранулематозні зміни |
Некротизуючий гломерулонефрит з екстракапілярною проліферацією епітеліальних клітин і формуванням півмісяців |
Сегментарний некротизуючий гломерулонефрит |
|
Ураження серця |
Іноді ураження клапанів |
Ураження виникають рідко |
Серцева недостатність |
|
Ураження периферичної нервової системи |
Невропатія, зумовлена васкулітом (10%) |
Невропатія, зумовлена васкулітом (58%) |
Невропатія, зумовлена васкулітом (78%) |
|
Гістологічні зміни |
Лейкоцитокластичний васкуліт, некротизуюче гранулематозне запалення |
Лейкоцитокластичний васкуліт, відсутність некротизуючого гранулематозного запалення |
Іфільтрація тканин еозинофілами, васкуліт, гранульоми |
|
Наявність АНЦА |
80–90% |
70% |
50% |
|
АНЦА до: |
Протеїнази-3>мієлопероксидази |
Мієлопероксидази>протеїнази-3 |
Мієлопероксидази>протеїнази-3 |
|
Еозинофілія |
Іноді легка |
Немає |
Завжди |
Також слід проводити диференційну діагностику з пурпурою Шенлейна — Геноха, ЕКВ, синдромом Гудпасчера (легенево-нирковий синдром), для якого характерним є утворення антитіл до базальної мембрани судин, СЧВ, саркоїдозом, сепсисом, грибковими інфекціями, злоякісними новоутвореннями та іншими хворобами, для яких характерне ушкодження багатьох органів.
Приклад формулювання діагнозу
МПА, підгострий перебіг, активність II ступеня. Ураження легень (легеневі інфільтрати, ексудативний плеврит, дихальна недостатність II ступеня), нирок (гломерулонефрит, артеріальна гіпертензія, хронічна ниркова недостатність II ступеня), суглобів (поліарталгії), нервової системи (поліневропатія).
Лікування
Метою лікування є досягнення ремісії, запобігання ураженню життєво важливих органів, підтримка ремісії, зниження ризику рецидиву, запобігання розвитку побічних ефектів лікарської терапії. Терапія має відповідати сучасним схемам, бути комплексною та індивідуальною.
У більшості випадків для індукції ремісії при МПА застосовують комбіновану терапію ГК і цитостатиками. У якості пригнічувальної терапії призначають преднізолон у дозі 1 мг/кг/добу (максимально 60 мг/добу) per os у кілька прийомів, при позитивній клініко-лабораторній динаміці через 7–10 днів дають повну дозу в один прийом вранці. Після досягнення ефекту, частіше через 3–4 тиж, дозу ГК знижують по 5 мг кожні 2 тиж з поступовим уповільненням швидкості зниження протягом 6 міс. Підтримувальна доза становить 0,15–0,2 мг/кг/добу (5–10 мг/добу). Циклофосфамід призначають (per os) у дозі 1–2 мг/кг/добу (максимально — 200 мг/добу) або у вигляді пульс-терапії в дозі 15 мг/кг/добу. Перші 3 курси пульс-терапії проводять з інтервалом 2 тиж, а наступні — через 3 тиж. Після досягнення ремісії (через 3–6 міс) дозу циклофосфаміду поступово знижують на 1,5 мг/кг/добу. При призначенні циклофосфаміду необхідно контролювати рівень лейкоцитів у периферичній крові, який має бути не нижчим 3–3,5•109/л, нейтрофілів — 1–1,5•109/л. На початку лікування контроль необхідно проводити через день, після стабілізації рівня лейкоцитів — не рідше ніж 1 раз на 2 тиж. При нирковій недостатності (креатинін >0,17 ммоль/л) і в осіб літнього віку доза циклофосфаміду має бути зниженою на 25–50%. Застосування циклофосфаміду більше 6 міс часто викликає побічні реакції (респіраторну патологію з морфологічними симптомами ураження легень).
Для підтримувальної терапії застосовують азатіоприн у дозі 1,5–2 мг/кг/добу або метотрексат (12,5–20 мг/тиж). Терапію ГК й цитостатиками доцільно проводити протягом не менш ніж 12 міс після досягнення ремісії.
При швидкопрогресуючому ураженні нирок проводять ескалаційну терапію. Призначають 7–10 процедур плазмаферезу протягом 14 днів (видалення плазми в обсязі 60 мл/кг маси тіла із заміщенням її рівнозначним обсягом 4,5–5% людського альбуміну) у комбінації з пульс-терапією метилпреднізолоном у дозі 15 мг/кг/ добу і циклофосфамідом у дозі 10 мг/кг/добу. Після пульс-терапії призначають в/в імуноглобулін у дозі 0,2–2 г/кг протягом 5 днів (можливе одноразове введення в дозі 2 г/кг).
При обмеженому ураженні судин і відсутності ознак прогресування захворювання призначають середні дози ГК (преднізолон 40–60 мг/добу) протягом 30–45 днів, при досягненні клінічно-лабораторного ефекту дозу поступово знижують до підтримувальної.
При рефрактерному перебігу, АНЦА-асоційованому варіанті призначають одну із наступних схем терапії: ГК з ритуксимабом або ГК з мофетилмікофенолатом або ГК з лефлуномідом, або ГК с інфліксимабом. В якості підтримувальної терапії в цьому випадку призначають ГК або ГК з ритуксимабом.
Також використовують лімфоцитаферез, імуносорбцію, проводять симптоматичну терапію.
Прогноз
Прогноз в основному зумовлений тяжкістю ураження нирок, рідше причиною смерті є легеневі кровотечі, інфекційні ускладнення, пов’язані із проведеною терапією. За умови застосування ГК і цитостатиків (циклофосфамід) 5-річна виживаність становить >65%.
Церебральний ангіїт (нейроваскуліт)
Церебральний ангіїт — захворювання, в основі якого лежить гранулематозне запалення дрібних і середніх артерій мозку. Синонімами терміну «церебральний ангіїт» є «ізольований ангіїт ЦНС», «первинний васкуліт (ангіїт) ЦНС», «ідіопатичний васкуліт (ангіїт) ЦНС».
Морфологія
При гістологічному дослідженні біоптатів виявляють запалення судин, лейкоцитарну інфільтрацію з переважанням лімфоцитів, нерідко — гранульоми, що свідчить про проліферативний характер запалення.
Епідеміологія
Ця форма васкуліту належить до рідкісних захворювань. Вік пацієнтів варіює від 7 до 70 років, частіше захворювання розвивається в період 40–60 років. Чоловіки і жінки захворюють однаково часто.
Етіопатогенез
У більшості випадків хвороби причина її невідома. У деяких хворих виявляють цитомегаловіруси, вірус Variolla zoster, піогенні бактерії. Визначено зв’язок з сифілісом, лімфогранулематозом. При люмбальній пункції у хворих реєструють ознаки хронічного менінгіту.
Клінічна картина
Захворювання дебютує сильним головним болем, вогнищевою неврологічною симптоматикою, парезами, фокальними епілептичними нападами, прогресуючим спастичним парапарезом, порушеннями чутливості. Основні прояви васкуліту включають: персистуючий сильний головний біль; порушення свідомості; психічні порушення; полівогнищеву неврологічну симптоматику; епілептичні напади. Захворювання характеризується одним із представлених нижче синдромів (табл. 21.27).
Таблиця 21.27
Синдроми, які проявляються при церебральному ангіїті
|
1 |
Багатовогнищева симптоматика, яка нагадує розсіяний склероз |
|
2 |
Енцефалопатія (гостра і підгостра), яка проявляється психопатологічними порушеннями |
|
3 |
Загальномозкова або вогнищева симптоматика, яка імітує пухлину головного мозку і швидко прогресує |
Нерідко уражаються черепні нерви. Багатовогнищеве ураження призводить до деменції з психотичними та маніакальними станами, зрідка — до паркінсонізму. Іноді переважають симптоми хронічного менінгіту або ураження спинного мозку. Виникають гострі порушення мозкового кровообігу, субарахноїдальні крововиливи. Периферична нервова система не уражається.
Перебіг захворювання гострий (з летальним наслідком протягом 1 року), підгострий або хронічний. При хронічному перебігу клінічні симптоми хвороби розвиваються в одних випадках поступово, в інших — після тривалого субклінічного перебігу настає різке загострення. Як правило, ознак ураження інших органів немає.
Діагностика
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих, наведено в табл. 21.28.
Таблиця 21.28
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих на церебральний ангіїт
|
Клінічний аналіз крові |
Характерним є лейкоцитоз, підвищення ШОЕ, у деяких хворих відзначають тромбоцитоз |
|
Імунологічні дослідження |
Виявляють антинуклеарні антитіла, можливе збільшення в крові вмісту комплементу |
|
Аналіз спинномозкової рідини |
Виявляють плеоцитоз, еритроцити, підвищується вміст білка, у половини пацієнтів відзначають олігоклональні антитіла |
|
МРТ; МРТ із контрастуванням |
Виявляють множинні або окремі вогнища в сірій або білій речовині головного мозку (лейкоареоз); набряк півкуль або частин мозку, гранульоми в периваскулярному просторі; ущільнення м’якої або павутинної оболонки мозку |
|
Ангіографія |
Виявляють аневризми судин, множинні сегментарні стенози, оклюзію судин, розвиток колатералей. Ці зміни відзначають у 37–90% пацієнтів з гістологічно верифікованим ізольованим ангіїтом ЦНС |
|
Гістологічне дослідження біоптатів |
У біоптатах паренхіми мозку, м’якої та павутинної оболонки виявляють характерні зміни (діагностична цінність не більше 50%) |
Чутливість комбінації дослідження спинномозкової рідини та МРТ становить 100%.
Діагностичні критерії ізольованого ангіїту ЦНС за Moore (1989) наведено в табл. 21.29.
Таблиця 21.29
Діагностичні критерії ізольованого ангіїту ЦНС (за: Moore R.A., 1989)
|
1. |
Головний біль, дрібновогнищева неврологічна симптоматика принаймні протягом 6 міс або раптова поява чи швидке прогресування неврологічних проявів |
|
2. |
Незвичайні результати церебральної ангіографії |
|
3. |
Виключення системної інфекції або запалення |
|
4. |
Гістологічні ознаки лептоменінгеального або паренхіматозного васкуліту |
Діагноз встановлюють (верифікують) на підставі результатів церебральної ангіографії та біопсії тканини мозку і мозкових оболонок. Також виключають інші причини стенозування мозкових артерій. У цілому діагностика хвороби є надзвичайно тяжкою, особливо у осіб похилого віку. Діагноз виключається у тому випадку, коли відзначають нормальна картина МРТ за відсутності змін спинномозкової рідини. Обов’язковою для підтвердження діагнозу є ангіографія, яка виявляє мультисегментарне звуження або оклюзію судин з постстенотичним розширенням, сповільнене виведення контрасту, наявність численних судинних анастомозів. Слід враховувати, що у 10–30% хворих ангіографія не виявляє змін. Діагностиці ангіїту мало сприяє МР-ангіографія, оскільки вона майже не виявляє змін в дрібних артеріях. При мультифокальному або дифузному ураженні мозку, яке розвинулося підгостро, завжди слід думати про ізольований ангіїт ЦНС.
Диференціація
Його диференціюють з великою кількістю захворювань:
- гострі порушення мозкового кровообігу;
- розсіяним склерозом;
- епілепсією;
- пухлинами головного мозку;
- абсцесом мозку;
- менінгітом;
- тромбоемболією;
- системними васкулітами (ВП, ГКА, артеріїтом Такаясу);
- СЧВ;
- первинним АФС;
- туберкульозом;
- саркоїдозом;
- токсоплазмозом.
Лікування
До препаратів, що дозволяють досягти ремісії, належать ГК і циклофосфамід (їх поєднання). Преднізолон (1 мг/кг/добу) і циклофосфамід (2 мг/кг/добу) застосовують протягом 3–12 міс з подальшим зниженням дози препаратів протягом 6 міс до підтримувальних (до 20 мг). При стабільному стані циклофосфамід відміняють після завершення 1 року лікування. При тяжкому гострому початку захворювання лікування починають з 3-денної пульс-терапії метилпреднізолоном або 3 процедур плазмаферезу. Надалі призначають преднізолон у поєднанні з азатіоприном (2 мг/кг/добу). У процесі лікування повторюють дослідження спинномозкової рідини та МРТ. Під час лікування кількість лейкоцитів в крові не має зменшуватися нижче 4000/мкл. При передчасній відміні терапії виникають рецидиви хвороби. Адекватна терапія дозволяє досягти повного одужання або тривалої ремісії.
Прогноз
Іноді виникають спонтанні ремісії. У цілому прогноз несприятливий, особливо за відсутності лікування та у випадку гострого перебігу хвороби.
Гранулематоз Вегенера
Гранулематоз Вегенера — захворювання, що характеризується гранулематозним запаленням респіраторного тракту (верхніх і нижніх відділів), некротизуючим васкулітом, що уражує судини дрібного та середнього калібру (капіляри, венули, артеріоли, артерії), некротизуючим гломерулонефритом. Для цього захворювання характерно:
- наявність некротизуючого артеріїту, що уражує дихальну систему і нирки;
- виразкування слизової оболонки носових пазух, носоглотки, гортані, трахеї;
- розвиток некротичних гранульом з ділянками некрозу, що нагадують географічну карту;
- ураження нирок у вигляді гломерулонефриту (з утворенням півмісяців);
- виявлення у 70% пацієнтів цитоплазматичних АНЦА;
- швидке прогресування хвороби за відсутності лікування, розвиток ниркової недостатності;
- висока ефективність циклофосфаміду.
Код за МКХ-10
М31.3 Гранулематоз Вегенера.
Епідеміологія
Поширеність захворювання становить 1–3 випадки на 100 тис. населення. Вважають, що гранулематоз Вегенера розвивається рідше, ніж СЧВ, РА, РПМ, скроневий артеріїт. Середній вік хворих — близько 40 років. Чоловіки і жінки хворіють з однаковою частотою.
Етіологія
У генезі та загостренні хвороби етіологічну роль відводять інфекції. Як можливі етіологічні чинники розглядаються Mycobacterium avium, Chlamydia, Helicobacter pylori, віруси гепатиту В і С, Herpes zoster, цитомегаловіруси, вірус Епштейна — Барр. Розвитку гранулематозу Вегенера часто передує бактеріальна інфекція верхніх дихальних шляхів. Визначається асоціація з парвовірусом В19 і стафілококами. Наслідком інфекції є імунне і токсичне ушкодження ендотеліоцитів, інших судинних структур. Тригерну роль відіграє гіперчутливість до антибіотиків, туберкулостатиків і противірусних засобів. Некротизуючий васкуліт з ерозивно-виразковим ураженням верхніх дихальних шляхів може виникнути після прийому сульфаніламідів, антибіотиків, протитуберкульозних засобів, дифенілгідантоїну, введення протиправцевої сироватки, тривалого прийому наркотиків (лікарські засоби — гаптени — при поєднанні з білками організму стають антигенами). Значення має також ідіосинкразія до сироваток, вакцин.
Патогенез
Патогенез також залишається значною мірою невідомим. Припускають надходження через верхні дихальні шляхи якогось антигена, що викликає розвиток гранулематозного запалення внаслідок алергічної реакції уповільненого типу. Роль імунних комплексів, які виявляють в крові та судинній стінці деяких хворих, не доведена. Не доведено також етіологічну роль антитіл до цитоплазми нейтрофілів (протеази-3), які визначають у крові хворих. Немає пояснень тому факту, що в змивній рідині з трахеобронхіального дерева переважно знаходять нейтрофіли (при інших гранулематозних захворюваннях — лімфоцити).
Патоморфологічні зміни
У стінках дрібних артерій і вен, а також в оточуючих тканинах виявляють запальні гранульоми. Некротичний васкуліт, гранульоми, порожнини утворюються в тканині легень. Внаслідок запалення, фіброзу і рубцювання розвивається обструкція бронхів. Гранулематозне запалення і некроз (з васкулітом або без) відмічають в слизових оболонках верхніх дихальних шляхів, додаткових пазухах носа, носоглотці. У нирках гранульоми виникають рідко, спочатку з’являються морфологічні ознаки вогнищевого гломерулонефриту, а потім екстракапілярного. Гранульоми в травному тракті та дихальних шляхах некротизуються (у них мало клітин фібробластичного ряду), у печінці й нирках — рубцюються. У слизовій оболонці верхніх дихальних шляхів розвивається гранулематозне запалення, васкуліт і некроз тканин (табл. 21.30).
Таблиця 21.30
Морфологічні зміни у верхніх дихальних шляхах при гранулематозі Вегенера
|
Порожнина носа |
У 70% хворих відзначають хронічне запалення з гнійними виділеннями, виразками і кровотечею. Можлива перфорація носової перегородки, руйнування хряща, деформація (сідлоподібна) носа |
|
Додаткові пазухи |
У 80% пацієнтів розвивається хронічний синусит |
|
Порожнина рота |
Хронічне запалення призводить до виразок |
|
Глотка |
Внаслідок хронічного запалення виникає євстахіїт, а потім — середній отит (гострий гнійний або хронічний) |
|
Гортань та трахея |
Наслідком хронічного гранулематозного запалення може стати стеноз гортані та трахеї зі стридорозним диханням і дихальною недостатністю |
У тканині легені виникає запальний процес, який розвивається гостро або хронічно (табл. 21.31).
Таблиця 21.31
Характер запального процесу в легенях при гранулематозі Вегенера
|
Гостре запалення |
У стінках судин, інтерстиціальній тканині, міжальвеолярних перегородках і альвеолярних порожнинах виявляють виражену інфільтрацію запальними клітинами з переважанням нейтрофілів. Гостре запалення супроводжується розвитком некротичного васкуліту дрібних судин, капіляритом альвеолярних перегородок |
|
Хронічне запалення |
У периваскулярному просторі інтерстиціальної тканини і перегородок альвеол утворюються запальні гранульоми (вузлики). Вони можуть розташовуватися в стінках судин і бронхах |
Гостре запалення переходить в хронічне гранулематозне. Як правило, у більшості хворих із гранулематозом Вегенера реєструють як гостре, так і хронічне запалення з переважанням одного з них. Вогнища гострого та хронічного запалення розсмоктуються без утворення рубця або з відкладенням в цих ділянках колагену.
При гістологічному дослідженні легень виявляють характерну тріаду ознак: некротичні поліморфно-клітинні гранульоми, васкуліт, вогнища некрозу (табл. 21.32).
Таблиця 21.32
Тріада морфологічних ознак в легенях при гранулематозі Вегенера
|
Гранульоми |
Мають неправильну форму, містять нейтрофіли, лімфоцити, плазматичні клітини, макрофаги, гігантські багатоядерні клітини, еозинофіли. Гістіоцити навколо вогнищ некрозу утворюють характерні «палісадні» структури |
|
Васкуліт |
Має деструктивний характер, розвивається в артеріях, венах і капілярах. У стінках судин виявляють некроз, тромбоз, інфільтрацію паличкоядерних лейкоцитів |
|
Вогнища некрозів |
Утворюються у гранульомах і легеневій тканині та нагадують географічну карту. У вогнищах некрозу за рахунок великої кількості зруйнованих ядер клітин виявляють базофілію. Вони нагадують мікроабсцеси. До прогнозованих причин некрозу відносять активацію нейтрофілів та ішемію |
У легенях бронхіальні артерії та вени уражуються більшою мірою, ніж судини легеневих артерій та легеневих вен. Процес розвивається білатерально, локалізується переважно в середніх та нижніх відділах легень, де виникають інфільтрація, геморагічні інфаркти, наявні порожнини розпаду (часто інфіковані). Крім гострого деструктивного процесу, розвивається пневмосклероз, адгезивний плеврит. У мікроциркуляторному руслі, у тому числі в альвеолярній тканині легень, виявляють капілярити, венуліти. Вони можуть супроводжуватися деструкцією, геморагічною інфільтрацією тканин. Відзначають ураження артерій середнього та дрібного калібру у вигляді мукоїдної дезорганізації сполучної тканини, фібриноїдного некрозу стінок з наступним склерозом та стенозуванням. Внаслідок деструкції стінок судин розвиваються аневризматичні розширення, які призводять до кровотеч. Таким чином, ураження судин носить системний характер із залученням вен усіх калібрів. У верхніх дихальних шляхах, бронхах, легенях виникають множинні артеріїти, панфлебіти, тромбофлебіти. При інфікуванні розвиваються септичні флебіти з гнійним розплавленням стінок.
Класифікація
Клінічну класифікацію гранулематозу Вегенера наведено в табл. 21.33.
Таблиця 21.33
Клінічна класифікація гранулематозу Вегенера (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічний |
|
|
Стадія |
Початкова, розгорнута, термінальна |
|
|
Ступінь активності |
0 (відсутня), I (мінімальна), II (помірна), III (висока) |
|
|
Клініко-морфологічна характеристика уражень |
Суглоби |
Артрити, артралгії |
|
Шкіра і м’язи |
Поліморфний висип, бульозні, геморагічні та папульозні висипи, виразково-некротичні зміни, некротичні вузлики, міопатія з розвитком атрофії |
|
|
Верхні дихальні шляхи |
Риніти, синусити (гнійно-геморагічні), виразково-некротичні ураження слизової оболонки глотки, гортані, трахеї, виразковий стоматит, глосит, хейліт, некротичне ураження мигдаликів |
|
|
Орган слуху |
Середній облітеруючий отит, осифікуючий лабіринтит |
|
|
Орган зору |
Одно- або двобічний екзофтальм з некрозом, склерит, кон’юнктивіт, кератит |
|
|
Легені |
Інфільтративне ураження легень з розвитком дихальної недостатності, інфаркти легень, плеврити, абсцеси, емпієма плеври, легеневі кровотечі, бронхообструктивний синдром |
|
|
Нирки |
Нефропатія з прогресуючою нирковою недостатністю, геморагічний цистит |
|
|
Серце |
Коронарит з розвитком інфаркту міокарда, порушеннями ритму і провідності; міокардит, перикардит, ендокардит з формуванням вади мітрального/трикуспідального клапанів |
|
|
ШКТ |
Гострий або хронічний панкреатит, інфаркти печінки і селезінки, виразково-некротичні зміни, кровотечі, гранульома шлунка |
|
|
Периферична нервова система |
Поліневропатія із залученням черепно-мозкових нервів |
|
|
ЦНС |
Інфаркти мозку, геморагічний інсульт, психози |
|
Умовно виділяють локальну, обмежену і генералізовану форми хвороби (табл. 21.34).
Таблиця 21.34
Умовні форми гранулематозу Вегенера
|
Форма |
Характеристики |
|
Локальна |
Ураження ЛОР-органів або очей. Можлива лихоманка, міалгії, артрити |
|
Генералізована |
Наявна тріада ознак ELK (E — ear, nose, throat; L — lung; K — kidney) |
|
Обмежена (лімітована) |
Ознаки ураження ЛОР-органів, легень. Інші ураження (шкіри, нервової системи, нирок) відсутні в дебюті захворювання. Гломерулонефрит може розвиватися в період розгорнутої клінічної картини |
Клінічна картина
Основні клінічні прояви гранулематозу Вегенера включають класичну тріаду:
- виразково-некротичні зміни у верхніх дихальних шляхах, трахеї та бронхах;
- ураження легень;
- ураження нирок.
Але можливе ураження (гранулематозне запалення, васкуліт, васкуліт + гранулематозне запалення) будь-якого іншого органа.
Характерні скарги пацієнтів із гранулематозом Вегенера:
- закладеність носа;
- стійкий нежить із серозно-кров’яними або гнійними виділеннями;
- носові кровотечі;
- біль у ділянці додаткових пазух;
- охриплість голосу;
- кашель із виділенням значної кількості гнійно-кров’яного мокротиння;
- підвищена температура тіла.
На початку хвороби розвивається ураження верхніх дихальних шляхів і легень, а в надалі приєднується ураження нирок. Рідше захворювання починається з виразкового стоматиту, ураження очей, вух. У дебюті захворювання відзначають ряд характерних проявів:
- виразково-некротичні ураження слизової оболонки носа, додаткових пазух, іноді — ротової порожнини;
- риніт, пансинусит (закладеність носа і сухість слизової оболонки переходять у стійкий нежить);
- серозно- або гнійно-кров’яні виділення з носа;
- носові кровотечі;
- порушення носового дихання, нюху;
- звуження носових ходів;
- перфорація носової перегородки;
- сідлоподібна деформація носа.
При огляді хворих виявляють сухі або гнійно-кров’яні кірочки на слизовій оболонці носа, набряклість спинки носа, біль при постукуванні пальцем у проекції додаткових пазух. Можливе виявлення виразкового стоматиту, глоситу, хейліту, а також однобічного екзофтальму (ураження орбіти), гнійного отиту.
Дещо пізніше, ніж ураження носа, ротової порожнини, розвивається виразково-некротичне ураження слизової оболонки глотки, гортані, трахеї, бронхів. З’являється охриплість голосу, біль у горлі, кашель із виділенням кров’яного мокротиння, кровохаркання. У разі розвитку стенозу гортані та трахеї виникає стридорозне дихання.
Генералізованого характеру патологічний процес набуває через декілька тижнів або місяців від початку хвороби. Уражуються легені і нирки, з’являється задишка, біль в грудній клітці. Бронхіт (іноді — бронхіальна астма), що розвивається, сприяє формуванню легеневого серця. При ураженні легень не відзначають повної відповідності між фізикальними проявами і рентгенологічними даними: рентгенологічні зміни виявляють у 100% пацієнтів, а фізикальні — тільки у 50%. Рентгенологічно визначаються:
- множинні вогнищеподібні тіні;
- тонкостінні порожнини;
- інфаркти легень;
- ателектази;
- плеврит.
Порожнини в тканині легень, що виникають внаслідок розпаду, можуть інфікуватися бактеріями і грибами. Клінічні варіанти ураження легень і відповідні морфологічні та рентгенологічні еквіваленти наведено в табл. 21.35.
Таблиця 21.35
Клінічні варіанти ураження легень і відповідні морфологічні та рентгенологічні еквіваленти
|
Варіант клінічного перебігу |
Морфологічні і рентгенологічні еквіваленти |
|
Кашель (епізодичний або постійний) |
Переважаючим є хронічне запалення, рентгенологічно визначаються вузли або інфільтрати стійкого характеру |
|
Пневмоніт (гострий або підгострий) |
Переважають ознаки гострого запалення. На рентгенограмах — транзиторні інфільтрати (інтерстиціальні або вогнищеві) |
|
Кровохаркання |
У легеневій тканині виявляють вогнища гострого запалення, капілярит перегородок альвеол, крововиливи в порожнину альвеол. Рентгенологічне дослідження підтверджує леткі вогнищеві інфільтрати |
|
Плеврит |
Фібринозний процес, ексудат. Ознаки ексудативного плевриту при рентгенологічному і ультразвуковому дослідженні |
|
Хронічна дихальна недостатність |
Хронічне гранулематозне запалення, фіброз легеневої тканини. На рентгенограмах — стійкі дифузні інфільтрати |
|
Безсимптомний перебіг |
Виявляють у ⅓ пацієнтів. Хронічне запалення є переважаючим, на ренгенограмах виявляють вузли або інфільтрати |
Можуть виникати легеневі кровотечі, збільшення лімфатичних вузлів середостіння, бронхоплевральні нориці. На КТ виявляють потовщення стінок бронхів (сегментарних, субсегментарних), міжальвеолярних перегородок, ателектази, стенози бронхів, трахеї, гортані. Знижується дифузійна здатність легень, порушується їх функція як за обструктивним типом (більш ніж в 50% випадків), так і рестриктивним (у 30–40% пацієнтів). Часто виникає колапс бронхів, пневмонія (постобструктивна), у 20% випадків прогресує легенева недостатність як результат фіброзу і пневмоніту, викликаних циклофосфамідом. У бронхоальвеолярному лаважі в активну фазу гранулематозу Вегенера виявляють макрофаги, нейтрофіли, еозинофіли, при помірній активності — лімфоцити (CD4+Th1-клітини), у разі альвеолярних геморагій (субклінічних, дифузних) — сидерофаги.
Симптоматика ураження нирок у хворих із гранулематозом Вегенера
У більшості хворих клінічні симптоми ураження нирок повністю відсутні, але наявні зміни в аналізах сечі (гематурія, піурія, протеїнурія, циліндрурія). Клінічні прояви виявляють у 15% пацієнтів. За відсутності нефропатії говорять про локальну (обмежену) форму гранулематозу Вегенера.
Варіанти ураження нирок при гранулематозі Вегенера:
- вогнищевий сегментарний некротичний гломерулонефрит;
- дифузний гломерулонефрит;
- гломерулонефрит з «півмісяцями»;
- некротичний гранулематозний васкуліт ниркових судин;
- розвиток гострої та хронічної ниркової недостатності при тяжкому ураженні нирок.
Ураження нирок у хворих на гранулематоз Вегенера не супроводжується підвищенням артеріального тиску.
Загальну характеристику клінічних проявів гранулематозу Вегенера наведено в табл. 21.36.
Таблиця 21.36
Клінічні прояви гранулематозу Вегенера
|
Загальні симптоми |
На початку хвороби і в період загострення відзначають лихоманку (у 25–30% пацієнтів), слабкість, схуднення, міалгії, артралгії, артрити. Причиною цих симптомів є посилений синтез цитокінів (ІЛ-1, -6, ФНП-α) у вогнищах запалення |
|
Шкіра |
У 50% хворих виявляють пурпуру, що пальпується, папули, везикули, гангренозну піодермію, виразки, підшкірні некротизовані вузлики в ділянці стегон, сідниць і великих суглобів, синдром Рейно. У біоптатах — вогнища екстраваскулярного запалення, некрози, некротичний васкуліт з гранулематозною інфільтрацією стінок судин (не завжди) |
|
Кістково-м’язовий апарат |
У дебюті хвороби і в розгорнутій стадії відзначають поліартралгію, можливий неерозивний, недеформуючий поліартрит дрібних і великих суглобів, моноолігоартрити. Ці зміни реєструють в 64% випадків, руйнування хрящів не розвивається |
|
Орган зору |
Ураження очей відзначають у 50% пацієнтів. На початку захворювання розвиваються симптом червоних очей, потім — запально-некротичні ураження увеального тракту, слізного мішка. Діагностують кератит, виразки рогівки, вузликовий некротичний склерит, епісклерит, увеїт, кон’юнктивіт, птоз, дакріоцистит, обструкцію носослізної протоки, неврит зорового нерва, псевдотумор орбіти (випинання очних яблук вперед і вниз у 15% хворих, порушення співдружності руху очних яблук), тромбоз артерій і вен сітківки, сліпота. У ретробульбарному просторі розвивається гранулематозне запалення і фіброзні зміни. Виникають запальна інфільтрація окорухових м’язів, стиснення зорових нервів |
|
ЛОР-органи |
У більшості хворих (70–95% хворих) відзначають виразково-некротичний риніт з гнійно-геморагічними виділеннями, перфорацією перегородки, деформацією носа (сідлоподібною), синусит. Розвивається серозний передній отит з можливим інфікуванням і втратою слуху, стоматит, гінгівіт. Можливий субфарингеальний стеноз гортані внаслідок гранулематозного запалення, обструкція дихальних шляхів, зміна тембру голосу. Необхідно врахувати, що перебіг ЛОР-патології у деяких хворих може бути безсимптомним |
|
Легені |
Розвиваються одиничні або множинні двобічні інфільтрати, схильні до розпаду і утворення каверн. Каверни, як правило, рідини не містять. Залучається легеневий інтерстицій, розвивається геморагічний альвеоліт, кровохаркання. Можуть виникати легеневі кровотечі, плеврит, збільшення медіастинальних лімфатичних вузлів, що має схожість з пухлинним ураженням, лімфогранулематозом. Швидко прогресує постгеморагічна анемія. У половини хворих ураження легень може проходити безсимптомно. Воно прогресує при ураженні нирок. Відзначають дихальну недостатність за змішаним типом. Легеневий процес ускладнюється пневмонією, розвивається фіброз. Індукувати розвиток пневмоніту може циклофосфамід. За клінічними ознаками ураження легень нагадує синдром Гудпасчера |
|
Серцево-судинна система |
Можливий розвиток (у 10%) гранулематозного міокардиту, коронариту, ендокардиту (вальвуліту), ураження мітрального і аортального клапанів, перикардиту (інфекційного або уремічного), інфаркту міокарда, АВ-блокад |
|
ШКТ |
Розвивається виразкове ураження кишечнику, виникають кровотечі, біль в животі, діарея |
|
Нирки |
Характерним є розвиток вогнищевого нефриту (на ранніх стадіях), дифузного швидкопрогресуючого гломерулонефриту з вираженою протеїнурією, гематурією, еритроцитарними циліндрами, порушенням функції (хронічна ниркова недостатність). Морфологічно виявляють гломеруліт з перигломерулярними гігантоклітинними гранульомами. Без лікування ниркова недостатність швидко прогресує. Терапія циклофосфаном може супроводжуватися геморагічним циститом |
|
Статеві органи |
Можливий розвиток орхіту, епідидиміту |
|
Нервова система |
Розвивається васкуліт, гранульоми головного мозку. У 50% пацієнтів у розгорнуту стадію виникають множинний мононеврит, дистальна симетрична поліневропатія. Уражуються черепно-мозкові нерви (II, VI, VII), розвивається енцефаліт, енцефалопатія, центральні паралічі, крововиливи в мозок, синдром Горнера, офтальмопарез, рецидивуюча болісна офтальмоплегія, епілептиформний синдром. Зрідка уражується гортанний нерв та хіазми. Ураження окорухових нервів та механічне обмеження рухомості очних яблук внаслідок орбітальних гранульом викликає офтальмоплегію. Початковим проявом хвороби може бути менінгіт та мієлопатія. Епілептичні напади є наслідком васкуліту, гранулематозу, легеневої та ниркової недостатності, сепсису, ДВЗ-синдрому. У 5% хворих виникають внутрішньочерепні крововиливи внаслідок васкуліту та інтрагранульомної геморагії. Інколи розвивається тромбоз мозкових вен |
У літературі є повідомлення про ураження в деяких випадках легеневої артерії, молочних залоз, привушних слинних залоз, шийки матки, піхви, яєчок, сечового каналу. Поява лихоманки у хворих на гранулематоз Вегенера зумовлена як гранулематозним запаленням, яке супроводжується посиленим синтезом цитокінів, так і приєднанням вторинної інфекції. Частоту клінічних проявів при гранулематозі Вегенера наведено (за: Malyak M., 1999) в табл. 21.37.
Таблиця 21.37
Частота клінічних проявів при гранулематозі Вегенера
|
Клінічні прояви |
Стадія хвороби |
|
| Дебют, % | Розгорнута стадія, % | |
|
Лихоманка |
25 |
30 |
|
Схуднення |
15 |
35 |
|
Ураження ЛОР-органів |
70 |
94 |
|
Стеноз (субфарингеальний) гортані |
– |
16 |
|
Ураження органа слуху |
25 |
60 |
|
Втрата слуху |
– |
15–40 |
|
Ураження легенів |
50 |
85 |
|
Безсимптомне ураження легенів |
50 |
50 |
|
Пневмонія (інфекція) |
– |
40 |
|
Гломерулонефрит |
11–18 |
77–85 |
|
Хронічна ниркова недостатність тяжкого ступеня |
– |
40 |
|
Кістково-м’язові ураження |
75 |
75 |
|
Ураження очей |
Рідко |
28–55 |
|
Сліпота |
– |
8 |
|
Кардіальна патологія |
– |
10 |
Порушення в нервовій системі відзначають у 33% пацієнтів, РФ виявляють у 50–60% випадків.
Характерні бактеріальні ускладнення у хворих на гранулематоз Вегенера наведено в табл. 21.38.
Таблиця 21.38
Бактеріальні ускладнення у хворих на гранулематоз Вегенера
|
Варіанти ураження |
Причина розвитку |
Збудники |
|
Бактеріальний синусит |
Порушення дренажу внаслідок закупорки слизом отвору додаткових пазух носа |
Staphylococcus aureus |
|
Бактеріальна гнійна пневмонія |
Порушення бронхіального дренажу в результаті закупорки слизом просвіту бронха |
S. aureus, Streptococcus pneumoniae |
|
Ускладнення імуносупресивної терапії (ГК, циклофосфамід) |
Streptococcus pneumoniae, Pneumocystis carinii, Legionella, герпесвіруси, мікобактерії, гриби |
|
|
Токсична дія ліків на легені (циклофосфамід) |
Приєднання вищенаведеної інфекції |
Слід враховувати, що циклофосфамід викликає розвиток легеневого фіброзу.
Діагностика
Діагностичні критерії гранулематозу Вегенера наведено в табл. 21.39.
Таблиця 21.39
Діагностичні критерії гранулематозу Вегенера (ACR, 1990)
|
1. |
Запалення носа і порожнини рота |
Гнійні або кров’яні виділення з носа |
|
2. |
Рентгенологічні зміни в легенях |
Вузлики, інфільтрати і порожнини в легенях |
|
3. |
Зміни сечі |
Мікрогематурія (>5 еритроцитів у полі зору) або скупчення еритроцитів в осадку сечі |
|
4. |
Біопсія |
У стінці артерії або периваскулярному (екстраваскулярному) просторі розвивається гранулематозне запалення |
Установити діагноз дозволяє наявність у хворого 2 і більше будь-яких критеріїв (чутливість — 88%, специфічність — 92%).
Зміни лабораторних показників
Відзначають неспецифічні зміни: нейтрофільний лейкоцитоз, тромбоцитоз, нормохромну анемію, підвищення показника ШОЕ, СРБ, зниження вмісту в крові альбуміну і підвищення — ЦІК (у окремих хворих), гамма-глобулінів (переважно за рахунок імуноглобуліну A), появу РФ. Для гранулематозу Вегенера не характерна лейкопенія і тромбоцитопенія, що слід враховувати при проведенні диференційної діагностики. Істотно не змінюється рівень комплементу в крові. Серологічним маркером хвороби є АНЦА до протеїнази-3. Їх визначення має основне значення для діагностики і оцінки активності гранулематозу Вегенера. Майже в усіх хворих на дану патологію визначають цитоплазматичні АНЦА і тільки у деяких — перинуклеарні. Цитоплазматичні АНЦА відображають присутність в крові антитіл до протеїнази-3. Титр цитоплазматичних АНЦА відповідає ступеню активності гранулематозу Вегенера в 60% випадків, він підвищується при загостренні хвороби і не змінюється при приєднанні гострих інфекцій.
Частоту виявлення цитоплазматичних АНЦА при васкулітах наведено в табл. 21.40 (Вест С.Дж., 1999 (ред.)).
Таблиця 21.40
Частота виявлення АНЦА при васкулітах
|
Захворювання |
Частота виявлення, % |
|
| Цитоплазматичні АНЦА | Перинуклеарні АНЦА | |
|
Гранулематоз Вегенера |
85 |
10 |
|
ВП |
5 |
15 |
|
Синдром Черджа — Стросс |
10 |
60 |
|
МПА |
15–45 |
45–80 |
|
Ідіопатичний гломерулонефрит з «півмісяцями» |
25 |
65 |
Слід зазначити, що виявлення цитоплазматичних АНЦА є характерним для обмеженої кількості захворювань. Водночас перинуклеарні АНЦА і атипові АНЦА (коли тип світіння визначити неможливо) зустрічаються при багатьох захворюваннях (СЧВ, РА, виразковий коліт, хвороба Крона, первинний склерозуючий холангіт та ін.).
Основа діагнозу гранулематозу Вегенера
Діагноз ґрунтується на наявності характерної клінічної картини (тріади), результатах біопсії (особливо тканин легень). Серологічні дослідження (визначення антитіл до протеази-3) малоінформативні, оскільки високий титр антитіл зберігається роками. Тільки у 66% хворих з 4-разовим підвищенням титру виникають рецидиви. Частіше усього біоптати для гістологічного дослідження беруть з уражених ділянок слизової оболонки назофарингеальної зони. У них виявляють ознаки васкуліту, паренхіматозного некрозу, гранулематозного запалення:
- фібриноїдний некроз судинних стінок (артерій, венул, капілярів), формування мікроабсцесів, дистрофію;
- периваскулярну лейкоцитарну інфільтрацію навколо некротичних вогнищ;
- гранульоми, що містять макрофаги, лімфоцити і гігантські багатоядерні клітини.
Найбільш інформативним є гістологічне вивчення біоптатів легень (табл. 21.41).
Таблиця 21.41
Інформативність інструментальних досліджень при гранулематозі Вегенера
|
Рентгенографія органів грудної клітки |
Виявляє в легенях солітарні або множинні двобічні інфільтрати, схильні до розпаду, формування порожнин, нерідко — плеврит, дифузні альвеолярні геморагії, збільшення лімфатичних вузлів, розвиток бронхоплевральних нориць. Рентгенологічні ознаки патології легень збігаються з клінічними тільки у 65% випадків |
|
КТ |
Дозволяє виявити стенози гортані, трахеї, бронхів, потовщення стінок бронхів (сегментарних, субсегментарних), міжальвеолярних перегородок, ателектази, а також ознаки ураження периферичних відділів легеневих артерій |
|
Дослідження функції дихання |
Знижується дифузійна здатність легень (її підвищення відзначають при розвитку дифузних альвеолярних геморагій). Функція легень у 50% хворих порушується за обструктивним типом, у 30–40% — за рестриктивним. Розвиток пнемонії або пневмоніту, фіброзу, індукованих циклофосфамідом, у 20% супроводжується прогресуючою легеневою недостатністю |
|
Бронхоальвеолярний лаваж |
В активну фазу хвороби переважають нейтрофіли, макрофаги, паличкоядерні лейкоцити, еозинофіли, при помірній активності — лімфоцити (СD4+Th1-клітини); при альвеолярних геморагіях (субклінічних або дифузних) визначаються сидерофаги |
|
Гістологічне вивчення біоптатів |
Біоптати легеневої тканини мають найбільшу діагностичну цінність. Високоінформативною є біопсія періорбітальної тканини. Цінність відкритої біопсії легень є вищою, ніж трансбронхіальної та дослідження тканин слизової оболонки носа і пазух. У легеневій тканині майже завжди виявляють ознаки гранулематозного запалення і васкуліту. У слизовій оболонці верхніх дихальних шляхів діагностують гранулематозне запалення, некроз, а васкуліт може не проявлятися. Біоптати нирок підтверджують розвиток гломерулонефриту |
Диференціація гранулематозу Вегенера
Досить складно диференціювати локальний та обмежений (лімітований) гранулематоз Вегенера. До захворювань, що супроводжуються розвитком легенево-ниркового синдрому, належать синдром Гудпасчера, МПА, синдром Черджа — Стросс, СЧВ, геморагічний васкуліт. Слід врахувати, що до розвитку гломерулонефриту може призвести пневмонія стрептококової та мікоплазмової етіології. Некротичними змінами на шкірі, в легенях, ураженням нирок можуть супроводжуватися лімфоми, пухлини підшлункової залози. Пухлини можуть індукувати імунні реакції, у тому числі пов’язані з АНЦА. Захворювання, з якими диференціюють гранулематоз Вегенера, наведено в табл. 21.42.
Таблиця 21.42
Захворювання, з якими диференціюють гранулематоз Вегенера
|
Синдром Черджа — Стросс |
Алергія в анамнезі, характерною є еозинофілія, відсутність гранульом, розпаду тканин верхніх дихальних шляхів і легеневої тканини |
|
МПА |
Немає руйнування тканин верхніх дихальних шляхів і легень, відсутні гранульоми |
|
Синдром Гудпасчера |
Визначаються антитіла до базальних мембран |
|
СЧВ |
Наявні антинуклеарні антитіла, антитіла до двоспіральної ДНК і Sm-антигену |
|
Ідіопатична серединна гранульома обличчя |
Належить до ангіоцентричних лімфом (групи Т-клітинних лімфом). Уражує верхні дихальні шляхи, включаючи додаткові пазухи носа, руйнуються м’які тканини і покривається виразками шкіра обличчя (що не характерно для гранулематозу Вегенера). Васкуліт має вторинний характер. Серединна гранульома може давати метастази, при біопсії виявляють запалення і некроз. На відміну від гранулематозу Вегенера, позитивний ефект дає опромінення пухлини |
|
Лімфоматоїдний гранулематоз |
Також належить до групи ангіоцентричних лімфом і уражує легені, шкіру, нирки, ЦНС, де відбувається інфільтрація стінок судин, навколишніх тканин атиповими лімфоцитами і плазматичними клітинами. Формуються запальні гранульоми, але не розвиваються. Перебіг хвороби злоякісний |
Досить складно диференціювати локальний та обмежений гранулематоз Вегенера. При проведенні диференційної діагностики визначають також титр антитіл до протеази-3. До захворювань, з якими диференціюють гранулематоз Вегенера, належать геморагічний васкуліт, стрептококова пневмонія з гломерулонефритом, ангіоцентрична злоякісна лімфома, лімфоматоїдний гранулематоз, саркоїдоз, туберкульоз, сифіліс, системні мікози, пухлини. Слід враховувати, що деструктивні процеси в дихальних шляхах можуть викликати пухлини, збудники сифілісу, туберкульозу, гриби, ліки (при інгаляціях). У таких випадках необхідно проводити культуральні дослідження. До числа захворювань, з якими необхідно диференціювати гранулематоз Вегенера, входять вторинні васкуліти і васкулопатії (табл. 21.43).
Таблиця 21.43
Вторинні васкуліти і васкулопатії
|
Псевдоваскулітні синдроми |
Етіологія |
Прояви |
|
Вторинні васкуліти |
Інфекційні (ВІЛ-інфекція, інфекційний ендокардит, вірусний гепатит) |
Можливі шуми в серці, лихоманка, піднігтьові геморагії, некрози пальців, порушення функції печінки. Необхідно з’ясувати анамнез (прийом наркотиків, численні статеві зв’язки, злоякісні новоутворення в анамнезі), провести ехокардіографію, посіви крові на стерильність, визначити наявність маркерів вірусних гепатитів і ВІЛ |
|
При злоякісних пухлинах, системні захворювання сполучної тканини, РА, саркоїдозі |
||
|
Васкулопатії |
АФС, токсична дія ліків |
|
Для паранеопластических васкулітів характерний хронічний перебіг, схильність до тромбоемболічних ускладнень, резистентність до терапії. Їх найчастіше виявляють при новоутвореннях легень, органів травлення, нирок.
Необхідне проведення у дітей та дорослих диференційної діагностики хвороби Вегенера, а також МПА, пурпури Шенлейна — Геноха, СКВ з синдромом Гудпасчера. Нерідко у хворих із цим синдромом відзначають артралгії, міалгії, можливий розвиток церебрального васкуліту, двобічного ретиніту, увеїту. Першим проявом церебрального васкуліту може бути епілептичний статус, при цьому ураження головного мозку піддається дії імуносупресантів, зокрема циклофосфаміду. При синдромі Гудпасчера, який дебютував у дитячому віці, окрім анти-БМК-антитіл (базальна мембрана капілярів), знаходять у крові АНЦА. АНЦА визначаються не менш ніж у 20% хворих дорослих із синдромом Гудпасчера. Іноді діагностують «перехрещення» з гранулематозом Вегенера. Нерідко анти-БМК-антитіла в крові з’являються пізніше, ніж АНЦА, при цьому підвищується тяжість ураження легень і нирок. Формування синдрому Гудпасчера можливе при таких аутоімунних захворюваннях, як пурпура Шенлейна — Геноха, міастенія, виразковий коліт. При первинному біліарному цирозі анти-БМК-антитіла визначаються в крові одночасно з антимітохондріальними антитілами та антитілами до мієлопероксидази. Синдром Гудпасчера може одночасно асоціюватися з цукровим діабетом 2-го типу, макроглобулінемією Вальденстрема. Вважаємо за необхідне привести клінічну характеристику цього синдрому.
Приклад формулювання діагнозу
Гранулематоз Вегенера, хронічний перебіг, активність II ступеня. Ураження верхніх дихальних шляхів (риніт, синусити), слизової оболонки порожнини рота (виразково-некротичний стоматит), легенів (інфільтрація, порожнини, дихальна недостатність II ступеня), нирок (гломерулонефрит, хронічна ниркова недостатність II ступеня).
Основні принципи лікування
Лікування включає призначення імуносупресорів і ГК (табл. 21.44).
Таблиця 21.44
Принципи терапії пацієнтів з гранулематозом Вегенера імуносупресорами і ГК
|
Імуносупресори |
Усередину циклофосфамід 2 мг/кг/добу + преднізолон 1 мг/кг/добу |
Циклофосфамід є препаратом вибору, дозволяє досягти ремісії і підтримувати її. Дозу слід підбирати так, щоб кількість лейкоцитів в крові була >3000 в 1 мкл (нейтрофілів >1500 в 1 мкл). Після досягнення ремісії хворі повинні приймати циклофосфамід ще 1 рік. Можна застосовувати його в/в (1 г/м2/міс) з нижчим ризиком виникнення ускладнень порівняно зі щоденним прийомом. Преднізолон в дозі 1–3 мг/кг/добу хворі отримують (одночасно з циклофосфамідом) протягом 1 міс, потім дозу поступово знижують. Загальна тривалість лікування становить не менше 6 міс. Така терапія дозволяє поліпшити стан 90% пацієнтів і досягти ремісії у 75%. Монотерапія ГК покращує стан хворих, але на прогноз мало впливає. У 50% пацієнтів у міру зниження дози ГК рецидивують зміни в органах |
|
Метотрексат усередину (не більше 15 мг/тиж) + преднізолон |
Ремісія може бути досягнута за допомогою метотрексату. Його призначають при непереносимості циклофосфаміду. У разі хорошої переносимості через 1–2 тиж дозу поступово підвищують до 20–25 мг/тиж |
|
|
Азатіоприн 1–2 мг/кг/добу + преднізолон |
Застосовують для підтримки ремісії, досягнутої циклофосфамідом |
|
|
Інші методи |
Трансплантація нирки |
Проводять у разі розвитку хронічної ниркової недостатності |
Сьогодні широко застосовують азатіоприн і метотрексат, які у поєднанні з преднізолоном дозволяють досягти швидкої і повної ремісії. Особливості терапії при різних варіантах перебігу хвороби (за: Насонов Е.Л., 2005; 2010 (ред.)) представлено в табл. 21.45.
Таблиця 21.45
Терапія при різних варіантах гранулематозу Вегенера
|
Стандартна терапія |
Циклофосфамід 2–3 мг/кг/добу в 2–3 прийоми до одержання ефекту (в середньому близько 4 тиж) + преднізолон всередину 1 мг/кг/добу в 1 прийом уранці після їди до одержання ефекту. Надалі дозу циклофосфаміду знижують по 25 мг кожні 2–3 міс |
Після отримання ефекту дозу преднізолону знижують на 5 мг кожні 2 тиж до підтримувальної. Доза циклофосфаміду залишається постійною протягом 1 року, а потім ще не менше 1 року після досягнення ремісії |
|
Швидкопрогресуючий перебіг |
Циклофосфамід всередину 3–5 мг/кг/добу в 2–3 прийоми до одержання ефекту (5–10 діб) + преднізолон всередину 2–15 мг/кг в 1 прийом уранці після їди до одержання ефекту (5–10 діб) |
Потім лікування проводять відповідно до схеми стандартної терапії |
|
Лікування за відсутності небезпечних для життя ускладнень |
Метотрексат всередину 0,15–0,3 мг/кг 1 раз на тиждень в 2 прийоми протягом тривалого часу + преднізолон всередину 1 мг/кг/добу тривалий час |
Тривалість лікування залежить від клінічного ефекту |
Високі дози ГК ефективні на ранніх стадіях хвороби, при тяжкому ураженні нирок і легень лікування ними безуспішне. Більш ефективним є застосування комбінації ГК та імуносупресорів (азатіоприн, метотрексат), а найбільший ефект дає циклофосфамід. Слід враховувати, що циклофосфамід викликає ускладнення: у 43% пацієнтів розвивається цистит, у 5% — рак сечового міхура, у 2% — мієлодиспластичний синдром. Поєднане лікування цим препаратом та преднізолоном дозволяє контролювати хворобу. При запізнілому початку лікування навіть цей засіб може не попередити розвиток тяжкого дифузного гломерулонефриту з нирковою недостатністю. При швидкопрогресуючому та вкрай тяжкому перебігу гранулематозу Вегенера циклофосфамід вводять в/в (5–10 мг/кг/добу) протягом 2–7 днів, потім продовжують лікування per os. При досягненні клініко-лабораторної ремісії переходять на підтримувальну дозу 50–25 мг/добу тривалістю 12–18 міс. Контроль за ефектом лікування включає динаміку ШОЕ, рівня сироваткового креатиніну, його кліренсу, систематичне дослідження крові, кількості лейкоцитів. Розвиток лейкопенії вимагає негайного зниження дози циклофосфаміду. Для попередження геморагічного циститу хворим рекомендують випивати до 2,5 л рідини на добу. У випадку появи висипу на шкірі, ураження очей та серозних оболонок (особливо перикарду) призначають короткий курс преднізолону (30–40 мг/добу) у поєднанні з циклофосфамідом.
При розвитку субглоткового стенозу гортані проводять негайну трахеотомію або планову хірургічну корекцію — механічну або лазерну дилатацію трахеї, ларинготрахеопластику. У випадку гострої дихальної недостатності (внаслідок дифузних альвеолярних геморагій) проводять штучну вентиляцію легень.
Критерії ефективності лікування
Вони включають позитивну динаміку клінічних симптомів, поліпшення/нормалізацію лабораторних показників, у тому числі ШОЕ, СРБ, РФ, білкових фракцій.
Після досягнення ремісії рецидиви виникають у 50% випадків. Позитивним є той факт, що ремісії можна досягти знову. Про персистенцію активності гранулематозу Вегенера (BVAS/WEG — Birmingham vasculitis activity score/WEG) мова йде тоді, коли присутні клінічні прояви хвороби і не поглиблюються протягом 28 діб з моменту попереднього огляду. До віддалених ускладнень належать хронічна ниркова недостатність, стеноз гортані, туговухість, зниження слуху, деформація носа, хронічний синусит.
Прогноз
Нелікована хвороба Вегенера — смертельне захворювання, найбільш тяжке з групи системних васкулітів. Водночас сучасна терапія гранулематозу Вегенера забезпечує задовільний прогноз. У разі пізньої діагностики хворі помирають протягом першого року від легенево-серцевої і ниркової недостатності, приєднання інфекції. При генералізованій формі смерть хворих настає протягом 5 міс. При лікуванні циклофосфамідом і ГК 4-річна виживаність становить 93% (Насонов Е.Л., 2005), 5-річна — більше 75% (Насонов Е.Л., 2010 (ред.)). Тяжкі ураження органів можуть виникати і через тривалий час. Причиною смерті можуть бути також серцево-судинні катастрофи, злоякісні новоутворення (зокрема рак сечового міхура).
Хвороба Бехчета
Хвороба Бехчета — системний васкуліт, для якого характерним є ураження шкіри, слизової оболонки порожнини рота, статевих органів, очей, а також нервової системи.
Код за МКХ-10
М35.2 Хвороба Бехчета.
Епідеміологія
У Європі і США щорічно реєструють 6 випадків хвороби на 100 тис. населення, в Японії — 10, в Туреччині — 190. Середній вік початку захворювання становить близько 40 років, з однаковою частотою хворіють чоловіки і жінки.
Етіологія і патогенез
Вони не з’ясовані. Припускають інфекційний (вірусний) генез хвороби Бехчета, трапляються її епідемічні спалахи.
Патоморфологічні зміни наведено в табл. 21.46.
Таблиця 21.46
Патоморфологічні зміни при хворобі Бехчета
|
Слизова оболонка порожнини рота |
Ознаки хронічного запалення з лімфо-макрофагальною інфільтрацією, вогнищами некрозу |
|
Шкіра, ділянка геніталій |
Прояви дифузного васкуліту з некрозом судинної стінки дрібних артерій і вен, крововиливами, периваскулярною інфільтрацією лімфоцитами і макрофагами |
Класифікація
Клінічну класифікацію хвороби Бехчета представлено в табл. 21.47.
Таблиця 21.47
Клінічна класифікація хвороби Бехчета (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічний |
|
|
Ступінь активності |
0 (відсутній), I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Слизові оболонки |
Афтозні виразки ротової порожнини |
|
Шкіра |
Герпетиформний висип, вузлувата еритема, псевдофолікуліт, папулопустульозні елементи, вугреподібні вузлики |
|
|
Статеві органи |
Виразки статевих органів, епідидиміт, рубцеві зміни |
|
|
Орган зору |
Передній і задній увеїт із порушенням зору |
|
|
Суглоби |
Артрит переважно колінних, рідше — гомілковостопних, променезап’ясткових суглобів та ін. |
|
|
ШКТ |
Абдомінальний синдром, гастродуоденіт, виразкове ураження кишечнику, кровотечі |
|
|
Нервова система |
Поліневропатія, менінгіт, параліч краніальних нервів, набряк сосочка зорового нерва |
|
|
Серце |
Перикардит, ендокардит, міокардит |
|
|
Судини |
Поверхневий тромбофлебіт, тромбоз глибоких вен, артеріальна оклюзія та/або аневризми |
|
Клінічна картина
У 25–75% пацієнтів першим проявом хвороби є афтозний стоматит. Афти розвиваються несподівано, локалізуються переважно на слизовій оболонці губ (не переходять за червону облямівку), ясен, щік і язика. Рідше уражуються піднебіння, мигдалики і глотка (на відміну від синдрому Рейтера або синдрому Стівенса — Джонсона). Афти мають жовтий колір в центрі та оточені віночком еритеми. Діаметр їх варіює від декількох міліметрів до 1,5 см. Виразки утворюють скупчення (кількість виразок може досягати 100) і заживають протягом 10 днів без утворення рубців. Появі виразки спочатку передує червона пляма, а потім бульбашка. Тривалість виразкового стоматиту становить від декількох днів до 1 міс. Він завершується утворенням невеликих рубців або проходить безслідно. Виразки схильні до рецидивування і різко болісні.
Комбінований афтоз
Йдеться про хворих, у яких немає системних проявів хвороби Бехчета, водночас спостерігається рецидивуюче афтозне ураження ротової порожнини і зовнішніх статевих органів або постійно наявні множинні афти на слизовій оболонці ротової порожнини. Слід врахувати, що ці ознаки можуть бути першим проявом хвороби Бехчета.
Частоту клінічних проявів наведено в табл. 21.48 (Enzenauer R.J., 1999).
Таблиця 21.48
Частота клінічних проявів при хворобі Бехчета
|
Клінічні симптоми |
Частота проявів, % |
|
Афтозне ураження ротової порожнини |
93–100 |
|
Виразки на зовнішніх статевих органах |
69–100 |
|
Симптоми ураження очей |
50–79 |
|
Артрит |
30–50 |
|
Ураження шкіри |
35–65 |
|
Ураження ЦНС |
10–30 |
|
Оклюзії/аневризми великих судин |
10–37 |
Особливості виразкового ураження статевих органів
Найчастіше уражуються мошонка і вульва, рідше — статевий член, слизова оболонка піхви, періанальні складки. На вигляд ці виразки аналогічні виразкам у ротовій порожнині, але глибші та залишають після себе рубці. Виразки у чоловіків відрізняються значною болісністю. При локалізації у піхві вони можуть бути безболісними. Можливі інші причини розвитку виразок на зовнішніх статевих органах (табл. 21.49).
Таблиця 21.49
Інші можливі причини розвитку виразок на зовнішніх статевих органах
|
Венеричні захворювання |
Сифіліс, м’який шанкр, венерична лімфогранульома, пахвова гранульома, простий герпес |
|
Ревматичні захворювання |
Синдром Рейтера, хвороба Крона |
|
Інші захворювання |
Механічні та хімічні ушкодження, непереносимість ліків, шкірні захворювання, які супроводжуються утворенням пухирів, інфекція, злоякісні пухлини (плоскоклітинний, базальноклітинний рак та ін.) |
Клінічні прояви з боку очей
До них належать передній/задній увеїт, кон’юнктивіт, виразка рогівки, папіліт зорового нерва й артеріїт. До сліпоти в основному призводить задній увеїт (в середньому сліпота розвивається через 4 роки від початку захворювання). Можливий розвиток гіпопіону — скупчення гною в передній камері ока, зміни кришталика, склоподібного тіла. Може розвиватися васкуліт сітківки. Необхідно враховувати, що гіпопіон реєструють при тяжкому увеїті, що асоціюється з HLA-B27. Увесь спектр очних уражень при хворобі Бехчета:
- іридоцикліт (частіше двобічний);
- увеїт;
- скупчення гною в передній камері ока (гіпопіон);
- глаукома;
- катаракта;
- хоріоретиніт;
- крововилив у склоподібне тіло;
- епісклерит;
- кератит, виразки рогівки;
- кон’юнктивіт;
- тромбофлебіт вен сітківки;
- інфаркти сітківки;
- неврит зорового нерва;
- втрата зору.
На ранній стадії хвороби Бехчета ураження очей (іридоцикліт, увеїт) не розвиваються, вони характерні для пізніших стадій.
Особливості артриту, що розвивається при хворобі Бехчета
Дуже часто виникають артралгії (частіше, ніж артрити), несиметричне ураження суглобів у вигляді мігруючого моно- або олігоартриту розвивається у 30–50% хворих. В основному до патологічного процесу залучаються колінні, гомілковостопні, ліктьові та променезап’ясткові суглоби, рідше уражуються плечові, кульшові, дрібні суглоби кистей і стоп, суглоби хребта. Можливий розвиток поліартриту, що нагадує перебіг РА, але ерозії суглобових поверхонь виникають вкрай рідко. У синовіальній рідині виявляються 5000–10 000 лейкоцитів в 1 мкл з переважанням нейтрофілів. Перебіг артриту підгострий (в більшості випадків суглобові прояви тривають не більше 3 міс) або хронічний, нерідко зустрічається сакроілеїт.
Шкірні прояви хвороби Бехчета. Відзначають гіперреактивність шкіри (патергія) на ін’єкцію або укол (що зв’язують з підвищеною хемотаксичною реакцією нейтрофілів), вугреподібний висип, папули, везикули, вузлики, вузлувату і багатоформну еритему. У 25–75% хворих через 48 год після підшкірного уколу стерильною голкою виникає еритематозна папула розміром більше 2 мм. Поява шкірного васкуліту характерна для раннього періоду хвороби, а надалі розвиваються його рецидиви. Шкірний висип переважно локалізується на гомілках.
Характер ураження судин. Ураження судин частіше зустрічається у чоловіків (4:1) і поєднується з вузлуватою еритемою. Основним судинним розладом при хворобі Бехчета є тромбоз вен. У пацієнтів виникає поверхневий тромбофлебіт нижніх кінцівок, у 30–40% спостерігаються тромбози глибоких вен кінцівок, верхньої та нижньої порожнистих вен, інтракраніальних судин. При цьому тромбоз глибоких вен часто поєднується з тробофлебітом поверхневих. Можливий розвиток синдрому Бадда — Кіарі.
Васкуліт частіше проявляється ураженням очей, ЦНС, легенів, нирок, але можуть уражуватися будь-які відділи судинної системи. При хворобі Бехчета можливий розвиток тромбозів великих вен і артерій з утворенням артеріальних аневризм. Відзначають формування тромбів в судинах, закупорку порожнистих, воротної та печінкових вен. Ураження артерій становить 12% від усіх судинних проявів, основним його типом є утворення аневризм (у 75% випадків). Аневризми артерій малого кола кровообігу, що розвиваються, можуть бути причиною смертельної кровотечі. Патологічний процес розвивається в судинах будь-якої локалізації та калібру, у тому числі коронарних. Найчастіше піддаються ураженню аорта, легенева, стегнова, підколінна, підключична та загальні сонні артерії. Можливий розрив аневризм аорти і легеневої артерії. Морфологічно в аневризматичних ділянках виявляються ознаки активного запалення. В уражених ділянках судин гістологічно виявляють периваскулярну інфільтрацію лімфоцитами і плазматичними клітинами, набряк і проліферація ендотелію, руйнування еластичної мембрани, дегенерацію середньої оболонки, васкуліт vasa vasorum. Призначення ГК й імуносупресивної терапії супроводжується зменшенням розмірів аневризм. Ураження артерій проявляється через 3–8 років від початку хвороби, тобто розвивається пізно. Гістологічні ознаки васкуліту знаходять в біоптатах шкіри, м’язів, кишечнику, серця. Прояви системного васкуліту, які виникають на пізніх стадіях хвороби Бехчета, наведено в табл. 21.50.
Таблиця 21.50
Прояви системного васкуліту на пізніх стадіях хвороби Бехчета
|
Органи і системи |
Характер ураження |
|
Судини |
|
|
Серце |
|
|
Легені |
|
|
Травний тракт |
|
|
Нирки |
|
|
Нервова система |
|
Неврологічні прояви хвороби Бехчета. Неврологічні прояви виникають у 20–40% хворих і можуть бути першим проявом хвороби. До них належать головний біль (у 52% хворих), атаксія внаслідок ураження мозочка, менінгоенцефаліт (у 28%), геміплегія/парапарез, паралічі черепних нервів (16%), псевдобульбарні паралічі, судомні напади (13%), екстрапірамідні симптоми, порушення зору (Enzenauer R.J., 1999). Загострення неврологічної симптоматики відзначають під час чергової хвилі виразкового ураження ротової порожнини і зовнішніх статевих органів, суглобового синдрому. Ураження ЦНС нагадує розсіяний склероз або васкуліт. Наслідком мультифокального ураження може бути псевдобульбарний параліч, когнітивні та емоціональні розлади. При гострому розвитку хвороби виникають сплутаність свідомості, часто спостерігається біль за типом мігрені та при напруженні. Зрідка причиною головного болю є серозний менінгіт, доброякісна внутрішньочерепна гіпертензія, тромбоз венозних синусів. Про інсульт свідчить раптова поява геміпарезу, афазії, геміанопсії. Причиною його буває тромбоз мозкових вен, оклюзія мозкової артерії. Зрідка розвивається сенсомоторна поліневропатія, множинна мононевропатія. Тяжке ураження ЦНС, що розвивається через 1–7 років від початку хвороби, призводить до смерті 40% хворих.
Ураження, що належать до MAGIC-синдрому. До нього відносять поєднання рецидивуючого поліхондриту з хворобою Бехчета (MAGIC-синдром — виразкове ураження рота і зовнішніх статевих органів із запаленням хряща — mouth and genital ulceration with inflamed cartilage).
Діагностика
Зміна лабораторних показників при хворобі Бехчета. Специфічних змін немає. У плазмі крові в період загострення підвищується вміст імуноглобулінів (IgG, IgA, IgM), СРБ, α2-глобулінів, відзначається лейкоцитоз, збільшення ШОЕ. У лікворі за наявності неврологічної симптоматики підвищується вміст білка і клітин (в основному лімфоцитів). У 55–85% визначається HLA-B5.
Основою діагнозу є клінічні прояви хвороби. Діагностичні критерії хвороби Бехчета наведено в табл. 21.51.
Таблиця 21.51
Діагностичні критерії хвороби Бехчета (Міжнародна група з вивчення хвороби Бехчета, 1990)
|
Основний діагностичний критерій |
|
|
Додаткові критерії |
|
|
|
|
Чутливість і специфічність цих діагностичних критеріїв становить відповідно до 95 і 98%.
Про наявність хвороби Бехчета можуть свідчити: підшкірний тромбофлебіт/тромбоз глибоких вен; епідидиміт; артеріальна оклюзія і/або аневризми; ураження ЦНС; артралгії/артрит; хвороба Бехчета у родичів; ураження травного каналу.
Діагностуванню хвороби Бехчета можуть сприяти наступні прояви:
- хвороба Бехчета у родичів;
- тромбофлебіт підшкірних вен;
- тромбоз глибоких вен;
- артеріальна оклюзія та/або аневризми;
- ураження ЦНС;
- артралгії/артрити;
- ураження ШКТ;
- епідидиміт.
Диференціація хвороби Бехчета. Слід проводити диференціацію з хворобою Крона, СЧВ, синдромом Рейтера, системними васкулітами. Афтозний стоматит може розвиватися у пацієнтів із запальними захворюваннями кишечнику, целіакією, дефіцитом вітаміну В12, фолієвої кислоти, заліза. У більшості випадків причина афтозного стоматиту залишається невідомою.
Слід зазначити, що А.С. Фаучі відносить хворобу Бехчета до так званих змішаних васкулітів. У цю групу він включив також вузлувату еритему, синдром Когана, стійку припідняту еритему, хворобу Ілза. Вузлувата еритема — це розповсюджене захворювання, яке вважають проявом гіперчутливості при ряді інших патологічних станів. Вона проявляється болісним вузлуватим ураженням дерми та підшкірних тканин. Синдром Когана характеризується інтерстиціальним кератитом у поєднанні з вестибулослуховими симптомами. Хвороба може бути зумовлена системним васкулітом, який уражує судини різного калібру, а також клапан аорти. Стійка припіднята еритема — хронічне шкірне захворювання невідомої етіології (припускають зв’язок хвороби зі стрептококовою інфекцією), яке характеризується розвитком постійних червоних, пурпурових, жовтуватих папул, плям, вузликів. Вони розташовуються симетрично на розгинальній поверхні кінцівок. При дослідженні біоптатів виявляють лейкоцитокластичний васкуліт, запалення венул з лейкоклазією в поєднанні з вираженим запальним інфільтратом дерми. Хвороба Ілза являє собою ретинальний васкуліт, на який хворіють переважно чоловіки на другому-третьому десятилітті життя. Хвороба характеризується синдромом рецидивуючих крововиливів у сітківку ока та склоподібне тіло.
Приклад формулювання діагнозу
Хвороба Бехчета, хронічний перебіг з ураженням шкіри і підшкірної клітковини (вузлувата еритема), слизових оболонок (виразковий стоматит, увеїт, афти на статевих органах), суглобів (поліартрит з ураженням колінних суглобів, недостатність функції суглобів I ступеня), нервової системи (поліневропатія).
Лікування
Рекомендації EULAR з лікування при хворобі Бехчета наведено в табл. 21.52.
Таблиця 21.52
Рекомендації EULAR з лікування пацієнтів з хворобою Бехчета
|
Усім пацієнтам із хворобою Бехчета, запаленням задніх сегментів очей слід призначати азатіоприн у поєднанні з ГК (системно) |
|
Хворим із тяжким ураженням очей та зниженням гостроти зору, васкулітом сітківки, залученням в процес жовтої плями рекомендується призначення азатіоприну у поєднанні з ГК, альтернативою може бути альфа-інтерферон (α-ІНФ) з/без ГК |
|
Не доведено ефективність антикоагулянтів, дезагрегантів, фібринолітичних препаратів у пацієнтів, які мають глибокий венозний тромбоз або артеріальні пошкодження |
|
Не розроблено рекомендацій з лікування пацієнтів з хворобою Бехчета, які мають шлунково-кишкові прояви. До хірургічного втручання можливе призначення сульфасалазину, ГК, азатіоприну, інгібіторів ФНП-α |
|
Лікування артриту може бути обмежене використанням колхіцину |
|
Контрольовані дослідження, які стосуються лікування при ураженні ЦНС, відсутні. При паренхіматозному ураженні застосовують ГК, α-ІНФ, при дуральному тромбозі синуса — ГК, азатіоприн, циклофосфамід, інгібітори ФНП-α |
|
Хворим з ураженням ЦНС циклоспорин А призначають тільки у випадках наявності внутрішньоочного запалення |
|
Локальне застосування ГК є лікуванням першої лінії при ізольованих виразках в роті або на геніталіях. При акнеподібних висипаннях використовують косметичні засоби. При домінуванні вузлуватої еритеми призначають колхіцин. У резистентних випадках виразок на ногах використовують азатіоприн, α-ІНФ, інгібітори ФНП-α |
Рекомендації з лікування при хворобі Бехчета (Насонов Е.Л., 2011 (ред.)) наведені також в табл. 21.53.
Таблиця 21.53
Рекомендації з лікування при хворобі Бехчета
|
ГК |
Локально |
Преднізолон (1%), дексаметазон (0,1%), тріамцинолон призначають у вигляді крапель 3–6 разів на день при загостреннях переднього увеїту. У тяжких випадках кількість інстиляцій може сягати 24 на добу з подальшим зниженням частоти застосування крапель та їх відміною (через 6–8 тиж). При тяжкому перебігу переднього увеїту та гіпопіону, загостренні заднього увеїту, ураженні склоподібного тіла, набряку макули застосовують локальні ін’єкції ГК |
|
|
Системно |
При задньому увеїті та панувеїті, васкуліті сітківки призначають преднізолон у дозі 1–2 мг/кг/добу або пульс-терапію — 1 г метилпреднізолону в/в протягом 3 днів. Пульс-терапія показана при ураженні очей (двобічному), ШКТ (кровотеча, гострий живіт), гострому асептичному менінгіті, нейропсихічних проявах, формуванні аневризм легеневих артерій. В таких випадках ГК поєднують з імуносупресорами, зокрема циклоспорином А. Монотерапія ГК вважається паліативним лікуванням. Тривале використання ГК у високих дозах може призвести до виникнення стероїдорезистентності, підвищення ризику тромбозів |
||
|
Колхіцин |
Досі продовжують розглядати як базисний препарат, який пригнічує активацію клітинної імунної відповіді |
||
|
Азатіоприн |
Його ефективність при хворобі Бехчета підтверджено контрольованими дослідженнями. У дозах 50–150 мг/добу знижує частоту рецидивів, пригнічує запалення при задньому увеїті, покращує прогноз хвороби, є безпечним при тривалому застосуванні. Доведено ефективність азатіоприну не тільки при васкуліті сітківки та задньому увеїті, але й при ураженнях шкіри, слизових оболонок |
||
|
Хлорамбуцил |
Застосовують (частіше в поєднанні з ГК) при ураженні очей, ЦНС у дозі 2 мг/добу з подальшим підвищенням її до 5–12 мг/добу. У процесі лікування рівень лейкоцитів в крові не має знижуватися нижче 3000/мм3 |
||
|
Циклофосфамід |
Лікування починають із дози 2 мг/кг/добу. При ураженні очей циклофосфамід призначають в/в у вигляді пульс-терапії по 200–400 мг/тиж у поєднанні з преднізолоном. Контрольовані дослідження не проводили |
||
|
Сульфасалазин |
Призначають протягом років в дозі 2–4 г/добу хворим з переважним ураженням кишечнику |
||
|
Метотрексат |
Призначають (7,5–10 мг/тиж) при ураженнях суглобів, шкіри, слизових оболонок, нервової системи |
||
|
Циклоспорин А |
Призначають (3–5 мг/кг/добу) при ураженні очей, знижує частоту та ступінь тяжкості загострень хвороби Бехчета. Поліпшує функцію зору, стан суглобів, шкіри, слизових оболонок. При досягненні ефекту дозу знижують до 3–1 мг/кг/добу. У випадку повної відміни препарату виникає загострення увеїту. Ефективність циклоспорину А підтверджено в багатьох рандомізованих контрольованих дослідженнях. Тривалість його застосування становить до 3 років. Її обмежує розвиток побічних ефектів: ураження нирок (при дозах 10 мг/кг/добу), підвищення артеріального тиску, шлунково-кишкові та неврологічні розлади, гіперплазія ясен |
||
|
α-Інтерферон |
Застосовують у дозі 3–18 млн МО 3 рази на тиждень (або 6 млн МО/добу) при тяжкому рефрактерному увеїті в поєднанні із ГК та імуносупресорами. Комбінація альфа-інтерферону з ГК в низьких дозах дозволяє досягти ремісії васкуліту сітківки |
||
|
Біологічні агенти |
Інфліксімаб |
Призначають в/в краплинно в дозі 5 мг/кг за схемою 1 раз в 0–2–6 тиж, а в подальшому кожні 8 тиж протягом 1 року — до досягнення ремісії увеоретиніту |
|
|
Етанерцепт |
У хворих із рефрактерним увеїтом покращується функція зору, зменшується кількість виразок у роті, загострень артриту, зникають папулопустульозні ураження шкіри |
||
Лікування пацієнтів із хворобою Бехчета наведено також в табл. 21.54.
Таблиця 21.54
Терапія при хворобі Бехчета
|
Імуносупресори |
Циклофосфамід (5 мг/кг/добу), хлорамбуцил (0,1–0,2 мг/кг/добу), азатіоприн (2–2,5 мг/кг/добу) |
Тривалість терапії становить не менше 6–12 міс. Препарати в підтримувальних дозах пацієнти приймають протягом 2–3 років |
|
Циклоспорин А 5 мг/кг/добу |
Тривале застосування призначають у разі тяжких проявів церебрального васкуліту і заднього увеїту |
|
|
ГК |
Преднізолон (метилпреднізолон) в дозі 10–15 мг/добу |
Призначають за наявності шкірно-слизового і суглобового синдрому в активну фазу хвороби |
|
Антикоагулянти |
Гепарин, низькомолекулярні гепарини, варфарин |
Застосовують з метою профілактики тромботичних ускладнень |
|
Симптоматична терапія |
НПЗП |
За наявності суглобового синдрому |
З метою зниження хемотаксичної активності нейтрофілів пацієнтам з хворобою Бехчета призначають колхіцин. У разі розвитку тяжкого менінгоенцефаліту або увеїту при хворобі Бехчета засобом вибору вважався хлорамбуцил. Проте, незважаючи на його застосування, сліпота розвивалася у 75% пацієнтів, а токсичний вплив на статеві залози відмічають у всіх хворих. Слід враховувати, що у пацієнтів із хворобою Бехчета в місцях ін’єкцій часто виникають гнійні ураження шкіри. При болісних виразках у порожнині рота застосовують полоскання розчинами анестезуючих речовин та аплікації ГК.
Критерії ефективності лікування
Про ефективність лікування свідчить позитивна клінічна симптоматика, поліпшення/нормалізація лабораторних показників (ШОЕ, СРБ, імуноглобуліни, α2-глобуліни, вміст лейкоцитів у крові).
Прогноз
Смертність у перші 5 років хвороби становить близько 16%. Її причиною є ураження ЦНС, судин, кишечнику (перфорація), легень (легенева кровотеча), серця.
Синдром Черджа — Стросс
Синдром Черджа — Стросс — гранулематозне, еозинофільне запалення респіраторного тракту і некротизуючий васкуліт дрібних та середніх судин, які часто поєднуються з бронхіальною астмою і еозинофілією.
Код за МКХ-10
М30.1 Поліартеріїт з ураженням легень (синдром Черджа — Стросс).
Перші свідчення про алергічний гранулематоз, клінічними проявами якого були бронхіальна астма в тяжкій формі, підвищення температури тіла, еозинофілія, а морфологічно визначався дисемінований некротизуючий артеріїт з міліарними вузликами за ходом артерій і в оточуючих тканинах, які складалися в основному з еозинофілів з багатоядерними гігантськими клітинами, з’явилися у 20-х роках ХХ ст. В 1951 році J. Churg і L. Strauss описали 23 випадки захворювання і включили його в рубрику ВП. Нові можливості імунології, а саме відкриття АНЦА та виявлення АНЦА до мієлопероксидази у хворих на ВП з переважним ураженням судин мікроциркуляторного русла і патологією легень, шкіри, серця, дозволили виділити з ВП синдром Черджа — Стросс. Зараз синдром Черджа — Стросс розглядають як окрему нозологічну форму — первинний системний АНЦА — опосередкований васкуліт. У нашій країні цю хворобу тривалий час розглядали в межах ВП як астматичний варіант. Синоніми синдрому Черджа — Стросс — алергічний гранулематоз і ангіїт, гіпереозинофільна астма з васкулітом, еозинофільний гранулематозний васкуліт.
Етіологія
Етіологія синдрому Черджа — Стросс невідома. Не відзначають зв’язку з інфекціями. Не прослідковується чіткого зв’язку з носійством певних імуногенетичних маркерів. Проте зараз є дані про підвищення частоти HLA-LR B1*0901 у хворих з АНЦА до мієлопероксидази, відзначається В-клітинна епітопна специфічність при АНЦА — опосередкованих васкулітах. Хворобу частіше діагностують у жителів сільської місцевості. У зв’язку з цим є думка, що тригерними можуть бути такі фактори оточуючого середовища, як пилок рослин, пестициди й інші хімічні речовини.
Поширеність, за даними епідеміологічних досліджень, становить 0,47 випадку на 100 тис. населення на рік. Захворюваність — 1–10 на 1 млн населення. За останні 10 років розповсюдженість синдрому Черджа — Стросс значно зросла і становить: в США — 4 нових випадки, у Франції — 11, у Німеччині — 12 на 1 млн населення на рік. Достовірних розбіжностей за статтю не виявлено. Можуть хворіти особи будь-якого віку, але пік захворювання припадає на вік 35–45 років.
Патогенез
Синдром Черджа — Стросс являє собою аутоімунний процес із залученням еозинофілів, Т-лімфоцитів, ендотеліальних клітин. Імовірно, найбільше значення мають Тh2 — тип імунної відповіді, яка зумовлює синтез цитокінів: ІЛ-3, -4, -5, -6, -9, -10, -13, ФНП-α, гранулоцитарно-макрофагального колонієстимулюючого фактора, хемокіну. Для патогенезу синдрому Черджа — Стросс важливим є механізм, пов’язаний з антитілами до лізосомальних ферментів нейтрофілів (АНЦА). У 50–70% пацієнтів з синдромом Черджа — Стросс виявляють АНЦА до мієлопероксидази. Їх присутність у сироватці крові корелює з наявністю у хворих бронхіальної астми і пурпури, легеневої кровотечі, некротизуючого гломерулонефриту, множинного мононевриту. У разі відсутності АНЦА у патогенезі важливу роль відіграють токсичні продукти дегрануляції еозинофілів, а в клінічній картині переважає поліпоз, гіперплазія слизової оболонки носа, легеневі інфільтрати, кардіоміопатія, поліневропатія, еозинофільний гастроентерит.
Патоморфологія
Синдром Черджа — Стросс займає проміжне місце між некротизуючими та гранулематозними васкулітами. Ураження характеризується наступною гістопатологічною тріадою: системний некротизуючий, гранулематозний сегментарний васкуліт артерій малого, іноді середнього, калібру та вен і венул, переважно легень, серця, шкіри, периферичних нервів; позасудинні еозинофільні гранульоми; інфільтрація тканин еозинофілами з переважною інтерстиціальною і периваскулярною локалізацією. У випадку наявності АНЦА до мієлопероксидази переважає ураження дрібних судин, за їх відсутності — інфільтрація тканин еозинофілами з відповідними клінічними проявами.
Клінічна картина
Синдром Черджа — Стросс — мультисистемний васкуліт, для якого характерним є наявність бронхіальної астми, алергічного риніту, еозинофілії. Починатися хвороба може раптово або поступово. В анамнезі у хворих відзначають сезонний алергічний риніт, харчову або контактну алергічну реакцію, поліноз.
У 1984 р. J.G. Lanham виділив у перебігу хвороби 3 періоди, що частіше розвиваються послідовно, але іноді симптоми, притаманні кожному періоду, з’являються одночасно. Починається хвороба з продромального періоду характерними алергічними проявами: риносинусопатією, бронхіальною астмою, полінозом. Другий період характеризується значною еозинофілією периферичної крові та тканин. Системний некротизуючий васкуліт з переважним ушкодженням судин легень, шкіри, серця, периферичних нервів, рідше — нирок розвивається в завершальний, третій, період хвороби.
Виділяють АНЦА-позитивний та АНЦА-негативний варіанти синдрому Черджа — Стросс. Клінічна класифікація синдрому Чержа — Стросс представлена в табл. 21.55.
Таблиця 21.55
Клінічна класифікація синдрому Черджа — Стросс
|
Перебіг |
Фаза, стадія активності |
Клініко-морфологічні ознаки ураження |
|
Гострий Рефрактерний Хронічний |
Неактивна Ремісія Часткова ремісія Активна:
|
|
Продромальний період звичайно триває 2–3 роки, але може продовжуватися 2–3 десятиліття. У дебюті хвороби реєструють симптоми ураження верхніх дихальних шляхів: алергічний риніт, який призводить до носової обструкції, поліпозу носа, рецидивуючого синуситу. У окремих хворих розвивається некротичний процес у порожнині носа, який нагадує гранулематоз Вегенера. Найбільш типовий прояв цього періоду — бронхоспазм, з часом розвиваються симптоми тяжкої форми бронхіальної астми, яка часто ускладнюється астматичним статусом. Бронхіальна астма стійка до бронхолітичної терапії і потребує призначення ГК. Бронхіальна астма у більшості пацієнтів є основним клінічним синдромом протягом декількох років. У подальшому може приєднуватися вторинна інфекція і бронхоектатична хвороба.
Про прогресування хвороби свідчать периферична і тканинна еозинофілія. Еозинофілію відзначають у 97% хворих, частіше у II стадії, але можуть діагностувати у будь-якій стадії хвороби. Вміст еозинофілів у периферичній крові перевищує 10% від всіх лейкоцитів (абсолютна кількість >1,5•109/л), у окремих хворих їх кількість досягає 30–85%. У деяких випадках периферична еозинофілія відсутня, а реєструється лише тканинна еозинофільна інфільтрація, що підтверджується даними біопсії. Це може бути пов’язано з природними коливаннями кількості еозинофілів у крові або з попередньою ГК-терапією.
Характерною ознакою другого періоду хвороби є також розвиток еозинофільного ураження різних органів, частіше легень, шлунка, кишечнику. У хворих у цей період діагностують: синдром Леффлера, еозинофільну пневмонію, еозинофільний гастроентерит.
У цій стадії у ⅔ хворих у легенях виявляють транзиторні еозинофільні інфільтрати, які швидко регресують при призначенні ГК. В окремих випадках відбуваються розпад інфільтратів і утворення порожнин, що потребує диференційної діагностики цього захворювання і гранулематозу Вегенера, туберкульозу, абсцесу. У мокроті хворих виявляють велику кількість еозинофілів. У ⅓ хворих відзначають еозинофільний плеврит, який клінічно проявляється болем у грудній клітці. У випадку ексудативного процесу в ексудаті виявляють велику кількість еозинофілів.
Перебіг еозинофільного гастроентериту характеризується періодичними загостреннями протягом декількох років. На початку хвороби відзначають гранулематозний еозинофільний гастроентерит, далі розвивається васкуліт судин кишкової стінки з відповідною симптоматикою. Клінічно проявляється болем у животі (60%), діареєю (33%), можливі шлунково-кишкові кровотечі (18%).
Розвиток наступного періоду хвороби часто провокується епізодом алергії. Для нього характерно зникнення еозинофілії, симптомів бронхіальної астми. У III стадії хвороби ураження легень діагностують у всіх хворих. У більшості з них посилюються наявні зміни і приєднуються симптоми, які свідчать про розвиток васкуліту. На ранньому етапі васкуліту виникають лише загальні конституціональні симптоми, які свідчать про системність ушкодження: слабкість, зменшення маси тіла, лихоманка з ознобами, артралгії, міалгії, у подальшому переважає клінічна картина тяжкого ураження різних органів, що часто загрожує життю хворого.
Одним із найбільш частих проявів цього періоду є ураження судин шкіри, яке відзначають у 60–80% хворих. Характерні геморагічні висипи — пурпура, еритема, кропив’янка, вузлики, некрози шкіри, сітчасте ліведо. Ураження суглобів у вигляді недеструктивного олігоартриту, поліартралгії виявляють у 50% хворих, при цьому уражаються як дрібні, так і великі суглоби.
У 35–60% хворих на синдром Черджа — Стросс виявляють різні варіанти ураження серця. Найбільш характерним є дифузне ураження міокарда, яке зумовлене інфільтрацією еозинофілами, з розвитком кардіоміопатії і серцевої недостатності в результаті загального і локального зниження скоротливості та дилатації порожнин серця. У ½ хворих виявляють різні зміни на ЕКГ. Рідше виявляють ексудативний або констриктивний перикардит, еозинофільний ендокардит. Є випадки патології клапанів з розвитком інфільтрації міокарда еозинофілами й ендоміокардіального фіброзу клапанів. У деяких хворих розвивається коронарит, що призводить до стенозу, іноді розвитку аневризм коронарних судин. Ці зміни ускладнюються розвитком клінічних симптомів гострого коронарного синдрому або інфаркту міокарда, артеріальної гіпертензії. Ураження серця є причиною смерті 50% хворих на синдром Черджа — Стросс.
Ураження периферичної нервової системи відзначають у 65–75% хворих з ушкодженням vasa nervorum, що призводить до ішемії, руйнування мієлінових і немієлінових волокон. Частіше діагностують периферичні мононеврити, які клінічно проявляються болем, слабкістю у кінцівках, зниженням чутливості в зоні іннервації ушкодженого нерва, рідше відмічається симетрична сенсорно-моторна поліневропатія за типом рукавичок та шкарпеток. Ураження черепно-мозкових нервів реєструють у 4–11% хворих у вигляді ішемічної офтальмопатії, описано поодинокі випадки розвитку парезів ІІ, ІІІ, ІV, VІ, VІІ, ХІІ пари, сенсорної невропатії V пари.
ЦНС залучається до патологічного процесу у 3–7% хворих, клінічно це проявляється симптомами енцефалопатії, інсультами, субарахноїдальними крововиливами, епілептиформними судомами, гіперкінезами або психічними розладами. Причиною ішемічного і геморагічного інсультів є васкуліт церебральних судин, а в ряді випадків артеріальна гіпертензія.
Ураження ШКТ в цей період перебігу синдрому Черджа — Стросс визначається у 30–62%. Це еозинофільний гастроентерит, дебют якого спостерігається частіше у ІІ стадії хвороби, а з її розвитком приєднується васкуліт мезентеріальних судин. Клінічно проявляється абдоміналгією, нудотою, блюванням, діареєю, кровотечею, меленою. Можливі виникнення кишкової непрохідності, перфорації кишечнику, перитоніту, іноді розвиток виразкового коліту.
Ураження нирок реєструють у 15–40% хворих. Частіше це вогнищевий гломерулонефрит. При дослідженні біоптату нирок виявляють фокально-сегментарний гломерулонефрит, а при імунофлуоресценції — фіксацію імуноглобуліну М і С3 фракції комплементу. Виявляють клінічні симптоми сечового синдрому з протеїнурією, еритроцитурією, артеріальну гіпертензію. У хворих з АНЦА до мієлопероксидази може розвиватися некротизуючий швидкопрогресуючий гломерулонефрит зі зниженням швидкості клубочкової фільтрації і розвитком ниркової недостатності.
У деяких випадках може спостерігатися васкуліт сечоводів і нижніх відділів сечової системи з розвитком обструктивної уропатії. Можливе ураження очей, спостерігалися випадки виразкування рогівки.
Лабораторні дослідження
Еозинофілія є характерною лабораторною ознакою хвороби, вміст еозинофілів у крові >1,5•109/л при підрахунку лейкоцитів. В активну стадію хвороби спостерігається лейкоцитоз, нормохромна нормоцитарна анемія, прискорена ШОЕ, збільшення вмісту СРБ. Можуть визначатися низькі титри РФ у сироватці, підвищення концентрації імуноглобуліну E, рівня ЦІК, гіпокомплементемія.
Для синдрому Черджа — Стросс характерно виявлення АНЦА, які реагують з мієлопероксидазою. Цей вид АНЦА виявляють у 50–70% хворих, в той час як АНЦА до протеїнази-3 — лише у 10%. Визначення АНЦА при синдромі Черджа — Стросс є важливим не лише для встановлення діагнозу, але й для вибору оптимальної терапевтичної тактики.
Морфологічні дослідження
Ці дослідження є обов’язковою складовою діагностики синдрому Черджа — Стросс. Для діагностування використовують біоптати шкіри, легень та нирок. Типовим є наявність некротизуючих гранульом, васкуліту дрібних, середніх судин, вен та еозинофільної пневмонії. У позасудинному просторі, інтерстиціальній тканині виявляють великі еозинофільні інфільтрати. Некротизуючі гранульоми частіше містяться поза судинами. Наявні еозинофіли та продукти їх життєдіяльності (кристали Шарко — Лейдена). Для уточнення діагнозу необхідно проводити біопсію слизової оболонки верхнього відділу дихальних шляхів і трансбронхіальну біопсію. У бронхах виявляють набряк, інфільтрацію слизової оболонки бронхів еозинофілами, гіпертрофію гладких м’язів, метаплазію келихоподібних клітин, потовщення базальної мембрани з формуванням слизових пробок у просвіті термінального відділу дихальних шляхів. У легенях розвиваються септальні капілярити з еозинофільною інфільтрацією, рідше некротизуючий альвеоліт із геморагічним синдромом. Важливе значення має дослідження бронхоальвеолярного лаважу і плеврального ексудату, в яких виявляють велику кількість еозинофілів. При дослідженні біоптату нирок спостерігають вогнищевий фокально-сегментарний гломерулонефрит, а при імунофлуоресценції — фіксацію імуноглобуліну М і С3 фракції комплементу.
Інструментальні дослідження
На рентгенограмах грудної клітки виявляють транзиторні інфільтрати, які розташовуються частіше у середній та нижній частках легень, іноді — збільшені лімфатичні вузли середостіння. При проведенні КТ реєструють периферичні інфільтрати, потовщення стінок бронхів, їх дилатацію, бронхоектази, розширені з гострокінцевими закінченнями судини легень. Плеврит може не виявлятися за допомогою рентгенологічних методів. Для діагностики субклінічного ураження легень проводять функціональні легеневі тести: спірометрію, бодіплетизмографію, визначення дифузійної здатності легень.
На ЕКГ знаходять зміни, відповідні пошкодженню серця: порушення ритму та провідності, ознаки метаболічних порушень у міокарді, гострих коронарних синдромів.
Діагноз
Наявність синдрому Черджа — Стросс можна припустити в пацієнта середнього віку, в анамнезі якого була бронхіальна астма, риніт, еозинофілія, а в подальшому розвинулися системні прояви, а також легеневі інфільтрати, кардіоміопатія, множинний поліневрит. Важливими для встановлення діагнозу є дані лабораторних та інструментальних досліджень, особливо визначення АНЦА і результати біопсії. Оцінку активності васкуліту проводять відповідно до загальноприйнятого індексу — BVAS (Birmingham vasculitis activity score). За допомогою індексу васкулітного пошкодження, який відображає незворотні зміни в органах і тканинах, оцінюють ступінь їх пошкодження.
Класифікаційні критерії синдрому Черджа — Стросс, запропоновані ACR (1990):
- утруднене дихання або дифузні хрипи під час вдиху;
- еозинофілія більше 10% при підрахунку лейкоцитів;
- алергія в анамнезі: сезонна алергія (алергічний риніт) або інша алергічна реакція (харчова, контактна), за винятком медикаментозної;
- мононевропатія, множинна мононевропатія або поліневропатія за типом рукавичок або шкарпеток;
- мігруючі або транзиторні легеневі інфільтрати, які виявляють при рентгенологічному дослідженні;
- синусит: біль в ділянці навколоносових пазух і рентгенологічні зміни;
- біопсія: скупчення еозинофілів у позасудинному просторі.
Діагноз синдрому Черджа — Стросс встановлюють за наявності у хворого 4 і більше будь-яких критеріїв (чутливість — 85%, специфічність — 99%).
Диференційний діагноз
Насамперед необхідно проводити диференційну діагностику синдрому Черджа — Стросс та інших системних васкулітів, а також станів, для яких характерна еозинофілія.
Для ВП, на відміну від синдрому Черджа — Стросс, не характерне ураження дрібних артерій, капілярів, вен и венул, не типовим є еозинофільне гранулематозне запалення тканин, периферична і тканинна еозинофілія, але притаманні мікроаневризми судин. При синдромі Черджа — Стросс може піддаватися ураженню будь-який орган, але в клінічній картині переважає ушкодження легень: бронхіальна астма, легеневі інфільтрати — ці симптоми не спостерігаються при ВП. Якщо при ВП при ураженні нирок розвиваються інфаркти, то при синдромі Черджа — Стросс — гломерулонефрит. При ВП частіше пошкоджуються: периферична нервова система, шкіра, травний тракт, нирки. Для синдрому Черджа — Стросс типовим є виявлення в сироватці крові АНЦА до мієлопероксидази у 67%, АНЦА до протеїнази-3 — у 10% хворих, для ВП АНЦА не характерні.
При МПА реєструють некротичний гломерулонефрит, який поєднується з екстракапілярною проліферацією епітеліальних клітин і формуванням півмісяців, а клінічно формується швидкопрогресуючий гломерулонефрит, у хворих на синдром Черджа — Стросс ураження нирок більш легкого ступеня, а превалює в клінічній картині ушкодження легень. При МПА не відзначають периферичної і тканинної еозинофілії.
Для хвороби Вегенера характерні епітеліоїдні гранульоми, схильні до розпаду в органах, які контактують із зовнішнім середовищем, а не еозинофільні, як при синдромі Черджа — Стросс. Розвиток бронхіальної астми, алергічного риніту, еозинофілії — не характерний для гранулематозу Вегенера. Важливим діагностичним маркером хвороби Вегенера є АНЦА до протеїнази-3, які виявляються в сироватці крові у 90% хворих в активну фазу. Основні причини смерті при синдромі Черджа — Стросс: інфаркт міокарда, застійна серцева недостатність, а при хворобі Вегенера — тяжке ураження легень і нирок.
Генез лейкемоїдної реакції еозинофільного типу у частини хворих важко пояснити, що викликає проблеми при проведенні диференційної діагностики. Основними причинами легеневих еозинофілій є лікарські препарати, паразитарна інвазія, алергічні стани. Ці еозинофілії є імуноглобулін Е-залежними і супроводжуються підвищенням його вмісту в периферичній крові. Слід пам’ятати, що при будь-якій з наведених гіпереозинофілій, в тому числі при синдромі Леффлера, може розвиватися ураження серця, але тільки при синдромі Черджа — Стросс відбувається інфільтрація еозинофілами тканин у поєднанні з васкулітом. Пухлинні еозинофілії відзначають в тому випадку, якщо еозинофільний росток є субстратом пухлини, і при пухлинах, які виділяють еозинофільний хемотаксичний фактор, — це неходжкінська лімфома, лімфогранулематоз, карцинома. Еозинофілії при пухлинах є імуноглобулін Е-незалежними, і вміст імуноглобуліну Е в периферичній крові не підвищується.
Лікування
Лікування хворого на синдром Черджа — Стросс має відповідати сучасним схемам терапії, бути комплексним та індивідуальним. Мета лікування — досягнення клініко-лабораторної ремісії, попередження ураження життєво важливих органів, підтримка ремісії, зниження ризику загострень, попередження розвитку побічних ефектів медикаментозної терапії, збільшення тривалості та покращення якості життя. Підхід до лікування різний у хворих з АНЦА-позитивним і АНЦА-негативним варіантами синдрому Черджа — Стросс.
Для індукції ремісії у випадку АНЦА-негативного варіанта синдрому Черджа — Стросс і відсутності ознак прогресування призначають монотерапію ГК. Преднізолон призначають в дозі 1 мг/кг/добу (не більше 60 мг) у декілька прийомів, через 8–10 днів при позитивній динаміці переходять на одноразовий прийом вранці. Після досягнення ефекту, через 3–4 тиж, дозу поступово знижують — по 5 мг кожні 2 тиж до підтримувальної (0,15–0,2 мг/кг/добу). Відміна ГК можлива не раніше ніж через 1 рік після початку лікування, але терапія може тривати 3–5 років.
При АНЦА-позитивному варіанті синдрому Черджа — Стросс, швидкопрогресуючому ураженні органів застосовують ГК с цитостатиками. Циклофосфамід призначають (per os) в дозі 1–2 мг/кг/добу (максимально 200 мг/добу) або у вигляді пульс-терапії в дозі 15 мг/кг/добу. Перші 3 курси пульс-терапії проводять з інтервалом 2 тиж, а потім через 3 тиж. Після досягнення ремісії (через 3–6 міс) дозу циклофосфаміду поступово знижують на 1,5 мг/кг/добу. При призначенні циклофосфаміду необхідно контролювати рівень лейкоцитів у периферичній крові, який має бути не меншим ніж 3–3,5•109/л, нейтрофілів — 1–1,5•109/л. На початку лікування контроль треба проводити через день, після стабілізації рівня лейкоцитів — не менше ніж 1 раз на 2 тиж. При нирковій недостатності (креатинін >0,17 ммоль/л) і у осіб похилого віку доза циклофосфаміду має бути знижена на 25–50%. Застосування циклофосфаміду >6 міс часто викликає побічні реакції (респіраторну патологію з морфологічними симптомами ураження легень).
При швидкопрогресуючому, тяжкому перебігу, з порушенням функції життєво важливих органів використовують ескалаційну терапію. Призначають 7–10 процедур плазмаферезу протягом 14 днів (видалення плазми в об’ємі 60 мл/кг маси тіла з заміщенням її відповідним об’ємом 4,5–5% людського альбуміну) у поєднанні з пульс-терапією метилпреднізолоном в дозі 15 мг/кг/добу і циклофосфамідом в дозі 10 мг/кг/добу.
Для підтримувальної терапії використовують ГК з азатіоприном у дозі 1,5–2 мг/кг/добу або циклофосфамід у дозі 1 мг/кг/добу. Терапію ГК і цитостатиками доцільно проводити не менш ніж протягом 24 міс після досягнення ремісії.
При рефрактерному перебігу АНЦА-асоційованого варіанта призначають одну із наступних схем терапії: ГК разом з ритуксимабом або ГК з мофетил мікофенолатом, або ГК с лефлуномідом, або ГК с інфліксимабом. В якості підтримувальної терапії в цьому випадку призначають ГК або ГК з ритуксимабом.
При синдромі Черджа — Стросс АНЦА-асоційованому варіанті без швидкопрогресуючого нефриту (креатинін <0,15 ммоль/л) і тяжкого ураження легень (локальна патологія верхніх дихальних шляхів) призначают ГК в поєднанні з метотрексатом у дозі 15–20 мг/добу. Якщо ця терапія ефективна, ГК з метотрексатом використовують (з корекцією дози) і в якості підтримувальної терапії протягом 2–5 років.
Приклад формулювання диагнозу: Синдром Черджа — Стросс, АНЦА-позитивний, гострий перебіг, разгорнута стадія, активність III ступеня. Ураження шкіри (ліведо, еритема), легень (легеневі інфільтрати, бронхіальна астма), серця (кардіоміопатія, серцева недостатність IIA, функціональний клас III), нирок (гломерулонефрит, артеріальна гіпертензія, хронічна ниркова недостатність I ступеня), нервової системи (поліневропатія).
Синдром Черджа — Стросс, АНЦА-негативний, хронічний перебіг, активність II ступеня, риніт, бронхіальна астма (дихальна недостатність II ступеня), поліневропатія.
Прогноз
ГК збільшують 5-річну виживаність хворих на синдром Черджа — Стросс до 50% і більше. При застосуванні циклофосфаміду і преднізолону в більшості випадків досягається ремісія. Прогноз при синдромі Черджа — Стросс більш сприятливий, ніж при гранулематозі Вегенера: 5-річна виживаність становить 75%. Основною причиною смерті є інфаркт миокарда і серцева недостатність. Можливий спонтанний развиток ремісії.
Геморагічний васкуліт
Геморагічний васкуліт (пурпура Шенлейна — Геноха) — це імунокомплексна системна запальна хвороба переважно капілярів, артеріол, венул головним чином шкіри, суглобів, черевної порожнини і нирок з відкладенням імуноглобуліну A.
Код за МКХ-10
М.31 Пурпура Шенлейна — Геноха.
J.L. Schonlein описав захворювання з геморагіями на шкірі та ураженням суглобів та виділив його в 1837 р. в окрему нозологічну форму. У 1861 р. E.N. Henoch доповнив клінічну картину хвороби, описавши випадки з абдомінальним синдром і ураженням нирок.
За частотою пурпура Шенлейна — Геноха займає перше місце серед системних васкулітів. Хвороба може виникнути в будь-якому віці, але найчастіше хворіють діти у віці від 2 до 10 років. Серед дітей до 14 років поширеність становить 23–25 випадків на 100 тис. населення. Частіше серед дітей хворіють хлопчики. Серед дорослих статевих відмінностей немає.
Етіологія, патогенез
Етіологія захворювання невідома. Вивчення анамнезу показало, що у значної частини хворих розвитку хвороби передують гострі інфекції дихальних шляхів, збудники яких, вочевидь, відіграють важливу роль у патогенезі пурпури Шенлейна — Геноха. Серед інфекцій, які передують васкуліту, найбільш часто відзначено стрептококову (група А), рідше інфекції, викликані мікоплазмами, легіонелами, обговорюється роль персистуючої вірусної інфекції, а саме, вірусу Епштейна — Барр, вірусу гепатиту B, аденовірусів, цитомегаловірусів, парвовірусу В19 та інших. Спостерігається зв’язок розвитку хвороби з вакцинацією, введенням сироваток. Хвороба частіше розвивається в осінньо-зимовий період і після переохолодження.
Пурпура Шенлейна — Геноха — це імунокомплексна хвороба. Конкретний антиген, що є пусковим чинником, невідомий, але патогенез хвороби вивчений досить глибоко. ЦІК, які відкладаються у стінці судини, фіксують комплемент, що викликає бурхливу імунну реакцію. Уражуються переважно шляхи мікроциркуляції: капіляри, артеріоли, венули. Імунні комплекси, які містять імуноглобулін A, виявляють на базальній мембрані судинної стінки різних органів і тканин. Найчастіше ушкоджуються судини шкіри, суглобів, черевної порожнини і нирок. Характерні деструктивні або деструктивно-продуктивні мікроваскуліти з периваскулярною інфільтрацією нейтрофілами, підвищенням судинної проникності, утворенням внутрішньосудинних пристінкових мікротромбів. Важливу роль у патогенезі пурпури Шенлейна — Геноха має порушення мікроциркуляції. Мікротромбози при цій патології нагадують порушення гемостазу при ДВЗ-синдромі, але на відміну від нього при пурпурі Шенлейна — Геноха відбувається пристінкове тромбоутворення, характерна вогнищевість ушкодження та пролонгована фаза гіперкоагуляції.
Клінічна картина
Початок хвороби частіше раптовий та гострий і, звичайно, хворий може точно вказати день і годину початку захворювання. Рідше відзначають субклінічні перші прояви хвороби, у такому разі пацієнт випадково виявляє висипи на шкірі та інші симптоми.
Для дебюту хвороби характерна тріада синдромів: шкірна пурпура, суглобовий і абдомінальний синдроми. У 50% випадків хвороба починається з ураження шкіри, у 25% хворих ураженню шкіри передують артралгії й артрит, рідше одночасно з’являються усі три синдроми, іноді першими симптомами є абдомінальні.
Ушкодження шкіри виявляють абсолютно у всіх хворих на пурпуру Шенлейна — Геноха. Типи шкірних уражень досить різноманітні, але найбільш типовим проявом є пурпура. Частіше спочатку з’являються еритематозні папули, які іноді трохи сверблять, або висипання за типом кропив’янки, які швидко трансформуються в пурпуру, яка є дрібнокрапчастим геморагічним висипом у вигляді петехій, плям розміром від 2 до 5 мм. Елементи пурпури піднімаються над поверхнею шкіри (пальпована пурпура) і не зникають при натисканні. Висипання симетрично локалізуються на розгинальних поверхнях нижніх кінцівок (на тильному боці ступні, гомілці, стегнах), а в тяжких випадках на сідницях, мошонці, попереку, однак висипання на тулубі рідкісні. Через кілька днів висипання бліднуть, набувають бурого відтінку (відкладення гемосидерину) і можуть зникати безслідно або залишати гіперпігментацію шкіри. Особливістю пурпури Шенлейна — Геноха є схильність до рецидивування і злиття елементів висипання, чому сприяє тривале перебування у вертикальному положенні. Зливна пурпура веде до утворення екхімозів, геморагічних пухирів, після руйнування яких утворюються глибокі ерозії. Висипання можуть супроводжуватися місцевими ангіоневротичними набряками та парестезіями. Слизові оболонки при пурпурі Шенлейна — Геноха не пошкоджуються.
Суглобовий синдром відзначають у ⅔ хворих. Він може передувати шкірним і абдомінальним проявам, але зазвичай розвивається одночасно з ними. Частіше спостерігаються артралгії, рідше олігоартрити. Характерне ураження великих суглобів нижніх кінцівок: гомілковостопних і колінних, рідше — променезап’ясткових і дрібних суглобів кисті. Ураження зазвичай симетричне. Біль в суглобах частіше помірний, посилюється при пальпації і рухах, досить часто виявляють мігруючий біль. Суглобовий синдром триває в середньому 1–2 тиж і проходить безслідно. У поодиноких випадках реєструють стійкий артрит з торпідним перебігом, що нагадує такий при РА, однак кісткової деструкції і стійких деформацій при пурпурі Шенлейна — Геноха не відбувається.
Шкірно-суглобовий синдром може супроводжуватися лихоманкою і явищами загальної інтоксикації.
Абдомінальний синдром (абдомінальну пурпуру) при пурпурі Шенлейна — Геноха відзначають у 70% дітей і у 50% дорослих. Симптоми ураження гастроінтестинальних судин розвиваються зазвичай через 1 тиж після появи шкірної пурпури. Частіше васкуліт локалізується в початковому і кінцевому відділах тонкого кишечнику, рідше уражаються інші відділи травної системи. Типовим є незначний больовий синдром, без чіткої локалізації, тривалістю 1–3 дні. Клінічно абдомінальна пурпура може проявлятися також раптовою появою нудоти, блювання, дифузним або переймоподібним, за типом кишкової кольки, болем у животі. Біль частіше локалізується в мезогастрії, але може виникати в правому підребер’ї, правій клубовій ділянці, симулювати гострий холецистит, панкреатит, апендицит. Може розвинутися картина «гострого живота», що вимагає виключення гострого апендициту, кишкової непрохідності, іншої патології. Іноді відзначають шлунково-кишкові кровотечі, інвагінацію, перфорацію кишечнику. У випадках, коли абдомінальний синдром передує висипанню, диференційна діагностика пурпури Шенлейна — Геноха і «гострого живота» складна.
Ураження нирок при пурпурі Шенлейна — Геноха виявляють у 10–50% хворих. Типовим є васкуліт капілярів клубочків, який призводить до розвитку гематуричного гломерулонефриту. Зазвичай він розвивається гостро — через 1–4 тиж після дебюту або під час одного з рецидивів хвороби. Украй рідко пурпура Шенлейна — Геноха дебютує з ураження нирок. У більшості хворих гломерулонефрит має легкий перебіг, клінічно проявляється сечовим синдромом, як правило, самовільно швидко усувається і закінчується повним видужанням. Дуже рідко на початку хвороби виявляють нефротичний синдром, симптоматичну артеріальну гіпертензію, ниркову недостатність. Хронічна хвороба нирок, яка призводить до артеріальної гіпертензії, зниження клубочкової фільтрації та розвитку хронічної ниркової недостатності, виникає у 5% хворих.
Ураження легенів зустрічається при пурпурі Шенлейна — Геноха рідко (6%). Інколи виникають судинні геморагічні пневмонії та плеврити. Клінічно це проявляється непродуктивним кашлем, задишкою, рідко виникає кровохаркання, можливі легеневі кровотечі. Зафіксовано випадки набряку гортані, трахеї.
Дуже рідко при пурпурі Шенлейна — Геноха уражається нервова система. У такому разі хворого турбує нападоподібний головний біль, запаморочення, можуть розвинутися енцефалопатія з психічними порушеннями, менінгеальні симптоми, можливий розвиток інфаркту мозку, субдуральної гематоми, судомного синдрому і поліневропатії. Зрідка у хворих виникають міокардити, серозити.
Залежно від ураженого органа виділяють наступні клінічні форми пурпури Шенлейна — Геноха: шкірну (просту, некротичну, з холодовою кропив’янкою та ангіоневротичними набряками), шкірно-суглобову, шкірно-вісцеральну (шкірно-абдомінальну, шкірно-ниркову, змішану) (табл. 21.56).
Таблиця 21.56
Клінічна класифікація пурпури Шенлейна — Геноха (АРУ, 2004)
|
Перебіг |
Блискавичний, гострий, рефрактерний, хронічно-рецидивуючий з тривалими ремісіями |
|
|
Фаза |
Активна, неактивна, ремісія, часткова ремісія, персистуюча активність |
|
|
Ступінь активності |
I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Шкіра |
Пурпура, виразки |
|
Суглоби |
Артрит, артралгії |
|
|
Нирки |
Гломерулонефрит з дифузною імунокомплексною та мезангіальною вогнищевою проліферацією, артеріальною гіпертензією |
|
|
ШКТ |
Абдомінальний синдром, кровотечі, кишкова непрохідність |
|
|
Легені |
Васкуліт, судинна пневмонія, геморагічний плеврит, набряк слизової оболонки трахеї та бронхів |
|
|
ЦНС |
Васкуліт судин головного мозку, енцефалопатія, менінгіальні симптоми, епілептиформні судоми |
|
Виділяють гострий, затяжний, хронічний рецидивуючий і персистуючий перебіг захворювання. При гострому перебігу хвороба починається раптово після інфекції, клінічна картина полісимптомна, яскраво виражена, з частим виникненням гломерулонефриту. Хронічний варіант частіше характеризується рецидивуючим шкірно-суглобовим синдромом. Рецидиви захворювання спостерігаються у 40% хворих.
Дебют і перебіг захворювання у дітей і дорослих мають ряд суттєвих відмінностей. Пурпура Шенлейна — Геноха у дітей відрізняється наявністю виразного зв’язку з інфекцією верхніх дихальних шляхів, а у 30% дітей спостерігається збільшення титру антистрептолізину-О, що свідчить про роль стрептококу як тригерного фактора. У 25% дітей артралгії і артрити передують ураженню шкіри, у ⅓ діагностують запалення, набряк мошонки, геморагії. У дорослих при пурпурі Шенлейна — Геноха рідше, ніж у дітей, виникають лихоманка, абдомінальний синдром, частіше — ураження суглобів, нирок. Якщо у дітей перебіг захворювання частіше легкий, то у дорослих зазвичай більш тривалий, з рецидивами шкірно-суглобового синдрому і розвитком хронічної хвороби нирок.
Лабораторні дослідження
В гострий період хвороби реєструють нейтрофільний лейкоцитоз, більш виражений при абдомінальному синдромі, помірну еозинофілію, збільшення ШОЕ і вмісту в плазмі крові білків гострої фази. У сироватці крові підвищується концентрація імуноглобуліну A, з’являються імуноглобулін A-вмісні імунні комплекси. У 30–40% хворих виявляють РФ. При ураженні нирок в сечі відзначають еритроцитурію, протеїнурію.
Морфологічні зміни
У стінці уражених судин методом прямої імунофлуоресценції виявляють відкладення імуноглобуліну A, що є характерною ознакою, при відповідній клінічній картині має діагностичне значення. Навколо капілярів знаходять скупчення нейтрофілів й еозинофілів, вогнища некрозу, у просвіті судин — агрегати тромбоцитів. У біоптаті шкіри відмічають лейкоцитокластичний васкуліт або некротичний васкуліт дрібних судин. При дослідженні слизової оболонки ШКТ виявляють ознаки капіляриту. У нирках розвивається гломерулонефрит з дифузною імунокомплексною і мезангіальною осередковою проліферацією, вираженість ознак варіює від мінімальних до тяжкого гломерулонефриту з «напівмісяцями». При електронному мікроскопічному дослідженні реєструють депозити у мезангії, субендотелії та субепітелії клубочків нирок, до складу яких входять імуноглобулін A, переважно першого і рідше другого субкласу, імуноглобуліни G, M, C3 фрагмент комплементу і фібрин. Морфологічне дослідження аутопсійного матеріалу тканини легень свідчить про некроз стінки капілярів з септальними та інтраальвеолярними геморагіями, з якими поєднуються депозити імуноглобуліну A.
Інструментальні дослідження
У хворих з ушкодженням ШКТ при рентгенологічному дослідженні виявляють множинні дефекти наповнення слизової оболонки і звуження просвіту тонкої кишки. При ендоскопічному дослідженні у пацієнтів з абдомінальною пурпурою відзначають геморагічний або ерозивний дуоденіт, гастрит, коліт. У разі лапароскопії виявляють геморагічні висипання або ділянки некрозу стінки кишки і очеревини.
Діагностика
Установити діагноз пурпури Шенлейна — Геноха нескладно за наявності класичної тріади: шкірної, абдомінальної пурпури і суглобового синдрому. Для встановлення діагнозу у дітей з легкими проявами хвороби достатньо даних анамнезу. У тяжких випадках або у разі сумнівів показано проведення біопсії уражених органів. Труднощі в діагностиці виникають у тому випадку, коли хвороба починається з абдомінального синдрому, або він переважає в клінічній картині хвороби. У таких випадках питання вирішується за участю хірурга, і часто виникає необхідність лапароскопії (табл. 21.57).
Наявність у хворого двох і більше будь-яких критеріїв дозволяє встановити діагноз з чутливістю 87,1% і специфічністю 87,7%.
Таблиця 21.57
Класифікаційні критерії геморагічного васкуліту (пурпури Шенлейна — Геноха) (ACR, 1990)
|
1. |
Пальпована пурпура |
Геморагічні висипання, які трохи піднімаються над шкірою, не пов’язані з тромбоцитопенією |
|
2. |
Вік <20 років |
Початок хвороби в віці <20 років |
|
3. |
Біль у животі |
Біль у животі дифузного характеру, що посилюється після прийому їжі, або ішемія кишечнику. Може розвинутися кишкова кровотеча |
|
4. |
Біопсія |
При гістологічному дослідженні виявляють гранулоцитарну інфільтрацію стінок артеріол та венул |
Диференційний діагноз
Проводять диференційну діагностику пурпури Шенлейна — Геноха та інших системних васкулітів, ЗЗСТ, інфекційного ендокардиту, хвороби Вальденстрема. На відміну від інших форм імунокомплексних захворювань, для пурпури Шенлейна — Геноха типовою є наявність яскравих відкладень ІgА в уражених судинах шкіри, мезангії клубочків нирок, які виявляють за допомогою методу прямої імунофлуоресценції. При відповідній клінічній картині ця ознака має діагностичне значення. Інші форми васкулітів дрібних судин також можуть супроводжуватися незначним відкладенням імуноглобуліну A в судинах, але в таких випадках він не буває основним імунореактантом.
Приклад формулювання діагнозу: геморагічний васкуліт, хронічний рецидивуючий перебіг, загострення (III рецидив), активність II ступеня. Шкірно-вісцеральна змішана форма з ураженням: шкіри (пурпура), суглобів (артрити колінних і гомілковостопних суглобів), порушення функції суглобів I ступеня, кишечнику (некротичний виразковий ентерит), нирок (гломерулонефрит, сечовий синдром, артеріальна гіпертензія II ступеня, хронічна хвороба нирок II ступеня), хронічна ниркова недостатність I ступеня, легенів (пневмоніт, кровохаркання), дихальна недостатність I ступеня.
Лікування
У гострий період перебігу пурпури Шенлейна — Геноха хворим необхідний постільний режим. Призначають дієту з виключенням продуктів, які можуть викликати алергічну реакцію: меду, какао, шоколаду, цитрусових, горіхів, риби та рибних продуктів, грибів, яєць, виробів зі здобного тіста, консервованих і копчених продуктів, майонезу та інших спецій. Забороняється вживати алкогольні напої.
У легких випадках пурпури Шенлейна — Геноха в медикаментозному лікуванні немає необхідності. Навіть при розвитку гломерулонефриту переконливих даних про те, що ГК або імуносупресори впливають на перебіг захворювання, не отримано. У більшості випадків прояви зникають спонтанно, тривалість хвороби становить від 5 до 16 тиж. Антибіотики призначають лише за наявності гострої або загостренні хронічної інфекції, а також при виявленні високого титру антистрептолізину-О. Оскільки прийом лікарських препаратів може служити пусковим чинником, поліпрагмазія не припустима.
При нетяжкому перебігу застосовують лише симптоматичну, антитромботичну терапію. Антиагреганти: тиклопідин по 0,25 г 2 рази на день під час прийому їжі впродовж 2–6 міс або дипіридамол 300 мг/добу з подальшим зниженням дози до 50–75 мг/добу і прийомом протягом декількох місяців, або пентоксифілін 300 мг/добу в/в краплинно протягом 10–14 днів з подальшим переходом на застосування всередину в дозі 600 мг/добу, підтримувальна доза — 300 мг/добу протягом 6 міс. При більш тяжкому перебігу призначають гепаринотерапію: 5000–10 000 од. через 6 год підшкірно в ділянку живота під контролем активованого часткового тромбопластинового часу або часу згортання крові, протягом періоду від 2 тиж до 1,5–2 міс. Ефективним є в/в введення свіжозамороженої плазми по 300–400 мл/добу № 3–5 у поєднанні з гепарином.
При ізольованому ураженні шкіри і суглобів засобом вибору є НПЗП у середніх дозах, тривалість прийому — не більше 4–6 тиж. Гідроксихлорохін призначають по 0,4 г/добу в разі хронічного перебігу при легких формах, шкірно-суглобовому синдромі.
При ураженні внутрішніх органів значної вираженості преднізолон призначають всередину в дозі 1 мг/кг/добу після прийому їжі (при абдомінальному синдромі доза може досягати 200–800 мг/добу, в/в), тривалість лікування залежить від клінічного ефекту, але має становити не менше 2 тиж.
Відповідно до рекомендацій KDIGO (Kidney Disease: Improving Global Outcomes, 2012) при нефриті на тлі пурпури Шенлейна — Геноха і наявності лише персистуючої протеїнурії >0,5–1,0 г/л/м2 призначають інгібітори АПФ або блокатори рецепторів ангіотензину з титруванням відповідно до рівня артеріального тиску та протеїнурії. При протеїнурії понад 1,0 г/л/м2 після спроби лікування інгібіторами АПФ або блокаторами рецепторів ангіотензину за умов швидкості клубочкової фільтрації >50 мл/хв/1,73 м2 пропонується 6-місячне лікування ГК. Починають терапію ГК з в/в введення пульсової дози 500 мг метилпреднізолону щоденно протягом 3 днів, або преднізолон призначають в одній добовій дозі 1 мг/кг (максимум 60 мг) упродовж мінімум 4 тиж та повільно знижують дозу протягом 6 міс після досягнення повної ремісії. Пропонується використовувати для лікування риб’ячий жир. Препарати системної ензимотерапії призначають в комбінованих програмах лікування.
За наявності пурпури Шенлейна — Геноха, асоційованої з нефритом з півмісяцями, і у разі швидкопрогресуючої втрати функції нирок для початкового лікування рекомендують ГК і циклофосфамід, який призначають кожні 3–4 тиж в/в у дозі 0,75 мг/м2 або в середину в дозі 1,5–2 мг/кг/добу в 2–3 прийоми с подальшим коригуванням дози до досягнення найнижчого рівня лейкоцитів >3000/мм3 у щодвохтижневому визначенні. Хворим, рефрактерним до стандартної імуносупресивної терапії зі швидким приростом вмісту креатиніну, або тим, які потребують діалізу, показано доповнення лікування плазмаферезом і в/в людським імуноглобуліном. Циклофосфамід відміняють після 3 міс терапії у пацієнтів, які є стабільно діаліззалежними, та тих, які вже не мають ніяких екстраренальних проявів хвороби. За наявності протипоказань до циклофосфаміду та пацієнтам без тяжких проявів хвороби як альтернативне початкове лікування рекомендують ритуксимаб і ГК. Пацієнтам, які досягли повної ремісії, необхідно призначати підтримувальну терапію впродовж щонайменше 18 міс: азатіоприн 1–2 мг/кг перорально, хворим з алергією або непереносимістю — мофетил мікофенолат до 1 г 2 рази на добу, у пацієнтів із непереносимістю азатіоприну і мофетил мікофенолату застосовують метотрексат у початковій дозі 0,3 мг/кг/тиж, максимум 25 мг/тиж, але якщо швидкість клубочкової фільтрації не <60 мл/хв/м2.
Про ефективність лікування свідчить нормалізація клінічних симптомів хвороби, нормалізація або позитивна динаміка лабораторних показників (ШОЕ, кількість лейкоцитів, показники гострої фази).
Хворі, які перенесли пурпуру Шенлейна — Геноха, підлягають диспансерному нагляду. Для профілактики рецидивів проводять санацію вогнищ хронічної інфекції, слід уникати вакцинації, переохолоджень, фізичних перевантажень.
Прогноз
У більшості випадків пурпури Шенлейна — Геноха прогноз сприятливий, реєструють швидке одужання. Протягом перших 2 років повне одужання відзначають у 93,9% дітей та у 89,2% дорослих. У цілому 5-річна виживаність становить 95%. У разі розвитку хронічного гломерулонефриту прогноз ускладнюється і 5-річна виживаність становить до 70%. Серед дітей, які потребують нирковозамісної терапії, питома вага випадків пурпури Шенлейна — Геноха становить 3–15%.
Гіперсенситивний васкуліт
Гіперсенситивний васкуліт (ГСВ) — імунокомплексний васкуліт судин малого калібру (капілярів, посткапілярних венул, рідше — артеріол).
Це найбільш поширена форма васкуліту, розвиток якої пов’язують із прийомом різних ліків або інфекцією. Ця форма об’єднує гетерогенні синдроми, при яких імунні комплекси, які утворюються у відповідь на введення ліків та інфекцію, відкладаються у капілярах, посткапілярних венулах, артеріолах, де фіксують комплемент, в наслідок цього виникає імунне запалення. До подібних синдромів відносять також сироваткову хворобу, медикаментозний васкуліт (рис. 21.1). Найчастіше ангіїт виникає внаслідок лікування сульфаніламідами, пеніцилінами, цефалоспоринами. Хворобі часто передує гостра вірусна інфекція з відповідними загальними симптомами (серед них вірус простого герпесу, вірус Епштейна — Барр, вірус імунодефіциту людини, вірус гепатиту С). Лікарські засоби, прийняті на початку хвороби, можуть служити фактором, який прискорює її розвиток. Але не завжди можливо встановити причину прогресування хвороби. ГСВ, що розвивається без наявності системних проявів з ушкодженням лише шкіри, іноді розглядають як васкуліт шкіри або шкірний лейкоцитокластичний ангіїт, який визначають як некротизуючий васкуліт із нейтрофільною інфільтрацією стінок судин, фібриноїдним некрозом і лейкоцитоклазією (фрагментацією ядер клітин).
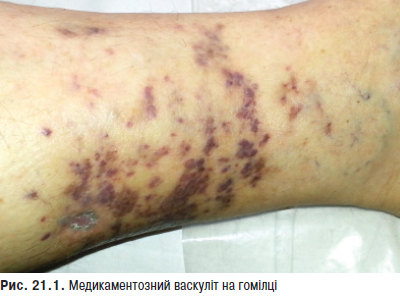
Клінічна картина
Зазвичай хвороба розвивається гостро. Якщо розвитку васкуліту передує гостра вірусна інфекція, то наявні відповідні симптоми. Найбільш частим проявом васкуліту дрібних судин шкіри є геморагічне висипання, яке локалізується переважно симетрично на розгинальних поверхнях дистальних відділів верхніх та нижніх кінцівок. У деяких випадках може відмічатися макулопапульозне висипання, яке представлене плоскими та підвищеними елементами різних розмірів. У тяжких випадках може бути уражений увесь зовнішній покрив. Висип пальпується і не блідне при натисненні, і не пов’язаний з тромбоцитопенією. Слизові оболонки, як правило, не пошкоджуються. Іноді наявні також артралгії та міалгії. Ураження нирок і ЦНС, зазвичай, відсутнє (табл. 21.58).
Таблиця 21.58
Клінічна класифікація ГСВ
|
Перебіг |
Гострий, рефрактерний, хронічно-рецидивуючий з тривалими ремісіями |
|
|
Фаза |
Активна, неактивна, ремісія, часткова ремісія, персистуюча активність |
|
|
Ступінь активності |
I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Шкіра |
Геморагічне висипання, макулопапульозне висипання |
|
Суглоби |
Артралгії, міалгії |
|
Морфологічні зміни
Для діагностики ГСВ має значення виявлення в біопсійному матеріалі гранулоцитів у периваскулярних і екстраваскулярних тканинах, а також ознак васкуліту судин дрібного калібру: діапедезу еритроцитів, каріорексису та лейкоцитоклазії.
Діагноз
Діагноз встановлюють на підставі ретельно зібраного анамнезу та клінічних проявів. З’ясовують можливий чинник: лікарський засіб, інфекція. Діагностичним методом вибору є біопсія шкіри.
Класифікаційні критерії гіперсенситивного васкуліту (АСR, 1990):
- Вік хворого на початку хвороби >16 років.
- Прийом відповідних ліків на початку хвороби
- Геморагічне висипання, що пальпується.
- Макулопапульозне висипання.
- При мікроскопічному дослідженні біопсійного матеріалу наявні нейтрофіли навколо артеріол та венул.
За наявності 3 і більше вищеперелічених критеріїв можна встановити діагноз ГСВ. Чутливість — 71%, специфічність — 83,9%.
Диференційний діагноз
Необхідно виключити васкуліти зі шкірними проявами: васкуліт Шенлейна — Геноха, гранулематоз Вегенера, МПА, синдром Черджа — Стросс, ЕКВ. Слід диференціювати ГСВ і ЗЗСТ, інфекційний ендокардит, хворобу Вальденстрема, хронічні вірусні гепатити та інші хвороби, які супроводжуються геморагічним ушкодженням шкіри.
Сироваткова хвороба виникає після введення білкових препаратів; клінічно характеризується наявністю кропив’янки (основним елементом висипання є пухир), артралгіями, лихоманкою, лімфаденопатією. Діагноз встановлюють за даними анамнезу, лабораторних тестів (високі показники гемолітичної активності комплементу, ЦІК) та біопсії.
Диференціювати ГСВ слід також від генералізованих алергічних дерматитів, в основі патогенезу яких лежать алергічні реакції негайного типу. Синдром Лайєлла — токсичний епідермальний некроліз — виникає за наявності алергії до лікарських засобів (сульфаніламідів, пеніциліну, тетрацикліну, анальгіну, ібупрофену та ін.). Характерне еритематозне, еритематозно-папульозне висипання, яке швидко трансформується в бульозну та після руйнування пухирів залишає під собою ерозивну поверхню. Ураження шкіри починається з обличчя, кінцівок і на 2–5-й день поширюється по всьому тілу. Пошкоджуються також слизові оболонки ротової порожнини, статевих органів, кон’юнктива, а також внутрішні органи: легені, міокард, печінка, нирки, мозок. Інша форма дерматиту, багатоформна ексудативна еритема, зазвичай починається гостро з лихоманки. Залежно від переваги тих чи інших первинних елементів розрізняють плямисту (яскраво-рожеві або червоні плями розміром від 2 мм до 2–3 см), папульозну (папули кільцеподібної форми розміром до 3–4 см) та бульозну (везикули з серозним і геморагічним вмістом, ерозії) форми. Ушкодження шкіри часто поєднується з ураженням слизової оболонки рота, губ. Синдром Стівенса — Джонса — це варіант перебігу багатоформної ексудативної еритеми з ураженням шкіри, слизової оболонки ротової порожнини, носа, очей, статевих органів з утворенням болісних ерозій, виразок, гнійно-кров’янистих кірок. Хворі скаржаться на біль у горлі; на слизовій оболонці ротової порожнини, щік, язиці, яснах виявляють болісні ерозії, виразки з сірувато-білим нальотом, ерозії, кров’янисті кірки виявляють на червоній облямівці губ. Також відзначають кон’юнктивіт, катарально-гнійний риніт, уретрит, ерозивний баланопостит, вагініт. Перебіг хвороби може ускладнюватися розвитком запалення легенів, міокарда, нирок.
Лікування
Найважливішим є виявлення фактора, який сприяв розвитку хвороби, та вилучення його. При медикаментозному васкуліті насамперед є необхідним припинення прийому усіх лікарських засобів до усунення симптомів хвороби. Потім їх знову можливо призначати по одному. Для лікування пацієнтів із тяжкими, рефрактерними формами ГСВ застосовують ГК, гепарин, антиагреганти.
Прогноз
У хворих на ГСВ прогноз залежить від чинника хвороби. Якщо визначено лікарські засоби, які спричинили хворобу, то відміна їх веде до видужання. Приблизно в половині випадків встановити причину васкуліту шкіри не вдається. У такому разі перебіг хвороби частіше рецидивуючий і не потребує призначення активної імуносупресивної терапії.
Гігантоклітинний артеріїт
Гігантоклітинний артеріїт (ГКА) — хвороба, яка характеризується гранулематозним запаленням аорти та її основних гілок — переважно позачерепних гілок сонної артерії, скроневої, хребетної або інших. Найчастіше уражується скронева артерія.
Код за МКХ-10
М31.5 Гігантоклітинний артеріїт з ревматичною поліміалгією.
М.35.3 Ревматична поліміалгія.
Епідеміологія
Розвивається у віці >50 років (трапляються окремі випадки хвороби у віці до 40 років і часто — у 50% — поєднується з РПМ). Хворіють переважно жінки, захворюваність становить 0,49–23,3 випадки на 100 тис. населення віком >50 років (Насонов Е.Л., 2010 (ред.)).
Етіологія і патогенез
Імуногенетичними маркерами ГКА є антигени HLA-DR4 і HLA-DRB1 (деякі алельні варіанти). Характерним є поєднання з РПМ. Дані, що підтверджують імунний характер порушень в патогенезі ГКА, включають: зміни синтезу цитокінів; наявність специфічних клонів Т-лімфоцитів; посилений синтез моноцитами ІЛ-1β, -6, лімфоцитами — ІЛ-2 і γ-інтерферону; проліферацію специфічних клонів Т-лімфоцитів. Проліферація в судинній стінці специфічних клонів Т-лімфоцитів свідчить про те, що в ній присутній антиген, який розпізнається лімфоцитами. Цей невизначений антиген є причиною виникнення гранулематозного запалення в стінці судин. У гранулематозному запаленні беруть участь лімфоцити CD4+/Th1 та макрофаги. Інтерферон-γ, який продукується субпопуляцією лімфоцитів Th1, стимулює макрофаги, викликає утворення в уражених судинах гігантських клітин.
При гістологічному дослідженні уражених артерій виявляють характерні зміни:
- вогнища некрозу у внутрішньому та м’язовому шарі стінки артерій середнього калібру;
- деструкцію еластичної мембрани;
- інфільтрацію лімфоцитами усіх шарів стінки артерій;
- гранульоми навколо вогнищ деструкції, які містять макрофаги, гігантські багатоядерні клітини, лімфоцити;
- потовщення інтими;
- пристінковий тромбоз;
- звуження/обтурацію просвіту артерій.
Запальний процес переходить у розвиток сполучної тканини, склероз стінок судин, стенозуванням їх просвіту, порушення кровопостачання органів. Можливим є виникнення продуктивного флебіту, тромбофлебіту.
Класифікація
Клінічна класифікація ГКА наведена в табл. 21.59.
Таблиця 21.59
Клінічна класифікація ГКА (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічний |
|
|
Ступінь активності |
0 (відсутній), I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Орган зору |
Зниження гостроти зору, випадання полів зору, тромбоз центральної артерії сітківки |
|
ЦНС |
Інсульти та інфаркти мозку |
|
|
Периферична нервова система |
Астеновегетативний синдром, психічні розлади |
|
|
Черепно-мозкові нерви |
Глосит, розлади мовлення, порушення смаку і нюху |
|
|
М’язи |
Синдром РПМ з синдромом переміжної кульгавості |
|
|
Шкіра |
Яскрава гіперемія над ураженими судинами шкіри обличчя, вузлики в результаті тромбозу дрібних артеріальних судин, некрози шкіри |
|
|
Аорта та великі артерії |
Звуження низхідного відділу аорти, аневризма грудного та черевного відділів, аортит, ураження скроневої артерії, аневризми сонної артерії |
|
|
Серце |
Інфаркт міокарда в результаті гранулематозного артеріїту коронарних артерій |
|
|
Судини ШКТ |
Абдомінальний синдром |
|
Клінічна картина
Характерні ознаки скроневого артеріїту:
- переважний розвиток хвороби у пацієнтів літнього віку (після 50 років);
- ураження артерій голови (переважно скроневих, рідше — очних і хребетних);
- виявлення при пальпації у звивистих скроневих артеріях щільних вузликів;
- скарги пацієнтів на головний біль, погіршення зору, лихоманку, загальну слабкість, зменшення маси тіла (до 10 кг протягом декількох місяців);
- сегментарне ураження судин, через що в 30% біоптатів специфічні морфологічні зміни відсутні;
- виявлення в біоптатах ознак гранулематозного запалення, гігантських багатоядерних клітин.
Виділяють 4 основні варіанти ГКА з:
- РПМ;
- краніальними симптомами;
- РПМ і краніальними симптомами;
- лихоманкою, іншими загальнозапальними проявами (без локалізованих симптомів).
Клінічну картину хвороби наведено в табл. 21.60.
Таблиця 21.60
Клінічна картина ГКА
|
Скарги хворих |
Пацієнти скаржаться на втрату апетиту, зменшення маси тіла (на 10 кг і більше протягом декількох місяців), субфебрильну температуру, загальну слабкість, підвищене потовиділення, безсоння, міалгії, артралгії. У міру прогресування хвороби з’являється сильний біль в тім’яній і скроневій ділянках, який посилюється під час їди (жування, ковтання), у другій половині дня та вночі. Найчастіше біль нападоподібного характеру, розповсюджується в чоло, потилицю, вуха, зуби. Виникає декілька разів на день, носить гострий, пульсуючий характер, його провокує прийом алкоголю. Характерним є синдром переміжної кульгавості — посилення болю в жувальних м’язах і язиці при розмові та під час їди |
|
Симптоматика залежно від ураженої артерії |
|
|
Внутрішньочерепні артерії |
|
|
Зовнішня сонна артерія |
|
|
Скронева артерія |
|
|
Артерії, які кровопостачають око |
Виникає набряк рогівки, епісклерит, склерит, ішемічний хоріоретиніт, ірит, кон’юнктивіт, передня ішемічна оптична невропатія, порушується зір. У тому числі розвивається раптова сліпота, яка може мати транзиторний характер. Уражуються окорухові м’язи (виникає їх параліч), з’являється диплопія, світлобоязнь, біль в очних яблуках, випадання полів зору. Розвивається набряк сітківки і диска зорового нерва, його атрофія, геморагії |
|
Верхньощелепна артерія |
Відзначають гіперестезію шкірних покривів голови |
|
Аорта та її гілки |
Наслідком їх ураження є аортит, аневризма аорти та великих артерій, аортальна недостатність, порушення кровообігу кінцівок, ішемічні інсульти, інфаркт міокарда |
|
Ниркові артерії |
Розвивається реноваскулярна артеріальна гіпертензія |

Сліпота у хворих на ГКА. Зміни на очному дні виникають через декілька місяців з моменту появи головного болю. Тому при своєчасному початку лікування сліпоти можна уникнути.
Симптоматика, подібна до хвороби Такаясу. У окремих хворих можуть відмічатися ураження коронарних, мезентеріальних, магістральних артерій кінцівок та ін. Може проявлятися клінічна симптоматика ураження інших артерій: інфаркт міокарда, інфаркти внутрішніх органів, розшаровуюча аневризма аорти.
Ураження верхніх дихальних шляхів. Відзначають у 10% хворих у вигляді кашлю (непродуктивного), лихоманки, іноді болю в грудній клітці та горлі (внаслідок ураження гілок зовнішньої сонної артерії).
Ураження суглобів. Ураження суглобів може розвиватися в дебюті хвороби у вигляді асиметричного моно- або олігоартриту великих суглобів (переважно колінних, гомілковостопних, променезап’ясткових). Проте можливий розвиток ревматоїдоподібного симетричного поліартриту з ураженням дрібних суглобів кистей та стоп (проксимальних міжфалангових, плеснофалангових суглобів).
Ураження нервової системи. Можливий розвиток множинних мононевритів, поліневропатії. Розвивається ішемічна невропатія зорового нерва, яка може призвести до втрати зору. Ураження черепних нервів носить транзиторний характер, регресує протягом декількох тижнів. Часто уражуються окорухові нерви. У 5% відзначають ішемічні інсульти. Розвиваються депресія, деменція, епілептичні напади та тремор. Виникає ішемія шийних нервових корінців, плечового сплетіння. Зустрічається симетрична сенсомоторна поліневропатія.
Діагностика
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих на ГКА, наведено в табл. 21.61.
Таблиця 21.61
Результати лабораторного та інструментального обстеження хворих на ГКА
|
I. Лабораторне дослідження |
|
|
Загальний аналіз крові |
Нейтрофільний лейкоцитоз, збільшення (не завжди) ШОЕ, анемія (нормо- або гіпохромна) |
|
Біохімічні дослідження |
Збільшується вміст СРБ в крові, серомукоїду, γ-глобуліну, фібронектину. Підвищується активність лужної фосфатази, АлАТ, АсАТ |
|
Імунологічні дослідження |
У крові підвищується рівень імуноглобуліну G, ІЛ-6 (>6 пг/мл) та активності комплементу |
|
Гістологічне дослідження біоптатів скроневої артерії |
У запальних гранульомах знаходять гігантські багатоядерні клітини |
|
Дослідження бронхоальвеолярного лаважу |
Ознаки Т-лімфоцитарного альвеоліту з переважанням CD4+-лімфоцитів |
|
II. Інструментальне дослідження |
|
|
Допплеросонографія |
Виявляють зниження артеріального тиску, пульсації артерій та кровотоку по сонних, скроневих, очноямкових і хребетних артеріях |
|
Реовазографія |
Відзначають асиметрію та порушення кровотоку по сонних, хребетних і скроневих артеріях |
|
Сфігмографія |
Характерним є зниження та деформація хвиль сфігмограми сонних і скроневих артерій |
|
Рентгенографія та КТ легенів |
Визначається базальний інтерстиціальний фіброз, дифузні ретикулярні зміни легеневого малюнка, множинні вузлики, аневризма грудного відділу аорти |
|
МРТ |
Виявляють запальні зміни у грудному відділі аорти, формування аневризми аорти |
При дослідженні бронхоальвеолярного лаважу виявляють ознаки Т-лімфоцитарного альвеоліту з переважанням CD4+-лімфоцитів. Виражене збільшення ШОЕ при ГКА реєструють не завжди: в 23% випадків показник нормальний. Чутливішим показником активності ГКА є підвищення в крові концентрації ІЛ-6.
Артерії, які рекомендується обстежувати. Необхідне обстеження артерій голови, шиї, тулуба та кінцівок (діаметр, болісність при пальпації, шуми, пульсація). Ангіографію судин проводять в діагностично тяжких випадках, коли виявляють ураження аорти та великих артерій. При цьому виявляють звуження судин.
Нижче наведено діагностичні критерії ГКА (табл. 21.62).
Таблиця 21.62
Діагностичні критерії ГКА (ACR, 1990)
|
1. |
Початок захворювання у віці >50 років |
Вперше симптоми з’являються у віці >50 років |
|
2. |
Поява «нового» головного болю |
Поява головного болю, який не спостерігався раніше, або зміна його характеру та/або локалізації |
|
3. |
Аномальні симптоми скроневої артерії |
Скронева артерія при пальпації чутлива, зменшення її пульсації не пов’язане з атеросклерозом артерій шиї |
|
4. |
Збільшення ШОЕ |
ШОЕ становить або перевищує 50 мм/год |
|
5. |
Зміни в біоптатах артерії |
Відзначено васкуліт з переважною інфільтрацією макрофагами або гранулематозним запаленням, гігантськими багатоядерними клітинами |
Установити діагноз дозволяє наявність 3 і більше будь-яких критеріїв (чутливість — 93,5%, специфічність — 91,2%).
Методи, що використовуються для підтвердження діагнозу ГКА. З цією метою проводять гістологічне вивчення біоптатів скроневої артерії, а також ангіографію. У цілому встановити діагноз дозволяють наступні дані:
- наявність лихоманки, анемії, підвищеного показника ШОЕ у пацієнтів літнього віку;
- характерні морфологічні зміни в серійних зрізах біоптатів скроневих артерій;
- різке поліпшення стану пацієнтів на тлі лікування ГК.
Досліджують сегмент скроневої артерії завдовжки 3–5 см, при негативних результатах рекомендується зробити біопсію на протилежному боці. Біопсія скроневої артерії необхідна в тих випадках, коли при терапії ГК відзначають недостатнє поліпшення стану хворого або показник ШОЕ мало змінюється. В артеріях хворих на ГКА виявляють наступні морфологічні зміни:
- панартеріїт;
- інфільтрацію стінки артерій лімфоцитами, макрофагами, гігантськими багатоядерними клітинами;
- проліферацію інтими;
- руйнування внутрішньої еластичної мембрани.
Негативні результати біопсії скроневої артерії не виключають діагнозу ГКА (можливе вогнищеве сегментарне ураження) та не є причиною непризначення ГК.
Проводять диференційну діагностику ГКА і атеросклерозу, РА, плечолопаткового періартриту, запальних міопатій, інфекції, злоякісних новоутворень, аутоімунного тиреоїдиту, системного амілоїдозу та ін.
Приклад формулювання діагнозу. ГКА (хвороба Хортона), хронічний перебіг, загострення, активність II ступеня. Ураження очей (тромбоз центральної артерії сітківки правого ока, втрата зору), черепно-мозкових нервів, РПМ.
Взаємозв’язок ревматичної поліміалгії та гігантоклітинного артеріїту
Припускають, що РПМ є проявом субклінічного перебігу ГКА. Симптоми РПМ присутні у 40–60% хворих на ГКА. Ознаки РПМ виникають одночасно з ураженням судин або приєднуються пізніше. У хворих з діагнозами ГКА і РПМ реєструють однакові морфологічні зміни в скроневих артеріях — запалення, кальцифікацію, атрофію. Відзначають взаємозв’язок скроневого артеріїту і РПМ за часом розвитку:
- РПМ і ГКА виникають одночасно.
- ГКА розвивається у хворих на РПМ (у 50% випадків).
- РПМ розвивається у хворих зі скроневим артеріїтом. Слід враховувати, що гістологічні ознаки скроневого артеріїту можна виявити у пацієнтів з РПМ, що не мають клінічних проявів артеріїту.
РПМ — захворювання опорно-рухового апарату запального характеру, яке розвивається після 50 років і проявляється стереотипним болем у ділянці шиї, плечового і тазового пояса, порушенням рухів, часто (у 50%) поєднується з хворобою Хортона. РПМ вважають клінічним синдромом — компонентом ГКА з субклінічним перебігом. Вона розвивається у осіб літнього і старечого віку і проявляється болем і скутістю в ділянці плечового і тазового пояса, високим показником ШОЕ. Як ми вже зазначали, у біоптатах скроневих артерій хворих з діагнозами РПМ та ГКА виявляють однакові зміни — запалення, кальцифікацію, атрофію. Симптоми, характерні для РПМ, виникають одночасно з судинними проявами або приєднуються пізніше. Ремісія настає при застосуванні ГК в невисоких дозах.
Епідеміологія
Частота виявлення нових випадків захворювання в різних країнах становить від 4,9 до 11,1 на 100 тис. населення. Частіше хворіють жінки (2:1), переважно хвороба розвивається у осіб літнього віку — на 7-му десятку життя. Симптомами РПМ є у 40–60% хворих на ГКА. З іншого боку, гістологічні ознаки скроневого артеріїту виявляють у 5–50% пацієнтів із симптомами РПМ.
Етіологія і патогенез
Досі не встановлено етіологію і патогенез РПМ. Згідно з існуючими двома гіпотезами патогенезу, симптоми хвороби зумовлені: 1) запаленням синовіальної оболонки в основному плечових і кульшових суглобів і/або періартикулярних тканин; 2) системним васкулітом. Запальні зміни виявляються в різних (не лише плечових і кульшових) суглобах. У судинах синовіальної оболонки запалення не виникає. У частини хворих клінічні ознаки запальної реакції відсутні, але відзначають морфологічну картину артеріїту. Виявляють ознаки генетичної схильності до розвитку захворювання: при РПМ реєструють підвищену частоту виявлення HLA-DR4.
Морфологічні зміни
Відхилень від нормальної м’язової тканини в біоптатах скелетних м’язів не виявляють. Але поєднання РПМ з хворобою Хортона дає змогу припускати, що основу захворювання становить ангіїт. Ознаки неспецифічного запалення виявляються в періартикулярних тканинах суглобів (плечових, колінних), відмічають картину вогнищевого синовіту. Зміни синовіальної рідини в більшості випадків носять незапальний характер, у окремих хворих можливий розвиток запального процесу з цитозом 10 000–20 000 в 1 мм3.
Клінічна картина
Клінічну картину РПМ наведено в табл. 21.63.
Таблиця 21.63
Основні клінічні прояви РПМ та їх характеристика
|
Симптоми |
Характеристика симптомів РПМ |
|
Больовий синдром |
Протягом перших 1–2 тиж біль може виникати в одній анатомічній ділянці (плече, тазовий пояс та ін.), до кінця місяця розгортається повна клінічна картина. Біль стереотипний та охоплює шию, плечі, плечові, кульшові суглоби, стегна. Біль двобічний (симетричний), постійний, посилюється при рухах, вираженість його зменшується у спокої. Біль носить ниючий характер, відчувається не як біль, а болісна втома. Відзначають уранішня скутість, обмеження рухливості в ділянці шиї, плечових і кульшових суглобах, більшою мірою порушені активні рухи. Може бути зменшеним обсяг пасивних рухів, особливо в плечових суглобах. Через біль порушується здатність до пересування, самообслуговування (хворим важко одягатися, роздягатися, умиватися, сідати і вставати та ін.). Пацієнти перестають виходити з дому, постійно лежать. При об’єктивному огляді можна виявити незначну болісність при пальпації горбків плечових кісток і великих вертлюгів стегнових кісток. Хворі скаржаться на м’язову слабкість, але справжня м’язова слабкість відсутня |
|
Ураження суглобів |
Артрит приєднується через декілька тижнів від моменту появи характерної клінічної картини хвороби (може з’являтися навіть через декілька місяців з моменту виникнення болю у м’язах). Але маніфестація суглобового синдрому (біль в кистях, синовіти, лігаментити, тендовагініти та ін.) можлива вже на початку хвороби. Для суглобового синдрому характерним є ураження невеликої кількості суглобів, слабка вираженість локального запалення, висока ефективність ГК і відсутність рентгенологічних змін. Він розвивається у ¼ хворих, частіше уражуються ключично-акроміальні, променезап’ясткові, колінні суглоби, рідко — дрібні суглоби кистей або стоп. Біль в суглобах менше виражений, ніж в м’язах плечового та тазового пояса. Симетричності ураження не спостерігається, запалюються 1–3 суглоби, припухлість їх незначна. Синовіальна рідина має незапальний або слабозапальний характер (до 1000–15 000 нейтрофілів на 1 мм3, в’язкість знижена). Найбільшу кількість ексудату відзначають в колінних суглобах. Артрит швидко купірується ГК, рецидиви не виникають. У ряду хворих (10–15%) розвивається слабовиражений синдром зап’ясткового каналу з характерними проявами (парестезіями, онімінням I–IV пальців, набряком кистей). У меншої кількості виявляють долонний фасцит (одно- або двобічний апоневрозит), тендовагініт сухожиль згиначів пальців з помірним набряком кисті, ущільненням і болісністю долонної фасції та сухожиль м’язів згиначів пальців, формуванням згинальних контрактур. Можуть розвиватися контрактури в плечових суглобах із залученням капсул, обмеженням рухів (активних та пасивних) і атрофією м’язів. Фасцит піддається ГК-терапії повільніше, ніж інші прояви |
|
Лихоманка |
Субфебрильну температуру тіла реєструють нерідко, іноді лихоманка досягає 38 °C і більше. Але підвищення температури тіла до високих значень для РПМ нехарактерне, як правило, приєднується в розпал клінічних симптомів хвороби, робить стан хворих тяжким і може бути стійким (тривати багато місяців) |
|
Інші прояви |
Досить швидко на тлі втрати апетиту зменшується маса тіла, з’являється загальна слабкість, погіршується настрій |
Основні клінічні прояви РПМ:
- сильний двобічний (симетричний) біль і скутість у м’язах плечового і тазового пояса та в ділянці шиї;
- уранішня скутість, яка зменшується у спокої, посилюється при рухах;
- симетричний серонегативний ревматоїдоподібний поліартрит із залученням колінних, променезап’ясткових, гомілковостопних, рідше — проксимальних міжфалангових і плеснофалангових суглобів;
- можливе ураження суглобів у вигляді моно- або олігоартриту;
- виражене збільшення показника ШОЕ (до 40–70 мм/год) і СРБ.
J.D. Singleton (1999) пояснює абревіатуру SECRET, яка описує РПМ (табл. 21.64).
Таблиця 21.64
Абревіатура SECRET
|
S |
Stiffness and pain |
Скутість та біль |
|
E |
Elderly individuals |
Літній вік хворих |
|
C |
Constitutional symptoms |
Загальні конституціональні симптоми у індоєвропейців |
|
R |
Arthritis (rheumatism) |
Артрит (ревматизм) |
|
E |
Elevated erythrocyte sedimentation rate (ESR) |
Підвищення ШОЕ |
|
T |
Temporal arteriitis |
Скроневий артеріїт |
Тривалість захворювання становить від 2 міс до кількох років. Виділяють так званий псевдопухлинний симптомокомплекс, який включає загальну слабкість, підвищену стомлюваність, анорексію, зменшення маси тіла, лихоманку. Тому в програму обстеження хворих включають фіброезофагогастродуоденоскопію, колоноскопію (іригоскопію), рідше — стернальну пункцію для виключення мієломної хвороби.
Характерний початок, варіанти дебюту хвороби. Хвороба, як правило, починається гостро. Підвищується температура тіла, з’являється постійний біль симетричного характеру в м’язах плечового пояса, шиї, сідниць, стегон. Однобічний характер болю можуть виявляти тільки на початку хвороби, надалі він двобічний і різко посилюється при рухах. Відзначають ранкову скутість (вона з’являється після періоду нерухомості), атрофія м’язів плечового і тазового пояса не розвивається, сила скелетних м’язів збережена. Повне розгортання клінічної картини хвороби відбувається протягом 2–3 тиж. Симптоми максимально проявляються через 1–2 міс хвороби. Сильний біль може призводити до знерухомлення хворих.
Можливі різні варіанти дебюту хвороби:
- поступове виникнення болю симетричного характеру;
- раптовий початок хвороби;
- дебют сильним больовим синдромом, який супроводжується різко вираженою скутістю;
- однобічне ураження в ранній період хвороби з подальшим поширенням на протилежний бік і набуттям симетричності;
- ураження на початку плечового пояса.
Характерні ознаки хвороби, які виявляють при фізикальному обстеженні хворих. У процесі огляду виявляють, що пацієнтам важко вставати зі стільця і присідати навпочіпки, а також одягатися, розчісуватися. Відзначають зміну ходи: хворі пересуваються дрібними кроками. При огляді хворих на РПМ також виявляють ряд змін:
- наявність загальної слабкості, депресії;
- підвищення температури тіла до субфебрильних значень (за наявності скроневого артеріїту можливі «температурні свічки»);
- зменшення маси тіла;
- болісність при пальпації в шийному відділі хребта;
- обмеження активних рухів в плечовому суглобі через біль;
- наявність контрактури плечового суглоба, капсуліту з обмеженням пасивних рухів, атрофією м’язів;
- біль, що виникає в проксимальних відділах, не проектується на суглоби, але посилюється при рухах в них;
- синовіти найчастіше колінних, променезап’ясткових, груднино-ключичних суглобів, рідше — плечових і кульшових;
- тунельний зап’ястковий синдром;
- збереження м’язової сили (за відсутності атрофії).
Біль і скутість при РПМ посилюються при рухах, зменшуються у спокої і поєднуються з різким збільшенням ШОЕ. Як правило, болісність м’язів при пальпації відсутня. М’язового болю дистальніше плечових та колінних суглобів не реєструють.
Біль посилюється в ранкові години, після тривалого періоду нерухомості. Уранішня скутість нагадує РА. У різних положеннях (лежачи, сидячи) найбільш сильний біль з’являється в м’язах, що піддаються стисканню масою тіла. У повному спокої зменшується вираженість болю і збільшується при рухах. М’язова сила у пацієнтів з РПМ зберігається, атрофії м’язів немає, активність так званих м’язових ферментів не змінюється. До загальних симптомів, характерних для РПМ, належать слабкість, підвищена стомлюваність, зниження апетиту і маси тіла, субфебрильна температура.
Ураження суглобів при РПМ. Артрит виникає у деяких хворих через декілька місяців з моменту появи болю у м’язах. Ураження великих суглобів олігоартикулярне, характерним є розвиток запалення, синовіту в плечових і кульшових суглобах. J.D. Singleton (1999) відзначає часте запалення при РПМ колінних, променезап’ясткових і груднино-ключичних суглобів. У синовіальній рідині виявляють нейтрофільний цитоз (кількість лейкоцитів варіює від 1000 до 20 000 в 1 мкл), її в’язкість знижується. Відзначають розвиток неспецифічного запалення в періартикулярних тканинах. Рентгенологічно визначаються ознаки ОА, ерозії суглобових поверхонь не утворюються. Можливий розвиток помірного набряку кистей (однієї або двох) без запалення дрібних суглобів, тунельного синдрому зап’ясткового каналу, тендиніту згиначів пальців з нестійкою контрактурою. Синовіти у хворих на РПМ іноді можна виявити тільки при сцинтиграфії, дослідженні синовіальної рідини, біопсії синовіальної оболонки.
У шийному відділі хребта, плечових і кульшових суглобах наявне обмеження рухів.
Тривалість РПМ становить від 2 міс до декількох років.
Діагностика
Зміни, які виявляють при лабораторному обстеженні хворих на РПМ, наведені в табл. 21.65.
Таблиця 21.65
Зміни, які виявляють при лабораторному обстеженні хворих на РПМ
|
Показники |
Характеристика порушень |
|
ШОЕ |
Підвищується з перших днів хвороби у всіх хворих, досягає рівня 40–70 мм/год. Значення показника прямо залежить від ступеня вираженості рухових порушень і больового синдрому |
|
СРБ, фібриноген, сіалові кислоти, серомукоїд |
Ці показники підвищуються, але чіткого зв’язку з клінічними проявами і застосуванням ГК не відзначено |
|
Активність трансаміназ, лужної фосфатази |
Ці показники підвищуються у 25% хворих. Причина підвищення невідома, через декілька днів після призначення ГК показники нормалізуються |
|
Еритроцити крові, гемоглобін |
У багатьох хворих виникає гіпохромна анемія |
У хворих іноді розвивається тромбоцитоз, підвищений вміст у крові γ-глобулінів. Усі показники запалення нормалізуються під дією преднізолону. У деяких пацієнтів з РПМ і ГКА показник ШОЕ може бути в межах нормальних значень. Високочутливим для визначення активності ГКА вважають показник вмісту в крові ІЛ-6. Наведено діагностичні критерії РПМ (табл. 21.66, 21.67).
Таблиця 21.66
Діагностичні критерії РПМ (за: Hamrin B., 1972)
|
1. |
Вік хворого >50 років |
|
2. |
Наявність болю в м’язах, принаймні, в 2 із 3 ділянок (шия, плечовий і тазовий пояс) |
|
3. |
Двобічна локалізація болю |
|
4. |
Переважання вказаної локалізації болю протягом активної фази хвороби |
|
5. |
ШОЕ >35 мм/год |
|
6. |
Тривалість симптомів хвороби не менше 2 міс |
|
7. |
Обмеження рухів в шийному відділі хребта, плечових і кульшових суглобах |
|
8. |
Загальна слабкість, підвищена втомлюваність, анорексія, зменшення маси тіла, лихоманка, анемія |
Примітка: перші 5 критеріїв є обов’язковими, інші — додатковими.
Таблиця 21.67
Діагностичні критерії РПМ (за: Bird H.A. et al., 1979)
|
Три або більше з представлених нижче ознак або позитивні результати біопсії скроневої артерії + мінімум одна з вказаних нижче ознак |
|
На Європейському конгресі ревматологів (2001, Прага) рекомендовано використання діагностичних критеріїв, запропонованих H.A. Bird, у поєднанні з таким додатковим критерієм, як швидке поліпшення стану після початку прийому ГК. Для діагностики РПМ необхідна наявність усіх перелічених ознак (чутливість — 99%). Для оцінки активності хвороби, досягнення ремісії та адекватності терапії, що проводиться, використовують спрощений індекс активності хвороби (SDAI PMR — simplified disease activity index polymyalgia rheumatica). Індекс розраховують наступним чином:
SDAI PMR = ВАШ пацієнта (0–10 см) + ВАШ дослідника (0–10 см) + (ранкова скутість (хв) × 0,1) + елевація верхніх кінцівок (3–0) + (ШОЕ (мм/год) × 0,1),
де ВАШ — візуальна аналогова шкала.
Інтенсивність больового синдрому розраховують за візуальною аналоговою шкалою, оцінюють пацієнт та дослідник. Тривалість ранкової скутості визначає пацієнт у хвилинах з моменту пробудження. Рівень елевації верхніх кінцівок відповідає значенням від 0 до 180 (поділяють на 3 ступені залежно від одержаних показників). Критерії оцінки активності РПМ: низька <7; середня — 7–17; висока >17.
Виділяють також діагностичні ознаки хвороби (табл. 21.68).
Таблиця 21.68
Діагностичні ознаки РПМ
|
1. |
Вік пацієнта на початку захворювання не менше 50 років |
|
2. |
Біль, принаймні, в 2 із 3 ділянок: плечовий, тазовий пояс і шия |
|
3. |
Двобічна локалізація больових відчуттів у плечовому і тазовому поясі |
|
4. |
Переважання вказаної локалізації больових відчуттів у плечовому і тазовому поясі |
|
5. |
Підвищення ШОЕ >35 мм/год |
|
6. |
Швидкий ефект при лікуванні преднізолоном в добовій дозі не більше 15 мг |
|
7. |
Відсутність ознак РА |
При опитуванні й огляді хворого слід виявити характерні прояви РПМ. Остаточне рішення про характер патології приймають після 2–3-тижневого застосування 15 мг (рідко 20 мг) преднізолону. Розвиток ремісії підтверджує діагноз РПМ.
Диференціація
Диференціюють РПМ і поліміозит, РА, ОА, плечолопатковий періартрит, остеохондроз шийного відділу хребта з радикулярним синдромом, хвороби м’яких тканин опорно-рухового апарату, фіброміалгію, міалгії на тлі інфекцій, гіперпаратиреоз, серонегативні спондилоартропатії. Відмітні ознаки деяких перелічених захворювань наведено в табл. 21.69.
Таблиця 21.69
Відмітні ознаки захворювань, які диференціюють з РПМ
|
Депресія |
Показник ШОЕ нормальний |
|
Гіпотиреоз |
ШОЕ нормальна, концентрація тиреотропного гормону в сироватці крові підвищена |
|
Фіброміалгія |
Пальпуються болісні точки, ШОЕ нормальна |
|
Плечолопатковий періартрит (двобічний) |
Характерними є біль в ділянці плечового пояса, обмеження рухів, але показник ШОЕ залишається нормальним, ефекту від преднізолону немає |
|
Початкові прояви РА |
Уражуються дрібні суглоби, визначається РФ, ерозивні зміни. У пацієнтів літнього віку можуть відмічати поширені міалгії на тлі лихоманки і ураження плечових суглобів, але водночас відзначають поліартикулярне ураження з раннім залученням дрібних суглобів кистей та стоп |
|
Поліміозит |
Переважає м’язова слабкість, підвищена активність КФК, альдолази, зміни на електроміограмі. Слабкість та біль виникають в м’язах плечового і тазового пояса, але характерним є переважання слабкості. Невеликі дози ГК неефективні |
|
ВП |
У дебюті хвороби може переважати міастенічний синдром з міалгіями, суглобово-м’язовий синдром (артрит, артралгії) |
|
Злоякісні пухлини |
Розвивається паранеопластичний поліміозит, який може бути розцінений як РПМ. Лікування преднізолоном дає незначний ефект. Спостерігаються клінічні ознаки пухлини |
|
Мієломна хвороба |
Проявляється генералізованим болем у м’язах, кістках, різким збільшенням ШОЕ. Низькі дози ГК (10–15 мг/добу преднізолону) ефекту не виявляють |
Аналогічний больовий синдром відзначають при серонегативних спондилоартритах, плечолопатковому періартриті, парапротеїнемічних гемобластозах. При ураженні в результаті ГКА артерій шиї, верхніх і нижніх кінцівок больовий синдром, характерний для РПМ, проявляється не завжди. У суперечливих випадках проводять пробне призначення ГК (терапія ex juvantibus).
У процесі проведення диференційної діагностики необхідне додаткове обстеження: визначення РФ, КФК, АсАТ, лужної фосфатази, кальцію, фосфору, сечової кислоти в крові, креатиніну в сечі, проведення електроміографії, рентгенографії, МРТ і УЗД суглобів, дослідження синовіальної рідини, біопсія м’язів, скроневої та інших артерій, сканування великих судин, обстеження для виключення онкопатології (фіброезофагогастродуоденоскопія, ректороманоскопія, колоноскопія, КТ органів черевної порожнини та ін.), мієломної хвороби (стернальна пункція).
Обстеження, необхідне для виключення асоційованого ГКА. За допомогою опитування слід виявити наявність скарг, характерних для хвороби Хортона, пальпаторно дослідити скроневі артерії, пульс на дистальних артеріях верхніх і нижніх кінцівок, провести аускультацію великих артерій шиї і в надключичних ямках, аорти. У разі приєднання хвороби Хортона з’являються головний біль, болісність, набряклість, почервоніння в ділянці скроні, відсутність пульсу на скроневій артерії. У тяжких випадках проводять інструментальні дослідження стану артерій, кровотоку в них, а також біопсію скроневої артерії.
Типові помилки в діагностиці РПМ. Помилкові висновки можуть бути, передусім, зроблені при застосуванні преднізолону в недостатній дозі (<15 мг), одноразовому його прийомі впродовж доби, малій тривалості лікування.
Лікування
Основним ефективним лікарським засобом при лікуванні є преднізолон (табл. 21.70).
Таблиця 21.70
Лікування при ГКА і РПМ
|
ГКА |
||||
|
ГК |
Початкова доза преднізолону зазвичай становить 40–60 мг/добу в декілька прийомів |
Застосовуються до нормалізації ШОЕ і зникнення симптомів. Знижують дозу по 2,5–5 мг кожні 2 тиж до досягнення дози 20 мг/добу, потім знижують на 10% кожні 2 тиж до дози 10 мг/добу, після цього добову дозу знижують на 1 мг кожні 4 тиж. Тривалість терапії встановлюють індивідуально у кожного хворого. Лікування може бути припинене, якщо протягом 6 міс на тлі прийому преднізолону в дозі 2,5 мг/добу відсутні клінічні прояви. Після завершення лікування кожні 8–12 тиж контролюють рівень ШОЕ |
||
|
Порушення зору і ураження великих судин відсутні |
Початкова доза преднізолону може становити 20 мг/добу |
|||
|
Тяжкий перебіг |
Дозу преднізолону збільшують до 60–80 мг/добу або проводять пульс-терапію метилпреднізолоном протягом 3 днів з переходом на дозу 20–30 мг/добу усередину 1 раз на добу. У такій дозі терапію проводять 1 міс і переходять на підтримувальну дозу, або до схеми лікування додають метотрексат 15–17,5 мг/тиж. Терапію припиняють, якщо протягом 6 міс на тлі прийому 2,5 мг/добу преднізолону клінічні прояви хвороби відсутні |
За відсутності ефекту протягом 2–3 тиж первинну дозу поступово підвищують. При застосуванні метотрексату можливий розвиток пневмоніту |
||
|
РПМ |
||||
|
ГК |
Початкова доза преднізолону становить 10–20 мг/добу в декілька прийомів |
Застосовується до нормалізації ШОЕ і зникнення симптомів. Знижують дозу на 1–2,5 мг/добу кожні 4 тиж до досягнення дози 10 мг/добу, після цього добову дозу знижують на 1 мг кожні 4 тиж. ШОЕ продовжують контролювати кожні 8–12 тиж протягом 12 міс після завершення лікування |
||
При рано розпочатому лікуванні преднізолоном клінічне поліпшення (зокрема зниження температури тіла) наступає в першу добу. Еволюція таких проявів, як головний біль, припухлість, болісність, поява пульсації скроневої артерії, покращення зору відбувається поступово. Сповільнений розвиток клінічного ефекту спостерігається і при залученні в патологічний процес великих артерій. Поліпшення при цьому наступає через 3–4 тиж, а у випадку тяжкого перебігу — через 8–16 тиж. Показники активності патологічного процесу (СРБ, α1-глобуліни та ін.) також нормалізуються повільно. У цілому морфологічні ознаки одужання значно відстають від клінічних. Так, гістологічні прояви активного васкуліту можна виявити в скроневій артерії у термін від 6 міс до 14 років від початку лікування. Слід враховувати, що передчасне зниження дози преднізолону або його відміна можуть призвести до загострення хвороби, залучення в запальний процес аорти та її гілок. У такому разі необхідно підвищити дозу ГК. При гострому перебігу скроневого артеріїту можливе в/в введення метилпреднізолону в масивних дозах, що призводить до швидкого відновлення зору. Одночасно призначають антикоагулянти непрямої дії. Прийом хворими на ГКА ацетилсаліцилової кислоти в дозі 100 мг/добу знижує ризик розвитку сліпоти та ішемічного інсульту. Слід враховувати, що втрата зору може розвинутися протягом декількох днів або навіть годин. Це вимагає невідкладного застосування ГК. Результати застосування у хворих на РПМ інфліксимабу, метотрексату, азатіоприну, циклоспорину неоднозначні.
При РПМ початкову дозу 15 мг преднізолону хворі приймають у 2–3 прийоми. Значне поліпшення стану відзначають уже через декілька днів застосування препарату. Якщо до 2–3-го тижня не встановлюється повна ремісія, дозу підвищують до 20 мг/добу. У разі артриту, апоневрозиту, синдрому зап’ясткового каналу ГК застосовують місцево (ін’єкції, фонофорез). Дози, яка дозволила досягти ремісії, дотримуються протягом 1 міс, а потім знижують по 1,25 мг кожні 7–10 днів за відсутності ознак загострення. У разі виникнення загострення дозу преднізолону тимчасово підвищують. Для досягнення повного одужання необхідно від 6 міс до 2–3 років. Для нормалізації показника ШОЕ під дією ГК необхідно декілька тижнів. Самопочуття хворих покращується вже через 1–2 доби. Значення ШОЕ необхідно контролювати кожний місяць протягом перших 3 міс лікування, а в подальшому — кожні 8–12 тиж протягом 1 року після завершення терапії. Повного одужання вдається досягти у всіх хворих. У більшості пацієнтів з РПМ через 10–15 міс преднізолон можна відмінити. Надто швидке зниження дози призводить до загострення хвороби, іноді рецидив виникає через декілька місяців після повного припинення лікування.
Внаслідок побічної дії ГК часто відзначають несиметричний біль у ділянці плечових і кульшових суглобів. Він виникає через декілька місяців від початку прийому ГК, імітує загострення хвороби та зумовлений дією препарату на м’язи і сухожилля, розвитком стероїдної міопатії. Вираженість побічних ефектів ГК зменшується після зниження дози препарату, призначення препаратів калію і засобів анаболічної дії (рибоксину, нандролону деканоату). На відміну від преднізолону, НПЗП не усувають больові відчуття і рухові обмеження.
До критеріїв ефективності лікування при ГКА відносять: позитивну динаміку клінічних симптомів; нормалізацію/позитивну динаміку загального аналізу крові, показників білка крові; нормалізацію температури тіла; позитивну динаміку при інструментальних дослідженнях — реовазографії, допплерографії, МРТ голови.
Профілактика ОП у хворих на РПМ. РПМ супроводжується локальним і системним ОП. Системний ОП пов’язаний з лікуванням ГК (яке може тривати декілька років), а також іммобілізацією, дефіцитом статевих гормонів, мікроелементів. ГК-терапія хворих на ГКА та РПМ має супроводжуватися призначенням з профілактичною метою комбінованих препаратів кальцію, вітаміну D3 і остеотропних мінералів. Препарати кальцію і вітаміну D3 призначають відразу ж при плануванні ГК-терапії. Випадки розвинутого ОП вимагають додаткового призначення протиостеопоротичних препаратів (стронція ранелату, бісфосфонатів, кальцитоніну та ін.).
Прогноз
При ГКА прогноз для життя сприятливий: 5-річна виживаність становить близько 100%. Можливий розвиток ускладнень — ураження артерій сітківки, часткова або повна втрата зору.
Перебіг РПМ носить хвилеподібний характер, спостерігаються періоди загострення та ремісії. У 20% хворих відзначаються рецидиви хвороби через проміжки часу, які становлять від декількох місяців до декількох років. Різка відміна ГК призводить до загострення хвороби. Дозу ГК починають знижувати не раніше ніж через 6–12 міс від початку лікування. Хвороба триває 1,5–2 роки, її результатом є склерозування скроневих артерій, наявність залишкових явищ з боку нервової системи і очей. Таким чином, прогноз відносно сприятливий. Більшість хворих з ізольованою РПМ одужують. Якщо вони не приймають ГК, то перебіг хвороби набуває хронічного хвилеподібного характеру. У деяких випадках можливе спонтанне (через 6–12 міс) одужання.
Артеріїт Такаясу
Патофізіологічною сутністю артеріїту Такаясу (неспецифічний аортоартеріїт, хвороба Такаясу, хвороба відсутності пульсу, гігантоклітинний аортит) є гранулематозне запалення (гранульоми складаються з мононуклеарів і гігантських багатоядерних клітин), яке уражує аорту і її основні гілки, призводить до оклюзії та відсутності пульсу на руках (одній або двох).
Код за МКХ-10
М31.4 Синдром дуги аорти (Такаясу).
Епідеміологія
Захворюваність становить 1,2–3,6 випадку на 100 тис. населення. Переважно хворіють жінки у віці 40–50 років.
Етіологія і патогенез
Етіологія та патогенез невідомі. Під впливом певних чинників в аорті та її основних гілках розвивається проліферативне гранулематозне запалення. У абсолютної більшості хворих (95%) виявляють антитіла до ендотелію судин, сенсибілізацію лімфоцитів до колагену II і III типу, порушення співвідношення субпопуляцій лімфоцитів, гормональної регуляції. До генетичних маркерів артеріїту Такаясу належать антигени HLA-A10, -B5, -Bw52, -DR2. Описано сімейні випадки захворювання, розвиток хвороби у однояйцевих близнюків.
Анатомічні типи артеріїту Такаясу представлено в табл. 21.71.
Таблиця 21.71
Анатомічні типи артеріїту Такаясу
|
Типи |
Характеристика |
|
I |
Характеризується ізольованим ураженням дуги аорти й артерій, які відходять від неї. Відзначається поєднання патології лівої підключичної артерії і загальної сонної |
|
II |
Відзначають ізольоване ураження грудного і/або черевного відділу аорти та їх гілок |
|
III |
Поєднується ураження дуги аорти (її гілок) з іншими відділами — грудним або черевним |
|
IV |
Відзначають ураження легеневої артерії, її гілок. Можливе поєднання з I, II і III типом |
Класифікація
Клінічну класифікацію хвороби представлено в табл. 21.72.
Таблиця 21.72
Клінічна класифікація артеріїту Такаясу (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічний |
|
|
Ступінь активності |
0 (відсутній), I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Аорта і судини |
Звуження низхідного відділу аорти, аневризми грудного і черевного відділів |
|
Серце |
Недостатність аортального клапана, кардіальні синдроми (інфаркт міокарда, серцева недостатність, артеріальна гіпертензія) |
|
|
Суглоби |
Артралгії |
|
|
ЦНС |
Інсульти та інфаркти мозку |
|
|
Орган зору |
Порушення зору |
|
|
ШКТ |
Абдомінальний синдром |
|
|
Судини легень |
Артеріїт легеневої артерії, головних або сегментарних артерій з переходом у хронічний бронхіт і хронічне легеневе серце |
|
Виділяють також стадії (фази) захворювання (табл. 21.73).
Таблиця 21.73
Стадії артеріїту Такаясу
|
Стадії |
Характеристика |
|
I (рання) |
Клінічна картина включає неспецифічні симптоми (лихоманка, слабкість, схуднення, артралгії, міалгії, біль в животі, сонливість), зумовлені запаленням, його системною фазою. Реєструють анемію, підвищену ШОЕ. У 10% пацієнтів скарги відсутні |
|
II (розгорнута) |
З’являються ознаки ішемії окремих органів і тканин внаслідок оклюзії. Уражені судини болісні при пальпації. У 40% хворих відзначають слабкість, втому, м’язовий біль (м’язи передпліччя, плеча), який посилюється при фізичному навантаженні. У 15–20% пацієнтів відсутній або сповільнений пульс на одній руці, відзначають асиметрію систолічного артеріального тиску на плечових артеріях. Виникають (у 7–15% випадків) біль у шиї, запаморочення, порушення зору, підвищення артеріального тиску, серцебиття, задишка. Вислуховується систолічний шум на загальних сонних артеріях (у 70% випадків), при пальпації вони болісні (у 15%). Нерідко реєструють шум і болісність при пальпації черевної аорти |
|
III |
Внаслідок розвитку фіброзу виникають стенози артерій, судинна недостатність. У 50–70% хворих відзначають симптоми переміжної кульгавості верхніх і нижніх кінцівок. Майже у ¼ пацієнтів є ознаки ураження легень: біль в грудній клітці, задишка, кашель, зрідка кровохаркання. У хворих відзначають біль у м’язах проксимальних відділів рук (частіше лівої), зменшення сили кисті. Біль може поширюватися на ліве плече, ділянку шиї, ліву половину грудної клітки, нижню щелепу. У більшості хворих (80–90%) зникає пульс на передпліччі, є різниця систолічного артеріального тиску на плечових артеріях. Критичну ішемію рук діагностують рідко, оскільки стеноз судин прогресує повільно, розвиваються колатералі |
Клінічна картина
Симптоми аортоартеріїту мають загальний і локальний характер (табл. 21.74).
Таблиця 21.74
Загальні і локальні симптоми артеріїту Такаясу
|
Загальні симптоми |
За багато місяців до виникнення симптомів ураження судин з’являються лихоманка, слабкість, нічне підвищене потовиділення, зниження апетиту, зменшення маси тіла, біль в суглобах |
|
Локальні симптоми |
Біль по ходу уражених судин, відсутність пульсу (особливо на підключичній артерії) |
Першим проявом хвороби Такаясу може бути генералізований біль, що нагадує РПМ, пізніше приєднуються ознаки ураження великих артерій (коронарних, верхніх кінцівок, можливе залучення судин легенів і брижі).
Особливості артеріїту Такаясу:
- хвороба зустрічається у жінок у віці до 40 років;
- патологічний процес частіше уражує аорту, її дугу і гілки дуги;
- можливе ураження легеневого стовбура;
- в уражених ділянках судин виявляють потовщення стінок і звуження просвіту;
- нерідко відсутній пульс на променевих артеріях, виявляють офтальмологічну і неврологічну симптоматику;
- при ураженні легеневої артерії і її гілок відзначають легеневу гіпертензію;
- при гістологічному дослідженні патологічно змінених судин реєструють гранулематозну гігантоклітинну реакцію (як при скроневому артеріїті);
- частіше перебіг хвороби може бути повільним, рідше фіксують летальні випадки через 1–2 роки з моменту встановлення діагнозу.
Характерні клінічні прояви артеріїту Такаясу наведено нижче в табл. 21.75.
Таблиця 21.75
Характерні клінічні прояви артеріїту Такаясу
|
Скарги |
Хворі скаржаться на слабкість, сонливість, наявність субфебрильної температури, зменшення маси тіла, біль в м’язах і крупних судинах |
|
Симптоми, пов’язані з порушенням кровотоку в різних органах |
|
|
Шкіра |
Спостерігаються вузликова еритема, гангренозна піодермія, іноді — шкірний васкуліт і феномен Рейно |
|
Очі |
Офтальмологічні розлади спостерігаються у ½ пацієнтів і характеризуються швидкою стомлюваністю очей, зниженням гостроти зору, звуженням полів зору, диплопією. Можлива раптова оклюзія центральної артерії сітківки і втрата зору, атрофія зорового нерва. Ретинопатія у хворих на АГ проходить стадії: 1) венозної дилатації; 2) формування мікроаневризм; 3) артеріовенозних анастомозів; 4) виникнення ускладнень — гіпертензивної ретинопатії, випадіння полів зору, катаракти, втрати очних рефлексів, атрофії зорового нерва та райдужної оболонки |
|
Нервова система |
Неврологічна симптоматика пов’язана з оклюзією сонних або хребетних артерій. Запальний процес в загальних сонних, артеріях хребта супроводжується запамороченням, головним болем, погіршенням пам’яті і уваги, нерідко — непритомністю, гострими порушеннями мозкового кровообігу. При аускультації сонних артерій вислуховується систолічний шум. Спостерігаються інсульти, ортостатичні запаморочення. Зрідка розвивається ішемічна мієлопатія. Внаслідок розриву аневризми виникають субарахноїдальні крововиливи. Ураження ниркових артерій викликає АГ з розвитком гіпертонічної енцефалопатії або внутрішньомозкових крововиливів. Розвивається також периферична поліневропатія |
|
Судини шиї і верхніх кінцівок |
На одній або двох руках виявляється відсутність пульсу на променевій, плечовій, підключичній артеріях, холодні кисті. Відзначаються біль в руках, слабкість, відчуття оніміння, що посилюється при фізичному навантаженні (синдром «переміжної кульгавості»). Біль може локалізуватися в ділянці шиї, лівого плеча і лівої половини грудної клітки. Відзначається різниця рівня артеріального тиску: він на руках вищий, ніж на ногах. Над ураженими судинами вислуховується систолічний шум, може відчуватися біль в крупних судинах, наприклад шиї |
|
Легені |
У 25% хворих розвивається легенева гіпертензія (з’являються ЕКГ-ознаки), відзначаються біль в грудній клітці, задишка. В цілому ураження легеневої артерії відзначається у 70% хворих, але у ¾ з них клінічні прояви відсутні (Насонов Е.Л., 2005). Розвиваються стенози та/або оклюзії лобарних і сегментарних гілок |
|
Висхідний відділ аорти, серце |
Відзначається ущільнення, дилатація висхідного відділу аорти, формування аневризми (зрідка). Можливий розвиток аортальної недостатності, нападів стенокардії, інфаркту міокарда. У половини хворих відзначають міокардит, який призводить до появи задишки, тахікардії, хронічної серцевої недостатності. В активну фазу хвороби міокардит виявляють у ½ хворих. При біопсії в міокарді виявляють інфільтрацію мононуклеарами, гранульоми, некроз міофібрил. Нерідко серце збільшується в розмірах, з’являється гіпертрофія лівого шлуночка, міжшлуночкової перегородки |
|
Черевний відділ аорти |
Ураження низхідного і черевного відділу аорти виявляють в 40–70% випадків. Вислуховується систолічний шум над аортою, знижується кровообіг в нижніх кінцівках, виникає біль при ходьбі. Ішемія органів черевної порожнини виникає рідко. Біль в животі може бути зумовлений запаленням аорти і аневризмою (у 9% випадків), що розшаровується |
|
Ниркові артерії |
Можливий тромбоз, наслідком ішемії є розвиток симптоматичної артеріальної гіпертензії (відзначається у 33–75% хворих). Ураження судин нирок спостерігається у 24–60% пацієнтів (Насонов Е.Л., 2005). У сечі виявляється білок, еритроцити. Ниркова недостатність розвивається рідко. АГ погано коригується антигіпертензивними препаратами. Підвищення інтрагломерулярного тиску призводить до гломерулосклерозу, зменшення кількості функціонуючих нефронів. Артеріальний тиск хворим слід вимірювати на 4 кінцівках |
Виділяють декілька основних синдромів артеріїту Такаясу (табл. 21.76).
Таблиця 21.76
Основні синдроми артеріїту Такаясу
|
Основні синдроми |
Характерні прояви |
|
Ішемічний |
Проявляється недостатністю кровопостачання рук, головного мозку, органа зору, нирок, серця, ніг, розвитком парестезій, занімінням, мерзлякуватістю, підвищеною втомлюваністю. Пульс на променевих, підключичних, сонних, скроневих артеріях поступово зникає. Кисті стають блідими, ціанотичними, з набряком, іноді — з гіперкератозом. Розвивається атрофія мязів кінцівок. Внаслідок ураження сонних артерій з’являються симптоми ішемії головного мозку, офтальмологічні симптоми. Ураження коронарних артерій призводить до появи симптомів ішемічної хвороби серця, інфаркту міокарда, ниркових — до реноваскулярної гіпертонії. Ураження артерій брижі призводить до абдомінальної ішемії, больового синдрому, розладів функції кишечнику, появи симптомів «гострого живота» |
|
Колатерального кровообігу |
Поверхневі вени в ділянці грудної клітки розширюються, з’являються узури на нижніх краях ребер (що видно на рентгенограмах). Ці зміни є наслідком оклюзії магістральних артерій. Вони сприяють збереженню кровотоку верхньої половини тулуба, зменшенню вираженості ішемічного синдрому |
|
Синокаротидний |
Проявляється памороченням голови, непритомністю, судомами, психічними порушеннями. Такі прояви виникають при поворотах голови, зміні положення тіла |
|
Гіпертонічний |
На неураженій руці або ногах підвищується артеріальний тиск, причиною чого є розвиток стенозу ниркових або сонних артерій |
Нижче в табл. 21.77 наведено клінічні прояви артеріїту Такаясу залежно від артерій, які зазнали ураження.
Таблиця 21.77
Клінічні прояви артеріїту Такаясу залежно від уражених артерій
|
Артерії, що уражуються |
Клінічна симптоматика |
|
Дуга аорти |
Недостатність аортального клапана з відповідними симптомами |
|
Черевний відділ аорти |
|
|
Сонні і хребетні артерії |
|
|
Підключичні артерії |
|
|
Коронарні артерії |
|
|
Мезентеріальні артерії |
|
|
Ниркові артерії |
|
|
Клубові артерії |
|
Частоту ураження різних артерій і симптоми при артеріїті Такаясу представлено в табл. 21.78.
Таблиця 21.78
Частота і симптоми ураження артерій при артеріїті Такаясу (Э. Фаучи, 2005)
|
Артерії |
Частота ураження, % |
Симптоми |
|
Підключичні |
93 |
Відсутність пульсу, біль за типом переміжної кульгавості, синдром Рейно |
|
Загальні сонні |
58 |
Транзиторна ішемія мозку, непритомність, порушення зору, інсульт |
|
Черевний відділ аорти |
47 |
Біль в животі, нудота, блювання |
|
Ниркові артерії |
38 |
Артеріальна гіпертензія, ниркова недостатність |
|
Хребетні |
35 |
Запаморочення, порушення зору |
|
Гілки черевного стовбура аорти, верхня брижова артерія |
18 |
Біль в животі, нудота, блювання |
|
Клубові |
17 |
Переміжна кульгавість |
|
Легеневі |
10–40 |
Задишка, атиповий біль в грудях |
|
Коронарні |
<10 |
Стенокардія, інфаркт міокарда |
Варіанти перебігу аортоартеріїту. Можливий блискавичний перебіг, повільне прогресування, стабілізація, спонтанна ремісія.
Шкірні висипання у хворих з артеріїтом Такаясу. Шкірні прояви (кропив’янка, вузлувата еритема, панікуліт) з’являються у разі гострого початку хвороби.
Ураження суглобів при артеріїті Такаясу. При гострому початку хвороби частіше відзначають артралгії та міалгії, але іноді уражаються великі суглоби. В окремих випадках можуть реєструвати ревматоїдоподібний синдром з ерозіями суглобових поверхонь.
Поєднання аортоартеріїту з іншими ревматичними хворобами. Можливе поєднання з АС, РА (синдромом Стілла) та ін.
Діагностика
Відхилення від норми, які виявляють при лабораторному та інструментальному обстеженні хворих на синдром Такаясу, представлено в табл. 21.79.
Таблиця 21.79
Відхилення від норми, які виявляють при лабораторному та інструментальному обстеженні хворих на артеріїт Такаясу
|
Клінічний аналіз крові |
Мають місце нейтрофільний лейкоцитоз, збільшення ШОЕ, анемія |
|
Біохімічні дослідження |
Відзначають зниження в крові рівня альбуміну, холестерину, ліпопротеїнових фракцій, гаптоглобіну, серомукоїду |
|
Імунологічні дослідження |
Можливе підвищення рівня в крові гаммаглобулінів, імуноглобулінів, зрідка — виявлення РФ |
|
УЗД судин, ангіографія |
Має місце зниження кровотоку у відповідних відділах |
|
Реоенцефалографія |
Відзначається зниження кровопостачання мозку |
Діагностичні критерії артеріїту Такаясу наведено в табл. 21.80.
Таблиця 21.80
Діагностичні критерії артеріїту Такаясу (ACR, 1990)
|
1. |
Вік <40 років |
Початок хвороби у віці <40 років |
|
2. |
Переміжна кульгавість кінцівок |
Слабкість і дискомфорт у м’язах кінцівок при рухах |
|
3. |
Ослаблення пульсу на плечовій артерії |
Зниження пульсації на одній або обох плечових артеріях |
|
4. |
Різниця артеріального тиску >10 мм рт. ст. |
При вимірюванні на плечових артеріях різниця систолічного тиску >10 мм рт. ст. |
|
5. |
Шум на підключичних артеріях або черевній аорті |
При аускультації виявляють шум над обома підключичними артеріями або над черевною аортою |
|
6. |
Зміни (фокальні, сегментарні) при ангіографії |
Звуження просвіту або оклюзія аорти, її великих гілок в проксимальних відділах верхніх і нижніх кінцівок, не пов’язані з атеросклерозом, фіброзно-м’язовою дисплазією та ін. |
Діагноз можливо встановити за наявності 3 і більше будь-яких критеріїв (чутливість — 90,5%, специфічність — 97,8%).
Основа діагнозу артеріїту Такаясу. Враховують характерні клінічні прояви, а також результати ангіографії, аортографії, МРТ (табл. 21.81). Біопсію проводять досить рідко. Характерними проявами хвороби, які реєструють при ангіографії, є: деформація контурів судин; стенозування; постстенотичне розширення; аневризми; ділянки оклюзії; посилення колатерального кровообігу.
Таблиця 21.81
Характерні клінічні прояви, результати лабораторного та інструментального обстеження хворих на артеріїт Такаясу
|
Характерні клінічні прояви |
Системні |
Лихоманка, загальне нездужання, швидка стомлюваність, лімфаденопатія |
|
Церебральні |
Головний біль, запаморочення, синкопальні стани, геміплегія та ін. |
|
|
Ішемічні |
Руки: дефіцит пульсу, швидка стомлюваність, втрата чутливості пальців, біль, зниження температури шкіри Ноги: переміжна кульгавість, слабкість, швидка стомлюваність |
|
|
Серцево-судинні |
Серцебиття, ангінальний біль, задишка, відчуття стиснення грудної клітки, аритмії, артеріальна гіпертензія |
|
|
Легеневі |
Задишка, кровохаркання |
|
|
Очні |
Транзиторне або персистуюче порушення зору, його втрата |
|
|
Шкірні |
Вузлувата еритема |
|
|
Больовий синдром |
Біль в шиї, спині |
|
|
Важливі клінічні симптоми |
Слабкий пульс або його відсутність на a. radialis та суттєва різниця між рівнем артеріального тиску на правій та лівій руці |
|
|
Слабкий пульс на стегнових артеріях, зниження артеріального тиску, різниця в рівнях артеріального тиску між верхніми та нижніми кінцівками |
||
|
Систолічні судинні шуми в ділянці шиї, спини, живота |
||
|
Недостатність аортальних клапанів (протодіастолічний шум на аорті та по лівому краю грудини) |
||
|
Підвищений артеріальний тиск у хворих молодого віку |
||
|
Зміни на очному дні, зниження гостроти зору |
||
|
Перфорація носової перегородки, атрофічні зміни на обличчі |
||
|
Лабораторні зміни |
Підвищений рівень ШОЕ, СРБ |
|
|
Анемія, лейкоцитоз |
||
|
Гіпергаммаглобулінемія, підвищений рівень IgG, A і С3, С4-компонентів комплементу |
||
|
Гіперкоагуляція (патологічний фібриноліз), підвищення активності тромбоцитів |
||
|
HLA-B52, -B39 |
||
|
Дані інструментальних методів |
Рентгенографія, КТ, МРТ |
Кальцифікація аорти, потовщення стінок грудного відділу |
|
Ангіографія, КТ, МРТ, ультрасонографія |
Стеноз (оклюзія) артерій: гілок дуги та абдомінальної аорти (атипова коарктація аорти), низхідної. Дифузна дилатація висхідного відділу аорти (з недостатністю аортальних клапанів), брахіоцефальних артерій, низхідного відділу аорти (комбінація з ділянками стенозу) |
|
|
Сцинтиграфія легень, ангіографія, КТ, МРТ |
Стеноз легеневих артерій |
|
|
Коронарографія |
Ураження коронарних артерій |
|
Патоморфологічні зміни в судинах
Макроскопічно аорта та уражені судини ригідні внаслідок розвитку фіброзу. Потовщення стінок судин призводить до звуження їх просвіту або оклюзії. Дилатація висхідного відділу аорти, потовщення аортальних комісур, стулок аортального клапана, їх фіброз є причиною розвитку аортальної регургітації. Просвіт судин перекривають організовані тромби. Коронарні артерії звужуються у ділянці устя, найбільш часто уражується ліва коронарна артерія. Стеноз (з можливою реканалізацією) розвивається в легеневих артеріях. Гранулематозне запалення локалізується в зовнішніх шарах медії і адвентиції. Наслідком постзапального потовщення інтими є розвиток тяжкого вторинного атеросклерозу. У гранульомах виявляються скупчення макрофагів, лімфоцитів і гігантських багатоядерних клітин. Запалення супроводжується фіброзом гранульом, склерозуванням і ушкодженням медії, руйнуванням еластичних мембран, проліферацією ендотелію, звуженням просвіту судин. Наслідком звуження просвіту судин є тромбоутворення. Уражуються vasa vasorum, порушується кровообіг в різних органах. Морфологічні зміни, які виявляють у пацієнтів з артеріїтом Такаясу:
- вогнища некрозу в середній оболонці судин, навколо них розташовуються клітинні інфільтрати, які складаються з макрофагів, гігантських багатоядерних, лімфоїдних клітин, плазмоцитів;
- клітинна інфільтрація поширюється на усі оболонки судинної стінки;
- розростання інтими, тромбози;
- прогресуючий склеротичний процес призводить до оклюзії просвіту артерій.
Описані зміни виражені в місцях відходження артерій від аорти, у тому числі від її черевного відділу. Внутрішньоорганні артерії подібним змінам не піддаються.
Відмінності патологоанатомічних змін у судинах при ГКА і хворобі Такаясу. Морфологічні зміни при цих двох захворюваннях ідентичні, відмінності полягають тільки в локалізації гранулематозного запалення. При ГКА переважно уражується сонна артерія, її гілки, хребетна артерія, при хворобі Такаясу — дуга аорти і судини, які відходять від неї.
Приклад формулювання діагнозу. Неспецифічний аортоартеріїт (артеріїт (хвороба) Такаясу), хронічний перебіг, активність II ступеня. Ураження аорти (недостатність аортального клапана, аневризма грудного відділу аорти, серцева недостатність IIА, функціональний клас III), порушення зору, гострі порушення мозкового кровообігу в анамнезі.
Лікування
Характер терапії, яку проводять при артеріїті Такаясу (за: Насонов Е.Л., 2005; 2011 (ред.)), описано в табл. 21.82.
Таблиця 21.82
Характер терапії, яку проводять при артеріїті Такаясу
|
Ранні стадії хвороби |
Преднізолон усередину 1 мг/кг/добу в 2–3 прийоми, 1 міс. Потім дозу поступово знижують до підтримувальної, яка не має перевищувати 10 мг/добу |
Лікування триває не менше 2–5 років. Досягти ремісії вдається у ⅔ хворих, проте у 50% пацієнтів після відміни ГК настає загострення. Монотерапія ГК не попереджує ураження клапанного апарату серця |
|
Загострення після відміни ГК |
Циклофосфамід в/в 500–700 мг/м2 1 раз на місяць протягом 9–12 міс + метилпреднізолон в/в 10 мг/кг 1 раз на місяць, 9–12 міс |
|
|
Відсутність стабілізації процесу |
Метотрексат усередину в початковій дозі 15 мг/тиж (максимальна — 25 мг/тиж) + преднізолон усередину 5–10 мг 1 раз на добу уранці після їди |
Тривалість терапії залежить від клінічного ефекту |
|
Наявність стенозу коронарних артерій |
Черезшкірна транслюмінальна ангіопластика, аортокоронарне шунтування |
|
|
Розшаровуюча аневризма аорти, стеноз/оклюзія сонних і ниркових артерій |
Хірургічне лікування |
|
Слід враховувати, що медикаментозна терапія ефективна на стадії гранулематозного судинного запалення (до розвитку фіброзної гіперплазії внутрішньої оболонки судин). ГК призначають хворим з активним перебігом хвороби (лихоманка, зменшення маси тіла), ознаками системного запального процесу (епісклерит, ірит, плеврит, перикардит, артрит, поліартрит), підвищеними гострофазовими показниками. При рефрактерності до преднізолону застосовують метотрексат, циклофосфамід, азатіоприн. Використання метотрексату (17,5 мг/тиж) в поєднанні з преднізолоном (у невисоких дозах) дозволяє досягти ремісії у 81% хворих (Насонов Е.Л., 2010 (ред.)) і довго її підтримувати. Тривала підтримувальна терапія (5–10 мг/добу) попереджує судинні ускладнення, збільшує тривалість життя хворих. Щомісячно також проводять пульс-терапію метилпреднізолоном та циклофосфамідом. Загальна тривалість лікування має становити не менше 6–9 міс.
За наявності суглобового синдрому призначають НПЗП — диклофенак натрію (100–150 мг/добу) або мелоксикам (7,5–15 мг/добу), або німесулід (100 мг 2 рази на добу), або целекоксиб (200 мг 1–2 рази на добу). При швидкому прогресуванні ішемічних розладів, небезпеці розвитку тромбозу призначають гепарин по 5000–10 000 ОД підшкірно кожні 6 год (підшкірне введення нерідко доповнюють в/в інфузією 10 000 ОД) протягом 4–8 тиж. Можливе використання низькомолекулярних гепаринів — еноксипарину (20 мг підшкірно 1 раз на добу), надропарину (0,3 мл підшкірно 1 раз на добу). Призначення антикоагулянтів поєднують із застосуванням дипіридамолу (225–400 мг/добу). За наявності тромбозу дипіридамол спочатку вводять в/в краплинно (2–4 мл 0,5% розчину), а надалі переводять хворих на пероральний прийом препарату протягом декількох місяців. Антикоагулянтну терапію проводять під клініко-інструментальним контролем стану кровообігу в зонах, в яких виявлена оклюзія. При артеріальній гіпертензії призначають гіпотензивні препарати. Необхідно проводити моніторинг ліпідного спектра крові, враховуючи, що атеросклеротичний процес може погіршувати стан судинної системи.
Хірургічні операції на аорті та магістральних артеріях бажано проводити в перші 5 років хвороби. До показань належать звуження просвіту артерій на 70% та більше у поєднанні з ішемічним синдромом. Операції проводять в неактивну стадію хвороби.
До критеріїв ефективності лікування відносять: позитивну динаміку клінічних симптомів; нормалізацію/позитивну динаміку загального аналізу крові, показників білків крові; нормалізацію температури тіла; позитивну динаміку при інструментальних дослідженнях — реовазографії, допплерографії, МРТ голови. Під впливом протизапальної терапії у пацієнтів з хворобою Такаясу зникають ознаки ішемії міокарда на ЕКГ.
Прогноз
Прогноз різко покращується при поєднанні преднізолону (40–60 мг/добу) з балонною ангіопластикою, хірургічним лікуванням стенозів. Операції проводять у період ремісії, досягнутої прийомом преднізолону. При неефективності преднізолону призначають метотрексат (до 25 мг/тиж), але порівняльні дослідження його ефективності не проводилися. До смерті призводять в основному порушення мозкового кровообігу і серцева недостатність. Летальність варіює від 10 до 70%.
У цілому прогноз визначається локалізацією уражених судин, тяжкістю ураження, характером терапії. У 20% хворих відзначають спонтанне одужання, у решти під впливом ГК-терапії наступає ремісія з подальшими загостреннями, прогресуванням хвороби. До несприятливих прогностичних факторів належать: ураження коронарних і ниркових артерій, розвиток коарктації аорти, аневризми, недостатності аортальних клапанів, ретинопатії.
Артеріїт Кавасакі
Хвороба Кавасакі є системним васкулітом (артеріїт), для якого характерне ураження великих, середніх та дрібних артерій і поєднання зі шкірно-слизовим лімфатичним синдромом. Він розвивається в основному у дітей в ранньому віці.
Код за МКХ-10
М30.3 Синдром Кавасакі.
Епідеміологія
Хвороба Кавасакі найбільш поширена серед представників монголоїдної раси. Розвивається у дітей віком до 5 років, пік хвороби припадає на перші 2 роки життя. Співвідношення хлопчиків та дівчаток становить 1,4:1,0. У Японії захворюваність дорівнює 67 випадкам на 100 тис. дітей, в США — 24,4 на 100 тис.
Етіологія і патогенез
Про інфекційну етіологію хвороби Кавасакі свідчать: 1) початок хвороби після інфекції верхніх дихальних шляхів; 2) епідемічні спалахи хвороби, циклічність; 3) повторні випадки хвороби в сім’ях хворих. Під впливом етіологічного чинника, до якого ймовірно відносять віруси Епштейна — Барр, герпесу, ретровіруси, рикетсії, виробляються антитіла до ендотелію перерахованих вище судин, розвивається імунокомплексне запалення. При цьому антитіла взаємодіють з антигеном Кавасакі, що з’являється на мембрані ендотелію. Імуногенетичним маркером хвороби Кавасакі є антиген HLA-Bw22.
Класифікація
Клінічну класифікацію хвороби Кавасакі наведено в табл. 21.83.
Таблиця 21.83
Клінічна класифікація хвороби Кавасакі (АРУ, 2004)
|
Перебіг |
Гострий, хронічний |
|
|
Ступінь активності |
0 (відсутній), I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Шкіра і слизові оболонки |
Двобічний кон’юнктивіт, увеїт, стоматит, глосит, псевдотонзиліт (збільшення і гіперемія мигдаликів), поліморфні висипання на шкірі (еритематозні бляшки, макулопапульозна еритема) |
|
Лімфатична система |
Лімфаденопатія |
|
|
Серцево-судинна система |
Міокардит з порушенням ритму, провідності й кардіомегалією, вальвуліт, дисфункція папілярних м’язів, перикардит |
|
|
Суглоби |
Артрит, поліартралгії |
|
|
ШКТ |
Абдомінальний синдром |
|
|
Сечостатева система |
Уретрит |
|
|
ЦНС |
Асептичний менінгіт |
|
Клінічна картина
Характерним початковим синдромом є лихоманка, яка у разі відсутності лікування зберігається до 2 тиж. Виділяють гострий, підгострий і період одужання. Тривалість гострого періоду становить 1–1,5 тиж, підгострого — 2–4 тиж, період одужання може тривати декілька років. У 3% випадків спостерігаються рецидиви захворювання. Клінічні особливості хвороби Кавасакі:
- хвороба виникає переважно (80% випадків) у дітей у віці молодше 4 років;
- уражуються артерії великого, середнього і малого калібру;
- характерними є виразки слизових оболонок, шкірні висипання, збільшення лімфатичних вузлів;
- у 20% випадків розвиваються ускладнення з боку серцево-судинної системи: залучаються коронарні артерії, утворюються аневризми;
- морфологічно визначається панартеріїт: запалення охоплює усі шари стінки судини;
- зазвичай захворювання проходить без лікування;
- летальний результат спостерігається у 1% хворих.
Симптоматику хвороби представлено в табл. 21.84.
Таблиця 21.84
Симптоматика хвороби Кавасакі
|
Уражені органи і системи |
Характер проявів |
|
Загальні симптоми |
Блідість шкірних покривів, занепокоєння |
|
Очі |
У перші тижні розвивається двобічний кон’юнктивіт, який може поєднуватися з увеїтом |
|
Органи слуху |
Може відзначатися зниження слуху |
|
ЦНС |
Іноді розвивається асептичний менінгіт. Відзначають збудливість високого ступеня |
|
Шкіра |
Характерна поява поліморфного макулопапульозного, уртикарного висипання в перші 5 тиж з моменту розвитку лихоманки. Вона локалізується на тулубі, проксимальних відділах кінцівок, в ділянці промежини. Потім приєднується еритема, болісне ущільнення шкіри в ділянці підошв і долонь. Шкіра на цих ділянках через 2–3 тиж починає лущитися |
|
Слизові оболонки |
Відзначають появу тріщин і кровоточивості на губах, почервоніння і сухість слизової оболонки рота. Язик набуває червоно-малинового кольору, мигдалини збільшуються в розмірах і стають гіперемованими |
|
Лімфатичні вузли |
Збільшуються шийні (з одного або двох боків) лімфовузли. Вони сягають 1,5 см і більше в діаметрі |
|
Легені |
За рентгенологічними даними ураження легень спостерігається у 15% дітей і виникає в перші 10 днів хвороби. Має місце посилення легеневого малюнка, дрібні вузлики, перибронхіальні потовщення, ателектази, ексудативний плеврит |
|
Серцево-судинна система |
Розвивається міокардит, який супроводжується збільшенням розмірів серця, аритміями, тахікардією, змінами сегменту ST і зубця T на ЕКГ, ритмом галопу з вислуховуванням III тону, клапанною регургітацією, розвитком гострої серцевої недостатності. Відзначають вальвуліт, дисфункцію папілярних м’язів, недостатність аортального і/або мітрального клапанів. Нерідко вислуховується шум тертя перикарда. У проксимальних відділах коронарних артерій у 15–25% пацієнтів з’являються аневризми, може розвиватися інфаркт міокарда, мітральна або аортальна недостатність. Аневризми можуть формуватися в підключичних, ліктьових, ниркових, стегнових і клубових артеріях |
|
Суглоби |
Хворі (близько 30%) скаржаться на біль в суглобах. Суглобовий синдром, що розвивається, з ураженням дрібних суглобів кистей та стоп, гомілковостопних і колінних суглобів, триває 3–4 тиж |
|
ШКТ |
У деяких хворих реєструють біль в животі, блювання, діарею |
|
Сечостатева система |
Зрідка розвивається уретрит |
Хвороба розпочинається гостро, з високої температури тіла (до 38–40 °С) переміжного характеру, тривалість лихоманки становить від 1 до 4 тиж та залежить від лікування: ацетилсаліцилова кислота, імуноглобулін для в/в введення зменшують її вираженість та тривалість. Тривала лихоманка сприяє розвитку аневризм коронарних артерій, підвищує ризик смерті. У 90% хворих виникає кон’юнктивіт з переважним ураженням бульбарної кон’юнктиви. Запальні зміни тривають 1–2 тиж. У 70% хворих в перші 5 днів виникають поліморфні висипання на шкірі тулуба, рук, ніг, в ділянці промежини. Виникає набряк, почервоніння шкіри долонь, підошв, які супроводжуються обмеженням рухів пальців кистей та стоп. У 50–70% хворих одночасно з лихоманкою розвивається лімфаденопатія. Інколи вона може передувати лихоманці. У 15–25% хворих в кінці 1-го тижня виникає стенокардія або інфаркт міокарда. Можлива поява покресленості нігтів, асептичних пустул на ліктях, над колінними суглобами, в ділянці сідниць.
Ускладнення гострої фази хвороби Кавасакі:
- гострий передній увеїт;
- артрит з ураженням дрібних суглобів кистей і стоп;
- міокардит, перикардит;
- артеріїт, який супроводжується розвитком аневризм (у 20% пацієнтів);
- нудота, діарея, біль у животі, жовтяниця (внаслідок водянки жовчного міхура).
У пацієнтів з хворобою Кавасакі є прояви, схожі на симптоми скарлатини: 1) шкірні висипання; 2) ураження слизових оболонок; 3) кон’юнктивіт; 4) лихоманка. У таких випадках доцільне проведення дослідження на стрептококову інфекцію. Для шкірного висипання, яке відзначають при хворобі Кавасакі, характерні деякі особливості:
- виникає у ⅔ хворих у перші 3 дні від початку лихоманки;
- проявляється уртикарним висипанням з великими еритематозними бляшками, макулопапульозними елементами;
- може нагадувати багатоформну еритему;
- локалізується на тулубі, в ділянці промежини, на проксимальних відділах кінцівок.
Окрім висипання через декілька днів від початку хвороби розвиваються еритема та індурація шкіри долонь і підошв, яка супроводжується різким болем і обмеженням рухливості пальців. Через 2–3 тиж в місцях локалізації еритеми з’являється лущення епідермісу.
Частота ураження коронарних артерій при хворобі Кавасакі. Ураження коронарних артерій, що призводить до розвитку аневризми, інфаркту міокарда, раптової смерті, спостерігається у 25% хворих. Аневризми (розширення коронарних артерій до 8 мм і більше в діаметрі) супроводжуються розвитком тромбів, що вимагає тривалої антикоагулянтної терапії. Вони піддаються зворотному розвитку: в перші 2 тиж аневризми відзначають у 20% хворих, через 1 рік — у 2%.
Діагностика
Зміни, які реєструють при лабораторному та інструментальному обстеженні пацієнтів, описано в табл. 21.85.
Таблиця 21.85
Зміни, які виявляють при лабораторному та інструментальному обстеженні пацієнтів з хворобою Кавасакі
|
Клінічний аналіз крові |
Нейтрофільний лейкоцитоз, зрушення лейкоцитарної формули вліво, тромбоцитоз (до 106/мм3 і більше), збільшення ШОЕ, анемія |
|
Біохімічні дослідження |
Підвищення в крові рівня СРБ, α-глобулінів, серомукоїду, активності АлАТ, АсАТ |
|
Імунологічні дослідження |
Збільшується зміст в крові імуноглобулінів, ЦІК. Відсутній антистрептолізин-О |
|
Загальний аналіз сечі |
Відзначають лейкоцитурію (переважають мононуклеарні клітини), протеїнурію |
|
Аналіз синовіальної рідини |
Відмічають цитоз за рахунок мононуклеарів |
|
ЕКГ |
АВ-блокади, аритмії, подовження інтервалу P–Q, розширення комплексу QRS, депресія ST, зниження вольтажу |
|
ехоКГ |
Збільшення розмірів серця, зниження фракції викиду лівого шлуночка |
|
Коронароангіографія |
Виявляються аневризматичні розширення коронарних артерій, їх звуження (обструкція). Ці зміни локалізуються в проксимальних ділянках судин |
|
Ренгтгенографія органів грудної порожнини |
Сітчастий характер легеневого рисунка (в 95% випадків) з дрібними вузликами, перибронхіальні потовщення, ателектази, ексудативний плеврит |
Діагностичні критерії хвороби Кавасакі наведено в табл. 21.86, 21.87.
Таблиця 21.86
Діагностичні критерії хвороби Кавасакі (за: Kawasaki T. et al., 1976)
|
1. Лихоманка |
Триває протягом 1–2 тиж і не піддається лікуванню антибіотиками |
|
2. Двобічний кон’юнктивіт |
|
|
3. Зміни на губах і в порожнині рота |
Еритема або тріщини губ Малиновий язик Дифузна еритема слизової оболонки порожнини рота і глотки |
|
4. Зміни в ділянці кінцівок |
Еритема долонь і/або підошв (початкова стадія) Індуративний набряк кистей і/або стоп (3–5-й день) Десквамація кінчиків пальців (2–3-й тиждень) |
|
5. Поліморфний висип на шкірі тулуба (без пухирів і кірочок) |
|
|
6. Лімфаденопатія шийних лімфатичних вузлів |
Гостра негнійна лімфаденопатія шийних лімфатичних вузлів (розмір одного лімфатичного вузла не менше 1,5 см) |
Таблиця 21.87
Діагностичні критерії хвороби Кавасакі (за: Hollister R.,1999)
|
1. |
Підвищена температура тіла, яка триває мінімум 5 днів, резистентна до лікування антибіотиками |
|
2. |
Двобічний кон’юнктивіт, часто тяжкого ступеня |
|
3. |
Почервоніння, сухість і набряк губ, утворення тріщин на губах і в кутах рота, малиновий язик, гіперемія мигдаликів, запалення слизової оболонки верхніх дихальних шляхів |
|
4. |
Еритема долонь і стоп з наростанням набряку і наступним лущенням шкіри |
|
5. |
Висипання поліморфного характеру (не везикулярне), розташоване переважно на тулубі та кінцівках |
|
6. |
Збільшення шийних лімфатичних вузлів |
Перераховані ознаки не мають бути пов’язані з іншою патологією. Достовірним діагноз вважається за наявності 5 або 4 з 6 критеріїв.
Діагностуванню хвороби можуть сприяти також наступні критерії: кардит (особливо міокардит, перикардит), діарея, артралгія/артрит, асептичний менінгіт, жовтяниця легкого ступеня, наявність лейкоцитозу, анемії, підвищення показників ШОЕ, СРБ, α2-глобуліну, відсутність антистрептолізину-О, незначне підвищення рівня сироваткових трансаміназ, протеїнурія та збільшення вмісту лейкоцитів у сечі. Вважають, що виявлені прижиттєво на коронарограмах або посмертно аневризми коронарних артерій є наслідком перенесеної в дитинстві хвороби Кавасакі.
Приклад формулювання діагнозу: хвороба Кавасакі, гострий перебіг, активність III ступеня. Ураження слизової оболонки очей (двобічний кон’юнктивіт, увеїт), губ і порожнини рота (хейліт, стоматит, глосит), мигдаликів (збільшення і гіперемія), шкіри (поліморфний висип на тулубі), лімфатичних вузлів (шийний лімфаденіт), серця (міокардит, АВ-блокада I ступеня, серцева недостатність IIA, функціональний клас III), суглобів (поліартралгії).
Діагноз хвороби Кавасакі встановлюють за наявності першого критерію і не менше 4 інших.
Лікування
Для проведення антикоагулянтної терапії в гостру фазу дітям призначають ацетилсаліцилову кислоту в дозі 100 мг/кг/добу в 3 прийоми, в період реконвалесценції — 10 мг/кг/добу. Лікування проводять до повного зворотного розвитку аневризм. Підтримувальна доза ацетилсаліцилової кислоти може становити 3–5 мг/кг/добу в 1 прийом і застосовуватися протягом 2–3 міс. Використовують також прямі і непрямі антикоагулянти.
Препаратом вибору є імуноглобулін для в/в введення, який застосовують в перші 10 днів хвороби. Уже через 12 год після його введення знижується температура тіла, настає регресія висипань, покращується загальний стан. Препарат не лише купірує гострі прояви хвороби, але і запобігає утворенню аневризм коронарних артерій. Для отримання ефекту досить одноразового введення препарату в дозі 2 г/кг/добу. За необхідності лікування повторюють через 5–7 днів. Можливі схеми застосування імуноглобуліну:
- по 0,4 г/кг/добу протягом 5 днів;
- 2 г/кг/добу одноразово з повторним введенням за необхідності через 5–7 днів;
- поєднання ацетилсаліцилової кислоти і в/в введення імуноглобуліну.
До побічних ефектів терапії імуноглобуліном належать головний і м’язовий біль, лихоманка. Вони мінімально виражені при повільному введенні препарату. Причиною розвитку побічних ефектів є присутність у препараті антитіл (проти різних токсинів, антистафілококових, антистрептококових та ін.). З метою запобігання тромбозу коронарних артерій додатково призначають ацетилсаліцилову кислоту з розрахунку 3–5 мг/кг/добу. При резистентності до приведеної вище терапії додають ГК.
При суглобовому синдромі призначають НПЗП: неселективні — диклофенак (100–150 мг/добу), селективні — целекоксиб (200 мг 1–2 рази на добу), мелоксикам (7,5–15 мг/добу), німесулід (100 мг 2 рази на добу), еторикоксиб 90 мг 1 раз на добу.
Прогноз
У більшості випадків він сприятливий. Смертність в перші 15–45 днів від початку хвороби становить 0,03–3%. Причиною смерті є міокардит, порушення ритму, розриви аневризм, тромбоз коронарних артерій, інфаркт міокарда, гостра серцево-судинна недостатність. Чинниками несприятливого прогнозу є:
- початок хвороби у віці старше 5 років;
- наявність лихоманки, яка триває більше 2 тиж;
- тромбоцитоз, лейкоцитоз, різке збільшення ШОЕ;
- підвищення концентрації в крові СРБ протягом 30 та більше днів;
- зниження гемоглобіну <100 г/л.
Облітеруючий тромбангіїт (хвороба Вінівартера — Бюргера)
Код за МКХ-10
I73.1 Облітеруючий тромбангіїт (хвороба Бюргера).
Облітеруючий тромбангіїт (ОТА) — хвороба, яка проявляється запаленням артерій середнього і дрібного калібру, вен і нервових стовбурів кінцівок. ОТА включено в групу васкулітів через супутню помірно виражену запальну реакцію стінки судин. Спочатку уражаються дистальні відділи судин кінцівок, потім у міру прогресування хвороби в патологічний процес залучаються і проксимальні. У гостру фазу хвороби в просвіті судин (уражаються переважно дрібні й середні артерії) утворюються гострозапальні тромби. Ці прояви виражені меншою мірою, ніж при інших типах васкулітів.
Епідеміологія
В основному хворіють чоловіки, які курять, середній вік яких становить 30 років. Жінки хворіють рідко і становлять менше 2% усіх хворих. Останніми роками збільшилася кількість випадків захворювання жінок, що також, очевидно, пов’язано з курінням. Даних про поширеність ОТА в загальній популяції немає. Серед осіб, які звертаються до медичних закладів, цю хворобу виявляють у 10 зі 100 тис. населення.
Етіологія і патогенез
Етіологія хвороби невідома, відзначається тісна асоціація з курінням і передбачається наявність генетичної схильності. У пацієнтів з цією патологією з підвищеною частотою виявляють антигени гістосумісності HLA-A9 та HLA-B5. До характерних особливостей, властивих ОТА, належать:
- виникнення хвороби у чоловіків молодого віку (20–35 лет), які курять;
- ураження судин (артерій середнього і дрібного калібру) нижніх кінцівок;
- виявлення при морфологічному дослідженні нейтрофільної і моноцитарної інфільтрації стінок судин, тромбів, проліферації ендотелію і фібробластів судинної стінки;
- поширення запалення зі стінок артерій на прилеглі вени та нервові стовбури;
- розвиток гангрени кінцівок, що нерідко вимагає ампутації;
- у разі відмови від куріння можливе припинення розвитку хвороби.
Клінічна картина
Клінічні прояви хвороби Бюргера наведено в табл. 21.88.
Таблиця 21.88
Клінічні прояви хвороби Бюргера
|
Серцево-судинна система |
Хворі скаржаться на біль в ногах, який посилюється вночі й у спокої, відчуття печіння, оніміння пальців стоп, мерзлякуватість (хворі не переносять холоду). У спокої біль відзначають у 33% пацієнтів, періодично вони з’являються в підйомі стопи. Виявляють стійкий ціаноз дистальних фаланг пальців стопи, набряки пальців, виразки і некрози (нерідко розвивається гангрена). Переміжну кульгавість відзначають не завжди (оскільки уражаються проксимальні відділи артерій), пульсація артерій зберігається. У 50–80% пацієнтів уражаються артерії верхніх кінцівок, у 10% — діагностують феномен Рейно. Наслідком ураження коронарних артерій може бути розвиток стенокардії та інфаркту міокарда. Розвивається мігруючий, рецидивуючий флебіт |
|
ШКТ |
Зрідка уражаються мезентеріальні артерії. У таких випадках відзначають біль у животі, картину «гострого живота», розвиваються виразки, некрози кишечнику і шлунково-кишкові кровотечі. Можливий розвиток кишкової непрохідності, абсцесів |
|
Нирки |
При тромбозі ниркової артерії розвивається інфаркт нирки, який проявляється болем, гіпертермією, гематурією, підвищенням артеріального тиску |
|
ЦНС |
Виникають порушення мозкового кровообігу (у тому числі повторні), ішемічні неврити зорових нервів, тромбоз судин сітківки, крововиливи на очному дні |
До перших проявів хвороби відносять симптом переміжної кульгавості, який є наслідком ішемії нижніх кінцівок. Можливе аналогічне ураження рук, двох і більше кінцівок. У 30–50% пацієнтів розвивається поверхневий тромбофлебіт, у 10% — феномен Рейно. Ураження починається з дистальних відділів кінцівок.
У тріаду, характерну для хвороби Бюргера, входять:
- оклюзія дистальних артерій;
- синдром Рейно;
- мігруючий тромбофлебіт поверхневих вен.
Розвитку гангрени та виразок сприяє тісне взуття, травма, але можливий і спонтанний процес.
Діагностика
Характерні зміни, які виявляють при лабораторно-біохімічному й інструментальному обстеженні пацієнтів з хворобою Бюргера, наведені в табл. 21.89.
Таблиця 21.89
Зміни, які виявляють при лабораторно-біохімічному та інструментальному обстеженні пацієнтів з хворобою Бюргера
|
Загальний аналіз крові |
Нейтрофільний лейкоцитоз, збільшення ШОЕ |
|
Біохімічні дослідження |
У крові збільшується вміст серомукоїду, сіалових кислот, гаптоглобіну, фібриногену, γ-глобулінів |
|
Коагулограма |
Виявляють підвищення агрегації тромбоцитів та здатності до згортання крові |
|
Імунологічні дослідження |
Виявляють антигени HLA-A9, -B5, -DR4 |
|
Радіоізотопне дослідження |
Відзначають порушення мікроциркуляції в ділянці нижніх кінцівок |
До інструментальних методів, які дозволяють діагностувати ОТА, належить ангіографія (виявляє нерівномірність просвіту артерій). Але зміни, які реєструють при ангіографії, не є специфічними для ОТА. Їх треба зіставляти з клінічними проявами хвороби.
На артеріограмах виявляють наступні ангіографічні ознаки хвороби:
- ураження дрібних та середніх артерій пальців кистей, стоп, долонних, підошовних, мало- та великогомілкових, ліктьових, променевих артерій;
- оклюзію або двобічні ділянки звуження просвіту артерій;
- відсутність патологічних змін у проксимальних судинах і ділянках судинного русла, розташованих між стенозованими;
- розвиток мереж колатералей різної форми навколо ділянок оклюзії;
- наявність незмінених ділянок в уражених фрагментах;
- відсутність ознак атеросклерозу й емболії.
Нерівномірне ущільнення, стовщення судинної стінки, звуження, оклюзію дистальних артерій кінцівок довзоляє виявити ультразвукова допплерографія.
Характерні зміни, які реєструють у біоптатах, залежать від періоду перебігу хвороби (табл. 21.90).
Таблиця 21.90
Зміни, які реєструють у біоптатах залежно від періоду перебігу хвороби
|
Гострий період |
Відзначають панваскуліт, виражену клітинну інфільтрацію, тромби. У гострий період не рекомендується брати біоптати, оскільки на місці їх узяття утворюються виразки |
|
Підгострий період |
Зменшується кількість клітин, відбувається процес реканалізації тромбу, розвиток периваскулярного фіброзу |
|
Пізня стадія |
Виявляють організований і реканалізований тромб, периваскулярний фіброз |
У біоптатах часто виявляють наявність венуліту (на відміну від інших васкулітів судин середнього калібру).
Клінічну класифікацію та діагностичні критерії ОТА наведено нижче (табл. 21.91, 21.92).
Таблиця 21.91
Клінічна класифікація ОТА (АРУ, 2004)
|
Стадії |
Початкова, розгорнута, термінальна (пізня) |
|
|
Ступінь активності |
0 (відсутній), I (мінімальний), II (помірний), III (високий) |
|
|
Клінічні варіанти |
Периферичний, вісцеральний, змішаний |
|
|
Клініко-морфологічна характеристика уражень |
Середні, дрібні артерії та вени |
Васкуліти переважно нижніх кінцівок |
|
Дрібні судини |
Синдром Рейно |
|
|
Шкіра |
Виразково-некротичний процес, гангрена |
|
|
Вени |
Мігруючий рецидивуючий флебіт |
|
|
Судини серця |
Стенокардія, інфаркт міокарда |
|
|
ШКТ |
Абдомінальний синдром, кишкова непрохідність, іноді шлунково-кишкова непрохідність |
|
|
ЦНС |
Повторні порушення мозкового кровообігу, ішемічний неврит зорового нерва |
|
|
Судини сітківки |
Тромбоз, крововиливи |
|
Таблиця 21.92
Діагностичні критерії ОТА (за: Moses M. et al., 1970)
|
Великий критерій |
Ішемія нижніх кінцівок у курців, у яких немає гіперліпідемії та цукрового діабету, системних захворювань сполучної тканини, гемопатій та ембологенної патології в анамнезі |
|
Малі критерії |
1. Рецидивуючий мігруючий тромбофлебіт |
|
2. Синдром Рейно |
|
|
3. Ішемія верхніх кінцівок |
Наявність у хворого 1 великого критерію і 2 малих дозволяє встановити діагноз хвороби Бюргера. Критерії не підтверджені. ОТА диференціюють з ВП, хворобою Такаясу, СЧВ, РА, ССД, АФС, васкулітами дрібних артерій.
Приклад формулювання діагнозу. ОТА (хвороба Вінівартера — Бюргера), розгорнута стадія, активність I ступеня. Ураження судин нижніх кінцівок, синдром Рейно, виразково-некротичний процес у ділянці правої гомілки, початкова стадія гангрени I пальця лівої стопи.
Лікування
Схеми лікування при ОТА наведено нижче (табл. 21.93).
Таблиця 21.93
Лікування при ОТА
|
Переважно периферична форма хвороби |
Антикоагулянти |
Прямі: гепарин (5000 ОД підшкірно 4 рази на добу), еноксапарин (20 мг підшкірно 1 раз на добу), надропарин (0,3 мл підшкірно 1 раз на добу). Непрямі: варфарин 3 мг/добу |
|
Антиагреганти |
Реополіглюкін (400 мл в/в краплинно через день у 10 прийомів), пентоксифілін (200–300 мг/добу в/в або 200–600 мг/добу усередину), дипіридамол (150–200 мг/добу усередину), клопідогрель (75 мг 1 раз на добу) |
|
|
НПЗП |
Неселективні (диклофенак натрію 100–150 мг/добу), селективні — целекоксиб (200 мг 1–2 рази на добу), мелоксикам (7,5–15 мг/добу), німесулід (100 мг 2 рази на добу) |
|
|
Швидкопрогресуюча вісцеральна патологія |
Преднізолон (метилпреднізолон) |
Доза становить 15–30 мг/добу. Після досягнення ремісії дозу ГК поступово знижують. У цілому тривалість терапії преднізолоном (метилпреднізолоном) становить декілька місяців. У вкрай тяжких випадках проводять комбіновану 3-денну пульс-терапію (метилпреднізолон + циклофосфамід) |
|
Імуносупресори |
При неефективності преднізолону призначають азатіоприн (початкова доза — 100 мг/добу, підтримувальна — 50–75 мг/добу) протягом 6–8 міс |
Використовують блокатори кальцієвих каналів, переважно дигідропіридинового ряду, ксантинолу нікотинат, ацетилсаліцилову кислоту (100 мг/добу). Також призначають інфузії алпростадилу — 1 ампулу (25 мкг) в 250 мл фізіологічного розчину в/в краплинно протягом 3 год щодня або через день у 5–15 прийомів (позитивний ефект лікування зберігається недовго). Пацієнтові слід припинити куріння.
Про ефективність лікування свідчать: позитивна динаміка клінічних симптомів, нормалізація/позитивна динаміка лабораторних показників (кількості лейкоцитів, ШОЕ, протеїнограми), позитивні зміни показників реовазографії і допплерографії.
Спроби шунтування стенозованих ділянок дають незадовільні результати. Ефективність симпатектомії становить 45–64% (вона не впливає на перебіг патологічного процесу та не призводить до стійкого симптоматичного поліпшення). У разі розвитку гангрени проводять ампутацію кінцівки.
Прогноз
Прогноз сприятливий для життя і значною мірою залежить від продовження куріння. Смертність хворих — на рівні популяційної, її причиною може бути тромбоемболія легенової артерії, інфаркт міокарда, ураження кишечнику, багатосудинне ураження декількох кінцівок.
Есенціальний кріоглобулінемічний васкуліт
Есенціальний кріоглобулінемічний васкуліт (ЕКВ), або змішана кріоглобулінемічна пурпура, — васкуліт з кріоглобулінемічними імунними депозитами, при якому ушкоджуються дрібні судини (артеріоли, капіляри, венули) переважно шкіри, клубочків нирок, який поєднується з кріоглобулінемією.
Код за МКХ-10
D89.1 Есенціальна кріоглобулінемія.
Вперше хворобу описано J. LoSpolluto та співавторами в 1962 р., в окрему нозологічну форму виділено M. Meltzel та співавторами в 1964 р.
Етіологія, патогенез
Кріоглобулінемія (КГ) — це наявність в сироватці крові імуноглобулінів (Ig) або імуноглобулінвмісних комплексів (кріоглобулінів), які є групою термолабільних білків або імунних комплексів, які мають здатність при низькій температурі (нижче 37 °С) спонтанно преципітувати або перетворюватися на гель, а при підвищенні температури знову трансформуватися в розчинний стан. Кріоглобуліни, що утворюються при окремих запальних хворобах, не завжди патогенні. У здорових людей кріоглобуліни визначаються в невеликій кількості, у осіб у віці старше 60 років. Спостерігаються випадки сімейної КГ.
J.C. Brouet, та співавтори, які проводили дослідження у великій групі хворих з КГ, залежно від виду кріоглобулінів, виокремили 3 основних її типи:
- I тип КГ — єдиний гомогенний, містить моноклональні IgМ, IgG або рідше IgА. Для цього типу характерним є, як правило, високий (5–30 мг/мл) вміст кріоглобулінів у сироватці крові та преципітат, який швидко утворюється на холоді. Серед осіб з КГ I тип відзначають у 10–25%, він частіше асоціюється з лімфопроліферативними захворюваннями;
- II тип — змішана КГ, складається з IgM і IgG, які мають антиглобулінову активність проти Fc-рецептора IgG (що має властивості РФ). У більшості випадків кріопротеїни складаються з моноклональних IgM і поліклональних комплексів — типу IgM-IgG, хоча можуть виявлятися також комплекси IgG-IgG і рідко IgA-IgG. Вміст кріоглобулінів у сироватці крові при II типі КГ у 40% випадків становить 1–5 мг/мл та ще у 40% — >5 мг/мл. II тип виявляють у 25% осіб з КГ. Він частіше асоційований з гепатитом С (90–96%);
- III тип — змішана КГ — містить поліклональні Ig одного або більше класів, інколи наявна домішка молекул, які не належать до Ig, наприклад білків комплементу, фібронектину або ліпопротеїнів. Велика їх частина — також кріоглобуліни типу імуноглобулін-антиімуноглобулін. Кріоглобуліни III типу виявити складніше, оскільки їх вміст у сироватці крові досить низький (0,1–1 мг/мл), тому їх преципітація протікає повільно. Спостерігаються такі зміни в 50% осіб з КГ. Развивається при аутоімунних захворюваннях, хронічних запаленнях, гепатиті С.
Залежно від типу КГ клінічні прояви мають 2 основні патогенетичні механізми. У разі охолодження крові в судинах шкіри і підшкірних тканин кінцівок, КГ через свої физико-хімічні властивості преципітують, осідають на поверхні еритроцитів, тромбоцитів, лейкоцитів, на стінках кровоносних судин, що викликає уповільнення кровотоку, переважно в капілярах, призводить до порушення мікроциркуляції. Саме процесами преципітації кріоглобулінів у просвіті судин пояснюються такі клінічні прояви, як симптоми судинної недостатності, що виникають на холоді, а гістологічними ознаками в цих випадках є обтурація судин різного калібру. У зв’язку з властивостями, описаними вище, для осіб з I і частково II типом КГ характерні ураження у вигляді оклюзії судин: акроціаноз, сітчасте ліведо, синдром Рейно, тромбози артерій, виразки, некрози шкіри та м’яких тканин дистальних фаланг пальців, а також синдром «підвищеної в’язкості крові», який може призводити до таких неврологічних проявів, як запаморочення, головний біль, інсульти. Другий патогенетичний механізм пов’язаний з відкладенням Ig, які входять до складу кріопреципітату, в судинній стінці, що веде до активації комплементу, і, як наслідок, розвивається імунокомплексне запалення судин. У сироватці крові хворих виявляють і ЦІК. Для осіб з II і III типом КГ характерними клінічними проявами є: пурпура, артралгії, міалгії. Такі ушкодження, як гломерулонефрит, периферична невропатія, які є проявами імунокомплексного ураження, частіше зустрічаються у пацієнтів зі змішаною КГ II і III типу.
ЕКВ є імунокомплексною хворобою, при якій утворюються імунні комплекси, які складаються головним чином з моноклонального ІgМ РФ і поліклонального IgG. Характерна активація В-клітинної ланки імунної системи з розвитком імунокомплексного запалення. Певне значення в патогенезі має гіперв’язкість крові та внутрішньосудинна холодова преципітація з утворенням мікротромбів та порушенням мікроциркуляції. Уражуються переважно дрібні судини шкіри, нирок. Хворобу діагностують у 15% осіб з II типом КГ.
Розвиток ЕКВ пов’язують з HCV-інфекцією, антигенами при ЕКВ є частини віріона віруса гепатиту С. Асоційовану з хронічним вірусним гепатитом С КГ виявляють у 34–54% хворих на ЕКВ, при цьому у 15–20% розвивається васкуліт тяжкого ступеня. У мононуклеарах і шкірі хворих на ЕКВ виявляють HCV-РНК геном. Вторинну змішану КГ вперше описано в 1977 р. як позапечінковий прояв хронічного вірусного гепатиту С. Нещодавні дослідження показали, що при змішаній КГ в 48% випадків виявляють HCV-інфекцію, ще в 28% випадків — разом з маркерами вірусу гепатиту B. На сьогодні більшість дослідників визнають HCV як етіологічний чинник КГ і як фактор позапечінкових проявів хронічного вірусного гепатиту С. Змішану КГ частіше відмічають в осіб у віці 40–60 років, її вважають маркером хронічної більше 10 років HCV-інфекції. Вважають, що при цій інфекції відбувається проліферація клонів В-клітин, що синтезують патологічний Ig, тому тривала персистенція HCV — необхідна умова еволюції КГ III типу, що містить 2 поліклональних Ig (частіше IgM і IgG), в II тип, який складається з поліклонального IgG і моноклонального компонента IgMк. Саме останньому, за сучасними уявленнями, належить ключова роль в розвитку ЕКВ, включаючи такий його прояв, як гломерулонефрит. Існує думка, що РФ, особливо моноклональний (IgМк), який володіє особливим ідіотипом WA-кросс, перехресно зв’язується з фібронектином мезангіального матриксу. Імунні комплекси, що утворюються в субендотеліальному просторі і мезангії клубочків та складаються з IgМк та анти-HCV IgG, призводять до розвитку ураження нирок.
Описані й інші інфекції, при яких може розвиватися змішана КГ. До таких належать інфекції, викликані вірусами (Епштейна — Барр, цитомегаловірус, аденовірус, ВІЛ), бактеріями (інфекційний ендокардит, лепра, стрептококовий гломерулонефрит, сифіліс, хвороба Лайма, Ку-лихоманка), найпростішими (кала-азар, малярія), а також грибкові захворювання (кокцидіоїдомікоз), паразитарні інфекції (токсоплазмоз, ехінококоз, шистосомоз).
У хворих на дифузні захворювання сполучної тканини досить часто діагностують змішану КГ, причому в ⅔ випадків — це КГ III типу. СЧВ і РА особливо часто поєднуються з КГ, в 50 і 25% випадків відповідно. У хворих на РА, який асоціюється з КГ, частіше розвивається ревматоїдний васкуліт, синдром Фелті. КГ може виникати: при ССД, синдромі Шегрена, поліміозиті, при системних васкулітах (пурпурі Шенлейна — Геноха, хворобі Бехчета, ВП), саркоїдозі, тиреоїдиті, хворобах нирок (проліферативний гломерулонефрит), а також при хронічних захворюваннях печінки, целіакії, легеневому фіброзі, вульгарній пухирчатці, первинних плазмоклітинних дискразіях, які включають плазмоклітинну лімфому, мієломну хворобу, макроглобулінемію Вальденстрема, первинний амілоїдоз, хворобу важких ланцюгів та ін. Отримано також експериментальну (поствакцинальну) КГ.
Клінічна картина
На ЕКВ хворіють переважно жінки у віці понад 50 років. Хвороба починається гостро. Найбільш характерною ознакою хвороби є ураження шкіри, яке спостерігається у 90% хворих і часто є приводом для звернення до лікаря. Пальпована пурпура — типовий шкірний прояв хвороби, інколи вона виникає на тлі папуло-нодозних висипань, найчастіше локалізується на шкірі стоп, гомілок, рідше на стегнах, супроводжується печією та свербежем. У тяжких випадках висипання з’являються на сідницях, животі, попереку, тулубі, верхніх кінцівках та обличчі. Висип може бути у вигляді невеликої кількості елементів, але може бути поширеним і зливним (рис. 21.3). Висипання стають ряснішими при охолодженні, тривалому стоянні, на стискуваних ділянках шкіри. Зазвичай висипання зберігаються 1–2 тиж, потім зникають, але можуть рецидивувати під впливом перерахованих провокуючих чинників. Рідше зустрічаються інші види шкірних проявів КГ: пігментація шкіри (40%), телеангіоектазії (11%), кропив’янка (4–10%), сітчасте ліведо (10–19%). Виразково-некротичні зміни шкіри, включаючи виразки гомілок і некроз дистальних ділянок кінцівок, виявляють у 10–30% випадків (табл. 21.94).
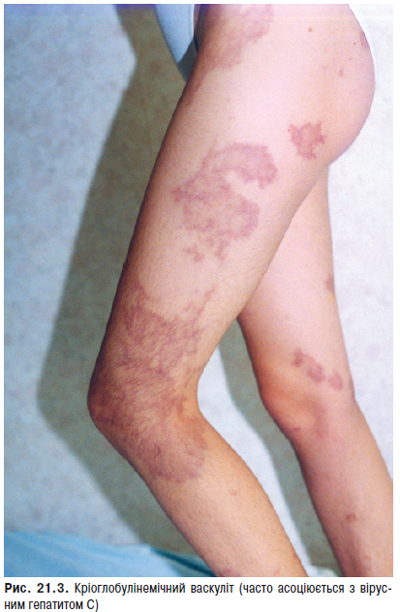
Таблиця 21.94
Клінічна класифікація ЕКВ (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічно-рецидивуючий із зазначенням кількості рецидивів |
|
|
Стадія |
Початкова, розгорнута, пізня (термінальна) Активна, неактивна, ремісія, часткова ремісія |
|
|
Ступінь активності |
I (мінімальний), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика уражень |
Шкіра |
Пурпура, некроз шкіри, кропив’янка, ліведо |
|
Дрібні судини |
Синдром Рейно |
|
|
Суглоби |
Артралгії, артрити |
|
|
Периферична нервова система |
Поліневропатія, астеновегетативний синдром |
|
|
Нирки |
Гломерулонефрит, нефротичний синдром, артеріальна гіпертензія |
|
|
Печінка |
Гепатит С |
|
|
ШКТ |
Абдомінальний синдром, спленомегалія |
|
|
Легені |
Васкуліт |
|
|
Серце |
Інфаркт міокарда |
|
|
Синдром Шегрена |
||
Для 33% хворих на ЕКВ характерна тріада Мельтцера, яка включає такі симптоми, як пурпуру, артралгії, немотивовану слабкість. Ці самі симптоми відзначали в осіб з симптоматичною КГ. Хворі скаржаться на слабкість м’язів, міалгії, симетричні мігруючі поліартралгії. Уже на ранніх стадіях хвороби у деяких пацієнтів суглобовий синдром проявляється симптомами артриту. Переважно до процесу залучаються проксимальні міжфалангові, плеснофалангові суглоби, рідше уражуються колінні, ліктьові, гомілковостопні суглоби. Інколи артрит носить мігруючий характер і нагадує ревматичне ураження суглобів або РеА. Артрит може рецидивувати, проте, навіть при рецидивах, деструкції тканин суглобів деформацій не спостерігається, рентгенологічні зміни відсутні.
При ЕКВ можна відзначати й інші симптоми. Феномен Рейно виявляють у 30–50% хворих, у 25% він є раннім проявом КГ. Генералізовану лімфаденопатію діагностують у 2–16% хворих, синдром Шегрена — у 14–40%. У 26% пацієнтів реєструють лихоманку.
Ураження нервової системи відзначають у 30% хворих у вигляді периферичної сенсорно-моторної поліневропатії, спочатку асиметричної, а потім симетричної, або розвивається гострий множинний мононеврит. У випадку ураження судин головного мозку можливі транзиторні порушення зору, геміпарези, гострий психоз та інші прояви.
Ураження печінки діагностують в 28–88% хворих. У частини пацієнтів (11%) виявляють виражені клінічні ознаки, у інших — лише зміни біохімічних показників (77%). Більш ніж у 70% пацієнтів в сироватці крові виявляють маркери вірусу гепатиту С та/або гепатиту В. У 28–88% випадків відзначають гепатомегалію, в 25–75% — спленомегалію.
Ушкодження нирок — гломерулонефрит є типовим проявом васкуліту і спостерігається у 50–80% хворих. Інколи хвороба дебютує з ураження нирок, що є несприятливою прогностичною ознакою. Клінічні прояви за ступенем тяжкості варіюють від ізольованого сечового синдрому до нефротичного синдрому з розвитком хронічної ниркової недостатності.
Частіше, більш ніж у 50% хворих на ЕКВ, ураження нирок клінічно проявляється помірним сечовим синдромом (протеїнурією й еритроцитурією), але можуть бути випадки з нефротичним синдромом, а також зрідка відзначають швидкопрогресуючий гломерулонефрит. У 10–30% випадків гломерулонефриту відзначають часткову або повну ремісію, в 20% — часті рецидиви. Для більшості хворих на ЕКВ з ураженням нирок характерна артеріальна гіпертензія з подальшим розвитком серцево-судинних ускладнень: інфаркту міокарда, церебрального інсульту, крововиливу в сітківку ока.
Ураження легень відзначають у 2–10% хворих, проте зазвичай не діагностують. За наявності пневмоніту хворі найчастіше скаржаться на задишку, рідше — на кашель, кровохаркання. У 74% хворих при рентгенографічному дослідженні виявляють помірне ураження інтерстиціальної тканини легень. Вимірювання параметрів функції зовнішнього дихання часто є корисним для виявлення ураження дистальних відділів повітроносних шляхів. Плеврит виявляють рідко.
Дуже рідко спостерігається абдомінальний біль, інфаркт міокарда.
Морфологічні зміни
Біопсія ураженого органа вкрай важлива для підтвердження діагнозу. У біоптаті елементів ураженої шкіри (пурпура) при світловому мікроскопічному дослідженні виявляють лейкоцитокластичний васкуліт дрібних і рідше середніх артерій. Методом прямої імунофлуоресценції виявляють преципітат IgG і IgM і компонентів комплементу.
При біопсії в тканині нирок частіше виявляють мезангіокапілярний (мезангіопроліферативний) гломерулонефрит з фібриноїдним некрозом дрібних судин і судин середнього калібру, знаходять внутрішньокапілярні тромби, які складаються з преципітату КГ, в яких при імунофлуоресцентному дослідженні виявляють IgМк, клубочки нирок, інфільтровані моноцитами, спостерігається потовщення базальної мембрани клубочків. Рідше відзначають екстракапілярний гломерулонефрит з півмісяцями та/або нирковим васкулітом.
Лабораторні дослідження
Для верифікації діагнозу важливо виявлення змішаної кріоглобулінемії та уточнення складу Ig (IgМ-діагностична ознака). При заборі крові для виявлення кріоглобулінів необхідно дотримуватись наступних правил: 1) кров беруть без антикоагулянтів у підігріті шприц і пробірку та поміщують у термостат при температурі 37 °С на 1–2 год; 2) після утворення згустку за допомогою центрифуги відокремлюють сироватку (при 37 °С) і витримують її при температурі 0–4 °С протягом 5–7 днів; 3) кількість кріоглобулінів визначають або прямим вимірюванням об’єму преципітату після центрифугування (кріокріт), або визначенням концентрації білків за допомогою спектрофотометра. Часті хибнонегативні результати визначення КГ, звичайно, пов’язані зі специфікою проведення дослідження і недостатнім досвідом лабораторій з проведення таких досліджень.
Важливою діагностичною ознакою ЕКВ є різке зниження вмісту деяких компонентів комплементу, а саме — С4, меншою мірою — С2, C1q при нормальній концентрації СЗ, зниження загальної гемолітичної активності комплементу (СН50). У 90–100% хворих виявляють РФ у високих титрах, це зумовлено тим, що моноклональний компонент КГ при II типі цієї патології практично завжди має активність РФ (тобто зв’язується з Fc-фрагментом IgG). У половині хворих виявляють АНФ.
У більшості хворих наявні лейкоцитоз, прискорена ШОЕ, підвищення рівня СРБ, гіпергаммаглобулінемія.
При ураженні нирок у сечі виявляють протеїнурію, еритроцитурію або гематурію, при порушенні функції нирок — креатинемію, зниження швидкості клубочкової фільтрації. Підвищення активності трансаміназ спостерігають у 50% хворих. Усім пацієнтам із ЕКВ необхідно проводити вірусологічне дослідження з метою виявлення маркерів вірусів гепатиту С і В з використанням імуноферментного аналізу (anti-HСV, HCV-RNA, HBs, HBe, anti-HBs, anti-HBe) та полімеразної ланцюгової реакції (визначення РНК HCV та ДНК HВV).
Діагностичні критерії ЕКВ, запропоновані H.H. Peter, W.L. Gross (1995): 1) підкреслені акрально-лейкоцитокластичні та/або набуті некротизуючі ураження шкіри; 2) поява або посилення їх під впливом холоду або вітру; 3) виявлення під час гістологічного дослідження васкуліту дрібних судин (артеріол, венул, капілярів); 4) виявлення кріоглобулінів.
Діагноз вважається достовірним за наявності усіх 4 критеріїв.
Диференційний діагноз
Оскільки при ЕКВ уражуються дрібні судини, диференціювати його необхідно перш за все з васкулітами з подібними ураженнями, до яких належать: геморагічний васкуліт, МПА, синдром Черджа — Стросс, гранулематоз Вегенера.
Слід пам’ятати, що при гранулематозі Вегенера, на відміну від ЕКВ, відзначають некротизуючий гранулематозний васкуліт, хвороба починається з ураження верхніх дихальних шляхів, ступінь ураження нирок і особливо легенів більш тяжкий, маркером активної фази хвороби при гранулематозі Вегенера є АНЦА до протеїнази-3.
Також необхідно диференціювати ЕКВ із хворобами, при яких розвивається вторинна КГ. Потрібно пам’ятати, що найчастішим проявом КГ є ураження шкіри, при цьому пальповану пурпуру відзначають у 60–70% хворих зі змішаною КГ (типи II і III) і лише в 15% пацієнтів із простою моноклональною КГ (тип I); у хворих з моноклональними кріоглобулінами частіше виникають виразки/некрози шкіри і м’яких тканин дистальних фаланг пальців, у 40–60% випадків відзначають феномен Рейно, артралгії/артрити — у 35% хворих (особливо при змішаних кріоглобулінеміях з низьким вмістом кріоглобулінів). Ураження нирок розвивається в 20–25% випадків, периферична невропатія — в 25%.
Існує взаємозв’язок аутоімунних і лімфопроліферативних хвороб. Їх асоціація зумовлена патогенетично і, можливо, пояснюється тривалою антигенною стимуляцією і терапією. Лімфаденопатія, гепатоспленомегалія, лихоманка належать до симптомів В-клітинної інтоксикації і можуть спостерігатися як при ревматичних, так і лімфопроліферативних захворюваннях. Лейкоцитокластичний васкуліт, рецидивуючі пурпури, виразкові ураження гомілок, виразково-некротичний васкуліт можуть виникати при плазматичних дискразіях.
Лікування
Головною умовою досягнення ремісії при ЕКВ є усунення причини виникнення КГ. Хворі повинні остерігатися переохолодження та тривалого перебування на ногах, оскільки це може посилити тяжкість перебігу васкуліту і сприяти розвитку рецидиву. Слід пам’ятати, що після досягнення клінічної ремісії при ЕКВ показники РФ і кріокріту часто залишаються підвищеними, тому їх не враховують при корекції лікування.
Лікувальні заходи залежать від того, які органи уражені, і тяжкості перебігу хвороби. Якщо у хворого наявні лише незначні шкірні прояви васкуліту, артралгії і не пошкоджені життєво важливі органи, часто буває достатнім призначення постільного режиму, НПЗП та заходів щодо запобігання переохолодженню.
Контрольованих досліджень методів лікування КГ не проводили, тому пропонуються різні схеми терапії. Для індукції ремісії за наявності пурпури і артриту, ушкодженні інших органів застосовують комплексну терапію протягом 3–6 міс: ГК, плазмаферез, противірусні препарати, при тяжкому перебігу васкуліту застосовують цитотоксичні препарати (циклофосфамід) та комбінацію цих методів або ГК з ритуксимабом.
За допомогою плазмаферезу можливо продовжити життя пацієнтам з гострими, загрозливими для життя формами хвороби, з проявами васкуліту, нефриту, злоякісною гіпертензією, тяжким пошкодженням ЦНС, судинною недостатністю (дистальні некрози) і синдромом підвищеної в’язкості крові. Після процедури плазмаферезу для запобігання утворенню нових антитіл паралельно необхідно призначити імуносупресивні препарати. Для підтримки ремісії протягом 2–5 років застосовують ГК з противірусними препаратами або, можливо, ритуксимаб.
При ураженні нирок і формуванні нефротичного синдрому або швидкопрогресуючому гломерулонефриті застосовують ГК, цитотоксичні препарати на тлі плазмаферезу або кріоаферезу. Лікування ГК починають з в/в введення пульсової дози (0,75–1,0 г) метилпреднізолону щоденно протягом 3 днів, після цього переходять на вживання препарату перорально, застосовують підтримувальну терапію з використанням препаратів у низьких дозах упродовж декількох місяців. Пацієнтам з тяжкою формою васкуліту, з ураженням нирок, ЦНС, шкіри або печінки потрібна більш агресивна імуносупресивна терапія: терапію циклофосфамідом з розрахунку 2 мг/кг — протягом 2–4 міс, плазмаферез (з обміном 3 л плазми) або кріосорбцію 3 рази на тиждень протягом 2–3 тиж.
При тяжкому перебігу васкуліту, в першу чергу до призначення противірусної терапії, призначають ГК та інші імуносупресивні засоби, оскільки у ряді випадків застосування на початку лікування противірусної терапії посилює перебіг васкуліту.
У хворих на ЕКВ з ураженням нирок і хронічним гепатитом С лікування починають з призначення преднізолону в дозі 1,0 мг/кг/добу і 5–7 сеансів кріоаферезу, які проводять через день. При зменшенні клінічних проявів васкуліту дозу преднізолону знижують і призначають противірусну терапію. Терапією вибору активного гепатиту С вважають комбіновану антивірусну терапію пегільованим інтерфероном і рибавірином.
Доведено, що лише елімінація HCV викликає стійку ремісію ЕКВ за наявності активного гепатиту С, тому сучасна противірусна терапія є основою лікування. Але не завжди лікування при ЕКВ починають з противірусної терапії.
Зараз при ЕКВ та у хворих з КГ з успіхом застосовують ритуксимаб. У 2008 р. M. Ramos-Casals та співавтори узагальнили дані використання генно-інженерних біологічних препаратів у 88 хворих на ЕКВ та встановили, що на тлі прийому ритуксимабу позитивного ефекту досягнуто в 87% випадках. Інгібітори ФНП-α у хворих на ЕКВ були неефективні або викликали високий відсоток побічних явищ. Анти-В-клітинна терапія ефективна у 75–90% пацієнтів при поєднанні ЕКВ і хронічного вірусного гепатиту В.
На сьогодні у хворих на ЕКВ і хронічний гепатит С французські дослідники рекомендують застосовувати схему лікування, вибір якої залежить від тяжкості стану: низька або помірна активність (пурпура, артралгії, сенсорна невропатія) — призначають пегільований інтерферон-α + рибавірин; тяжкий перебіг (прогресуюче ураження нирок, множинний мононеврит, виразково-некротичний ангіїт) — ритуксимаб — пегільований інтерферон-α + рибавірин; потенційно смертельний перебіг (швидкопрогресуючий гломерулонефрит, ушкодження ЦНС, ШКТ, легеневий васкуліт) — ГК, плазмаферез, циклофосфамід і/або ритуксимаб, а потім пегільований інтерферон-α + рибавірин.
Прогноз
Прогноз хвороби більшою мірою визначає наявність ураження нирок. У перші 7 років після дебюту хвороби помирають близько 70% хворих з пошкодженням нирок і лише 31% з інтактними нирками. Прогноз при ураженні нирок визначається активністю васкуліту, наявністю артеріальної гіпертензії, прогресуванням серцевої недостатності. Причинами смерті є також ураження печінки, інфекційні захворювання, хвороби серцево-судинної системи і лімфопроліферативні захворювання. Відсутність адекватного лікування хворих на ЕКВ, асоційований з хронічним гепатитом С, призводить до смерті.
Лікування
Етапи, які виділяють у лікуванні системних васкулітів. Відповідно до активності патологічного процесу виділяють декілька фаз перебігу васкулітів (табл. 21.95).
Лікування системних васкулітів включає індукцію ремісії (3–6 міс), тривалу (2–5 років) підтримувальну терапію з досягненням стійкої повної ремісії хвороби, а також лікування в період загострення. У дебюті хвороби та при загостреннях досягають швидкого пригнічення імунної відповіді за допомогою короткого курсу агресивної (ескалаційної) терапії. Подальша підтримувальна терапія імуносупресорами дозволяє досягти клінічної та лабораторної ремісії.
Таблиця 21.95
Фази васкулітів залежно від активності запалення
|
Фаза хвороби |
Коротка характеристика фази |
|
|
Загострення |
«Велике» |
У запальний процес залучено життєво важливі органи. Необхідна ескалаційна терапія циклофосфамідом у поєднанні з підвищенням дози ГК до 30 мг/добу. Проводять пульс-терапію метилпреднізолоном, застосовують Ig людини для в/в введення, плазмаферез |
|
«Мале» |
Збільшення суми балів від 0–1 до 5, яке вимагає підвищення дози ГК до 30 мг/добу |
|
|
Персистуюча (низька) активність |
Персистують загальні симптоми запалення, які купірують підвищенням дози ГК. Ескалація терапії не потрібна |
|
|
Часткова ремісія |
На фоні лікування загальна сума балів зменшується на 50% порівняно з початковою |
|
|
Ремісія |
Ознаки активності відсутні (загальна сума 0–1 бал). Вміст СРБ нормальний на тлі стабільної підтримувальної терапії |
|
|
Неактивна фаза |
Відзначають ремісію, необхідність в підтримувальній терапії відсутня |
|
Показання і тактика призначення лікарських засобів при різних васкулітах наведена в табл. 21.96.
Таблиця 21.96
Лікарські засоби, які застосовують при системних васкулітах
|
Лікарські засоби |
Васкуліти, при яких їх застосовують |
|
|
ГК |
Як основний метод лікування |
ГКА, артеріїт Такаясу, а також синдром Черджа — Стросс, кріоглобулінемічний васкуліт, тяжкі форми геморагічного васкуліту (з ураженням ШКТ та нирок). Швидкий ефект ГК розглядають як діагностичну ознаку ГКА та РПМ |
|
Не використовують у вигляді монотерапії |
Гранулематоз Вегенера, МПА, ВП |
|
|
Циклофосфамід |
Є препаратом вибору при гранулематозі Вегенера, МПА, ВП (за відсутності маркерів реплікації вірусу гепатиту В), тяжких формах геморагічного васкуліту та синдрому Черджа — Стросс з ураженням нирок |
|
|
Метотрексат |
У поєднанні з ГК при гранулематозі Вегенера без тяжкого ураження легень та швидкопрогресуючого нефриту при непереносимості циклофосфаміду або для підтримання ремісії хвороби |
|
|
Азатіоприн |
Для підтримання ремісії при некротизуючих васкулітах |
|
|
Циклоспорин |
Пригнічує прогресування увеїту при хворобі Бехчета більшою мірою, ніж колхіцин та пульс-терапія циклофосфамідом. При неефективності інших лікарських засобів можуть застосовувати для підтримання ремісії при гранулематозі Вегенера |
|
|
Ig людини нормальний для в/в введення |
Позитивний ефект дає в поєднанні з плазмаферезом при синдромі Черджа — Стросса. При інших васкулітах його ефективність не доведена |
|
|
Мікофенолату мофетил |
Застосовується для підтримання ремісії у хворих на гранулематоз Вегенера |
|
|
Ритуксимаб |
Дозволяє досягти клінічного ефекту при АНЦА-асоційованих васкулітах у 90% хворих |
|
|
Лефлуномід |
У дозі 40 мг/добу в поєднанні з преднізолоном (10 мг/добу) застосовують для підтримання ремісії при гранулематозі Вегенера |
|
|
Колхіцин |
Знижує частоту та тяжкість загострення хвороби Бехчета, її прогресування |
|
|
Ко-тримоксазол (сульфаметоксазол + триметоприм) |
У ранню фазу гранулематозу Вегенера та при лімітованих формах хвороби можна застосовувати для підтримання ремісії. Призначають для профілактики інфекційних ускладнень на тлі імуносупресивної терапії метотрексатом в період підтримання ремісії хвороби. Для підтримання ремісії при генералізованій формі гранулематозу Вегенера препарат неефективний (як у вигляді монотерапії, так і в поєднанні з преднізолоном) |
|
|
Противірусні препарати |
Призначають за наявності маркерів реплікації вірусу гепатиту В та інфікування вірусом гепатиту С при кріоглобулінемічному васкуліті |
|
|
Плазмаферез |
Включають у комбіновану терапію при гострому прогресуючому перебігу (тяжкому) васкулітів з нефритом. У поєднанні з ГК застосовують при лікуванні ЕКВ та ВП, асоційованого з вірусом гепатиту В |
|
|
Хірургічне лікування |
Проводять при артеріїті Такаясу, ОТА, гранулематозі Вегенера (при субглоточному стенозі) |
|
Терапію системних васкулітів за Е.Л. Насоновим (2006; 2011 (ред.)) описано в табл. 21.97.
Таблиця 21.97
Терапія системних васкулітів
|
Препарати |
Патологія |
Показання |
Тактика призначення |
|
ГК |
ГКА, артеріїт Такаясу |
Індукція ремісії (ГК — основний метод лікування) |
На початку хвороби доза ГК становить 1 мг/кг/добу (не >60 мг/добу) у декілька прийомів; через 7–10 днів переходять на одноразовий прийом вранці. Пригнічувальну терапію проводять 3–4 тиж. Підтримувальна терапія (0,15–0,2 мг/кг/добу) починається після досягнення ефекту (початкову дозу знижують поступово — по 5 мг кожні 2 тиж) і продовжується 1–5 років. Показання до пульс-терапії включають:
|
|
Синдром Черджа — Стросс без АНЦА, кріоглобулінемічний васкуліт |
Призначають при відсутності ознак прогресування |
||
|
Геморагічний васкуліт |
При тяжких формах з ураженням ШКТ і нирок |
||
|
Гранулематоз Вегенера, МПА, ВП |
Монотерапію ГК не використовують |
||
|
ГКА, РПМ |
Відзначається швидкий ефект, який розглядають як діагностичну ознаку |
||
|
Циклофосфамід |
Гранулематоз Вегенера, МПА, ВП (за відсутності реплікації вірусу гепатиту В) |
Є препаратом вибору для індукції ремісії |
У перші 10–14 днів призначають у дозі 1–2 мг/кг/добу. Надалі дозу знижують залежно від вмісту лейкоцитів у периферичній крові. При швидкому прогресуванні початкова доза становить 4 мг/кг/добу протягом 3 днів, потім 7 днів по 2 мг/кг/добу або проводиться пульс-терапія (15 мг/кг) з інтервалами між першими 3 курсами 2 тиж, а в подальшому — через 3 тиж. Після досягнення повної ремісії лікування продовжують не менше 12 міс. Потім дозу поступово знижують по 20–50 мг протягом 2–3 міс. Ефективною може бути інтермітуюча терапія циклофосфамідом у високих дозах у поєднанні з ГК, але при такому режимі у хворих із гранулематозом Вегенера частіше виникають загострення. Після досягнення ремісії, лікування циклофосфамідом необхідно проводити не менше 1 року |
|
Тяжкі форми геморагічного васкуліту, синдрому Черджа — Стросс |
Швидкопрогресуюче ураження судин і нирок, незважаючи на позитивний початковий ефект від ГК |
||
|
Азатіоприн |
Рефрактерний перебіг ГКА, артеріїт Такаясу |
Досягнення ремісії |
У дозі 2–3 мг/кг/добу застосовують у випадку непереносимості метотрексату або циклофосфаміду |
|
Гранулематоз Вегенера, МПА, ВП, синдром Черджа — Стросс |
Підтримка ремісії |
Оптимальна доза становить 1–3 мг/кг/добу, підтримувальна — 50 мг/добу. Побічні ефекти реєструють рідше, ніж при застосуванні циклофосфаміду |
|
|
Метотрексат |
Гранулематоз Вегенера без швидкопрогресуючого нефриту (креатинін <150 ммоль/л) та важкого ураження легень |
1. При непереносимості циклофосфаміду 2. Для підтримки ремісії хвороби |
Призначають по 12,5–17,5 мг/тиж у поєднанні з ГК |
|
Артеріїт Такаясу, ГКА |
Індукція ремісії |
У дозі 17,5 мг/тиж (7,5 мг/тиж — неефективна) у поєднанні з ГК в низьких дозах призначається при рефрактерному перебігу |
|
|
Імуноглобуліни |
Синдром Черджа — Стросс |
Індукція ремісії |
Застосовуються щомісяця в дозі 2 г/кг протягом 6 міс |
|
Хвороба Кавасакі |
Індукція ремісії |
В\в імуноглобулін є препаратом вибору, він попереджує розвиток ускладнень, ураження коронарних артерій |
|
|
Інші випадки |
Індукція ремісії |
Призначають при рефрактерному перебігу інших васкулітів у випадках, коли має місце протипоказання до застосування цитостатичних препаратів (вагітність, інфекційні ускладнення, до і після хірургічних операцій) |
|
|
Мікофенолату мофетил |
Гранулематоз Вегенера |
Підтримка ремісії |
У дозі 2 г/добу в поєднанні з преднізолоном (10 мг/добу) застосовують при непереносимості, неефективності азатіоприну або метотрексату |
|
Циклоспорин |
Хвороба Бехчета |
Пригнічення прогресування |
Початкова доза 5 мг/кг/добу з подальшим зниженням до 2 мг/кг/добу |
|
Гранулематоз Вегенера |
Підтримка ремісії |
Доза така сама. Призначається при неефективності іншої терапії. Варто враховувати нефротоксичність циклоспорину |
|
|
Лефлуномід |
Гранулематоз Вегенера |
Підтримка ремісії |
Доза 40 мг/добу у поєднанні з 10 мг/добу преднізолону. Застосовують при непереносимості, неефективності азатіоприну або метотрексату |
|
Ко-тримоксазол/ триметоприм |
Гранулематоз Вегенера |
Підтримка ремісії в ранню стадію хвороби, при обмеженому ураженні (ЛОР-органів) |
Доза (160/800 мг) 2 рази на день |
|
Профілактика інфекційних ускладнень, викликаних пневмоцистою на тлі лікування метотрексатом |
Доза 160/800 мг 3 рази на день 3 рази на тиждень |
||
|
Колхіцин |
Хвороба Бехчета |
Загострення, прогресування хвороби |
Доза 0, 5–1,5 мг/добу |
|
Пентоксифілін |
Системні васкуліти |
Вазоспастичний та ішемічний синдром, ураження шкіри й нирок |
Доза варіює від 300 до 1200 мг/добу |
|
Плазмаферез |
Есенціальна кріоглобулінемія; ВП, асоційований з вірусом гепатиту В |
1. Гострий прогресуючий перебіг. 2. Швидкопрогресуючий нефрит (рівень креатиніну >500 мкмоль/л). 3. Тяжкий перебіг васкуліту, у тому числі альвеолярні геморагії |
Призначають у складі комбінованої терапії |
|
Хірургічне лікування |
Артеріїт Такаясу, ОТА |
Стеноз, оклюзія артерій, критична ішемія, некроз |
|
|
Гранулематоз Вегенера |
Субглоточний стеноз |
Механічна дилатація трахеї в поєднанні з локальним введенням ГК |
Серед ГК препаратом вибору є преднізолон (початкова доза 1 мг/кг/добу, усередину). З імуносупресорів віддають перевагу циклофосфаміду (початкова доза 2 мг/кг/добу). При тяжких системних васкулітах циклофосфамід приймають протягом 1 року після досягнення ремісії. Потім дозу поступово знижують і через кілька місяців відміняють препарат. Інші імуносупресори при васкулітах не настільки ефективні, як циклофосфамід. При його непереносимості, небажанні приймати (через ризик побічних ефектів) застосовують метотрексат, азатіоприн (з гіршими результатами). Імуносупресори поєднують із плазмаферезом, переливанням донорської плазми. Ефективність циклоспорину при васкулітах невисока. При неуточнених системних васкулітах призначають ГК, а імуносупресори додають при недостатній ефективності ГК. Після досягнення ремісії переходять на прийом ГК через день. При усіх васкулітах цитостатики призначають у тих випадках, коли неможливо знизити дозу ГК без погіршення стану хворих. Усім, у кого проводять тривале (>3 міс) лікування ГК у дозі 5 мг/добу і більше, призначають комбіновані препарати кальцію (1000–1500 мг/добу), які додатково містять остеотропні мінерали, і вітаміну D (800 МО/добу).
Ритуксимаб призначають в період індукції ремісії в дозі 375 мг/м2 4 рази щотижнево в поєднанні з преднізолоном (1 мг/кг/добу) або без нього хворим з АНЦА-асоційованим васкулітом, а також ЕКВ, які раніше не відповідали на терапію.
Комбіновану терапію системних некротизуючих васкулітів за Е.Л. Насоновим (2006; 2011 (ред.)) представлено в табл. 21.98.
Таблиця 21.98
Комбінована терапія системних некротизуючих васкулітів
|
Терапія |
Препарати |
Дози й тактика |
|
Індукційна |
Циклофосфамід |
2 мг/кг/добу (максимум 150 мг/добу) протягом 1 міс у поєднанні із преднізолоном 1 мг/кг/добу (максимально 80 мг/добу) (хворим у віці більше 60 років дозу циклофосфіміду знижують на 25 мг). Дозу преднізолону знижують щотижня до 10 мг/добу |
|
Підтримувальна |
Азатіоприн |
Доза становить 2 мг/кг/добу у поєднанні із преднізолоном 5–10 мг/добу |
|
Циклофосфамід |
1 мг/кг/добу у поєднанні з преднізолоном 5–10 мг/добу |
|
|
Ескалаційна |
Плазмаферез або пульс-терапія метилпреднізолоном + циклофосфамід |
При некротизуючих васкулітах з ураженням нирок (креатинін >500 мкмоль/л) та легень (геморагії) проводять 7–10 процедур плазмаферезу протягом 14 днів (з видаленням плазми 60 мл/кг маси тіла та заміщенням її відповідним об’ємом 4,5–5% альбуміну) в поєднанні з пульс-терапією метилпреднізолоном (15 мг/кг/добу) та циклофосфамідом (10 мг/кг/добу) |
Показання до проведення пульс-терапії при системних васкулітах за Е.Л. Насоновим (2005) подано в табл. 21.99.
Таблиця 21.99
Показання до проведення пульс-терапії при системних васкулітах
|
Назва васкуліту |
Показання |
|
ВП |
Периферична гангрена, поліневропатія, ураження ШКТ, загострення хвороби |
|
МПА |
Швидкопрогресуючий гломерулонефрит, загострення хвороби |
|
Гранулематоз Вегенера |
Ураження нирок, легень, загострення хвороби |
|
Синдром Черджа — Стросс |
Загострення хвороби |
|
ГКА |
Офтальмологічні ускладнення |
|
Артеріїт Такаясу |
Період індукції ремісії, загострення хвороби, передопераційна підготовка в активну фазу |
|
Геморагічний васкуліт |
Ураження ШКТ, нирок, загострення хвороби |
|
Есенціальна кріоглобулінемічна пурпура |
Ураження нирок, загострення хвороби |
Примітки: при ВП, МПА, гранулематозі Вегенера, синдромі Черджа — Стросс, артеріїті Такаясу, геморагічному васкуліті пульс-терапію проводять у поєднанні з циклофосфамідом, при есенціальній кріоглобулінемічній пурпурі — з плазмаферезом
Схеми інтенсивної терапії, які застосовують при різних васкулітах (табл. 21.100).
Таблиця 21.100
Схеми інтенсивної терапії, які застосовують при різних васкулітах
|
ВП |
Комбіновану терапію метилпреднізолоном (1000 мг в/в 3 дні) і циклофосфамідом (1000 мг у 1-й день) призначають при прогресуванні хвороби, активному тяжкому перебігу з підвищенням рівня креатиніну в крові >500 мкмоль/л, легеневими кровотечами. Такі курси пульс-терапії повторюють з інтервалом 6 міс. До застосування пульс-терапії доцільне проведення курсу плазмаферезу (7–10 сеансів) |
|
МПА |
Комбіновану терапію (курси плазмаферезу + пульс-терапія метилпреднізолоном і циклофосфамідом + в/в введення імуноглобуліну) призначають пацієнтам зі швидким прогресуванням ниркової недостатності, легеневими геморагіями |
|
Синдром Черджа — Стросс |
Використовують пульс-терапію метилпреднізолоном у комбінації з циклофосфамідом і плазмаферезом |
|
Гранулематоз Вегенера |
Пацієнтам із генералізованою формою хвороби, гломерулонефритом, увеїтом, неврологічними розладами призначають комбіновану пульс-терапію метилпреднізолоном і циклофосфамідом (в/в або усередину). У перші місяці хвороби пульс-терапію проводять щомісяця |
|
Геморагічний васкуліт |
Призначення пульс-терапії (з наступним переходом на прийом ГК усередину) доцільне при тяжкому перебігу хвороби (за наявності абдомінального синдрому, кровохаркання, ураження нирок). Застосовують також плазмаферез, азатіоприн, циклоспорин, гепарин, дезагреганти |
|
ГКА |
Пульс-терапія показана при тяжкому прогресуючому перебігу хвороби, зниженні зору, раптовому розвитку сліпоти. Терапію ГК доповнюють метотрексатом, циклофосфамідом, циклоспорином А |
|
Неспецифічний аортоартеріїт |
Щомісячні курси пульс-терапії метилпреднізолоном і циклофосфамідом проводять протягом 9–12 міс при тяжкому рецидивуючому перебігу хвороби |
|
ОТА, ЕКВ, шкірні ангіїти |
Застосовують пульс-терапію метилпреднізолоном у комбінації з циклофосфамідом і плазмаферезом |
Побічні ефекти й ускладнення, які виникають при проведенні пульс-терапії, наведено в табл. 21.101.
Таблиця 21.101
Побічні ефекти і ускладнення пульс-терапії
|
Частота побічних ефектів і ускладнень |
Характер |
Примітки |
|
|
Побічні ефекти |
|||
|
Часті |
Тахікардія |
Відзначають у більшості хворих під час інфузії та після неї (до 48 год). Купірують седативними засобами і блокаторами β-адренорецепторів |
|
|
Гіперемія обличчя |
|||
|
Рідкісні |
Емоційне порушення (безсоння) |
Усувається седативними, снодійними препаратами |
|
|
Гіпотонія |
Відзначають у 1–2% пацієнтів під час інфузії та протягом 2 год після неї. Усуваються кардіотонічними засобами й дексаметазоном |
||
|
Брадикардія |
|||
|
Гикавка |
Може зберігатися декілька діб, купірується атропіном, прокінетиками |
||
|
Ускладнення |
|||
|
Часті |
Інтеркурентні інфекції (вірусні, бактеріальні) |
Виникають у ослаблених хворих після пульс-терапії з включенням циклофосфаміду |
|
|
Рідкісні |
Артрити великих суглобів |
Розвиваються в період від кількох годин до кількох діб після інфузії менше ніж у 1% хворих і купіруються НПЗП |
|
|
Украй рідкісні |
Анафілаксія |
Відзначають в одиничних випадках. У таких хворих застосовують адреналін, дексаметазон (16–40 мг) в/в |
|
|
Раптова смерть |
Відомі декілька випадків |
||
Випадки раптової смерті описано на тлі шлуночкових аритмій, що виникли в результаті гострих метаболічних порушень у міокарді. Проведення пульс-терапії не рекомендується за наявності вираженої серцевої недостатності, неконтрольованої артеріальної гіпертензії, тяжких аритмій, необхідне також дотримання обережності у осіб літнього та старечого віку. У зв’язку з підвищеним ризиком розвитку кардіальних ускладнень не рекомендується поєднувати пульс-терапію із введенням фуросеміду.
Випадки, в яких у хворих із системним васкулітом проводять противірусну терапію. Противірусна терапія показана за наявності маркерів реплікації вірусів гепатиту В та інфекції вірусом гепатиту С у пацієнтів із кріоглобулінемічним васкулітом (Насонов Е.Л., 2006 (ред.)) (табл. 21.102). У першому випадку призначають інтерферон-α + ламівудин 100 мг/добу до 6 міс у поєднанні із ГК і плазмаферезом, у другому — інтерферон-α + рибавірин + ГК, цитостатики і плазмаферез.
Таблиця 21.102
Терапія при інфікуванні вірусом гепатиту С пацієнтів із кріоглобулінемічним васкулітом залежно від ступеня тяжкості перебігу хвороби
|
Ступінь тяжкості |
Індукційна терапія |
Підтримувальна терапія |
|
Помірно тяжкий |
Призначають протягом 6 міс 3×106 ОД 3 рази на тиждень інтерферон-α + ГК менше 7,5 мг/добу |
Застосовують в тих самих дозах протягом 6–12 міс |
|
Тяжкий |
Протягом 6 міс призначають циклофосфамід в/в 0,5–1 г/м2 кожні 3 тижні + ГК 0,5–1,0 мг/кг в/в, потім перорально |
Протягом 6–12 міс призначають інтерферон-α 3×106 ОД 3 рази на тиждень + рибавірин |
|
Потенційно смертельний |
Протягом 6 міс циклофосфамід призначають по 2 мг/кг/добу усередину + ГК 500–1000 мг в/в протягом 3 днів з переходом на підтримувальну дозу + плазмаферез по 3 л плазми 3 рази на тиждень |
Протягом 6–12 міс призначають інтерферон-α 3×106 ОД 3 рази на тиждень + рибавірин |
При поєднанні метотрексату в дозі 17,5 мг/тиж з ГК в низьких дозах ремісія досягається у 80% пацієнтів з артеріїтом Такаясу, прогресуванню хвороби можна запобігти в 50%. Позитивний ефект застосування в/в імуноглобуліну відзначають тільки при синдромі Черджа — Стросс. Поєднання плазмаферезу з гемосорбцією в основному використовують при ВП, гранулематозі Вегенера, КГ. Включення плазмаферезу в комплексну терапію хворих на ВП і синдром Черджа — Стросс не підвищує 5-річну виживаність. Терапія з застосуванням плазмаферезу не має переваг порівняно зі стандартною схемою (ГК + цитостатики). Неефективним є застосування ко-тримоксазолу/триметоприму в якості монотерапії або в поєднанні із преднізолоном для підтримки ремісії при генералізованій формі гранулематозу Вегенера. Колхіцин знижує частоту та тяжкість загострень, прогресування хвороби Бехчета.
Препарати, які включають у комплексну терапію васкулітів. Додатково до основної терапії (ГК, імуносупресори) призначають:
- ацетилсаліцилову кислоту у низьких дозах (80–100 мг/добу);
- тиклопідин, клопідогрель;
- гепарин, надропарин кальцію та ін;
- пентоксифілін;
- алпростадил;
- ксантинолу нікотинат;
- низькомолекулярні декстрани;
- судинорозширювальні;
- інгібітори АПФ;
- інтерферон-α;
- похідні амінохіноліну;
- препарати системної ензимотерапії.
Але клінічна ефективність антиагрегантів, антикоагулянтів, вазодилататорів, ангіопротекторів, інгібіторів АПФ при системних васкулітах не підтверджена в клінічних випробуваннях.
При васкулітах з переважним ураженням шкіри досить симптоматичного лікування, короткого курсу ГК, тому що внутрішні органи уражуються рідко, а імуносупресори можуть завдати шкоди. При вторинних васкулітах проводять лікування основної хвороби.
Васкулопатії
Васкулопатії — патологія судин, яка не супроводжується явними морфологічними ознаками запальної клітинної інфільтрації стінки та периваскулярного простору (Баранов А.А., Насонов Е.Л., 1999). Відзначають дистрофію, мікротромбози, некроз, десквамацію ендотеліальних клітин, ураження базальної мембрани (набряк, фібриноїдний некроз, фрагментація). Клінічні прояви, результати інструментальних та морфологічних досліджень нагадують зміни при васкулітах.
Код за МКХ-10
М31.9 Некротизуюча васкулопатія неуточнена.
Васкулопатії у хворих зі злоякісними пухлинами
Васкулопатії є частим паранеопластичним синдромом, вони частіше розвиваються при гемобластозах (мієло-, лімфопроліферативних захворюваннях, мієломі), рідше — при солідних пухлинах. Реєструють макулопапульозні висипання, петехії, виразки, гангрену м’яких тканин кінцівок. При волосато-клітинному лейкозі розвиваються васкуліти шкіри, скроневих та коронарних артерій (ураження нагадує ВП). При гістологічних дослідженнях виявляють периваскулярні інфільтрати, до складу яких входят клітини пухлини.
При мієломній хворобі відзначають лейкоцитокластичний васкуліт, кріоглобулінемію. У хворих із IgA-мієломою виявляють запальні зміни в шкірі, кріокристалоглобуліни відкладаються в кістковому мозку, рогівці ока, міокарді, синовіальній оболонці, судинах легень, печінки, селезінки, нирок. При цьому запальні зміни судин (які характерні для васкулітів) не реєструють. Відкладання кристалічної речовини (депозитів) у просвіті малих і середніх судин та периваскулярно призводить до ішемічних змін в органах і тканинах, геморагічних висипань, утворення некротичних виразок шкіри гомілок, носової перетинки. Ішемія судин нижніх кінцівок може закінчитися ампутацією ніг.
Зрідка діагностують внутрішньосудинний лімфоматоз (злоякісний ангіоендотеліоматоз), який характеризується ураженням судин кори головного мозку, швидким прогресуючим порушенням його функції. При цьому розвивається оклюзія малих артерій, геморагічні та ішемічні інфаркти. Ураження судин ЦНС може бути дифузним (злоякісна форма) або ізольованим (доброякісна форма). У першому випадку хворі помирають протягом 6 міс від початку хвороби, у другому — прогноз більш сприятливий. Початок злоякісного варіанта хвороби гострий з прогресуванням порушень психіки, розвитком деменції, реєструють дифузний головний біль, транзиторне порушення зору. Можливий дебют у вигляді гострого порушення мозкового кровообігу. Зрідка відзначають лихоманку, міалгії, артралгії, схуднення, ураження шкіри (геморагічні, негеморагічні підшкірні утворення, бляшки, телеангіоектазії, некротичні виразки). Відмічаються різке збільшення ШОЕ, не виявляються кріоглобуліни, ЦІК, АНФ, РФ, зниження концентрації комплементу. При КТ-дослідженні виявляють дифузну атрофію мозку, інфаркти; при ангіографії судин головного мозку — ділянки звуження артерій, їх оклюзію. Слід враховувати, що внутрішньосудинний лімфоматоз (злоякісний ангіоендотеліоматоз) клінічно і ангіографічно нагадує первинний васкуліт ЦНС. Найбільш інформативною є біопсія головного мозку з морфологічним та імуногістологічним дослідженням біоптатів. Клітини, які закривають просвіт судин, експресують антигенні детермінанти лімфоцитів (CD45+, LN-1, LN-2).
Міксома
У хворих з передсердною міксомою відзначають широкий спектр імунологічних порушень, які нагадують аутоімунні захворювання. Констатують розвиток мікроаневризм (подібно до ВП), мікроемболів, які пенетрують в ендотелій з наступною деструкцією внутрішньої еластичної мембрани, середньої оболонки. Патологічний процес продовжується в судинах навіть після видалення пухлини.
Хіміотерапія злоякісних пухлин
Хіміотерапія з приводу злоякісних утворень супроводжується підвищенням частоти виникнення артеріальних та венозних тромбозів.
Тромботична тромбоцитопенічна пурпура (хвороба Мошкович)
Код за МКХ-10
М31.1 Тромботична мікроангіопатія. Тромботична тромбоцитопенічна пурпура
характеризується мікроангіопатичною гемолітичною анемією, тромбоцитопенією, неврологічними порушеннями, лихоманкою, ураженням нирок, розвитком легеневої недостатності. Вторинна форма ТТП ускладнює перебіг інфекції (вірусної та бактеріальної), онкологічних та ревматичних захворювань, прийом медикаментів. Прийом лікарських засобів, ВІЛ-інфекція, вагітність можуть бути причиною як ТТП, так і гемолітико-уремічного синдрому (ГУС). Ці захворювання можуть починатися з внутрішньосудинної агрегації тромбоцитів внаслідок тромботичної мікроангіопатії.
Морфологія. В усіх органах та системах виявляють сегментарні оклюзії артеріол, капілярів, венул гіаліновими тромбами. Відбувається відкладання гіаліну в субендотелії, з’єднання його з тромбами у просвіті судин. Тромботичні маси містять РАS-позитивні ендотеліальні фібрили, рештки тромбоцитів, фібронектин, фібрин. Наслідком оклюзії судин є розвиток ішемії органів, інфарктів. Найбільш значні зміни відбуваються у головному мозку, серці, селезінці. Запальну інфільрацію клітинами в стінках судин та периваскулярному просторі не реєструють.
Патогенез. У плазмі крові хворих визначається Са-залежна протеаза, яка розщеплює мультимери фактора Віллебранда на фрагменти. Пул фактора Віллебранда в плазмі крові продукується переважно ендотеліальними клітинами (ЕК). Під час загострення ТТП відбувається інтенсивна стимуляція ЕК, підвищена продукція великих форм фактора Віллебранда, які взаємодіють з рецепторами тромбоцитів, викликають їх агрегацію та прикріплення до субендотелію. Мультимери фактора Віллебранда зникають з кровотоку при розвитку тромбоцитопенії, прогресуванні хвороби. Якщо вони з’являються в крові в період ремісії, то це свідчить про продовження стимуляції ЕК та прогнозує подальше загострення хвороби. В основі рецидивуючої форми ТТП, можливо, лежить спадковий дефект продукції мультиформ фактора Віллебранда. Встановлено, що екзотоксини шигел можуть стимулювати ЕК (без їх пошкодження), викликати збільшення продукції ними фактора Віллебранда. До інших механізмів відносять зниження синтезу ЕК простацикліну та речовини, яка стимулює його синтез. В основі розвитку мікроангіопатичної гемолітичної анемії при ТТП (та ГУС) лежать: 1) пошкодження еритроцитів при проходженні через оклюдовані судини; 2) руйнування різних форм еритроцитів внаслідок їх прикріплення до ЕК через мультимери фактора Віллебранда або тромбоспондин; 3) індукція апоптозу мікросудинного ендотелію.
Клініка. Перебіг ТТП у більшості хворих (90%) має гострий характер. При цьому відзначають слабкість, лихоманку, геморагії, жовтуху, артрити або артралгії. Головним проявом є ураження головного мозку: реєструють головний біль, парестезії, судоми, порушення зору та мовлення, психози, розвиток коми. Такі прояви тривають не більше 2–3 діб, у 50% випадків вони повторюються. Їх причиною є порушення кровотоку по судинах головного мозку, розвиток інфарктів та геморагій. Виявляють ураження очей — набряк сітківки, крововиливи в неї. Анемія супроводжується шизоцитозом, сфероцитозом, ретикулоцитозом, значною тромбоцитопенією (8–40•109/л), зменшенням тривалості життя тромбоцитів. У 70% хворих відзначають протеїнурію, мікрогематурію, зрідка гостру ниркову недостатність. Розвиваються дифузні зміни в міокарді, з’являється біль у животі, шлунково-кишкова кровотеча, картина гострого живота. Ураження судин легень та печінки не є характерним.
Діагностика. Реєструють тромбоцитопенію, Кумбс-негативну гемолітичну анемію, фрагментацію еритроцитів, ретикулоцитоз, нейтрофільний лейкоцитоз (у 60% випадків). Помірно підвищується концентрація в крові непрямого білірубіну, ЛДГ. Зміни з боку згортання крові мають вторинний характер. Зрідка в плазмі виявляють продукти деградації фібронектину, розчинні фібрин-мономерні комплекси.
Лікування. Проводять трансфузію свіжозамороженої плазми (20–40 мл/кг протягом перших днів) до відновлення нормальної кількості тромбоцитів, припинення гемолізу еритроцитів, нормалізації рівня ЛДГ, відновлення функції ЦНС. Трансфузії плазми крові можуть тривати до 30 днів. Проводять плазмаферез (починають з видалення 50–60 мл/кг плазми крові на добу). Повторні процедури виконують за наявності гострої ниркової недостатності. Можливе використання інфузій простацикліну (2–5 нг/кг/добу). Монотерапія дезагрегантами є неефективною. У деяких випадках застосовують преднізолон (1 мг/кг/добу), пульс-терапію, азатіоприн (100–150 мг/добу). Протягом перших 6 міс від початку хвороби призначають ацетилсаліцилову кислоту, дипіридамол.
Прогноз. Без лікування 80% хворих помирають протягом 3 міс від початку лікування. Плазмаферез, трансфузії плазми дозволяють досягти одужання 80—90% хворих.
Гемолітико-уремічний синдром
Основними проявами є характерні для ТТП гематологічні порушення у поєднанні з ураженням нирок. Епідемічну форму ГУС, яка супроводжується діареєю, діагностують у дітей у віці до 5 років. Варіант ГУС без ураження кишечнику розвивається у дорослих осіб (співвідношення жінок і чоловіків 3:1). До етіологічних факторів ГУС, асоційованого з діареєю, відносять екзотоксини 1-го типу Shigella dysenteria та серотипу 0157:Н7 E. Colli. В організмі продукуються антитіла, які нейтралізують ці токсини. Цитотоксини чинять негативний вплив на ЕК. Також продукуються антитіла до пошкоджених структур ендотелію, активуються нейтрофіли. Друга форма ГУС може розвинутися під час вагітності, на тлі лікування хінідином, мітоміцином, після вірусної інфекції, ВІЛ-інфекції.
Морфологія. При асоціації ГУС з діареєю не виявляють ознак активації, проліферації та відшарування ендотелію. Відзначають внутрішньосудинні тромбоцитарні тромби, оклюзію капілярів, артеріол клубочків нирок. При другій формі ГУС тромби виявляють тільки в артеріолах і дрібних артеріях нирок.
Патогенез. В основі патогенезу ГУС лежать патологія ендотелію та агрегація тромбоцитів у мікросудинах нирок. Активація згортання крові, утворення фібрину є вторинними. При повній закупорці судин виникає некроз клубочків і канальців з розвитком артеріальної гіпертензії та хронічної ниркової недостатності. Патогенетичні механізми ГУС подібні до таких ТТП. При обох хворобах виявлено зниження концентрації вільного білка S, збільшення продукції інгібітора тканинного активатора плазміногену, локальні порушення фібринолізу.
Клініка. Клінічна картина подібна до тієї, яку реєструють при ТТП. У дітей можливий дебют з лихоманки та шлунково-кишкових порушень. Неврологічна симптоматика виражена меншою мірою, можливе ураження судин легень. Частіше уражуються нирки з розвитком гострої або хронічної ниркової недостатності, артеріальної гіпертензії.
Діагностика. Див. діагностику ТТП.
Лікування. Терапія ГУС майже не відрізняється від лікування ТТП. Застосовують плазмаферез у поєднанні з замісною терапією (плазма, еритроцитарна маса). ГК, гепарин, дезагреганти у дітей неефективні. Хворим зі стійким порушенням функції нирок проводять програмний гемодіаліз або пересадку нирок.
Прогноз. При своєчасному лікуванні прогноз сприятливий у 90% випадків.
Лікування препаратами ріжків
При тривалому лікуванні хворих з мігренню препаратами ріжків виникає хронічний ерготизм з розвитком стенозів та оклюзій артерій головного мозку, серця, нижніх кінцівок. Таке ураження нагадує ВП, артеріїт Такаясу, ОТА. Лікувальний ефект у таких випадках дають спазмолітики (антагоністи кальцію та ін.), а ГК та цитостатики неефективні та, навпаки, можуть сприяти прогресуванню стенозів.
Мукоїдна васкулопатія
Ця хвороба невідомої етіології зустрічається переважно у чоловіків молодого віку. Патологічний процес розвивається практично в усіх судинах (артеріях, венах, v. nervorum). У внутрішній та середній оболонках артерій виявляють депозити кислих мукополісахаридів, глікозаміногліканів, гіперплазію клітин стінки судини. Накопичення мукополісахаридів відбувається також в адвентиції судин (в ділянці v. nervorum), в ділянці нервових пучків. Наслідком цього процесу є концентричне потовщення інтими, середньої оболонки артерій, звуження їх просвіту. Відбувається процес дистрофічної мінералізації внутрішньої еластичної мембрани, артерії стають дуже щільними при пальпації (за консистенцією нагадують хрящ). Запальний процес (інфільтрація клітинами, деструкція еластичних та колагенових волокон) не виявляють. Ущільнюються стінки вен, які на вигляд і за гістологічною структурою не відрізняються від артерій.
Дифузна ангіокератома (хвороба Андерсона — Фабрі)
В основі хвороби лежить генетичний дефект обміну сфінголіпідів. В ендотелії, перителії, гладком’язових клітинах судин накопичуються гліколіпіди (церамідтригексозид, цераміддигалактозид). Причиною цього накопичення є зниження активності ферментів лізосомної α-галактозидази або церамідтриоксозид-α-галактозидази. У результаті порушується структура та функції багатьох органів та систем (нервова система, серце, легені, печінка, нирки, м’язи, шкіра). Дебют хвороби реєструють у віці 6–7 років. На шкірі ніг, в крижово-поперековій ділянці, навколо пупка, на сідницях з’являються висипання (ангіокератоми) червоного, синюшного кольору, які нагадують геморагічний васкуліт. Але вони м’які на дотик, піднімаються над рівнем шкіри, стають блідими при натискуванні, кровоточать при травмуванні. На деяких вузликах виявляють ділянки гіперкератозу. Уражується нервова система, виникають напади акропарестезій у пальцях рук та ніг (часом больового характеру), лихоманка, підвищується ШОЕ. Відзначають атаксію, ністагм, полінейропатію (демієлінізуюча та аксональна), менінгеальні симптоми, дистрофію рогівки ока, параорбітальні набряки, аневризматичні розширення вен кон’юнктиви (двобічні). На 2–3-му десятилітті життя розвивається ураження внутрішніх органів: серця (дилатаційна кардіоміопатія зі серцевою недостатністю, патологія клапанного апарату, блокади, інфаркт міокарда), нирок (протеїнурія, ліпідурія, хронічна ниркова недостатність), ЦНС (інсульти).
Діагноз ґрунтується на характерних клінічних проявах (ангіокератоми, акропарестезії, ураження внутрішніх органів), наявності зниження активності α-галактозидази в плазмі крові та лейкоцитах, результатах біопсії шкіри.
Радіаційне ураження судин
У ділянках судин середнього та великого калібру, на які впливала радіація, розвивається потовщення інтими, пристінковий тромбоз, оклюзія, стеноз, аневризми з їх розривом. Через 10 і більше років після дії іонізуючого опромінення настає фіброз стінки судини, периваскулярного простору, з’являються також ознаки атеросклеротичного ураження судин.
Емболія судин кристалами холестерину
Емболія судин кристалами холестерину може симулювати клінічні прояви ВП, ОТА, шкірного васкуліту. Вона розвивається у хворих, у яких проводять ангіографію (в 3% випадків), операції на магістральних судинах. Ураження малих та середніх судин ЦНС, нирок, нижніх кінцівок, шкіри викликають фрагменти атеросклеротичних бляшок (гранульом). Атероемболія спостерігається у осіб похилого віку з тяжкими атеросклеротичними ураженнями після реконструктивних операцій на судинах. У таких випадках розвивається гостра ниркова недостатність, виникають ураження шкіри. Наслідком емболізації може бути розвиток підшкірних вузликів, сітчастого ліведо, висипів геморагічного характеру, виразок, гангрени м’яких ткан, пальців ніг. Можливе ураження підшлункової залози, стінок кишечнику з перфорацією. У випадку ураження нирок у хворих відзначають мікрогематурію, протеїнурію, збільшення в крові концентрації креатиніну. Може відбуватися зниження рівня комплементу, підвищення концентрації ЦІК. У 40–90% хворих з атеротромболізацією збільшується в крові кількість лейкоцитів, еозинофілів, підвищується показник ШОЕ. Іноді зменшується кількість тромбоцитів. При біопсії уражених органів, тканин виявляють кристали холестерину в малих та середніх артеріях.
Фібромускулярна дисплазія
Хворіють переважно жінки, патологічні зміни локалізуються в ділянках відходження ниркових артерій від черевної аорти. Можливі множинні ділянки дисплазії у вигляді стенозів і мікроаневризм ниркових артерій, які чергуються. Такий процес нагадує ВП, його наслідком є реноваскулярна артеріальна гіпертензія. Для підтвердження діагнозу необхідне проведення ангіографії.
Синдром Гудпасчера
Синдром Гудпасчера — це системний капілярит з переважним ураженням легень (геморагічний пневмоніт) і нирок (гематуричний гломерулонефрит). Синдром Гудпасчера не є васкулітом як таким. Але він перехрещується з гранулемотозом Вегенера, пурпурою Шенлейна — Геноха.
Код за МКХ-10
Розділ «Системні ураження сполучної тканини» (М30–М36).
М31.0 Синдром Гудпасчера.
Епідеміологія
Частіше хворіють чоловіки у віці 20–30 років. Але розвиток синдрому Гудпасчера можливий у жінок, дітей, осіб літнього віку. Точна поширеність захворювання невідома.
Етіологія і патогенез
Етіологія синдрому Гудпасчера невідома. Відзначають зв’язок з інфекцією (вірусною та бактеріальною), непереносимістю ліків, переохолодженням. Ініціювати та викликати загострення синдрому Гудпасчера можуть віруси, пневмоцисти, грибки, D-пеніциламін, вуглеводні, вживання кокаїну. Синдром Гудпасчера може проявляти паранеопластичний характер.
Припускають спільність аутоантигенів базальних мембран альвеол і капілярів клубочків нирок. Мішенню анти-БМК-антитіл, що утворюються при синдромі Гудпасчера, є α-3 ланцюг колагену IV типу. Порушень у гені α-3 ланцюга колагену IV типу при синдромі Гудпасчера не виявлено. Існує достовірний зв’язок з носійством HLA-DRB1–15, HLA-DQB1–6. Фактором, що спричиняє розвиток синдрому Гудпасчера, є HLA-DRW-2. Ураження легень частіше виявляють у курців, перебіг захворювання у них більш тяжкий. Зв’язок анти-БМК-антитіл з α-3 ланцюгом коллагену IV типу відбувається через так званий неколагеновий домен. Існує 4 типи анти-БМК-антитіл (А, В, АВ, Х), які взаємодіють з епітопами, розташованими на неколагеновому домені. А і В типи взаємодіють відповідно з А і В епітопами, АВ — з обома. З якими епітопами взаємодіють антитіла типу Х, не з’ясовано. В експерименті індукування синдрому Гудпасчера можна викликати введенням антилегеневої сироватки шляхом імунізації мишей, у яких відсутні В-лімфоцити. Розвиток синдрому Гудпасчера може бути зумовлений порушеннями регуляції експресії імуноглобуліну G, моноклональною проліферацією імуноглобуліну А.
Відкладення уздовж базальних мембран імунних комплексів супроводжується розвитком імунозапального процесу — альвеоліту і гломерулонефриту. У запаленні беруть участь Т-лімфоцити, моноцити, ендотеліоцити, паличкоядерні лейкоцити, альвеолярні макрофаги, а також цитокіни (ІЛ-1, ФНП-α, фактори росту — тромбоцитарний, інсуліноподібний, β-трансформуючий), метаболіти арахідонової кислоти, вільні радикали, протеолітичні ферменти, адгезивні молекули.
Морфологічні зміни
У пацієнтів із синдромом Гудпасчера виявляють:
- антитіла (циркулюючі та фіксовані в тканинах) до базальних мембран капілярів нирок, які перехресно реагують із антигенами базальних мембран легень;
- зміни базальних мембран альвеолярних перегородок, легеневих і ниркових капілярів;
- у легенях виявляють некротизуючий васкуліт із крововиливами в порожнину альвеол, у нирках — проліферативний або некротичний гломерулонефрит.
Відзначають також інші морфологічні зміни:
- порушення мікроциркуляції в легенях і нирках;
- розвиток у легенях венулітів, артеріолітів, капіляритів, деструкції цих судин;
- розвиток альвеоліту з геморагічним ексудатом в альвеолах внаслідок ураження капілярів у міжальвеолярних перегородках;
- розвиток екстракапілярного проліферативного гломерулонефриту з формуванням гіалінозу і фіброзу, ниркової недостатності;
- розвиток гемосидерозу, пневмосклерозу внаслідок альвеоліту, внутрішньоальвеолярних крововиливів.
Клінічна картина
Характерні клінічні прояви хвороби представлено в табл. 21.103.
Таблиця 21.103
Характерні клінічні прояви синдрому Гудпасчера
|
Початок захворювання |
Хвороба починається гостро, після грипу |
|
Зовнішній вигляд |
Блідість шкіри, набряклість обличчя, ціаноз слизових оболонок |
|
Загальні симптоми |
Слабість, зниження працездатності, схуднення, підвищення температури тіла до фебрильних значень, зменшення м’язової сили |
|
Характерні симптоми |
Кровохаркання або легенева кровотеча, задишка, лихоманка, артралгії, міалгії |
|
Серцево-судинна система |
Відзначають артеріальну гіпертензію, збільшення розмірів серця, ослаблення тонів. Вислуховується систолічний шум над верхівкою і у точці Боткіна. У випадку розвитку тяжкої ниркової недостатності може з’явитися шум тертя перикарда. При вираженій артеріальній гіпертензії може розвинутися гостра серцева недостатність, набряк легень. Особливо це стосується термінальної стадії хвороби |
|
Легені |
Виникають кашель, біль у грудній клітці, кровохаркання (або легенева кровотеча), задишка, що посилюється при фізичному навантаженні. Аускультативно вислуховується велика кількість сухих та вологих хрипів у нижніх і середніх відділах легень (кількість вологих хрипів збільшується в період кровохаркання). Перкусія виявляє вкорочення перкуторного звуку над ділянками легені, де відбулися крововиливи. Рентгенологічно проявляється множинна вогнищева та зливна інфільтрація в обох легенях. Рецидивуючі легеневі кровотечі призводять до анемії. Можливе ураження легень за типом фіброзуючого альвеоліту, що розвивається при сполученні анти-БМК-антитіл з АНЦА. Інтерстиціальна пневмонія може передувати легеневим кровотечам та швидкопрогресуючому нефриту |
|
Нирки |
Одночасно з легеневою симптоматикою або трохи пізніше розвивається швидкопрогресуючий гломерулонефрит, може виникнути нефротичний синдром і ниркова недостатність. Симптоматичну артеріальну гіпертензію реєструють рідко. Ниркова недостатність, що розвинулася, посилює прояви анемії. При морфологічному дослідженні кількість клубочків з півмісяцями може перевищувати 85%. Відзначають лінійні або гранулярні (рідше) відкладення IgG і С3. Макрогематурію діагностують більш ніж в 30% хворих, у більшості випадків анемія помірна. Функція нирок знижується дуже швидко: протягом декількох діб з моменту початку хвороби може досягти термінального рівня. Артеріальна гіпертензія виникає тільки при розвитку термінальної ниркової недостатності |
|
ЦНС |
Нерідко розвивається церебральний васкуліт, епілептичний статус. При ураженні головного мозку ефективним є циклофосфамід |
|
Орган зору |
До рідкісних проявів синдрому Гудпасчера належать двобічний ретиніт, увеїт, зниження гостроти зору |
Найчастіше синдром Гудпасчера починається із клінічних проявів ураження легень. Початок захворювання із клінічних ознак ураження нирок виявляють в 10–15% випадків. У таких випадках відзначають клінічну картину гломерулонефриту — олігурію, набряки, артеріальну гіпертензію. Симптоми ураження легень приєднуються пізніше.
Перебіг хвороби. У більшості випадків хвороба неухильно прогресує, розвивається легенева й ниркова недостатність, які є найбільш частою причиною смерті хворих. Тривалість життя хворих варіює від декількох місяців до 3 років.
Діагностика
Результати лабораторного та інструментального обстеження хворих представлено в табл. 21.104.
Таблиця 21.104
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих із синдромом Гудпасчера
|
Лабораторні дослідження |
|
|
Загальний аналіз крові |
Виникають лейкоцитоз зі зрушенням лейкоцитарної формули вліво, гіпохромна залізодефіцитна анемія, значне підвищення ШОЕ. Залізодефіцитна анемія може розвитися раніше за ознаки поразки легенів і нирок, але частіше вона пов’язана із втратою крові |
|
Біохімічні дослідження |
Зниження концентрації в крові заліза, збільшення вмісту креатиніну, сечовини, α2— і γ-глобулінів, серомукоїду, гаптоглобіну |
|
Імунологічні дослідження |
Виявляють ЦІК, антитіла до БМК клубочків нирок і альвеол (метод непрямої імунофлуоресценції, радіоімунологічний метод). Можливе зниження вмісту в крові Т-лімфоцитів. У 20% пацієнтів виявляють АНЦА |
|
Дослідження мокротиння |
Виявляють еритроцити, гемосидерин, сидерофаги |
|
Інструментальні дослідження |
|
|
Рентгенографія легень |
Легеневі інфільтрати, розміщені в прикореневій ділянці, середніх і нижніх відділах легень. Інфільтрація має прогресуючий характер |
|
Спірографія |
Відзначають порушення функції зовнішнього дихання спочатку за рестриктивним типом, надалі — за змішаним |
|
Газовий склад крові |
Розвивається артеріальна гіпоксемія |
|
ЕКГ |
Зниження амплітуди зубців Т, інтервалу ST, ознаки гіпертрофії міокарда лівого шлуночка |
Показання до проведення відкритої біопсії легеневої тканини і нирок
Біопсію проводять у випадках, коли неможливо встановити діагноз за допомогою неінвазивних методів. Зміни у біоптатах виявляють за допомогою гістологічного та імунофлуоресцентного методів (табл. 21.105).
Таблиця 21.105
Зміни у біоптатах, виявлені за допомогою гістологічного та імунофлуоресцентного методів
|
Гістологічний метод |
Ознаки геморагічного альвеоліту, гемосидерозу, альвеолярного фіброзу в легеневій тканині й гломерулонефриту (частіше екстракапілярного) — у нирках |
|
Імунофлуоресцентний метод |
Лінійні відкладення імуноглобуліну G і С3-компонента комплементу на базальних мембранах легеневих альвеол і ниркових клубочків |
У процесі проведення діагностики потрібно враховувати:
- молодий вік пацієнтів;
- виникнення хвороби після грипу;
- задишку;
- наявність геморагічної пневмонії;
- наявність гематуричного гломерулонефриту, нефротичного синдрому (рідше) з хронічною нирковою недостатністю;
- наявність у сироватці крові антитіл до базальних мембран нирок, анемії;
- артеріальну гіпертензію (рідко).
Діагностичні критерії синдрому Гудпасчера:
- Сполучення легеневого кровохаркання/кровотечі з симптоматикою гематуричного гломерулонефриту або нефротичним синдромом.
- Відсутність симптомів ураження інших органів.
- Прогресуючий перебіг хвороби з розвитком дихальної й ниркової недостатності.
- Анемія, зумовлена легеневими кровотечами й гематурією.
- Артеріальна гіпертензія (не завжди).
- Виявлення в легенях при рентгенологічному обстеженні двобічної інфільтрації, сітчастої деформації легеневого малюнка.
- Виявлення в сироватці крові високих титрів антитіл до БМК альвеол і ниркових клубочків.
- Виявлення в біоптатах депозитів імуноглобуліну G і С3-компонента комплементу.
Синдром Гудпасчера доводиться диференціювати з системними васкулітами, геморагічними діатезами, захворюваннями, які супроводжуються легеневою гіпертензією, туберкульозом легень, бронхоектазами та ін. Можливе сполучення синдрому Гудпасчера з іншими захворюваннями, зокрема розсіяним склерозом, пурпурою Шенлейна — Геноха, міастенією, виразковим колітом, первинним біліарним цирозом, цукровим діабетом II типу, макроглобулінемією Вальденстрема.
Лікування
Терапію, яку проводять при синдромі Гудпасчера, наведено в табл. 21.106.
Таблиця 21.106
Терапія, яку проводять при синдромі Гудпасчера
|
Терапія |
Дози препаратів, методи лікування |
Примітки |
|
ГК |
Преднізолон 100 мг/добу (або метилпреднізолон) |
У сполученні з цитостатиками |
|
Пульс-терапія метилпреднізолоном по 1,0 г протягом 3 днів |
Можлива комбінація з плазмаферезом |
|
|
Імуносупресори |
Циклофосфамід 150–200 мг/добу |
У сполученні з преднізолоном або метилпреднізолоном |
|
Азатіоприн 150–200 мг/добу |
У сполученні з преднізолоном або метилпреднізолоном |
|
|
Еферентні та інші методи |
Плазмаферез, лімфоцитоферез, імуносорбція |
|
|
Гемодіаліз, трансплантація нирок |
У випадках розвитку хронічної ниркової недостатності |
|
Контрольовані дослідження не проводили через рідкісність патології. Плазмаферез, імуносорбцію в поєднанні з активною імуносупресивною терапією застосовують і у випадках сполучення анти-БМК-антитіл з АНЦА. Ефективним є поєднання високих доз ГК та циклофосфаміду, особливо при легеневих ураженнях. Імуносупресори меншою мірою ефективні при ураженні нирок. У деяких випадках більш ефективним є мікофенолова кислота. Пересадження нирок не попереджає загострення легеневого процесу.
Прогноз
Методи лікування, застосовувані сьогодні, не дозволяють досягти повного одужання (рецидиви хвороби можуть виникнути навіть через 10 років від її початку). Фактори ризику розвитку рецидивів не ідентифіковані. Прогноз несприятливий: протягом 6–12 міс наступає смерть внаслідок ниркової або легенево-серцевої недостатності. Факторами ризику несприятливих наслідків при синдромі Гудпасчера є:
- ураження нирок, що передує легеневим кровотечам;
- олігурія або анурія в момент установлення діагнозу;
- сироватковий креатинін >600 мкмоль/л;
- півмісяці в >50–85% клубочків.
Феномен Рейно
Феномен Рейно — вазоспастичний розлад, для якого характерні періодичні напади відмежованої зміни кольору пальців, які супроводжуються болем і відчуттям задубіння при перебуванні на холоді.
Код за МКХ-10
I73.0 Синдром Рейно.
Епідеміологія
Поширеність феномену Рейно становить близько 3–4%, у країнах з холодним кліматом вона може сягати 30%. Істинна поширеність невідома, оскільки хворі з легкою формою хвороби, як правило, не звертаються за медичною допомогою.
Етіопатогенез
Феномен Рейно може мати первинний і вторинний характер. Первинний феномен Рейно частіше відзначають у жінок (4–9:1). Вторинний — у хворих з різною патологією:
- системними ревматичними захворюваннями;
- професійними хворобами (вібраційна хвороба),
- облітеруючим ураженням артерій;
- захворюваннями, викликаними хімічними речовинами і лікарськими засобами;
- синдромами підвищеної в’язкості крові та ін.
Регуляція периферичного кровотоку в організмі людини забезпечується:
- тонусом самих судин;
- активністю симпатичної нервової системи;
- реологічними факторами, у тому числі в’язкістю крові;
- дією циркулюючих гормонів.
У пальцях рук існують тільки симпатичні вазоконстрикторні волокна. Вазоконстрикція розвивається у відповідь на охолодження і зумовлена посиленням симпатичного тонусу. Механізм вазоспазму як при первинному, так і вторинному феномені Рейно не з’ясований. Артеріальний кровотік у пальці залежить від градієнта тиску, який у свою чергу визначається в’язкістю крові, довжиною радіуса судини. Радіус судини змінюється і залежить від товщини її стінки, тонусу гладких м’язів, активності симпатичної нервової системи. Відповідно до закону Пуазейля кровотік по судині зворотно пропорційний IV ступеню його радіусу. Розвиток феномену Рейно пояснюють:
- гіперактивністю симпатичної нервової системи;
- порушенням місцевої регуляції судинного тонусу;
- підвищеною периферичною рецепцією α-адренергічної стимуляції;
- дефіцитом кальцитонін-гензв’язаного пептиду, судинорозширювального нейропептиду;
- високою концентрацією ендотеліну.
При вторинному феномені Рейно припиненню кровотоку по артеріях пальців сприяють судинні аномалії. Спровокувати припинення кровотоку по артерії пальця можуть:
- проксимальна обструкція;
- оклюзія судини;
- порушення реології (підвищена в’язкість, поліцитемія).
Колір ділянки ураження при феномені Рейно послідовно змінюється від білого до синього, потім до червоного. Збліднення настає при початковому спазмі артерії пальця. Деоксигенація венозної крові, що наступила, призводить до забарвлення пальця в синій колір, реактивна гіперемія — до почервоніння (фінальна стадія):
- Перша фаза. Порушується кровообіг, пальці біліють.
- Друга фаза. Розвивається ціаноз, зумовлений локальною акумуляцією відновленого гемоглобіну.
- Третя фаза. Кровообіг поновлюється, пальці червоніють.
Клінічна картина
Клінічні прояви при первинному і вторинному феномені Рейно дещо відрізняються (табл. 21.107).
Таблиця 21.107
Клінічні прояви первинного та вторинного феномену Рейно
|
Первинний |
Первинний феномен Рейно починається у жінок у віці 15–45 років. Частіше уражуються пальці верхніх кінцівок, у 40% пацієнтів — і нижніх. Можуть уражуватись також ніс, язик і губи. Великі пальці з незрозумілих причин до патологічного процесу не залучаються. Зміни кольору поєднуються з відчуттям задубіння на ішемічній стадії і пульсуючим болем на стадії реактивної гіперемії. Тяжкість, частота і тривалість нападів широко варіюють. У деяких пацієнтів реєструють декілька нападів за день, у інших — 2–3 протягом зими. У результаті повторних нападів виникають виразки пальців, тріщини та гангрена |
|
Вторинний |
Початок вторинного феномену Рейно відзначають у чоловіків і жінок на 3–4-му десятилітті життя. Хоча симптоми вазоспазму ідентичні первинному, трофічні ускладнення виникають значно частіше |
Усі 3 фази феномену Рейно відзначають рідко. Найхарактернішою ознакою вважають збліднення, ціаноз може відбуватися без спазму судин у здорових людей. Тому для діагностики феномену Рейно необхідно бачити хоч би 2 фази — збліднення і гіперемію. Основним провокуючим чинником при феномені Рейно є холод, особливо за умови стиснення пальцями різних предметів на холоді. До інших провокуючих факторів відносять прийом гормонів, травми, емоції. Спазм судин при феномені Рейно не обмежується тільки артеріями пальців. Тому деякі автори вважають феномен Рейно системним судинним захворюванням, оскільки ця патологія супроводжується спазмом артерій різної локалізації: стравоходу, трахеї, міокарда, легень, нирок. В осіб з первинним феноменом Рейно реєструють часту мігрень і біль у грудній клітці.
Діагноз
При фізикальному обстеженні пацієнтів з первинним феноменом Рейно відхилень від норми часто не виявляють. Метою огляду є виключення вторинного характеру феномену Рейно на тлі інших хвороб і передусім системних захворювань сполучної тканини. Необхідно провести мікроскопію капілярів навколонігтьового валика пацієнтів. Діагноз встановлюють досить легко, коли пацієнти звертаються у клініку в момент нападу. Провокація нападу зануренням рук пацієнта в холодну воду не завжди дає результати. Вивчають стан капілярного русла навколонігтьових валиків і сітківки ока. На навколонігтьовий валик наносять краплю імерсійної олії і поміщають його під офтальмоскоп (+40 діоптрій). Капілярне русло потрапляє у фокус на відстані декількох міліметрів від лінзи. При нормальному стані капілярного русла видно рівномірно розподілені капіляри. Системні захворювання сполучної тканини характеризуються розширеними, звитими, нерівномірно розподіленими капілярами з ділянками «безсудинної» тканини. Патогенетичних лабораторних тестів для діагностики феномену Рейно сьогодні немає. При первинному феномені Рейно у більшості випадків лабораторні показники нормальні, у ⅓ пацієнтів у сироватці крові виявляють низький титр антинуклеарних антитіл. У ряду хворих (менше 25%) надалі розвиваються аутоімунні процеси. Пацієнти з ознаками системних захворювань сполучної тканини потребують відповідного обстеження. До маркерів можливого вторинного феномену Рейно належать:
- чоловіча стать;
- початок нападів спазму судин пальців після досягнення віку 30 років;
- однобічне ураження або зміна кольору шкіри на усій кінцівці;
- наявність трофічних змін;
- патологічні зміни при мікроскопічному обстеженні капілярів навколонігтьового валика;
- наявність склеродактилії, стоншення та сухості шкіри;
- виявлення аутоантитіл (особливо антицентромерних, антинуклеарних).
Лікування
Медикаментозна терапія показана хворим з тяжкими, частими і тривалими нападами, що виникають при мінімальній провокації та дотриманні профілактичних заходів. У більшості хворих її проводять тільки в зимові місяці. Досить хороший ефект дають:
1. Блокатори кальцієвих каналів (дигідропіридинові похідні — ніфедипін, ісрадипін, амлодипін).
2. Симпатолітичні засоби (празозин і феноксибензамін, що виявляють пряму судинорозширювальну дію).
3. Антагоністи серотонінових рецепторів (серотонінзалежну вазоконстрикцію і агрегацію тромбоцитів блокує кетансерин).
4. Аналоги простагландину (гальмування агрегації тромбоцитів і пряму вазодилатацію викликає ілопрост).
5. Гідралазин і нітрати для локального застосування.
Дози та ефекти деяких препаратів наведено в табл. 21.108.
Таблиця 21.108
Дози препаратів, які застосовують при лікуванні феномену Рейно
|
Група препаратів |
Препарат |
Примітки |
|
Блокатори кальцієвих каналів |
Ніфедипін у дозі 30–60 мг/добу за 3–4 прийоми |
Знижує частоту, інтенсивність, зменшує тривалість епізодів вазоспазму, прискорює загоєння дигітальних виразок. До негативних ефектів належать рефлекторна тахікардія, головний біль, запаморочення, гіперемія обличчя. Артеріальна гіпотензія, негативна хронотропна дія призводять до набряку гомілок |
|
Амлодипін у дозі 5–20 мг/добу |
Знижує частоту та інтенсивність вазоспастичних приступів. Може викликати набряк гомілок (у 50% випадків) |
|
|
Ісрадипін у дозі 5 мг/добу за 2 прийоми |
При недостатньому ефекті дозу можна підвищити до 10 мг/добу |
|
|
Фелодипін у дозі 5–10 мг/добу |
Знижує частоту та інтенсивність вазоспазму |
|
|
Дилтіазем у дозі 90–240 мг/добу |
Знижує частоту та інтенсивність вазоспазму (не у всіх хворих) |
|
|
Блокатори зворотного захвату серотоніну |
Флуоксетин у дозі 20 мг/добу |
Знижує частоту та інтенсивність вазоспазму |
|
Блокатори α-адренорецепторів |
Празозин у дозі 1–6 мг/добу |
Достовірно знижує частоту та інтенсивність вазоспазму у хворих з первинним феноменом Рейно |
|
Блокатори рецепторів ангіотензину II |
Лозартан у дозі 50 мг/добу |
Знижує частоту та інтенсивність вазоспазму, особливо при первинному феномені Рейно (більш ефективно, ніж ніфедипин) |
|
Простагландини |
Алпростадил (простагландин Е1) щоденно в/в 20–60 мкг в 100–200 мл фізіологічного розчину хлориду натрію протягом 15–20 днів |
Сприяє зниженню частоти та інтенсивності епізодів вазоспазму, загоєнню виразок на пальцях (стимулює репарацію трофічних виразок кінцівок). Терапевтичний ефект зберігається протягом 4–9 міс |
|
Синтетичні простацикліни |
Ілопрост |
Пригнічує агрегацію та адгезію тромбоцитів. Зменшує кількість та інтенсивність епізодів вазоспазму, прискорює загоєння виразок на пальцях. Не має переваг перед алпростадилом (виникає багато побічних ефектів) |
|
Інгібітори фосфодіестерази 5-го типу |
Силденафіл |
Викликає вазодилатацію, зменшує кількість та інтенсивність епізодів вазоспазму, прискорює загоєння виразок на пальцях |
|
Неселективні антагоністи ендотелінових рецепторів |
Бозентан |
Знижує частоту розвитку нових дигітальних виразок |
|
Антиагреганти |
Пентоксифілін усередину 600–1200 мг/добу, в/в 300 мг в ізотонічному розчині натрію хлориду або в 5% розчині глюкози |
Пригнічує агрегацію тромбоцитів |
|
Низькомолекулярні декстрани |
Декстран |
В/в інфузії 200–600 мл щоденно в комплексі з антиагрегантами |
У деяких випадках проводять оперативне втручання. Показаннями до хірургічного лікування є:
- відсутність ефекту від консервативної терапії;
- ішемічні ускладнення, які регулярно повторюються;
- загроза розвитку некрозу пальців.
Перед операцією проводять блокаду зірчастого ганглія або епідуральну блокаду. Якщо тимчасова симпатична блокада супроводжується тривалим клінічним поліпшенням, проводять шийну або поперекову симпатектомію. Надалі застосовують підтримувальну терапію вазодилататорами.
Профілактика нападів при феномені Рейно. Необхідно припинити паління, попереджати зниження температури усього тіла (використовувати теплий одяг, хімічні й електричні обігрівачі для рук і ніг та ін.).
Прогноз
Прогноз сприятливий у пацієнтів з первинним феноменом Рейно. У 10% з них напади повністю проходять, у ⅓ кількість і тяжкість нападів з часом зменшуються, ще у ⅓ — істотних змін не виявляють. У більшості пацієнтів з первинним феноменом Рейно супутні хвороби ніколи не розвиваються, тести на аутоімунні антитіла негативні.
Глава 22. Ревматоїдний артрит
РА — аутоімунна хвороба з невідомою етіологією, для якої характерним є симетричний ерозивний артрит (синовіт) та широкий спектр позасуглобових (системних) проявів.
Код за МКХ-10
М05 Серопозитивний ревматоїдний артрит
М05.0 Синдром Фелті
Ревматоїдний артрит зі спленомегалією і лейкопенією
М05.1 Ревматоїдна хвороба легень (J99.0)
М05.2 Ревматоїдний васкуліт
М05.3 Ревматоїдний артрит із залученням інших органів і систем
Ревматоїдний(-а):
- кардит (I52.8)
- ендокардит (I39.-)
- міокардит (I41.8)
- міопатія (G73.7)
- перикардит (I32.8)
- поліневропатія (G63.6)
М05.8 Інші серопозитивні ревматоїдні артрити
М05.9 Серопозитивний ревматоїдний артрит неуточнений
М06 Інші ревматоїдні артрити
М06.0 Серонегативний ревматоїдний артрит
М06.1 Хвороба Стіла, що виникла у дорослих
Виключена: хвороба Стіла БДУ (М08.2)
М06.2 Ревматоїдний бурсит
М06.3 Ревматоїдні вузлики
М06.4 Запальна поліартропатія
Виключений: поліартрит БДУ (М13.0)
М06.8 Інші уточнені ревматоїдні артрити
М06.9 Ревматоїдний артрит неуточнений
Епідеміологія
Розповсюдженість РА становить близько 0,5–1,5% населення у всьому світі, а економічні втрати від РА для суспільства можна зіставити з такими при ішемічній хворобі серця. Через 5 років від початку хвороби, незважаючи на лікування базовими препаратами, 16% пацієнтів втрачають працездатність, а через 20 років — 90%, у третини з яких розвивається повна інвалідність.
В Україні налічується понад 118 тис. хворих на РА, серед них близько 54 тис. — працездатного віку та перебувають на диспансерному обліку.
Згідно з даними Державної статистичної звітності МОЗ України за 2009 р. показник захворюваності серед працездатного населення становить 15,2 на 100 тис. населення (в абсолютних показниках — 4226 випадків).
Жінки хворіють частіше за чоловіків у 2–5 разів, співвідношення чоловіків і жінок становить у середньому 1:2,5–3. РА може виникати в будь-якому віці (в дитячому — ювенільний РА), але найчастіше хворобу діагностують у віці 40–50 років. Серед осіб у віці молодше 35 років поширеність РА становить 0,38%, а 55 років і старше — 1,4%. Високу частоту розвитку РА відзначають у близьких родичів хворих (3,5%), особливо у осіб жіночої статі (5,1%).
Життєвий прогноз у пацієнтів із РА такий самий несприятливий, як при лімфогранулематозі, цукровому діабеті 2-го типу, трьохсудинному ураженні коронарних артерій.
Витрати, пов’язані з ревматоїдним артритом:
- США: 0,5–1% = 2 млн хворих. Витрачається 65 млрд дол. США.
- Канада: 300 тис. хворих. 6,2 млрд дол.
- Росія: 275 тис. хворих. 650 тис. дол.
Загальні витрати у Великобританії, пов’язані з РА, оцінюють у 1,3 млрд фунтів стерлінгів, а в Швеції ці витрати перевищують 3 млрд шведських крон (близько 423 млн дол.) на рік, що відповідає витратам на лікування хворих онкологічного профілю. Згідно з даними фармакоекономічного аналізу, проведеного в США, встановлено, що роботодавець з 5000 працівниками за рік витрачає 1,2 млн дол. за рахунок працівників з РА, що приблизно відповідає 15,9 тис. дол. на одного працівника з РА в рік.
Етіологія
Етіологія РА невідома. Вивчають тригерну роль широкого спектра екзогенних (наприклад куріння), інфекційних (вірус Епштейна — Барр, парвовірус В19, ретровіруси, суперантигени і стресорні білки бактерій) і ендогенних (цитруліновані білки і пептиди) чинників.
Крім того, потенційно артритогенні стимули умовно поділяють на дві категорії.
- Ад’ювантні. До цієї групи відносять мікробні (глюкани, ліпополісахариди, бактеріальну ДНК) і зовнішьосередовищні (куріння, компоненти мінеральних масел, вугільний пил) чинники.
- Аутоантигенні. До них належать білки колагену (колаген типів II, IX, X, XI, олігомерний матриксний білок хряща, протеоглікани).
Найважливішим фактором ризику розвитку РА вважають куріння, яке часто асоціюється з більш тяжким перебігом захворювання (серопозитивність за РФ, поява ревматоїдних вузликів, швидке ерозування суглобів). Наприклад, у жінок, які курять більше 25 сигарет на день, ризик розвитку РА протягом 20 років становить 1,4.
Припускають, що потенційні етіологічні чинники, взаємодіючи з генетичною схильністю, беруть не пряму, а опосередковану участь у розвитку РА.
Генетична схильність. Існування генетичної схильності в розвитку РА підтверджують наступні факти.
- у монозиготних близнюків виявляють більш високу конкордантність у розвитку РА, ніж у дизиготних (12–15% проти 3,5%);
- у родичів першої лінії спорідненості ризик захворіти на РА становить 1,5 порівняно з популяцією.
Ризик розвитку РА асоційований з носійством антигену головного комплексу гістосумісності класу II НLА-DR4 (і DR1), який включає більше 22 алелей. При вивченні індивідуальних алелів ідентифіковано два, найбільш тісно асоційованих з РА: DRВ1*0401 і DRВ1*0404. Характерною особливістю цих алелей вважають амінокислотну послідовність (лейцин-глютамін-лізин-аргінін-аланін в положеннях відповідно 67, 70, 71, 72 і 74) в третій гіперваріабельній області НLА-DR3-ланцюгів, яка отримала назву «загальний епітоп» (shared epitope — SЕ). Носійство НLА-DRВ1*0401 виявляють у 50–61% пацієнтів з РА, а НLА-DRВ1*0404 — у 27–37%.
Існує декілька гіпотез, які пояснюють зв’язок між носійством SЕ і розвитком РА. Нагадаємо, що функція НLА-DR, експресується на поверхні так званих антиген-презентуючих клітин, полягає у зв’язуванні та поданні (презентуванні) пептидних антигенів СD4+ Т-лімфоцитам. Оскільки поліморфізм амінокислотних залишків головного комплексу гістосумісності класу II визначає структуру пептиду, який може зв’язуватися з високою авідністю, яка визначає характер активації СD4+ Т-лімфоцитів, припускають, що SЕ може ефективно презентувати артритогенний пептид Т-лімфоцитам. Відповідно до іншої моделі, при виникненні молекулярної мімікрії між амінокислотною послідовністю SЕ і артритогенного антигену сам SЕ може індукувати активацію Т-клітин. Виявлена гомологія амінокислотних послідовностей SЕ і деяких артритогенних антигенів, таких як глікопротеїн вірусу Епштейна — Барр, білок теплового шоку, Еscherichia coli та ін. Крім того, носійство SЕ асоційоване з тяжкістю перебігу РА і прогресуванням ерозивного процесу. За даними метааналізу (10 досліджень, 1027 пацієнтів з РА), носійство одного або двох алелей DR4+ пов’язано з дворазовим підвищенням ризику ерозії суглобів. Ризик розвитку позасуглобових проявів (ревматоїдний васкуліт, інтерстиціальні хвороби легень, синдром Фелті) також асоційований з носійством SЕ [0401,0404 і (або) 0408]. Нарешті, носійство двох алелів DR («доза» гена), що мають SЕ, сильніше пов’язане з тяжкістю РА, ніж носійство однієї копії. Таким чином, SЕ асоційоване не з ризиком виникнення РА в цілому, а з розвитком тяжкого варіанта перебігу хвороби.
Окремий інтерес становлять дані про зв’язок між SЕ, факторами ризику розвитку РА і аутоімунними порушеннями (РФ і АЦЦП (антитіла до циклічного цитрулінового пептиду)), характерними для цієї хвороби. Нагадаємо, що серопозитивність за РФ пов’язана з тяжким перебігом і несприятливим прогнозом хвороби. Імунні комплекси, що містять РФ, мають виражений патогенний потенціал.
Особливу увагу привертають АЦЦП. Встановлено, що утворення цитруліну з аргініну є посттрансляційною конверсією однієї амінокислоти в іншу в результаті дезамінування. Процес регулюють ферменти, які кодуються сімейством генів пептидиларгініндезамінази (ПАДI1-ПАДI4 і ПАДI16). Оскільки аргінін — позитивно заряджена амінокислота, а цитрулін — нейтрально, поява останньої призводить до зміни структури і підвищення імуногенності модифікованих білків. Зростає їх афінність до DR4 і здатність активувати Т-лімфоцити. Цитрулювання білків — універсальний процес, асоційований з розвитком запалення, а також із впливом факторів зовнішнього середовища, в першу чергу, куріння. В якості артритогенних цитрульованих білків можуть виступати фібриноген, віментин, фібронектин, α-енолаза, антиген-1 і ядерні білки вірусу Епштейна — Барр, а також аутоепітопи, локалізовані в антигензв’язуючих ділянках рецепторів Т- і В-лімфоцитів.
У недавніх дослідженнях було переконливо показано, що поєднання носійства SЕ з курінням (а також зі зловживанням кофеїну і прийомом контрацептивів) істотно підвищує ризик розвитку РФ-позитивного і особливо АЦЦП-позитивного РА. Примітно, що за відсутності носійства SЕ фактори зовнішнього середовища не підвищують ризик розвитку цієї хвороби. Обговорюють роль та інших генетичних факторів, безпосередньо не пов’язаних з НLА-DR. До них відносять поліморфізм генів пептидиларгініндезамінази, білка тирозинфосфатази N22 (protein tyrosine phosphatase N22 — PTPN2), суtotoxic-Т-lymphocyte-associated antigen-4 — СТLА-4.
Припускають, що при РА пептидиларгініндезаміназа стає стабільнішою, що, в свою чергу, може призводити до підвищення здатності ферменту цитрулювати білки або до зростання кількості модифікованих білків з вираженою артритогенною активністю.
Ген РТРN22 кодує синтез тирозинфосфатази — ферменту, який регулює поріг активації Т-клітинних рецепторів. У пацієнтів з АЦЦП-позитивним РА виявлено підвищення частоти носійства алелів гена РТРN22, яке асоційоване зі зниженням порога активації Т-клітин, а отже, з їх гіперактивністю. Важливу роль може грати поліморфізм гена СТLА-4, продукти якого пригнічують надлишкову активацію Т-лімфоцитів. Нагадаємо, що для оптимальної активації Т-лімфоцитів необхідно, як мінімум, два сигнали. Один з них може бути реалізований в процесі взаємодії Т-клітинних рецепторів з комплексом «пептид — головний комплекс гістосумісності», експресованим на мембрані антиген-презентуючих клітин, інший — за рахунок взаємодії так званих кістковостимулюючих рецепторів на Т-клітинах і відповідних лігандів на антиген-презентуючих клітинах. Ключовий кістковостимуляторний сигнал забезпечує взаємодію СD28 на Т-лімфоцитах і СD80 та СD86 на антиген-презентуючих клітинах. При отриманні обох сигналів Т-лімфоцити проліферують і синтезують цитокіни, які активують інші клітини імунної системи (в першу чергу макрофаги). За відсутності кістковостимуляторного сигналу Т-лімфоцити втрачають здатність ефективно відповідати на антигенні стимули і гинуть шляхом апоптозу. Зв’язування СТLА-4, що являє собою високоавідних ліганд для СD80 і СD86, запобігає появі кісковостимулюючого ефекту активації СD28 на Т-лімфоцити (кістковостимулюючий сигнал 2) і таким чином пригнічує активацію Т-клітин. Носійство певних алелів СТLА-4 асоційоване з розвитком АЦЦП-позитивного РА.
Отже, результати проведених досліджень свідчать про існування принаймні двох клініко-генетичних субтипів РА.
- АЦЦП-позитивний субтип. Розвиток асоційований з SЕ, НLА-DRВ1, визначеними алелями генів РТРN22, СТLА-4, пептидиларгініндезамінази і вираженою взаємодією генетичних чинників і факторів зовнішнього середовища, в першу чергу куріння.
- АЦЦП-негативний субтип. Його розвиток пов’язують з носійство НLА-А1, В8, DRВ1*03 гаплотипу.
Фактори ризику розвитку ревматоїдного артриту
У великому проспективному дослідженні встановлено, що якщо маса тіла при народженні >4,54 кг, то ризик розвитку РА у зрілому віці підвищується в 2 рази.
У дослідженні P. Stolt та співавторів встановлено зв’язок куріння з розвитком РА тільки у серопозитивних хворих і епітопними алелями за системою HLA.Також було визначено зв’язок між курінням і епітопними алелями і АЦЦП.
АЦЦП субтип альфа-енолаза більш чітко корелює з виявленням РА.
У курців з хронічною обструктивною хворобою легень зростає ризик розвитку аутоімунних захворювань (ССД, СЧВ та ін.), у тому числі РА.
Встановлено ризик розвитку цукрового діабету у пацієнтів з РА, ПсА і псоріазом. Підтвердженням цього є те, що ефективна протиревматична терапія може нормалізувати вуглеводневий обмін (повернути рівень глюкози до нормальних значень).
Патогенез
З точки зору патогенетичних механізмів, РА — гетерогенне захворювання. В основі розвитку лежить складний взаємодоповнюючий вплив генетично детермінованих та набутих дефектів нормальних імунорегуляторних механізмів, які обмежують патологічну активацію імунної системи у відповідь на потенційно патогенні, а нерідко й фізіологічні стимули. Це визначає різноманіття клінічних та імунологічних проявів, що робить РА більш схожим на клініко-імунологічний синдром, ніж на одну гомогенну хворобу.
Основу патологічного процесу при РА становить системне аутоімунне запалення, яке найбільше зачіпає синовіальну оболонку суглобів. Саме розвиток прогресуючого неконтрольованого синовіального запалення відрізняє РА від інших хронічних запальних захворювань людини як ревматичної, так і неревматичної природи.
Провідна морфологічна ознака ревматоїдного запалення — формування ектопічного вогнища гіперплазії синовіальної тканини. Інвазивний ріст цієї структури (пануса) призводить до руйнування суглобового хряща й субхондральної кістки. У синовіальній тканині відзначають збільшення кількості сіновіоцитів типу А (клітин, що нагадують макрофаги) і В (клітин, які нагадують фібробласти), товщини інтими (з 2–3 до 10 і більше шарів вистільних клітин), інфільтрацію імунними та запальними клітинами (макрофагами, Т- і В-лімфоцитами, плазматичними і дендритними клітинами), утворення фолікулів, які складаються з лімфоїдних клітин і нагадують центри росту лімфатичних вузлів. Рання ознака ревматоїдного синовіту — утворення нових судин (неоангіогенез). Цей процес асоційований з транссудацією та міграцією лімфоцитів у синовіальну тканину і лейкоцитів — в синовіальну рідину.
Розвиток РА пов’язаний з генетично детермінованою Т-клітинною імунною відповіддю на широкий спектр потенційно патогенних (артритогенних) антигенів. При РА переважає Тh1-тип імунної відповіді. Для нього характерна гіперпродукція прозапальних цитокінів, таких як ФНП-α, ІЛ-1, -12, -7, -17, -18, а також ІЛ-2 і гамма-інтерферон. Важливий наслідок поляризації імунної відповіді за Тh1-типом — переважання синтезу прозапальних цитокінів над антизапальними (Тh2-типу). У широкому спектрі прозапальних цитокінів центральне місце займають ФНП-α, ІЛ-1 та ІЛ-6, які індукують синтез медіаторів, що підтримують запалення і сприяють руйнуванню суглобів.
Патогенез РА представлений на рис. 22.1.
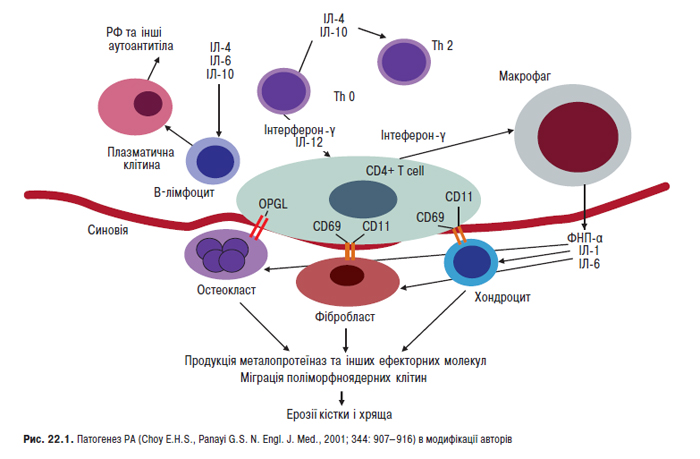
Використання нових моделей дозволило краще зрозуміти прогресування РА і зв’язок між запаленням і пошкодженням. Механізми зв’язку між запаленням та ерозією при РА.
- Аутоімунні В-лімфоцити не руйнуються в кістковому мозку і мігрують в суглоб, вони мігрують до синовіальної оболонки, де взаємодіють з Т-лімфоцитами та іншими клітинами запалення, що викликають синовіт.
- В-клітини так само рухаються до субхондральної кістки, викликаючи остеїт.
- Ерозії кістки так само викликаються ФНП-α і RANKL (Receptor activator of nuclear factor kappa-B ligand), через активацію остеобластів.
Ці механізми можуть бути заблоковані таким чином:
- Ритуксимаб виснажує В-лімфоцити, які запускають механізм розвитку РА, перериваючи запалення і розвиток ерозій.
- Анти-ФНП-α блокують розвиток синовіту і остеїту, отже блокують розвиток ерозій, а також опосередковану RANKL активацію осеокластів.
- Анти-RANKL і золедронова кислота безпосередньо блокують розвиток ерозії самі собою.
Блокування цих механізмів представлено на рис. 22.2.
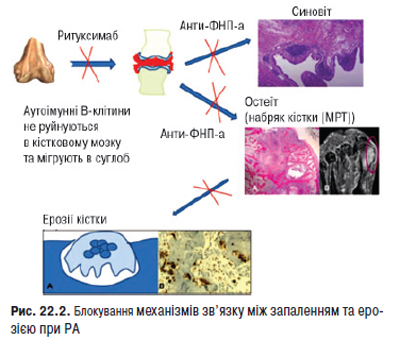
Запалення — головний фактор ризику для системної втрати кісткової маси. Прозапальні цитокіни, такі як ФНП-α, ІЛ-6, -1 порушують кістковий гомеостаз та індукують втрату кісткової маси.
Ключовими в запальній втраті кісткової маси є також DKK-1, склеростин, які так само викликають потужне інгібування формування остеобластів. Нейтралізація DKK-1 в експерименті повністю захищає від запальної втрати кісткової маси, знижуючи активність ФНП-α, тим самим знижуючи активність остеокластів і підвищуючи активність остеобластів у зв’язку з експресією Wnt β-катеніну, остеокальцину і остеопротегерину. Слід зазначити, що Wnt β-катеніновий шлях є головним у формуванні кісткової тканини.
Окремі цитокіни відіграють важливу роль у розвитку запального процесу і залежно від їх особливостей впливають і на інші процеси, виявляючи так звані плейотропні ефекти. Це властиво, зокрема, ІЛ-6. Плейотропну активність ІЛ-6 описано на рис. 22.3.
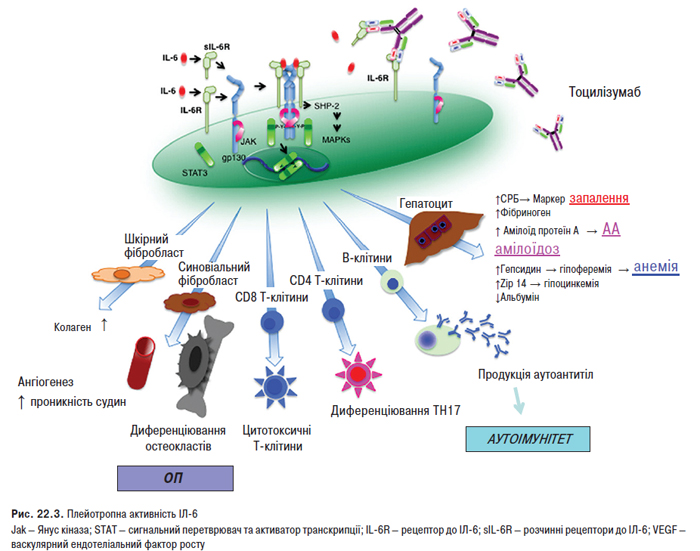
ІЛ-6 активує янус-кіназу, викликає гомодемінералізацію gp130 і активує сімейство тирозинових кіназ. Крім того, янус-кінази індукують фосфорилювання тирозину gp130 сигнального перетворювача і активатора транскрипції (STAT3) через SH2 домен. Це супроводжується фосфорилюванням STAT3 за допомогою янус-кінази і транслокацією в ядро регулятора активації генів.
Гуманізовані антитіла до рецепторів ІЛ-6 (ІЛ-6Р), тоцилізумаб, зв’язуються з розчинною (ІЛ-6Р) та мембранною фракцією ІЛ-6Р і конкурентно блокують зв’язування ІЛ-6 з його рецепторами.
ІЛ-6 індукує диференціювання клітин або специфічну експресію генів, виробництво білків гострої фази в гепатоцитах, зниження концентрації заліза і цинку, індукцію імуноглобуліну у В-клітинах, цитотоксичну Т-клітинну диференціацію та Th17. ІЛ-6 також впливає на синовіальні фібробласти для виробництва RANKL і VEGF, які сприяють диференціації остеокластів і ангіогенезу.
Крім того, ІЛ-6 стимулює шкірні фібробласти для виробництва колагену.
Слід підкреслити, що прогресування РА — процес, що динамічно розвивається, який (як з точки зору патогенетичних механізмів, так і на підставі клініко-інструментальних і лабораторних проявів) умовно поділяють на кілька стадій.
Стадії ревматоїдного артриту:
- Рання (безсимптомна) стадія характеризується судинною та клітинною активацією
- Розгорнута (швидка хронізація запалення) стадія проявляється порушенням ангіогенезу, активацією ендотелію, клітинною міграцією; інфільтрацією активованими СD4+ Т-лімфоцитами синовіальної тканини, утворенням РФ та інших аутоантитіл, імунних комплексів; синтезом прозапальних цитокінів, простагландинів, металопротеаз, колагенази
- Пізня стадія характеризується соматичними мутаціями та дефектами апоптозу синовіальних клітин
Т-лімфоцити. Основну роль у розвитку РА відіграють Т-лімфоцити, які становлять >50% клітинного інфільтрату сполучної тканини, в той час як частка В-клітин і плазматичних клітин не перевищує 5%.
Переважна більшість Т-лімфоцитів належить субпопуляції СD4+ Т-клітин з фенотипом, характерним для клітин пам’яті, і маркерами, притаманними для активованих лімфоцитів (НLА-DR і СD27). При цьому СD27+ СD4+ Т-лімфоцити проявляють активність хелперних клітин, що стимулюють синтез аутоантитіл В-клітинами. Крім того, в синовіальній тканині при РА відзначають накопичення в надмірній кількості СD4+, СD8+, СD28 Т-клітин, які володіють активністю аутореактивних природних кілерних клітин. Збільшення кількості цих клітин корелює з розвитком ерозій в суглобах і підвищеним ризиком розвитку кардіоваскулярних ускладнень.
Важливим компонентом активації Т-клітин при РА вважають кісткову стимуляцію. Відомо, що надання антигену Т-лімфоцитів антиген-презентуючми клітинами (дендритними клітинами, макрофагами, В-клітинами), у чому бере участь система рецептор-корецептор (СD28/СD80/86, міжклітинні молекули адгезії, антиген-1, асоційований з функцією лімфоцитів), призводить до анергії і загибелі Т-клітин. У ревматоїдній синовіальній тканині виявляють гіперекспресію кістковостимуляторних молекул, що може призводити до активації і проліферації Т-клітин за відсутності специфічних аутоантигенних стимулів, які підтримують розвиток аутоімунного процесу.
Регуляторні Т-клітини. Нагадаємо, що, за сучасними уявленнями, розвиток аутоімунних захворювань пов’язаний з порушенням супресорних механізмів, які контролюють толерантність Т- і В-лімфоцитів до аутоантигенів. У свою чергу, активовані аутореактивні Т- і В-клітини індукують запалення і пошкодження власних тканин організму.
Нещодавно був відкритий новий механізм імунологічної толерантності, у реалізації якого беруть участь СD4+, СD25+ Т-клітини (субпопуляція СD4+ Т-лімфоцитів), які отримали назву регуляторних Т-клітин. Вони мають здатність пригнічувати аутоантигенспецифічну проліферацію і ефекторні функції аутореактивних лімфоцитів. В активації, розвитку і реалізації функцій регуляторних Т-клітин бере участь ядерний фактор транскрипції, пов’язаний з Х-хромосомою (Fохр3). Цей фактор розглядають як унікальний мембранний маркер регуляторних Т-клітин. Роль СD4+, СD25+ Т-клітин у розвитку РА була переконливо показана на моделях експериментальних артритів, індукованих колагеном та іншими аутоантигенами. У пацієнтів з РА відносна кількість СD4+, СD25+ Т-клітин істотно не змінена, однак їх функціональна активність помітно знижена. При такому стані виявляють підвищення концентрації ІЛ-15 та -7, що пригнічують регуляторну активність СD4+, СD25+ Т-клітин. Великий інтерес становлять дані про відновлення функцій СD4+, СD25+ Т-клітин на тлі лікування інфліксимабом.
Порушення апоптозу. Істотну роль у прогресуванні РА відіграє порушення апоптозу (програмованої загибелі) Т-клітин. У синовіальній тканині при РА виявляють гіперекспресію гена пухлинної супресії р53. Мутації в цьому гені призводять до порушень нормальної репарації ДНК і процесу апоптозу.
В-клітини. Результати експериментальних досліджень свідчать про значну роль В-лімфоцитів у регуляції Т-клітинної імунної відповіді при РА. На моделі експериментального артриту у мишей з тяжким комбінованим імунодефіцитом (NOD-SCID), що розвивається при перенесенні синовіальної тканини від пацієнтів з активним РА, показано, що В-лімфоцити беруть участь в активації СD4 Т-клітин за Тh1-типом у запальній синовіальній тканині, виконуючи функцію специфічних антиген-презентуючих клітин. В-клітини, які синтезують РФ, мають унікальну здатність взаємодіяти з імунними комплексами і презентувати широкий спектр аутоантигенів, а активовані В-клітини експресують кістковостимуляторні молекули (В7 і СD40), необхідні для повноцінної активації Т-клітин. Обговорюють ефекторну роль В-клітин у розвитку суглобової деструкції. Цей процес реалізується за рахунок синтезу прозапальних цитокінів (ФНП-α, ІЛ-1 і лімфотоксину), а також ІЛ-6 та -10, які здійснюють додаткову стимулюючу дію на В-лімфоцити.
Гладкі клітини. Отримано дані про участь гладких клітин у розвитку ревматоїдного синовіту. Встановлено, що їх активація, в індукції якої беруть участь компоненти комплементу, аутоантитіла і цитокіни, призводить до синтезу широкого спектра медіаторів запалення (гістаміну, ФНП-α, триптази, хімази і деяких інших), що стимулюють хондроцити, синовіальні фібробласти і макрофаги. Ці клітини, в свою чергу, також синтезують прозапальні медіатори, які викликають набряк (гістамін) і деструкцію кісткової тканини.
Ангіогенез. Одним з найбільш ранніх етапів розвитку ревматоїдного синовіту вважають ріст нових судин у синовіальній оболонці. Цей процес асоційований з накопиченням рідини в порожнині суглоба та трансміграцією лімфоцитів і лейкоцитів у синовіальну рідину. Істотне збільшення обсягу ревматоїдної синовіальної тканини створює умови для розвитку відносної гіпоксії, що вважають стимулом для синтезу фактора транскрипції-1, індукованого гіпоксією. Цей фактор активує транскрипцію генів молекул, які беруть участь в ангіогенезі, і в першу чергу, судинного ендотеліального фактора росту. До інших важливих ангіогенних субстанцій відносять гепаринзв’язуючий фактор росту, макрофагальний ангіогенний фактор, простагландини Е1 і Е2, ІЛ-8, ангіопоетин-1, а також молекули адгезії: інтегрин a5bЗ. На ангіогенез опосередковано впливає ФНП-α, який стимулює експресію рецепторів для ангіопоетину-1.
Цитокіни. Ключову роль у розвитку хронічного запального процесу відіграють цитокіни, синтезовані клітинами, які інфільтрують синовіальну оболонку суглоба. Цитокіни, що мають важливе значення в розвитку РА, потенційно можуть бути мішенями для фармакотерапії (табл. 22.1).
Таблиця 22.1
Загальна характеристика основних цитокінів, які беруть участь у розвитку РА
|
Гранулоцитарно-макрофагальний колонієстимулюючий фактор |
Макрофаги |
Експресія HLR-DR на антиген-презентуючих клітинах, активація T-клітинної імунної відповіді |
Моноклональні антитіла до гранулоцитарно-макрофагального колонієстимулюючого фактора; фаза II |
|
ФНП-α |
Макрофаги, Т- и В-лімфоцити |
Основний прозапальний цитокін при РА |
Інгібітори ФНП-α; зареєстровані для лікування при РА |
|
ІЛ-1 |
Макрофаги, Т-клітини, В-клітини |
Основний прозапальний цитокін при РА |
Інгібітор ІЛ-1; зареєстрований для лікування при РА |
|
ІЛ-6 |
Макрофаги, Т- и В-лімфоцити, фібробласти |
Лейкоцитоз, лихоманка, синтез судинного ендотеліального фактора росту, диференціювання остеокластів |
Гуманізовані моноклональні антитіла до ІЛ-6-рецептора (тоцилізумаб); фаза III |
|
ІЛ-7 (сімейство ІЛ-2) |
Макрофаги, фібробласти та ін. |
Активація, проліферація, ріст та дозрівання; індукує Тh1-клітини, цитотоксичність СD8+ клітин та моноклональних клітин; індукує синтез прозапальних цитокінів моноцитами; основний цитокін остеокластогенезу при РА |
Немає даних |
|
ІЛ-15 |
Макрофаги |
Індукція синтезу ФНП-α (за допомогою активації Т-клітин); утворення Th-17-клітин |
Моноклональні антитіла до ІЛ-15; фаза II |
|
Суперсімейство ІЛ-12 (ІЛ-17, -12, -23) |
|||
|
ІЛ-17 |
Т-клітини (Th-17), макрофаги, дендритні клітини |
Індукція синтезу ФНП-α, ІЛ-6, ІЛ-1, ІЛ-8, макрофагального запального білка 1, матриксних металопротеїназ, RANKL, ЦОГ 2-го типу, активація остеокластів, пригнічення остеобластів |
Моноклональні антитіла до ІЛ-17; фаза І–II |
|
ІЛ-12 |
Макрофаги, дендритні клітини |
Індукує Тh1-тип імунної відповіді (активація наївних СD+ Т-клітин в ефекторні клітини), индукує синтез цитокінів NК-клітинами |
Моноклональні антитіла до ІЛ-12/23; фаза IIІ (при псоріазі) |
|
ІЛ-23 |
Макрофаги, дендритні клітини |
Індукує (поряд з ІЛ-15) утворення Тh17-клітин |
Моноклональні антитіла до ІЛ-15; фаза IIІ (при псоріазі) |
|
ІЛ-18 (структурна схожість з ІЛ-1в) |
Макрофаги, Т-клітини, В-клітини |
Активація Т-клітин за Тh1-типом, індукція синтезу прозапальних цитокінів |
Немає даних |
|
ІЛ-32 |
Т-клітини, NК-клітини, епітеліальні клітини |
Синергічний ефект з іншими прозапальними цитокінами, включаючи ФНП-α |
Немає даних |
Синовіальні вистільні клітини. Під впливом прозапальних цитокінів синовіальні вистільні клітини набувають так званого трансформаційного фенотипу. Хоча, на відміну від пухлинних, синовіальні вистільні клітини не метастазують, вони набувають здатності до інвазії сполучної тканини хряща і зв’язок та стимулюють активацію і диференціювання остеокластів, які викликають резорбцію кісткової тканини.
Механізми деструкції хряща і субхондральної кістки. Важливу роль у деструкції хрящової і кісткової тканини відіграють матриксні металопротеїнази, такі як стромелізин (матриксна металопротеїназа-3), матриксна металопротеїназа-13, макрофагальна еластаза (матриксна металопротеїназа-12) та ін. Їх синтезують під впливом прозапальних медіаторів макрофаги і синовіальні фібробласти. Характерною особливістю ураження суглобів при РА вважають прогресування деструкції кістково-суглобової тканини за відсутності характерного для запалення репараційного компонента. На відміну від інших хвороб суглобів (наприклад спондилоартритів), коли процеси деструкції супроводжує адекватне формування нової кісткової тканини, при РА локальна кісткова резорбція істотно переважає над процесами відновлення.
В основі цього феномену лежать декілька механізмів:
- у зоні запалення відзначають гіперпродукцію ФНП-α. Цей цитокін має здатність збільшувати кількість остеокластів і зменшувати кількість остеобластів в осередку ураження;
- гіперекспресія ліганду рецептора активатора NK-kВ (receptor activator NF-kB ligand-RANKL) сприяє деструкції кістки за рахунок підвищення активності остеокластів.
Роль аутоантитіл. Припускають, що РФ і АЦЦП не тільки є діагностичним маркером, а й мають велике значення в розвитку РА. Ці антитіла виявляють на ранніх стадіях і навіть до клінічної маніфестації хвороби. Появу антитіл пов’язують із більш активним перебігом захворювання і більш вираженою деструкцією суглобів. Пасивне введення АЦЦП ускладнює перебіг експериментального колагенового артриту.
Інші фактори. До інших важливих стимуляторів запалення і тканинної деструкції відносять продукти активації комплементу (анафілотоксинів С3а, С5а). Їх утворення пов’язане з імунними комплексами, що містять РФ, і АЦЦП, а також широким спектром «неімунних» медіаторів, включаючи оксид азоту, нейропептиди (субстанція Р), метаболіти арахідонової кислоти, фактори згортання крові та фібринолізу.
Таким чином, РА вважають гетерогенною, мультифакторіальною хворобою. При цьому фактори зовнішнього середовища і генетичні чинники ризику вносять різний внесок при формуванні окремих варіантів хвороби.
Предиктори прогресування ревматоїдного артриту
Раннім предиктором, який прогнозує підвищений ризик рентгенологічного прогресування, є екскреція з сечею С-телопетиду колагену 2-го типу (СТХ-2). Критерієм розвитку структурних змін при РА є набряк кістки з розвитком остеїту за даними МРТ та УЗД. Предиктори відповіді на метотрексат у хворих на ранній РА, які раніше не отримували базисних препаратів: куріння, жіноча стать, більша тривалість симптомів і молодий вік. Вони передбачають найгіршу відповідь на метотрексат у хворих з уперше виявленим РА.
Класифікація
1. Основний діагноз
- Серопозитивний РА (М05.8)
- Серонегативний РА (М06.0)
Особливі клінічні форми РА:
- синдром Фелті (М05.0);
- хвороба Стілла, що розвинулася у дорослих (М06.1).
Ймовірний РА (М05.9, М06.4, М06.9)
2. Клінічні стадії за тривалістю хвороби (ACR 2008)
- Дуже ранній — до 3 міс
- Ранній — до 6 міс
- Середньої тривалості — 6 міс — 24 міс
- Тривалий — більше 24 міс
3. Ступінь активності хвороби
- 0 = ремісія (DAS28 <2,6).
- I = низька (DAS28 = 2,6–3,2).
- II = середня (DAS28 = 3,3–5,1).
- III = висока (DAS28> 5,1).
4. Системні та екстраартикулярні ознаки РА
Неспецифічні ознаки: втома, втрата маси тіла, лихоманка, анемія (при хронічному перебігу).
Екстраартикулярні ознаки:
Шкіра: підшкірні вузлики (ревматоїдні вузлики) виявляють в середньому у 25% хворих.
Серце: ураження перикарду зустрічається часто, але рідко буває клінічно вираженим.
Легені: ураження плеври зазвичай клінічно не виражено. У легенях можуть з’являтися вузлики, котрі можуть розпадатися, кальцифікуватися, бути джерелом запалення.
Судини: васкуліти дрібних судин можуть викликати інфаркти, виразково-некротичний васкуліт, ліведо-ангіїт. Також можуть виникати піодермія, гангрена.
Очі: можуть виникати сухі кератокон’юнктивіти, склерити, епісклерити, васкуліт сітківки.
Нерви: можуть розвиватися тунельні синдроми, поліневропатії. Мононеврити частійше бувають пов’язані з васкулітами.
Селезінка, елементи крові: спленомегалію та нейтропенію інколи відзначають у хворих з тяжким РА (синдром Фелті). Часто такі хвори є РФ-позитивними.
5. Інструментальна характеристика
- Наявність або відсутність ерозій (за даними рентгенографії, МРТ, УЗД):
- неерозивний;
- ерозивний.
- Рентгенологічна стадія (за Штейнброкером):
- I стадія. Невеликий навколосуглобовий ОП. Поодинокі гроноподібні просвітлення кісткової тканини. Незначне звуження суглобових щілин в окремих суглобах.
- II стадія. Помірний (виражений) навколосуглобовий ОП. Множинні гроноподібні прояснення кіскової тканини. Звуження суглобових щілин. Поодинокі (1–4) ерозії суглобових поверхонь. Незначні деформації кісток.
- III стадія. Помірний (виражений) навколосуглобовий ОП. Множинні гроноподібні просвітлення кісткової тканини. Звуження суглобових щілин. Множинні (5 і більше) ерозії суглобових поверхонь. Множинні виражені деформації кісток. Підвивихи та вивихи суглобів.
- IV стадія. Помірний (виражений) навколосуглобовий (поширений) ОП. Множинні гроноподібні просвітлення кісткової тканини. Звуження суглобових щілин. Множинні ерозії кісток і суглобових поверхонь. Множинні виражені деформації кісток. Підвивихи і вивихи суглобів. Поодинокі (множинні) кісткові анкілози. Субхондральний остеосклероз. Остеофіти на краях суглобових поверхонь.
6. Додаткова імунологічна характеристика — АЦЦП
- АЦЦЛ — присутні (+).
- АЦЦЛ — відсутні (-).
7. Функціональний клас
- I — повністю збережені можливості самообслуговування, займатися непрофесійною і професійною діяльністю.
- II — збережені можливості самообслуговування, займатися непрофесійною діяльністю, обмежена здатність займатися професійною діяльністю.
- III — збережені можливості самообслуговування, обмежені можливості займатися непрофесійною і професійною діяльністю.
- IV — обмежені можливості самообслуговування, займатися непрофесійною і професійною діяльністю.
Ускладнення:
- вторинний системний амілоїдоз;
- вторинний артроз;
- ОП (системний);
- остеонекроз;
- тунельні синдроми (синдром карпального каналу, синдроми стискання ліктьового, великогомілкового нервів);
- підвивих в атланто-аксіальному суглобі, у тому числі з мієлопатією, нестабільністю шийного відділу хребта;
- атеросклероз.
Клінічна картина
Розвитку артриту може передувати продромальний період тривалістю від декількох тижнів до декількох місяців. До його основних проявів відносять підвищену стомлюваність, втрату маси тіла, періодичний біль у суглобах (часто при зміні атмосферного тиску), зниження апетиту, підвищене потовиділення, субфебрильну температуру тіла, помірну анемію, збільшення ШОЕ.
Початок хвороби. Варіанти початку хвороби різноманітні. Зазвичай починається з поліартриту, рідше переважають артралгії, ранкова скутість в суглобах, погіршення загального стану, слабкість, втрата маси тіла, субфебрильна температура тіла, лімфаденопатія. Ці симптоми можуть передувати клінічно вираженому ураженню суглобів.
Умовно виділяють наступні варіанти дебюту РА.
- Симетричний поліартрит з поступовим (протягом декількох місяців) наростанням болю та скутості, переважно в дрібних суглобах кистей.
- Гострий поліартрит з переважним ураженням суглобів кистей і стоп, вираженою ранковою скутістю (зазвичай відзначають ранню появу РФ в крові).
- Моно- або олігоартрит колінних або плечових суглобів з наступним швидким залученням в процес дрібних суглобів кистей і стоп. Гострий моноартрит великих суглобів, що нагадує септичний або мікрокристалічний артрит.
- Гострий оліго- або поліартрит з системними явищами (фебрильна лихоманка, лімфаденопатія, гепатоспленомегалія). Найчастіше відзначають у пацієнтів молодого віку (нагадує хворобу Стілла дорослих).
- Паліндромний ревматизм. Характерні рецидивуючі атаки гострого симетричного поліартриту суглобів кистей, рідше — колінних і ліктьових суглобів, тривалістю до декількох годин або днів з наступним повним одужанням.
- Рецидивуючий бурсит і тендосиновіт зазвичай променезап’ясткових суглобів.
- Гострий поліартрит в осіб похилого віку з множинним ураженням дрібних і великих суглобів, вираженим болем, дифузним набряком і обмеженням рухливості суглобів. Отримав назву «RS3PE-синдром» (remiting seronegative symmetrical synovitis with pitting edema — ремітуючий серонегативний симетричний синовіт з подушкоподібним набряком).
- Генералізована міальгія, скутість, депресія, двосторонній синдром зап’ястного каналу, зменшення маси тіла. Зазвичай реєструють у літньому віці. Нагадує РПМ. Появу характерних клінічних ознак РА відзначають пізніше.
Варіанти перебігу. За характером прогресування деструкції суглобів і позасуглобових (системних) проявів виділяють декілька варіантів перебігу РА.
- Тривала спонтанна клінічна ремісія (<10% випадків).
- Інтермітуючий перебіг (15–30% випадків). Періодично виникає повна або часткова ремісія (спонтанна або індукована лікуванням), яка переходить у загострення із залученням до процесу раніше не уражених суглобів.
- Прогресуючий перебіг (60–75% випадків). Збільшується вираженість деструкції суглобів, відзначають ураження нових суглобів, розвиток позасуглобових (системних) проявів.
- Швидкопрогресуючий перебіг (10–20% випадків). Активність хвороби постійно висока, відзначають тяжкі позасуглобові (системні) прояви.
Ураження суглобів. Клінічні ознаки ураження суглобів можуть бути умовно розділені на 2 категорії: потенційно зворотні (зазвичай ранні) (синовіт) та незворотні структурні (більш пізні) (ерозії, анкілоз). Такий поділ має значення для встановлення стадії захворювання, прогнозу і вибору тактики лікування. Структурні пошкодження можуть розвиватися швидко (іноді протягом перших місяців хвороби). Найбільш яскрава ознака запалення синовіальної оболонки суглобів при РА — ранкова скутість. Її тривалість зазвичай корелює з вираженістю синовіту і становить не менше 1 год.
Характерним для РА вважають стійке симетричне поліартикулярне запалення п’ястково-фалангових, проксимальних міжфалангових і променезап’ясткових суглобів обох кистей. Досить рано можна спостерігати порушення функції кисті (пацієнтові важко або неможливо стиснути руку в кулак).
До відносно ранніх ознак відносять атрофію міжкісткових (червоподібних) м’язів. При розвитку цього стану відзначають своєрідне схуднення тилу кисті. При великій тривалості хвороби часто відзначають атрофію м’язів інших локалізацій: передпліччя, надпліччя, стегон, гомілок. Атрофія пов’язана з обмеженням рухливості в відповідних суглобах.
При тривалому перебігу хронічного артриту фіброзні зміни в тканинах суглоба сприяють зморщуванню капсули, зв’язок, сухожиль, руйнуванню суглобової поверхні і, як наслідок, розвитку вираженої деформації суглобів, підвивихів і контрактур. Відзначають обмеження обсягу рухів, а в міру розвитку анкілозів настає повне знерухомлення одного або декількох суглобів (рис. 22.4).
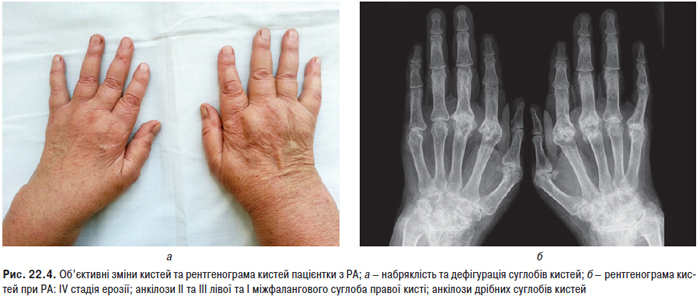
Подальше прогресування РА призводить до формування різних типів підвивихів. Найбільш типовою для цієї хвороби вважають ліктьову девіацію кисті з відхиленням пальців у бік ліктьової кістки, зумовлену підвивихами у п’ястково-фалангових суглобах і слабкістю м’язів. Така форма кисті отримала назву «плавець моржа». Деформація пальців за типом «шиї лебедя» відбувається при формуванні згинальної контрактури в п’ястково-фалангових суглобах, перерозгинанні проксимальних і згинанні дистальних міжфалангових суглобів. Деформація суглобів може мати вигляд ґудзикової петлі. При цьому відбувається виражене згинання в п’ястково-фалангових суглобах при перерозгинанні дистальних міжфалангових суглобів. Ці зміни кисті різко обмежують її можливості, утруднюють виконання звичайних рухів. Пацієнту важко тримати чашку, ложку і т.д. Цьому сприяє також розвиток контрактури великого пальця кисті в результаті ураження п’ясно-трапецієподібного суглоба (рис. 22.5).
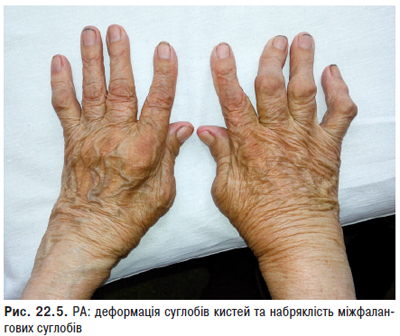
Артрит суглобів зап’ястя призводить до деструкції дрібних кісток з подальшим утворенням єдиного кісткового блоку і формуванням анкілозу з зап’ястно-п’ясткових суглобів. Анкілозування самого променезап’ясткового суглоба зустрічається рідко. Запальний процес у ньому виявляють за появою припухлості, хворобливості при русі. При артриті променеліктьового суглоба пацієнти відзначають болісність при пронації та супінації. Нерідкісне ускладнення артриту цього суглоба — виникнення заднього підвивиху головки ліктьової кістки через її рухливість.
Суглоби стоп бувають уражені у третини пацієнтів вже в ранній період хвороби. Процес переважно локалізований в плеснофалангових суглобах пальців II–IV. До основних проявів артриту цієї локалізації відносять біль при ходьбі та особливо при підстрибуванні або стоянні на пальцях. Відзначають поступовий розвиток деформації стопи з опусканням переднього склепіння, формуванням hallux valgus і молотоподібної деформації інших пальців з підвивихами в плеснофалангових суглобах. Головки плеснових кісток виступають з підошовного боку стопи, що часто призводить до появи «натоптишів» і некрозів шкіри над суглобами.
Ураження ліктьового суглоба частіше відзначають при більш тривалому перебігу РА. Патологічний процес у суглобі призводить до обмеження згинання та розгинання з подальшим утворенням контрактури в положенні напівзгинання та напівпронації. При цьому нерідко відбувається стискання ліктьового нерва з розвитком парестезії в ділянці іннервації. Ураження плечового суглоба зумовлено не тільки синовітом, але й залученням дистальної третини ключиці, синовіальних сумок і м’язів плечового пояса, шиї та грудної клітки. При появі запального процесу в плечовому суглобі відзначають припухлість і болісність при пальпації як з зовнішньої поверхні, так і з боку пахвової ямки.
Ураження гомілковостопного суглоба спостерігають рідше, як правило, при тяжкому прогресуючому поліартриті. Основний клінічний прояв цього стану — набряк в ділянці кісточки. При залученні в патологічний процес зв’язкового апарату і м’язів гомілки може виникати нестабільність гомілковостопного суглоба з частими підвивихами.
Колінні суглоби — часта локалізація ревматоїдного ураження. Випіт у суглобі визначають за балотуванням при пальпації в напрямку з передньолатерального відділу до медіальної сторони.
Кульшовий суглоб зазвичай буває залучений в патологічний процес на пізніх стадіях хвороби. При істинному ураженні кульшового суглоба пацієнти відчувають біль, який ірадіює в пахвову ділянку або нижній відділ сідничної ділянки.
Ураження суглобів хребта зустрічаються рідко і на пізніх стадіях. Уражується в основному шийний відділ внаслідок артриту атланто-аксіального суглоба. При цьому виникають біль і скутість. Залучення дуговідросткових суглобів СIII–СV може призводити до їх зсуву і внаслідок цього до стискання спинного мозку.
При РА також буває уражений скронево-нижньощелепний суглоб, обмежуючи відкриття рота і ускладнюючи прийом їжі.
Позасуглобові (системні) прояви. Для РА характерний розвиток різноманітних системних (позасуглобових) проявів. Іноді вони можуть переважати в клінічній картині хвороби. Розвиток позасуглобових проявів (васкуліт, плеврит, перикардит, синдром Фелті, периферична невропатія, ураження очей, гломерулонефрит) вважають фактором ризику смерті, в першу чергу від кардіоваскулярної патології.
Ревматоїдні вузлики вважають типовим проявом РА, хоча їх рідко виявляють на ранній стадії. Їх появу зазвичай реєструють через 3–5 років від початку хвороби (приблизно 30% тривало хворіють РА). Характерні ознаки ревматоїдних вузликів: щільна консистенція, безболісність, відсутність змін шкіри над ними, відсутність спаяності з прилеглими тканинами, локалізація в ділянці ліктів (зовнішня поверхня ліктьового відростка), сухожиль кисті, ахіллових сухожиль, волосистої частини голови, крижів.
Васкуліт — також один із характерних системних проявів РА. В основі його винекнення лежить запалення судинного русла — панартеріїт. Розвиток васкуліту частіше відзначають у чоловіків, у пацієнтів з високими титрами РФ, вираженими ерозивними змінами в суглобах, ревматоїдними вузликами та іншими системними проявами РА, високим рівнем кріоглобулінів.
Характерними ознаками ревматоїдного васкуліту вважають дигітальний артеріїт, виразки шкіри (включаючи гангренозну піодермію), периферичну невропатію, артеріїт внутрішніх органів (серця, легень, кишечнику, нирок), пурпуру, яка пальпується.
Ураження легень відносять до числа найбільш частих проявів позасуглобового РА (у 10–20% пацієнтів в дебюті хвороби). Зазвичай ця патологія виникає в перші 5 років від початку хвороби. У процес можуть бути залучені всі анатомічні відділи легень.
Первинні ураження дихальної системи при РА
Захворювання плеври:
- плеврит;
- фіброз плеври.
Хвороби дихальних шляхів:
- крикоаритеноїдний артрит;
- бронхоектази;
- фолікулярний бронхіоліт;
- облітеруючий бронхіоліт;
- дифузний панбронхіоліт.
Інтерстиціальні хвороби легень:
- інтерстиціальна пневмонія (звичайна, неспецифічна, організована, лімфоцитарна);
- гостра еозинофільна пневмонія;
- дифузне пошкодження альвеол;
- апікальне фібробульозне захворювання;
- амілоїдоз;
- ревматоїдні вузли.
Судинні захворювання легень:
- легенева гіпертензія;
- васкуліт;
- дифузні альвеолярні геморагії з капіляритами. Вторинні ураження дихальної системи при РА.
Опортуністичні інфекції:
- легеневий туберкульоз;
- атипова мікобактеріальна інфекція;
- нокардіоз;
- аспергільоз;
- цитомегаловірусний пневмоніт.
Токсичні ураження легень у результаті лікування:
- метотрексатом;
- солями золота;
- пеніциламіном;
- сульфасалазином.
Ураження легень у результаті впливу інгібіторів ФНП-α (підвищення ризику розвитку туберкульозу та інших опортуністичних інфекцій).
Ураження серця відносно рідко бувають клінічно значущими при РА і включають перикардит, міокардит, ендокардит, дуже рідко — коронарний артеріїт, гранулематозний аортит.
Характерним проявом РА вважають ранній прискорений розвиток атеросклерозу та його ускладнень: гострого інфаркту міокарда та інсульту.
При активному ревматоїдному процесі іноді виявляють міозит з вогнищами некрозу м’язових волокон, підвищенням рівня КФК, трансаміназ.
Найбільш частою причиною ураження нирок при РА вважають вторинний амілоїдоз (реактивний, пов’язаний із запаленням, АА-амілоїдоз), характерний для пацієнтів з великою тривалістю хвороби (7–10 років і більше) при високій активності процесу. Нефротичний синдром при РА майже завжди пов’язаний з амілоїдозом нирок.
Амілоїдоз при РА вважають однією з основних причин смерті. Утворення амілоїдних фібрил при цій хворобі пов’язано з хронічним запаленням і буває результатом системного відкладення сироваткового амілоїдного білка А (SАА — serum amyloid A protein). SАА, як і СРБ, вважають білком гострої фази запалення. Синтез SАА відбувається в печінці під впливом прозапальних цитокінів: ІЛ-1 і ФНП-α, а його концентрація в крові швидко підвищується на тлі запалення. Однак гіперпродукція SАА — необхідна, але недостатня умова розвитку амілоїдозу. Обговорюють значення інших факторів, включаючи носійство певних алелів гена SАА, аполіпопротеїну Е, DR-антигенів, РФ, поліморфізм амілоїдного Р-компонента, рецепторів ФНП-α та ін. Відзначено кореляцію між підвищенням сироваткової концентрації ФНП-α та відповідних рецепторів і виникненням амілоїдної нефропатії і супутньої анемії.
Розвитку клінічно вираженого амілоїдозу передує стадія безсимптомного амілоїдозу (за даними біопсії). Такий стан виявляють значно частіше, ніж клінічно виражений амілоїдоз. Наприклад, в процесі тривалого (14 років) спостереження за великою групою пацієнтів з тривалим (>5 років) РА клінічно виражений амілоїдоз був відзначений в 5% випадків, а безсимптомний — в 11%.
У цілому розвиток амілоїдозу реєструють у пацієнтів, що довго хворіють на РА, з неконтрольованою високою активністю хвороби. Найбільш характерним проявом амілоїдозу вважають нефропатію (протеїнурія і ниркова недостатність), рідше відзначають ураження кишечнику (проноси, перфорації), селезінки (підвищення ризику розвитку інфекційних ускладнень), серця (серцева недостатність).
Рідко відзначають мембранозний і мембранозно-проліферативний гломерулонефрит на фоні, як правило, високої активності РА. Для цього ураження характерна поява слідової протеїнурії, мікрогематурії. Ураження очей при РА виявляють відносно рідко — у вигляді епісклериту або склериту. Особливо характерний розвиток сухого кератокон’юнктивіту (синдром Шегрена).
Синдром Фелті діагностують лише у пацієнтів з тривалим поточним серопозитивним РА. Прояви синдрому: хронічний артрит зі спленомегалією та стійкою гранулоцитопенією (<2000/мм3). Справжня поширеність синдрому Фелті невідома. Вважають, що він розвивається приблизно у 3% пацієнтів з РА. Більше 75% хворих становлять жінки у віці 50–70 років і з тривалістю хвороби >10 років. У деяких випадках спленомегалія і гранулоцитопенія виникають до появи ознак артриту.
Сімейна агрегація і носійство НLА-DR4 (95% випадків) свідчать про участь генетичних факторів у розвитку цього синдрому. У третини пацієнтів з синдромом Фелті виявляють клональну експансію СD3+, СD8+ великих гранулярних лімфоцитів. Вони становлять близько 5% лімфоцитів периферичної крові, мають активність природних кілерних клітин і беруть участь в антитілозалежній клітинній цитотоксичності. Однак чітких клінічних та імуногенетичних відмінностей у пацієнтів з великими гранулярними лімфоцитами і без них не відзначено.
Розвиток гранулоцитопенії при синдромі Фелті пов’язують з кількома механізмами.
- Надмірне утворення імунних комплексів (або антигранулоцитарних антитіл). Зв’язуючись з мембраною гранулоцитів або потрапляючи всередину, вони сприяють руйнуванню гранулоцитів клітинами ретикулоендотеліальної системи.
- Придушення гранулоцитопоезу гуморальними медіаторами або супресорними Т-лімфоцитами.
- Синтез антитіл до гранулоцитарного колонієстимулюючого фактора. Утворення факторів (Fals та ін.), які індукують апоптоз нейтрофілів.
Особливості ураження суглобів при синдромі Фелті в цілому відповідають таким при серопозитивному РА, але у деяких пацієнтів клінічна активність артриту може бути мінімальною і не відповідати високій лабораторній активності хвороби (збільшення ШОЕ та рівня СРБ).
Спленомегалія зазвичай значна (розмір селезінки в середньому в 4 рази більший за норму), але у 5–10% ця ознака виражена мінімально.
У цілому частота (і тяжкість) позасуглобових проявів при синдромі Фелті вищі, ніж при серопозитивному РА.
Характерна особливість синдрому Фелті — підвищення чутливості до інфекційних ускладнень. При цьому чіткої кореляції між ризиком розвитку інфекцій і вираженістю гранулоцитопенії (за винятком зменшення кількості гранулоцитів до <1000/мм3) не простежують. Інфекційні ускладнення звичайно пов’язані з широко розповсюдженими мікроорганізмами (стафілококами, стрептококами, грамнегативними бактеріями). Вогнища інфекції найчастіше бувають локалізовані в шкірі та легенях і добре відповідають на антибактеріальну терапію. Водночас зі спленомегалією у більшості пацієнтів виявляють гепатомегалії, а у деяких відзначають синдром нодулярної регенеративної гіперплазії. Характерне підвищення ризику розвитку неходжкінської лімфоми.
Діагностика
Діагностичні критерії РА представлено в табл. 22.2.
Таблиця 22.2
Класифікаційні критерії РА (ACR/EULAR, 2010)
|
Кількість залучених суглобів (0–5) |
|
|
1 великий суглоб |
0 |
|
2–10 великих суглобів |
1 |
|
1–3 дрібних суглоби (великі суглоби не враховуються) |
2 |
|
4–10 дрібних суглобів (великі суглоби не враховуються) |
|
|
Серологія (0–3) |
|
|
Негативний РФ і негативний АЦЦП |
0 |
|
Слабопозитивний РФ і слабопозитивний АЦЦП |
2 |
|
Різко позитивний РФ і різко позитивний АЦЦП |
3 |
|
Тривалість симптомів (0–1) |
|
|
<6 тиж |
0 |
|
≥6 тиж |
1 |
|
Гострофазові показники (0–1) |
|
|
Нормальні СРБ і нормальна ШОЕ |
0 |
|
Позитивний СРБ і збільшена ШОЕ |
1 |
Примітка: ≥6 — веріфікований РА.
Значення ранньої діагностики. Розвиток імунопатологічного процесу з субклінічним перебігом відбувається за багато місяців (або років) до появи явних ознак РА. За даними біопсії синовіальної оболонки суглобів, ознаки хронічного синовіту виявляють вже на самому початку хвороби не тільки в запалених, а й «нормальних» суглобах. В умовно здорових людей, у подальшому хворих на РА, виявляють різні імунологічні порушення, характерні для РА (підвищення рівня РФ, АЦЦП, СРБ), задовго до появи перших клінічних симптомів хвороби.
У ⅔ пацієнтів структурні зміни (ерозії) суглобів виникають дуже швидко, вже протягом перших 2 років з моменту початку хвороби. Встановлено, що запобігання структурним пошкодженням в дебюті РА сприяє збереженню функціональної активності пацієнтів у довготривалій перспективі. Проте проміжок часу, коли активна терапія БПЗП може ефективно зменшувати прогресування ураження суглобів (так зване вікно можливості), досить короткий та іноді становить всього декілька місяців від початку хвороби.
Очевидно, що РА — яскравий приклад хвороби, при якій віддалений прогноз багато в чому залежить від того, наскільки рано встановлено діагноз і розпочато активну фармакотерапію. У цьому відношенні РА певною мірою нагадує такі захворювання, як цукровий діабет і артеріальну гіпертензію. Однак якщо рання діагностика артеріальної гіпертензії і цукрового діабету в переважній більшості випадків не становить труднощів, оскільки вона заснована на оцінці добре відомих лікарям загальної практики клінічних проявів і використанні доступних лабораторних та інструментальних методів, то діагностика РА в дебюті хвороби — значно важче завдання (іноді нерозв’язне). Це пов’язано з низкою об’єктивних і суб’єктивних обставин. По-перше, симптоми раннього РА часто неспецифічні, їх можна спостерігати при надзвичайно широкому колі як ревматичних, так і не ревматичних захворювань, а загальноприйняті класифікаційні критерії достовірного РА не підходять для ранньої діагностики. По-друге, для встановлення такого діагнозу необхідні спеціальні знання та навички в оцінці клінічних та рентгенологічних ознак ураження суглобів, а також уміння інтерпретувати лабораторні (імунологічні) тести, з якими погано обізнані лікарі загальної практики.
Таким чином, одна з причин несприятливого прогнозу при РА — тривалий період між початком хвороби і надходженням пацієнта під спостереження ревматолога. Очевидно, що важливий фактор, який сприяє поліпшенню прогнозу у пацієнтів з РА, — активне виявлення цього захворювання на поліклінічному етапі лікарями загальної практики.
Група європейських та американських ревматологів (під егідою EULAR) розробила алгоритм, який дозволяє більш активно виявляти ранній РА на поліклінічному етапі. Як діагностичну ознаку раннього РА (а також показника активності хвороби) враховують тривалість ранкової скутості (>30 хв), а при огляді пацієнтів — «тест бічного стискання» п’ястково-фалангових і плеснофалангових суглобів. Позитивні результати тесту відображають розвиток суглобового запалення. Незважаючи на те що швидке прогресування ураження суглобів більш імовірно при високих титрах РФ, збільшенні ШОЕ та підвищенні рівня СРБ, слід пам’ятати, що ці показники на ранній стадії хвороби часто відповідають нормі. У зв’язку з цим негативні результати лабораторних тестів не виключають діагнозу РА, а отже, передбачають необхідність направлення пацієнтів на консультацію до ревматолога.
Лабораторні дослідження
Цілі проведення лабораторних досліджень: підтвердження діагнозу; виключення інших захворювань; оцінка активності захворювання; оцінка прогнозу; оцінка ефективності лікування; виявлення ускладнень захворювання.
Зміни лабораторних показників, які виявляються при РА
Анемія (рівень гемоглобіну <140 г/л у чоловіків і 120 г/л — у жінок) — показник активності хвороби. Анемію діагностують у 30–50% випадків. Виникають будь-які форми анемії, але найчастіше анемія хронічного запалення і рідше — залізодефіцитна анемія. При виявленні даного стану необхідно виключити шлунково-кишкову кровотечу.
Збільшення ШОЕ та підвищення рівня СРБ —критерій для диференційної діагностики РА та незапальних захворювань суглобів. Дозволяє оцінити активність запалення, ефективність лікування, тяжкість захворювання, ризик прогресування деструкції суглобів.
Гіпоальбумінемія. Часто зумовлена нефротоксичністю препаратів, які застосовують для лікування пацієнтів з РА.
Підвищення рівня креатиніну. Зумовлене нефротоксичністю препаратів, які застосовують для лікування хворих на РА.
Лейкоцитоз (тромбоцитоз, еозинофілія). Показник тяжкого перебігу РА, часто з позасуглобовими (системними) проявами. Відзначають поєднання з високим рівнем РФ. Вважають показанням до призначення ГК. При виявленні цього стану необхідно виключити розвиток інфекційного процесу.
Нейтропенія. Ознака розвитку синдрому Фелті.
Підвищення рівня печінкових ферментів. Показник активності хвороби. Зміна також може бути зумовлена гепатотоксичністю препаратів, які застосовуються для лікування, або пов’язане з інфікуванням вірусами гепатиту В або С.
Підвищення рівня глюкози. Пов’язано із застосуванням ГК.
Дисліпідемія. Пов’язана із застосуванням ГК, але може бути зумовлена активністю запалення.
Підвищення рівня РФ. Виявляють у 70–90% пацієнтів. Високі титри в дебюті хвороби корелюють з тяжкістю, швидкістю прогресування патологічного процесу та розвитком системних проявів. Однак динаміка титрів не завжди відображає ефективність ікування. Проте рівень РФ — недостатньо чутливий і специфічний маркер на ранній стадії РА (у дебюті виявляють приблизно у 50% пацієнтів). Специфічність низька також в осіб похилого віку.
Підвищення рівня АЦЦП. Більш специфічний маркер РА, ніж рівень РФ. Збільшення титрів і РФ, і АЦЦП дозволяє діагностувати РА з більш високою чутливістю і специфічністю, ніж підвищення рівня тільки одного з цих показників. Виявлення АЦЦП вважають критерієм для диференційної діагностики РА на ранній стадії з іншими захворюваннями, перебіг яких характеризується поліартритом (первинний синдром Шегрена, СЧВ, вірусний гепатит В і С та ін.) Крім того, за підвищенням рівня АЦЦП прогнозують ризик розвитку деструкції суглобів у пацієнтів з раннім РА.
Підвищення рівня АНФ. Виявляють в 30–40% випадків, зазвичай при тяжкому перебігу РА.
Підвищення рівня імуноглобулінів (IgG, IgМ, IgA), концентрації компонентів комплементу, ЦІК. Зміни неспецифічні, у зв’язку з цим не рекомендують використовувати визначення цих показників як рутинних досліджень.
Визначення НLА-DR4. Маркер тяжкого перебігу РА і несприятливого прогнозу.
Виявлення маркерів вірусу гепатиту В, С і ВІЛ. У цьому випадку необхідно уникати призначення гепатотоксичних препаратів.
Зміни в синовіальній рідині (зниження в’язкості, пухкі муцинові згустки, лейкоцитоз (більше 6×109/л), нейтрофільоз (25–90%). Дослідження має допоміжне значення. Використовують для диференційної діагностики РА з іншими захворюваннями суглобів, в першу чергу мікрокристалічним і септичним артритами.
Зміни в плевральній рідині: білок >3 г/л (ексудат), глюкоза >5 ммоль/л, ЛДГ >1000 од./мл, рН = 7,0, титр РФ >1:320, рівень комплементу (СН50) знижений, лімфоцити (нейтрофіли, еозинофіли). Дослідження необхідне для диференційної діагностики з іншими захворюваннями легень і плеври. Необхідно пам’ятати, що лабораторних досліджень, специфічних для діагностики РА, не розроблено.
Інструментальні дослідження
Інструментальні методи дослідження мають важливе значення для підтвердження діагнозу та диференційної діагностики РА.
Рентгенологічне дослідження суглобів. Рентгенографія кистей і стоп необхідна для підтвердження діагнозу РА, встановлення стадії і оцінки прогресування деструкції суглобів (рис. 22.6–22.8). Характерних для РА змін в інших суглобах (принаймні на ранніх стадіях хвороби) не відзначають. Для оцінки прогресування суглобової деструкції за рентгенологічними ознаками використовують модифікований метод Шарпа і метод Ларсена.
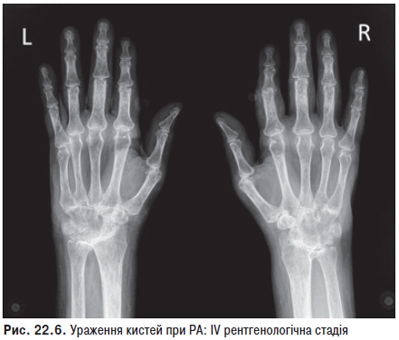
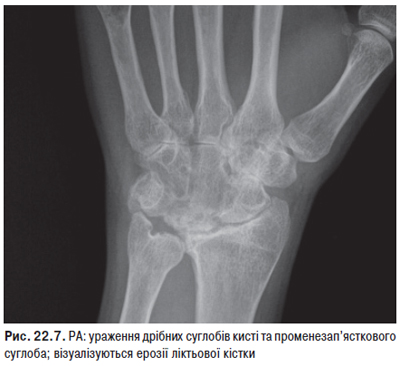
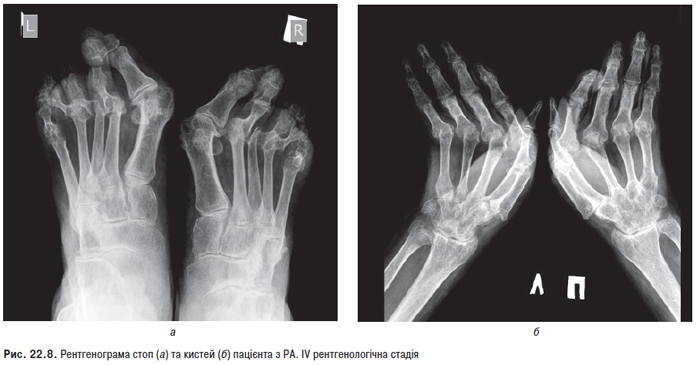
Експерти EULAR рекомендують метод Ларсена, коли зміни оцінюють кілька дослідників. Якщо оцінку деструкції проводить один фахівець, краще застосовувати модифікований метод Шарпа (більш чутливий).
Для виявлення підвивиху в атланто-аксіальному з’єднанні або шийного спондилолістезу доцільно виконати рентгенографію шийного відділу хребта.
Допплерівська ультрасонографія. Більш чутливий метод для виявлення синовіту колінного суглоба, ніж рентгенографія, але не для діагностики синовіту дрібних суглобів кистей і стоп.
МРТ. Більш чутливий метод виявлення синовіту в дебюті РА, ніж рентгенографія. Зміни, які виявляють за допомогою МРТ (синовіт, набряк й ерозії кісткової тканини), дозволяють прогнозувати прогресування деструкції суглобів (за даними рентгенологічного дослідження). Проте подібні зміни іноді виявляють у клінічно «нормальних» суглобах, тому значення МРТ для ранньої діагностики та прогнозування наслідків РА вимагає подальшого вивчення. Крім того, МРТ можна використовувати для ранньої діагностики остеонекрозу.
КТ. Для виявлення уражень легень доцільно використовувати КТ з високою роздільною здатністю.
Артроскопія. Необхідна для диференційної діагностики РА з вільозно-нодулярним синовітом, артрозом, травматичними ушкодженнями суглоба та ін.
Рентгенографія органів грудної клітки. Застосовують для виявлення та диференційної діагностики ревматоїдного ураження органів грудної клітки з саркоїдозом, пухлинами цієї локалізації, туберкульозом та іншими інфекційними процесами.
Езофагогастродуоденоскопія. Виконують у пацієнтів, які отримують НПЗП, і при виявленні анемії.
ЕхоКГ. Застосовують для діагностики ревматоїдного перикардиту і міокардиту, уражень серця, пов’язаних з атеросклеротичним процесом.
Біопсія. На дослідження беруть зразок тканин (слизової оболонки ШКТ, підшкірного жирового шару, ясен, нирки та інших органів) при підозрі на амілоїдоз.
Рентгенівська абсорбціометрія. Метод використовують для діагностики ОП. З його допомогою визначають МЩКТ. Дослідження МЩКТ доцільно за наявності таких факторів ризику розвитку ОП:
- вік (жінки старше 50 років, чоловіки — 60 років);
- висока активність хвороби (стійке підвищення рівня СРБ >20 мг/л або ШОЕ >20 мм/год);
- відповідний функціональний статус — стадія за Штейнброкером III–IV або значення індексу НАQ (Неаlth Assesment Questioaire) >1,25;
- маса тіла <60 кг;
- прийом ГК.
Чутливість (при виявленні 3 з 5 критеріїв) для діагностики ОП при РА становить у жінок 76%, у чоловіків — 83%, а специфічність відповідно — 54 і 50%.
Приклади формулювання діагнозів РА, поліартрит, серопозитивний варіант, активна фаза, активність III ступеня з переважним ураженням суглобів кистей, променезап’ясткових, плечових, колінних суглобів; синдром Рейно, рентгенологічна стадія III, суглобова функціональна недостатність II–III.
РА, серопозитивний варіант, поліартрит з переважним ураженням щелепних суглобів, кистей і колінних суглобів; ревматоїдна хвороба легень (альвеоліт), поліневропатія, активна фаза, ІII ступінь, рентгенологічна стадія II, суглобова функціональна недостатність II.
РА, серонегативний варіант, поліартрит з переважним ураженням колінних суглобів, кистей і стоп, активна фаза, активність II ст., рентгенологічна стадія I, суглобова функціональна недостатність I.
Диференційна діагностика
Захворювання, з якими необхідно диференціювати РА, представлено в табл. 22.3. Найчастіше необхідність у диференційній діагностиці виникає в дебюті захворювання при ураженні суглобів у вигляді моно- і олігоартриту. При цьому необхідно, в першу чергу, звертати увагу на такі типові ознаки РА, як симетричність артриту, переважне ураження суглобів кистей з порушенням їх функцій, розвиток ерозивного процесу в суглобах кистей, виявлення РФ і особливо АЦЦП.
Таблиця 22.3
Критерії диференційної діагностики РА
|
Хвороба |
Характеристика хвороби |
Критерії |
|
ОА |
Виявляють дегенерацію суглобового хряща з залученням дистальних (вузлики Гебердена) та проксимальних (вузлики Бушара) міжфалангових суглобів кистей, зап’ястно-п’ясткових, колінних, кульшових, плеснофалангових суглобів, шийного та поперекового відділів хребта. Збільшення вираженості болю при русі, остеофіти та звуження суглобових щілин на рентгенограмах. Лабораторні порушення відсутні |
Незначна припухлість м’яких тканин із залучення дистальних міжфалангових суглобів, відсутність вираженої вранішньої скутості, збільшення вираженості болю до кінця дня |
|
СЧВ |
Аутоімунні захворювання, пов’язані з продукцією антитіл до багатьох різноманітних клітинних та внутрішньоклітинних білків. Характерне симетричне ураження дрібних суглобів кистей, променезап’ясткових та колінних суглобів, розвиток неерозивного деформуючого артриту (Жаку). Можливий виражений набряк м’яких тканин, але кількість внутрішньосуглобового випоту зазвичай мінімальна |
Артрит недеформуючий (за винятком артриту Жаку). Виявляють АНФ (але ця ознака присутня лише у 30% пацієнтів з РА), рідко — невисокі титри РФ. На рентгенограмах відсутні ознаки кісткових ерозій |
|
Подагра |
Виявляють відкладення кристалів моноурату натрію в тканинах суглоба і навколо. Перша атака захворювання моноартикулярна, з переважним ураженням плеснофалангового суглоба I. При хронічній формі може бути симетричне ураження дрібних суглобів кистей і стоп з появою тофусів. На рентгенограмах виявляють ознаки субкортикальних ерозій |
Діагноз встановлюють при виявленні кристалів з характерним негативним подвійним заломленням променя при поляризаційній мікроскопії в синовіальній рідині або тофусах. У 30% випадків виявляють РФ |
|
ПсА |
Моноартрит, асиметричний олігоартрит, симетричний поліартрит, мутилюючий артрит, ураження осьового скелета |
Часто спостерігають ураження дистальних міжфалангових суглобів, веретеноподібну припухлість пальців. Виражені характерні для псоріазу зміни шкіри і нігтів |
|
АС |
Хронічне системне запальне ураження осьового скелета. Можливе залучення периферичних суглобів. Відзначають розвиток апікального легеневого фіброзу, появу болю в спині |
Характерний асиметричний моно- або олігоартрит великих суглобів (кульшових, колінних, плечових), хребетного стовпа, попереково-клубових з’єднань. Виявляють носійство НLА-В27 |
|
Синдром Рейтера |
Пов’язаний з інфікуванням різними мікроорганізмами (Chlamydia, Escherichia coli, Salmonella, Саmpylobacter, Yersinia). Симптомокомплекс включає уретрит, кон’юнктивіт і артрит. Пацієнти відзначають появу болю в п’яткових ділянках, розвиток ентезитів, кератодермії на долонях і підошвах і циркулярного баланіту |
Артрит олігоартикулярний і асиметричний, з переважним ураженням нижніх кінцівок. Виявляють носійство НLА-В27 |
|
Бактеріальний ендокардит |
Відзначають ураження великих суглобів, лихоманку з лейкоцитозом, появу серцевих шумів |
Необхідно робити посів крові у всіх пацієнтів з лихоманкою і поліартритом |
|
Септичний артрит |
Зазвичай виявляють моноартикулярний, рідше олігоартикулярний, артрит з переважним ураженням великих суглобів. Відзначають гіпертермію, гіперемію і припухлість суглобів з обмеженням обсягу рухів. Артрит може бути мігруючим |
Необхідно зробити посів крові, вивчити (використовуючи забарвлення препарату за Граммом) клітинний склад рідини, отриманої з порожнини суглоба, провести її культуральне дослідження. Проте слід пам’ятати, що септичний артрит виникає також і у пацієнтів з РА |
|
Вірусні артрити |
Характерна ранкова скутість, симетричне ураження суглобів кистей і променезап’ясткових суглобів, вірусна екзантема, іноді виявляють РФ. Причиною розвитку даної патології можуть бути парвовірус В19, вірус Епштейна — Барр, аденовіруси, вірус краснухи, гепатиту В і С |
Звертають увагу на характерний анамнез. Вірусну природу хвороби підтверджують серологічними реакціями. Вірусний артрит в більшості випадків спонтанно проходить через 4–6 тиж (за винятком артриту, пов’язаного з парвовірусною інфекцією) |
|
ССД |
Характерні васкулопатія (з ураженням дрібних судин) і фіброз. Рідко виникає артрит, зазвичай виявляють артралгії, обмеження рухів, пов’язане з набряком шкіри і підшкірного жирового шару |
Феномен Рейно та ущільнення шкіри |
|
ІЗМ |
Виявляють запалення м’язів, проксимальну м’язову слабкість, підвищення рівня КФК і альдолази, артралгії та міалгії, патологічні зміни на електроміограмі |
Артрит з вираженим синовітом виникає рідко |
|
ЗЗСТ |
Характерні ознаки СЧВ, ССД та міозиту. У 60–70% випадків виникає артрит (може бути деформуючим і ерозивним) |
Виявляють антитіла до U1-рибонуклепротеїну |
|
Хвороба Лайма |
Системне запальне захворювання, що викликається Borrelia burgdferi. На ранніх стадіях виникає мігруюча еритема і кардіальна патологія, на пізніх — інтермітуючий моно- або олігоартрит (у 15% пацієнтів може бути хронічним і ерозивним), енцефалопатія та невропатія |
В анамнезі є вказівка на укус кліща. Діагноз підтверджують за допомогою серологічних реакцій (Western Blot) на бореліоз. Проте у 5% здорових людей серологічні реакції дають псевдо позитивні результаті |
|
РПМ |
Характерна дифузний біль і ранкова скутість в осьових суглобах і проксимальних групах м’язів. Припухлість суглобів виявляють рідше. Спостерігають виражене збільшення ШОЕ. Захворювання рідко виникає у віці молодше 50 років |
У 10–15% поєднується з ГКА. Симптоми швидко проходять при використанні ГК |
|
Хвороба Бехчета |
Виникають рецидивуючі хворобливі виразки на слизових оболонках рота і статевих органів, ураження ЦНС, увеїт, в 50% випадків — моноартикулярний артрит із переважним залученням в процес великих суглобів. Позитивний тест «патергіі» |
Необхідний диференційний діагноз з ураженням очей (склерит) при РА |
|
Амілоїдоз |
Виявляють періартикулярне відкладення амілоїду, іноді випіт у порожнині суглоба |
Фарбування аспірованої суглобової рідини конго червоним |
|
Гемохроматоз |
Аутосомно-рецесивне захворювання. Спостерігають порушення всмоктування і відкладення заліза, збільшення кісткових структур п’ястково-фалангових суглобів II і III, підвищення рівня заліза та феритину в сироватці крові зі зниженням трансферинзв’язуючої здібності. На рентгенограмах виявляють хондрокальциноз |
Діагностують патологію за допомогою біопсії печінки. Захворювання асоційоване з цукровим діабетом, серцевою недостатністю і цирозом |
|
Саркоїдоз |
Хронічне гранулематозне захворювання. У 10–15% випадків виникає хронічний симетричний поліартрит (при поєднанні з вузлуватою еритемою формується синдром Лефгрена) |
Характерна клінічна картина і результати біопсії (виявлення гранульоми) |
|
Сімейна середземноморська лихоманка |
Відзначають рецидивуючі атаки гострого синовіту (моно- або олігоартикулярного) великих суглобів, асоційовані з лихоманкою, плевритом і перитонітом |
Сімейний анамнез, національні особливості, амілоїдоз (пізнє ускладнення) |
|
Рецидивуючий поліхондрит |
Поширене прогресуюче запалення і деструкція хрящової і сполучної тканини. Виникає мігруючий асиметричний, неерозивний артрит дрібних і великих суглобів, запалення і деформація хряща вушної раковини |
У 30% випадків може бути при інших ревматичних хворобах |
|
Фіброміалгія |
Поширений м’язово-скелетний біль і скутість, парестезії, непродуктивний сон, втома, множинні симетричні тригерні точки (для встановлення діагнозу досить виявлення 11 з 18). Патології суглобів за результатами лабораторних та інструментальних досліджень не виявляють |
Зрідка необхідний диференційний діагноз з ранньою стадією РА |
Лікування
При виборі лікування необхідно враховувати фактори ризику несприятливого прогнозу і тривалість періоду між появою симптомів і початком прийому БПЗП. До факторів несприятливого прогнозу, що зумовлює необхідність проведення більш активної терапії, відносять наступні. Серопозитивність за РФ і АЦЦП в дебюті хвороби. Високу запальну активність. Залучення в патологічний процес багатьох суглобів. Розвиток позасуглобових проявів. Збільшення ШОЕ та рівня СРБ. Виявлення певних алелів НLА-DR (0101, 0401, 0404/0408, 1402). Виявлення ерозій в суглобах у дебюті хвороби. Молодий чи літній вік початку хвороби. Погані соціально-економічні умови життя.
Немедикаментозне лікування
В основі лікування РА лежить мультидисциплінарний підхід, заснований на використанні нефармакологічних і фармакологічних методів, залучення фахівців інших медичних спеціальностей (ортопедів, фізіотерапевтів, кардіологів, неврологів, психологів та ін.).
За відсутності серйозних деформацій суглобів пацієнти продовжують працювати, однак значні фізичні навантаження їм протипоказані. Пацієнтам слід уникати факторів, які потенційно можуть провокувати загострення хвороби (інтеркурентні інфекції, стрес та ін.). Рекомендують припинити курити і обмежити прийом алкоголю. Підтримання ідеальної маси тіла сприяє зменшенню навантаження на суглоби, зниженню ризику смерті і розвитку ОП. Для цього необхідно дотримуватися збалансованої дієти, яка включає їжу з високим вмістом поліненасичених жирних кислот (риб’ячий жир, оливкова олія), фрукти, овочі. Вживання цих продуктів потенційно знижує інтенсивність запалення.
Важливе значення мають програми навчання пацієнтів (зміна стереотипу рухової активності), ЛФК, спеціальні вправи (1–2 рази на тиждень), спрямовані на зміцнення м’язової сили, фізіотерапевтичні методи (при помірній активності РА). Ортопедичні методи, спрямовані на профілактику та корекцію типових деформацій суглобів і нестабільності шийного відділу хребта.
Санаторно-курортне лікування рекомендують лише пацієнтам з мінімальною активністю РА або в стадії ремісії.
Протягом усього періоду хвороби необхідна активна профілактика та лікування супутніх захворювань, в першу чергу кардіоваскулярної патології.
Слід особливо підкреслити, що немедикаментозні методи чинять помірну і короткочасну дію. Вплив на прогресування захворювання не доведено. Описані заходи підвищують ефективність симптоматичної терапії і допомагають у корекції стійких деформацій суглобів.
Медикаментозне лікування
Лікування РА — відповідно до адаптованих рекомендацій EULAR i ACR (2010) із застосування синтетичних та біологічних БПЗП. Алгоритм діагностики РА (ACR/EULAR 2010), 15 рекомендацій щодо лікування пацієнтів із РА, ортопедичне лікування при РА та ACR-критерії якості лікування див. розділ І, глава 2.
НПЗП мають пряму протизапальну дію. Мета призначення НПЗП при РА — зменшення вираженості симптомів хвороби (болю, скутості, припухлості суглобів).
НПЗП не впливають на активність запалення, не здатні вплинути на перебіг захворювання і прогресування деструкції суглобів. Проте НПЗП вважають основним засобом для симптоматичного лікування РА і засобом першого ряду при призначенні в комплексі з БПЗП. Лікування НПЗП обов’язково потрібно поєднувати з призначенням БПЗП, оскільки частота розвитку ремісії на тлі монотерапії НПЗП істотно нижча, ніж на тлі лікування будь-яким БПЗП.
ГК. Застосування ГК в низьких дозах (преднізолон <10 мг/добу) дозволяє ефективно контролювати клінічні прояви РА, пов’язані з запаленням суглобів. При ранньому РА лікування ГК (у поєднанні з БПЗП) має більш виражений клінічний ефект (за критеріями ACR) і частіше призводить до розвитку стійкої ремісії, ніж монотерапія БПЗП. ГК потенційно можуть посилювати дію БПЗП щодо уповільнення прогресування деструкції суглобів при ранньому РА. При цьому ефект ГК зберігається після завершення їх прийому.
При РА ГК не слід використовувати як монотерапію. Їх необхідно застосовувати в комбінації з БПЗП (бетаметазон 0,25–2 мл внутрішньосуглобово або періартикулярно в ділянку ураженого суглоба не частіше 1 разу на 1–3 міс в той самий суглоб залежно від дози та розміру суглоба). За відсутності особливих показань доза ГК не має перевищувати 10 мг/добу (у перерахунку на преднізолон).
При призначенні ГК при РА слід пам’ятати, що їх прийом приводить до розвитку великої кількості побічних ефектів. Побічні ефекти частіше відзначають при неадекватному використанні препаратів (тривалий прийом у високих дозах). При цьому необхідно мати на увазі, що деякі побічні ефекти (наприклад тяжке ураження ШКТ, печінки та інших органів) виникають рідше, ніж при лікуванні НПЗП і БПЗП. Крім того, для запобігання деяким небажаним ефектам (наприклад ГК-ОП) розроблено ефективні заходи профілактики.
Показання до призначення ГК в низьких дозах
- Пригнічення запалення суглобів до початку дії БПЗП («bridge»-терапія).
- Пригнічення запалення суглобів при загостренні хвороби або розвитку ускладнень лікування БПЗП.
- Неефективність НПЗП і БПЗП.
- Протипоказання до призначення НПЗП (наприклад, у осіб похилого віку з «виразковим» анамнезом і (або) порушенням функцій нирок).
- Досягнення ремісії при деяких варіантах РА (наприклад при серонегативному РА в осіб похилого віку, що нагадує РПМ).
ГК в середніх та високих дозах усередину (15 мг/добу і більше, зазвичай 30–40 мг/добу в перерахунку на преднізолон) застосовують для лікування тяжких системних проявів РА (випітний серозит, гемолітична анемія, шкірний васкуліт, пропасниця та ін.), а також особливих форм хвороби (синдром Фелті, синдром Стілла у дорослих). Тривалість лікування визначають за часом, необхідним для пригнічення симптомів. Курс зазвичай становить 4–6 тиж, після чого поступово знижують дозу і переходять на лікування ГК в низьких дозах.
Рутинне застосування ГК при РА не рекомендують. Призначати препарати з цієї групи повинен лікар-ревматолог.
Пульс-терапію ГК застосовують у пацієнтів з тяжкими системними проявами РА. Цей метод дозволяє досягти швидкого (протягом 24 год), але короткочасного пригнічення активності запалення суглобів.
Оскільки позитивного впливу пульс-терапії ГК на прогресування деструкції суглобів і прогноз не доведено, їх застосування (без особливих показань) не рекомендують.
Локальне (внутрішньосуглобове) введення ГК в поєднанні з прийомом БПЗП ефективно пригнічує запалення суглобів на початку хвороби або при загостренні процесу, але не впливає на прогресування деструкції суглобів. При проведенні локальної терапії слід дотримуватися загальних рекомендацій.
У пацієнтів зі стійким і (або) ерозивним артритом лікування БПЗП потрібно починати якомога раніше (в межах 3 міс від моменту появи симптомів хвороби), навіть якщо вони формально не відповідають діагностичним критеріям РА (недиференційований артрит). Ранній початок лікування БПЗП сприяє поліпшенню стану пацієнта і уповільнює прогресування деструкції суглобів. Пізніше призначення БПЗП (через 3–6 міс від початку хвороби) знижує ефективність монотерапії. Чим більша тривалість хвороби, тим нижча ефективність БПЗП. При недиференційованому артриті призначення метотрексату знижує ризик трансформації хвороби в достовірний РА, особливо у пацієнтів, у чиїй крові виявляють АЦЦП.
На тлі лікування необхідно ретельно оцінювати динаміку активності захворювання (індекс DAS) не рідше 1 разу на 3 міс. Коректний підбір БПЗП залежно від активності хвороби істотно підвищує ефективність лікування при ранньому РА.
Прийом БПЗП слід продовжувати навіть при зниженні активності хвороби та досягненні ремісії, оскільки скасування препарату часто призводить до загострення і прогресування деструктивних змін у суглобах. При досягненні ремісії можна знизити дозу БПЗП, якщо при цьому не настає загострення.
Прогноз
У кінці XX ст. у середньому близько половини пацієнтів втрачали працездатність протягом перших 10 років, до 15-го року хвороби приблизно 80% хворих ставали інвалідами групи I і II. У пацієнтів з РА відзначали зменшення тривалості життя порівняно із загальною популяцією на 5–10 років. Найпоширенішими причинами смерті були кардіоваскулярні хвороби (інсульт, гострий інфаркт міокарда), виникнення яких пов’язують з інтенсивним розвитком атеросклерозу і схильністю до тромбозів внаслідок хронічного імунного запалення. Нерідко реєстрували летальні випадки через вторинний амілоїдоз, супутні інфекції (пневмонії, нагноєння м’яких тканин та ін.).
Сучасне активне лікування, особливо на ранній стадії РА, дозволяє дійсно поліпшити результати, пов’язані зі збереженням працездатності, досягти клінічної ремісії у 40–50% пацієнтів, довести очікувану тривалість життя до популяційного рівня.
Інформація для пацієнта. РА — аутоімунна рематична хвороба. Для неї характерний розвиток ерозивного артриту і системне ураження внутрішніх органів. Симптоми РА зазвичай бувають стійкими і за відсутності лікування неухильно прогресують.
Медикаментозну терапію вважають основним методом лікування пацієнтів з РА. Це єдиний спосіб, який дозволяє загальмувати розвиток запального процесу і зберегти рухливість у суглобах. Інші методи лікування: фізіотерапія, дієта, ЛФК мають допоміжне значення і не здатні суттєво вплинути на перебіг хвороби.
В основі лікування хворих на РА лежить застосування БПЗП. До них відносять велику кількість різноманітних за хімічною структурою та фармакологічними властивостями лікарських засобів, таких як метотрексат, лефлуномід, сульфасалазин та ін. Їх об’єднує здатність більшою чи меншою мірою і за рахунок різних механізмів пригнічувати запалення і/або патологічну активацію імунної системи. Новим методом лікування пацієнтів з РА вважають застосування так званих біологічних агентів. Біологічні агенти (не плутати з біологічно активними добавками!) — це білкові молекули, які вибірково впливають на окремі речовини або групи клітин, які беруть участь у процесі хронічного запалення. До біологічних препаратів відносять інфліксимаб, ритуксимаб, адалімумаб.
Лікування РА зазвичай починають з призначення метотрексату або лефлуноміду. Біологічні агенти (інфліксимаб, адалімумаб і ритуксимаб), як правило, додають до цих препаратів при недостатній ефективності монотерапії. Швидкий протизапальний ефект можуть давати ГК. НПЗП являють собою важливий компонент лікування РА, оскільки можуть зменшити вираженість болю і скутість в суглобах. Найбільш часто застосовують диклофенак, німесулід, мелоксикам, кетопрофен, целекоксиб.
Медикаментозне лікування РА може давати дуже хороші результати, але вимагає ретельного контролю. Контроль повинен проводити кваліфікований фахівець-ревматолог і сам пацієнт. Пацієнту необхідно відвідувати лікаря не рідше ніж 1 раз на 3 міс на початку лікування. Крім огляду, призначають аналізи крові, щороку проводять рентгенологічне дослідження суглобів для оцінки перебігу захворювання. Слід пам’ятати про обмеження, пов’язані з лікуванням. На тлі терапії метотрексатом і лефлуномідом не рекомендують вживання алкоголю. Доцільно уникати контактів з інфекційними хворими. Застосування більшості протизапальних засобів потребує запобігання вагітності (вона можлива після відміни лікування).
Глава 23. Ювенільний артрит
Гетерогенна група хворих на ювенільний артрит (ЮА) включає ЮРА, ювенільний спондилоартрит, ПсА, а також випадки, коли тривалий час нозологічна приналежність артриту залишається неуточненою. В останньому випадку використовують термін «ювенільний хронічний артрит». Загальним для перелічених захворювань є хронічне прогресування патологічного процесу, рання інвалідизація. Синонімом ЮА є ювенільний ідіопатичний артрит — ЮІА (класифікація ILAR, 1997 р.) і ювенільний хронічний артрит — ЮХА (класифікація EULAR). Що стосується терміну ЮРА, то правильним є вживання його як еквіваленту РА дорослих (з урахуванням особливостей, властивих дитячому віку). Але його нерідко використовують для групового позначення хронічних запальних захворювань, які виникають у дитячому віці (до 16 років), тобто в таких випадках ЮРА вважають самостійною нозологічною формою, а не варіантом РА.
Код за МКХ-10
М08 Юнацький (ювенільний) артрит.
М08.0. Юнацький (ювенільний) ревматоїдний артрит (РФ(+) і РФ(–)).
М08.1. Юнацький (ювенільний) анкілозуючий спондиліт.
М08.2. Юнацький (ювенільний) артрит з системним початком.
М08.3. Юнацький (ювенільний) поліартрит (серонегативний).
М08.4. Юнацький (ювенільний) пауціартикулярний артрит.
М08.8. Інші ювенільні артрити.
М08.1. Юнацький неуточнений.
Епідеміологія
Захворюваність ЮРА становить 2–16 випадків на 100 тис. дітей у віці до 16 років, поширеність — 0,05–0,6% (Насонов Е.Л., 2006 (ред.)). Чинники, що провокують розвиток ЮА, включають інфекцію (бактеріальну, вірусну, гостру респіраторну вірусну інфекцію), травми, у тому числі психологічні, інсоляцію, вакцинацію.
Профілактика
Первинну профілактику ЮА не проводять.
Класифікації
Єдиного погляду на термінологію і класифікацію ЮА немає. Класифікацію ЮА надано нижче (табл. 23.1).
Таблиця 23.1
Класифікації ЮА
|
ACR |
EULAR |
Міжнародна ліга ревматологічних асоціацій (ILAR) |
|
ЮРА:
|
ЮХА:
ЮРА (РФ+):
Ювенільний ПсА Ювенільний АС |
ЮІА:
ПсА Артрит, що поєднується з ентезитом Некласифіковані артрити |
Ситуація з термінологією і класифікацією ювенільних артритів складна. Представлені вище в трьох класифікаціях ЮРА, ЮХА і ЮІА, незважаючи на деякі відмінності, є еквівалентами (Насонов Е.Л., 2006 (ред.)). Нижче подано характеристику кваліфікаційних критеріїв ЮА (табл. 23.2).
Таблиця 23.2
Характеристика класифікаційних критеріїв ЮА
|
Класифікації ЮА |
ACR |
EULAR |
ILAR |
|
Вік початку захворювання |
<16 |
<16 |
<16 |
|
Мінімальна тривалість артриту |
6 тиж |
3 міс |
6 тиж, але класифікують за варіантами >6 міс |
|
Системний варіант |
+ |
+ |
+ |
|
Поліартикулярний варіант РФ(-) |
+ |
+ |
+ |
|
Поліартикулярний варіант РФ(+) |
+ |
+ + → діагноз ЮРА |
+ |
|
Олігоартикулярний варіант |
+ |
+ |
+ |
|
Артрит, що поєднується з ентезитом |
+ |
||
|
ПсА |
+ |
||
|
Недиференційований артрит |
+ |
||
|
Наявність псоріазу у пацієнта або родичів |
Виключають |
Допускають |
Допускають для певних категорій |
|
Наявність ознак спондилоартропатії |
Виключають |
Допускають |
|
|
Наявність HLA-B27 |
Допускають |
||
У табл. 23.3 представлено класифікацію ЮІА, прийняту на конгресі ILAR (2001).
Таблиця 23.3
Класифікація ЮІА (ILAR, 2001)
|
Класифікаційна категорія |
Визначення |
Виключення |
|
Системний артрит |
Артрит 1 і більше суглобів і/або попередня лихоманка не менше 2 тиж, які реєструють протягом 3 днів щодня у поєднанні з одним або більше з наступних ознак: |
1, 2, 3, 4 |
|
||
|
||
|
||
|
||
|
Олігоартрит |
Артрит, що уражує 1–4 суглоби в перші 6 міс хвороби. Виділяють 2 субкатегорії: |
1, 2, 3, 4, 5 |
|
1) персистуючий артрит, що уражує не більше 4 суглобів упродовж усієї хвороби; |
||
|
2) артрит, що поширився, уражує більше 4 суглобів після закінчення 6 міс хвороби |
||
|
Поліартрит РФ(-) |
Артрит, що уражує 5 і більше суглобів протягом перших 6 міс хвороби. Тест на РФ(-) |
1, 2, 3, 4, 5 |
|
Поліартрит РФ(+) |
Артрит, що уражує 5 і більше суглобів протягом перших 6 міс хвороби. Два і більше позитивних тести на РФ протягом 3 міс |
1, 2, 3, 5 |
|
ПсА |
Артрит і псоріаз або артрит і як мінімум 2 з наступних ознак: |
2, 3, 4, 5 |
|
||
|
||
|
||
|
Артрит, що поєднується з ентезитом |
Артрит і ентезит, або артрит, або ентезит в поєднанні, як мінімум, з двома з наступних ознак: |
1, 4, 5 |
|
||
|
||
|
||
|
||
|
Недиференційований артрит |
Артрит, що не відповідає жодній з категорій або відповідає критеріям 2 і більше категорій |
|
|
Критерії виключення |
||
|
1. Псоріаз або наявність псоріазу у родичів I ступеня спорідненості |
||
|
2. Артрит у HLA-B27-позитивних хлопчиків з початком захворювання у віці старше 6 років |
||
|
3. АС, артрит з ентезитом, сакроілеїт при запальних захворюваннях кишечнику, синдром Рейтера, гострий передній увеїт або наявність одного з цих захворювань в анамнезі у родичів I ступеня спорідненості |
||
|
4. Виявлення IgM-РФ, принаймні дворазове, в дослідженнях, виконаних протягом 3 міс |
||
|
5. Наявність системного артриту у пацієнта |
||
Класифікація ЮІА ILAR дозволяє за необхідності провести рекласифікацію пацієнтів з діагнозами ЮРА і ЮХА. Нижче приведено варіанти ЮА (табл. 23.4).
Таблиця 23.4
Варіанти ЮА
|
Системний |
Ним зазвичай починається класичний ЮРА, такий, що асоціюється з поняттям «хвороба Стілла». Проте у невеликого відсотка хворих можливий розвиток надалі інших нозологічних форм, наприклад ювенільного спондилоартриту. Системний варіант характеризується несприятливим прогнозом, частою деструкцією суглобів, асептичних некрозів, амілоїдозу, ургентних станів. Цей варіант неоднорідний і представлений двома субтипами |
|
|
А. Деструктивний поліартрит генералізований |
Найбільш тяжкий клінічний варіант ЮА, рефрактерний до протиревматичної терапії. Він вимагає агресивної лікувальної тактики |
|
|
Б. Відстрочений, нестійкий і/або лімітований суглобовий синдром |
Прогноз відносно сприятливий |
|
|
Поліартритичний |
Як і олігоартикулярний, поширений, він властивий дебюту істинного ЮРА. Проте надалі не виключається розвиток інших нозологічних форм — ювенільного спондилоартриту, ПсА |
|
|
Олігоартикулярний |
Вкрай рідко призводить до розвитку класичного ЮРА, асоціюється з частими тривалими ремісіями, розвитком надалі серонегативних спондилоартритів |
|
Подана в таблиці інформація підтверджує, що усі 3 варіанти ЮА (ЮРА, ЮІА, ЮХА) — гетерогенні, й найбільшою гетерогенністю з них виділяється олігоартикулярний.
Клінічна картина
До продромальних явищ відносять погіршення загального стану, зменшення маси тіла, слабкість, артралгії, уранішню скутість, субфебрильну температуру тіла.
Відзначають варіанти дебюту ЮА: системний, поліартикулярний, олігоартикулярний з ураженням очей або без нього. Клінічні особливості ЮА, які відрізняють його від аналогічних хвороб у дорослих, представлено в табл. 23.5.
Таблиця 23.5
Клінічні особливості групи ЮА, які відрізняють його від аналогічних хвороб у дорослих
|
|
|
|
|
|
|
|
|
|
|
|
Біль в суглобах у дітей виражений значно меншою мірою, ніж у дорослих, іноді біль і скутість можуть бути повністю відсутніми. Характеристику варіантів ЮА подано в табл. 23.6.
Таблиця 23.6
Коротка характеристика варіантів ЮА
|
Системний варіант |
Вік хворих |
Частіше розвивається у віці 1–7 років |
|
|
Характер суглобового синдрому |
Найчастіше уражуються колінні, гомілковостопні і променезап’ясткові суглоби, рідше — дрібні суглоби кистей і стоп. До процесу залучається шийний відділ хребта, кульшові і щелепно-скроневі суглоби (нерідко развивається мікрогнатія — «пташина щелепа»). Також нерідко утворюються множинні ерозії, асептичні некрози (частіше кульшових суглобів), порушується функціональна здатність |
||
|
Інтенсивність запалення |
Має місце виражений ексудативний компонент, відчуття скутості суглобів |
||
|
Лабораторні дані |
Лейкоцитоз, нейтрофільоз зі зрушенням вліво, значне збільшення ШОЕ, анемія |
||
|
Характер синовіальної рідини |
Цитоз досягає 50 000–100 000 в 1 мкл |
||
|
Фізичний розвиток |
Пацієнти відстають у фізичному розвитку через високу активність захворювання, імуносупресивну терапію і нейроендокринні порушення |
||
|
Поліартикулярний варіант |
Характер дебюту |
Гострий варіант |
Симптоматика близька до синдрому Стілла (за винятком висипу і вісцеральних проявів). Швидко развивається кісткова деструкція і функціональна недостатність |
|
Підгострий варіант. Діти дошкільного і молодшого шкільного віку |
Симптоми хвороби і показники запалення менш виражені. Віддалений прогноз серйозний відносно розвитку функціональних порушень |
||
|
Підгострий варіант, що проходить за дорослим типом (частіше у дівчаток старшого віку) |
Початок субманіфестний, є уранішня скутість, конфігурація суглобів змінюється безболісно, переважно уражуються кисті. Запальна активність помірна або мінімальна, часто виявляється РФ в сироватці крові. Нерідко розвивається деструкція суглобів, порушується їх функція |
||
|
Підтипи |
I РФ(+) підтип |
Цей варіант має місце в 5–10% випадків хвороби; частіше хворіють дівчатка (80%) у віці 8–15 років; структурні зміни в суглобах розвиваються в перші 6 міс хвороби; поліартрит (5 і більше суглобів) симетричний, уражуються колінні, промене-зап’ясткові, гомілковостопні і дрібні суглоби кистей і стоп; ерозії й анкілози дрібних кісток зап’ястя розвиваються до кінця першого року хвороби; деструктивний артрит розвивається у 50% пацієнтів; асоційований з РФ(+), як мінімум в 2 тестах протягом 3 міс |
|
|
II РФ(-) підтип |
Цей субтип розвивається у менше 10% усіх випадків ЮА і має низку особливостей: виникає в будь-якому віці (період від 1 до 15 років) і в більшості випадків має доброякісний перебіг; частіше (90%) хворіють дівчатка; уражуються симетрично великі і дрібні суглоби (5 і більше суглобів в перші 6 міс), а також суглоби шийного відділу хребта і щелепно-скроневі; в більшості випадків артрит прогресує, але стани, що загрожують життю, не розвиваються; деструкція суглобів (переважно кульшових і дрібних суглобів кистей) розвивається у 10% пацієнтів при неадекватному лікуванні; можливий розвиток увеїту |
||
|
Пауци(оліго)артикулярний варіант |
Вік |
Відзначається в усіх вікових групах |
|
|
Характер дебюту |
Підгострий початок з повільним прогресуванням або доброякісний перебіг з поступовим (через 6–12 міс) залученням суглобів |
||
|
Типи |
Персистуючий |
Кількість уражених суглобів протягом хвороби не перевищує 4 |
|
|
Поширений |
Олігоартикулярний тип ураження відзначають тільки в перші 6 міс хвороби. Потім до патологічного процесу залучається більша кількість суглобів |
||
|
Наслідки хвороби |
В деяких випадках формується ЮРА або розгортається один з варіантів спондилоартритів. Може наступити багаторічна повна клініко-лабораторна ремісія |
||
|
Моноолігоартикулярний варіант |
Вік |
Розвивається у дівчаток раннього віку |
|
|
Характер ураження очей |
Субклінічно (частіше) розвивається увеїт. Він може призводити до інвалідизації. Маркером активності і ризику ураження очей є АНФ |
||
|
Наслідки хвороби |
1) Уражується тільки орган зору, настає повна регресія суглобового синдрому; 2) зберігається симптоматика недиференційованого хронічного артриту; 3) розвивається один з варіантів серонегативних спондилоартритів |
||
Інші варіанти ЮА. Відповідно до класифікації ILAR до ЮІА також відносять артрит, що поєднується з ентезитом, і ПсА (табл. 23.7).
Таблиця 23.7
Інші варіанти ЮІА
|
Артрит, що поєднується з ентезитом |
У цю категорію включають дітей, у яких артрит поєднується з ентезитом або не менше ніж з 2 із наступних критеріїв: наявність HLA-B27; біль в хребті запального характеру; біль в крижово-клубових з’єднаннях; передній увеїт, що супроводжується больовим синдромом, почервонінням очного яблука або світлобоязнь; наявність в сімейному анамнезі переднього увеїту з больовим синдромом, спондилоартропатією або запального захворювання кишечнику |
|
ПсА |
У цю категорію включають дітей з псоріазом; з артритом і сімейним анамнезом, обтяженим псоріазом у родичів 1-ї лінії спорідненості, з дактилітом, ураженнями нігтьової пластинки. Для характеристики артриту використовують критерії віку початку, характеру артриту (симетричний, асиметричний), перебігу артриту (оліго- або поліартрит), наявність увеїту, АНФ |
Артрит, що поєднується з ентезитом, представляє групу запальних захворювань суглобів, які є ранньою стадією (до ураження хребта) серонегативних спондилоартритів (ювенільний спондилоартрит). Термін «ювенільні спондилоартрити» об’єднує: 1) ювенільний АС; 2) спондилоартритичний варіант ПсА; 3) артрит, асоційований із запальними захворюваннями кишечнику (хвороба Крона, виразковий коліт); 4) синдром Рейтера. Перебіг ювенільного спондилоартриту доброякісний, рецидивуючий, можливі тривалі ремісії (Никишина И.П., 2010). У міру збільшення віку дитини частота периферичного артриту і ентезиту знижується, а вираженість симптомів ураження хребта збільшується. Після 20 років формується типова клінічна картина АС. У дітей у віці до 7 років ПсА розвивається у вигляді олігоартриту, що поєднується з дактилітом. При цьому можуть уражатися суглоби верхніх і нижніх кінцівок. У дітей, старших 7 років, може розвиватися поліартрит (при загальному переважанні олігоартритичного варіанта), деструктивний мутилюючий артрит. Для підлітків характерним є розвиток спондилоартритичного варіанта. Дактиліт (патогномонічний симптом ПсА) може призвести до періоститу. Шкірні прояви псоріазу можуть з’явитися через багато років після початку артриту.
Особливості, властиві системному варіанту ЮРА, наведено також в табл. 23.8.
Таблиця 23.8
Особливості, властиві системному варіанту ЮРА
|
Частота розвитку системного варіанта |
Відзначають в 10–20% усіх випадків ЮРА |
|
Вік пацієнтів |
Розвивається в будь-якому дитячому віці |
|
Стать |
Однаково часто хворіють дівчатка і хлопчики |
|
Характеристика системних проявів |
|
|
Характерний початок |
Гострий або підгострий |
|
Лихоманка |
Фебрильна або гектична (переважно в ранкові години), озноб, підвищене потовиділення |
|
Характер висипу |
Плямистий, плямисто-папульозний, уртикарний, геморагічний; без свербежу (зрідка може відзначатися); нестійкий (зникає протягом короткого часу); з’являється і посилюється при лихоманці |
|
Ураження серця |
Міокардит, перикардит |
|
Ураження легень |
Пневмоніт, плевропневмоніт, фіброзуючий альвеоліт (рідко) |
|
Васкуліт |
Долонний або підошовний капілярит Акроціаноз Ліведоподібні прояви |
|
Лімфаденопатія |
+ |
|
Гепатоспленомегалія |
+ |
|
Характеристика суглобового синдрому |
|
|
Варіанти ураження суглобів |
А) Поліартикулярне ураження з самого початку хвороби із залученням шийного відділу хребта, швидким розвитком деформацій, контрактур, атрофії м’язів |
|
Б) Симетричний олігоартрит або поліартрит (рідше) з ураженням великих суглобів — колінних, гомілковостопних, кульшових |
|
|
В) Розвиток суглобового синдрому через декілька місяців або навіть років після початку системних проявів |
|
|
Г) Переважання артралгії і міалгії з посиленням при лихоманці |
|
Особливості перебігу різних варіантів ЮРА наведено в табл. 23.9.
Таблиця 23.9
Особливості перебігу різних варіантів ЮРА
|
Моно- або олігоартикулярний тип ураження |
Уражуються великі (колінні і гомілковостопні) суглоби. У дівчаток з наявністю ЮРА у віці до 2 років виявляється АНФ, розвивається увеїт. Кореляції між вираженістю суглобових проявів і ураженням очей немає. У 20% пацієнтів протягом тривалого часу зберігається моно- або олігоартрит (уражується 2–4 суглоби), у інших надалі ураження набуває поліартикулярного характеру |
|
Поліартрит |
Розвивається у 35–40% хворих (частіше у дівчаток), при цьому має місце спондилоартрит шийного відділу, а кількість уражених суглобів менша, ніж при РА у дорослих. Початок може бути як гострим, так і поступовим. Частіше уражуються великі суглоби, рідше — дрібні кистей і стоп. З позасуглобових проявів відзначають генералізовану лімфаденопатію, пневмоніт, перикардит, збільшення печінки і селезінки. Перебіг хвороби хвилеподібний, у деяких дітей можлива повна ремісія, у інших — стійко активний поліартрит, який закінчується розвитком контрактур |
|
Синдром Стілла і Вісслера — Фанконі |
Характерна інтермітуюча лихоманка з ознобами (піддається лікуванню ГК і НПЗП). При синдромі Вісслера — Фанконі лихоманка може зберігатися тижнями. При синдромі Стілла лихоманка супроводжується шкірними висипаннями, які локалізуються на тулубі і проксимальних відділах кінцівок. Синдром Стілла супроводжується позасуглобовими проявами: пневмонітом, ексудативним перикардитом, міокардитом, ендокардитом, перитонітом (іноді), генералізованою лімфаденопатією, збільшенням печінки, селезінки. Може розвинутися недостатність аортального і мітрального клапанів, гломеруліт |
На наш погляд, синдром Вісслера — Фанконі — це системний варіант ЮРА, який відрізняється від хвороби Стілла відсутністю виразного суглобного синдрому. Характерні прояви синдрому Вісслера — Фанконі описано в табл. 23.10.
Таблиця 23.10
Характерні прояви синдрому Вісслера — Фанконі
|
Прояви |
Характеристика |
|
Вік пацієнтів |
Від 1–3 до 24 та більше років (у дорослих осіб розвиваються рецидиви синдрому, який почався у дитинстві) |
|
Провокуючі фактори |
Інфекція, вакцинація, введення білків, лікування сульфаніламідними препаратами та ін. |
|
Загальний стан |
Не завжди порушується |
|
Лихоманка |
Місяці та роки триває інтермітуюча гіпертермія (від 36 до 41 оС). Добове коливання температури тіла может становити 3–4 оС, іноді підвищена температура зберігається протягом декількох годин. Вона може нормалізуватися та бути нормальною декілька днів. Антибіотики на температуру не впливають, водночас її знижують анальгетики, жарознижувальні та ГК |
|
Характер висипань на шкірі |
Дрібноплямиста скарлатиноподібна, великоплямиста кіроподібна, уртикарна, папульозна, кільцеподібна еритема, багатоформна ексудативна еритема. Кіроподібна висипка локалізується на обличчі, передпліччях, в ділянці променезап’ясткових суглобів, на гомілках. Висип може покривати все тіло. Висипання можуть триматися декілька годин, зрідка затримуються на декілька днів. Можливі набряки за типом Квінке, шкірні крововиливи |
|
Стан суглобів, кісток, м’язів |
Суглобний синдром не характерний, мають місце артралгії, біль в усьому тілі. Можливі невеликі набряки, в тому числі пальців |
|
Лімфатичні вузли |
Помірне генералізоване збільшення |
|
Легені |
Можливий розвиток плевритів |
|
Серце |
Перикардит або міокардит (ендокардит не развивається) |
|
Печінка та селезінка |
Характерне ураження печінки з жовтяницею, збільшення селезінки. Розміри селезінки збільшуються при підвищенні температури тіла та зменшуються при її нормалізації |
|
Аналізи крові |
Посіви на стерильність негативні. Анемія нормо- або гіпохромна (гемоглобін может знижуватися до 49 г/л, еритроцити — до 2,5х1012/л), кількість лейкоцитів в крові варіює від 6500 до 53 000 в 1 мкл, відзначають зрушення формули вліво, можлива токсична зернистість нейтрофілів. Вміст ретикулоцитів нормальний або збільшений, ШОЕ підвищена, незважаючи на нормалізацію температури тіла та поліпшення загального стану |
|
Біохімічний аналіз крові |
Загальний вміст білка в крові нормальний або збільшений. Кількість альбумінів в крові зменшується, α2— та γ-глобулінів — збільшується |
|
Перебіг хвороби |
Характеризується ремісіями та загостреннями. Перший напад найбільш тяжкий, наступні рецидиви більш легкі та короткотривалі, але може бути і навпаки. Ремісії можуть бути багаторічними і тоді рецидиви виникають вже у молодих дорослих осіб |
|
Причини смерті |
Смерть може бути результатом ускладнень, інтеркурентних причин |
Спільні риси поліартикулярного і системного варіантів ЮРА. При поліартикулярному і системному варіантах ЮРА відзначають:
- уповільнення фізичного розвитку, зростання окремих сегментів скелета;
- відставання в рості;
- недорозвинення нижньої щелепи;
- вкорочення або подовження кісток (наприклад фаланг пальців).
Окрім персистуючого і поширеного типу, олігоартикулярного (класифікація ILAR), виділяють субтипи з раннім і пізнім початком олігоартикулярного варіанта ЮА. Їх особливості продемонстровано в табл. 23.11.
Таблиця 23.11
Порівняльна характеристика субтипів олігоартикулярного варіанта ЮА
|
Ознаки |
Субтип з раннім початком |
Субтип з пізнім початком |
|
Клінічні особливості |
||
|
Частота розвитку |
50% випадків |
10–15% випадків |
|
Вік дітей |
1–5 років |
8–15 років |
|
Стать |
85% дівчатка |
90% хлопчики |
|
Характер ураження суглобів |
Часто асиметричний |
Асиметричний |
|
Суглоби, що уражуються |
Колінні, гомілковостопні, ліктьові, променезап’ясткові |
Переважно суглоби нижніх кінцівок (стоп, кульшові) |
|
Розвиток сакроілеїту |
– |
Можливе ураження крижово-клубових з’єднань |
|
Частота розвитку деструкції суглобів |
У 25% випадків |
– |
|
Частота розвитку іридоцикліту |
У 30–50% пацієнтів |
У 5–10% хворих |
|
Лабораторні дані |
||
|
РФ |
РФ(-) |
РФ(–) |
|
СРБ |
Запальні зміни в крові можуть бути відсутні |
Концентрація в крові підвищена |
|
ШОЕ |
Помірно підвищена |
Вище 40 мм/год |
|
АНФ |
Виявляється у 80% хворих |
Титр невисокий |
|
HLA-B27 |
– |
Виявляється часто |
Узагальнені дані з особливостей суглобового синдрому і позасуглобових проявів у пацієнтів з ЮРА наведено в табл. 23.12.
Таблиця 23.12
Узагальнені дані особливостей суглобового синдрому і позасуглобових проявів у пацієнтів з ЮРА
|
1. Суглобовий синдром |
|
|
Артралгія, зміна контурів, припухлість за рахунок випоту, запалення в періартикулярних тканинах. Суглоб теплий, але гіперемія спостерігається рідко. Біль при рухах, чутливість при пальпації, відсутність болю (чи мінімум) у спокої. Залучаються колінні, гомілковостопні, променезап’ясткові, дрібні суглоби кистей, ліктьові й кульшові. Симетричність при поліартикулярній формі. Дистальні міжфалангові суглоби уражуються у 10% дітей. Ліктьова девіація кистей. Залучається шийний відділ хребта (у 60% при поліартикулярному і системному варіантах), зменшується обсяг рухів, розвивається анкілоз, знижується розгинальна здатність. Уражаються щелепно-скроневі суглоби, розвивається «пташина щелепа» (мікрогнатія), але анкілоз виникає рідко. Акроміально-ключичні, груднино-ключичні уражуються рідко, крижово-клубові — у 10% (у тяжких лежачих хворих). Можуть розвинутися синовіальні кісти плечового, ліктьового, колінного (кіста Бейкера) суглобів |
|
|
2. Екстраартикулярні прояви |
|
|
Лихоманка |
Спостерігається у 62–90% хворих, стійкий підйом до 39–40 °С відзначається тільки при системному варіанті. Лихоманка класична інтермітуюча — 2 піки в пообідній і вечірній час. Супроводжується висипом, лімфаденопатією, гепатолієнальним синдромом. Лихоманка з висипом — кардинальна ознака системності, що випереджає інші симптоми |
|
Висипання |
Виявляють тільки при системному варіанті у 20–70% дітей. Плямисто-папульозні шкірні зміни, зазвичай без свербежу, 2–6 мм в діаметрі, рожевого забарвлення з блідим центром, зникають при легкому натисканні. Висип завжди виникає на тлі лихоманки, його локалізація — груди, живіт, руки і ноги. Гістологічно: набряк колагенових волокон, периваскулярна інфільтрація |
|
Генералізоване порушення росту |
Порушується процес росту, виявляється асиметрія розвитку тіла. Впливає кортикостероїдна терапія: ГК порушують процес росту, посилюють його гальмування, зумовлене хворобою (мінімально значуща доза 5 мг/добу протягом 6 міс). В період ремісії порушення росту відбувається за відсутності змін в епіфізах. Максимальне порушення росту відзначають при системному варіанті |
|
Локальне порушення росту кісткових структур |
Мікрогнатія (гіпоплазія нижньої щелепи) часто поєднується з аналогічними змінами в шийному відділі хребта. Вона максимально виражена при розвитку хвороби у віці до 4 років. Однобічний процес веде до зміщення щелепи вбік. Реєструють брахідактилію (короткопалість), затримку розвитку долонь і стоп, укорочення кінцівки за рахунок змін в епіфізі, можливе її подовження в результаті посиленої васкуляризації паросткових зон |
|
Підшкірні вузлики |
Відзначають в 5–10% випадків і, як правило, за наявності РФ. Місце локалізації: дистальніше за ліктьовий відросток, сухожильні піхви згиначів пальців рук, сухожилля п’ят, перенісся. Вузлики зникають через декілька тижнів лікування |
|
Ураження м’язів |
М’язова гіпотрофія, вкорочення м’язів і зв’язок, в результаті — згинальні контрактури і міотонія. Можливий неспецифічний міозит з периваскулітом, лімфоцитарною інфільтрацією |
|
Ураження серця |
Перикардит з випотом, можливий безсимптомний перебіг, тампонада. Зрідка може передувати суглобовому синдрому і триває від 1 до 8 тиж. Рідше розвивається міокардит без ураження перикарда |
|
Ураження легень |
Рідкісне явище у дітей (розвивається у 4%). Пневмоніт і плеврит можуть перебігати безсимптомно |
|
Лімфаденопатія |
Часто зустрічається (пахвова, ліктьова, пахова ділянка). Помилково діагностують гематологічну і онкологічну патологію. Можливий біль у животі |
|
Спленомегалія |
Відзначають у 25% дітей при системному варіанті. Значна спленомегалія — ознака амілоїдної інфільтрації |
|
Ураження печінки |
Одна з перших ознак системності. Стійка гепатомегалія — результат іншої патології або медикаментозної терапії |
|
Ураження нирок |
Перші симптоми — протеїнурія, гематурія. Можлива медикаментозна нефропатія (НПЗП, метотрексат, солі золота, пеніциламін). У 10% померлих констатують амілоїдоз нирок. Амілоїдоз розвиватися і при поліартикулярній формі |
|
Ураження очей |
Найбільш тяжкі і нез’ясовані прояви. Типовий хронічний увеїт розвивається у дівчаток, які захворіли в ранньому віці, з олігоартикулярним синдромом і наявністю АНФ. Уражується райдужка, війкове тіло — розвивається іридоцикліт. Початок безсимптомний, може бути присутнім больовий компонент, гіперемія, підвищення світлочутливості, порушення зору. Увеїт за декілька років може передувати артриту. У 65–70% випадків увеїт двобічний. Процес може призвести до злипання райдужки і кришталика з втратою реакції зіниці. До пізніх ускладнень належать стрічкоподібна кератопатія, вторинна катаракта і глаукома |
Ускладнення при різних варіантах ЮРА наведено в табл. 23.13.
Таблиця 23.13
Ускладнення при різних варіантах ЮРА
|
Варіанти перебігу ЮРА |
Ускладнення |
|
Системний |
Синдром активації макрофагів; серцево-легенева недостатність; затримка росту; амілоїдоз; приєднання інфекції (бактеріальний сепсис, генералізована вірусна інфекція) |
|
Поліартикулярний |
Затримка росту; згинальні контрактури; рання інвалідизація |
|
Олігоартикулярний |
Асиметрія довжини кінцівок, катаракта, глаукома, сліпота як наслідок увеїту, інвалідизація внаслідок ураження очей і суглобів |
Симптоми, які відзначають у хворих з системним варіантом ЮРА і належать до проявів «синдрому активації макрофагів», включають:
- лихоманку гектичного типу;
- геморагічний висип, кровотечу зі слизових оболонок;
- збільшення розмірів лімфатичних вузлів, печінки, селезінки;
- зменшення кількості в крові лейкоцитів, тромбоцитів;
- зниження показника ШОЕ;
- збільшення в крові рівня трансаміназ, фібриногену, продуктів деградації фібрину, тригліцеридів;
- зниження концентрації II, VII, X факторів згортання крові;
- наявність в кістковому мозку великої кількості макрофагів, що фагоцитують гемопоетичні клітини;
- розвиток поліорганної недостатності, порушення свідомості, летальний результат.
Розвиток синдрому активації макрофагів може викликати інфекція (вірусна, бактеріальна), НПЗП, золото, інші лікарські засоби. Симптоми синдрому активації макрофагів може посилити застосування метотрексату і, навпаки, позитивний ефект дають в/в ГК і циклоспорин А.
Діагностика
Діагноз ґрунтується на застосуванні класифікаційних критеріїв ЮРА (АCR), ЮХА (EULAR) або ЮІА (ILAR), які представлені вище, а також використовують східноєвропейські діагностичні критерії (табл. 23.14). Відповідно до класифікації ЮІА (ILAR, 2001), щоб ідентифікувати варіант артриту, потрібно 6 міс, оскільки протягом цього часу формується клінічна картина (до цього артрит вважають некласифікованим). При формулюванні діагнозу ЮРА/ЮХА вказують: 1) клінічний варіант (системний — поліартрит, олігоартрит, поліартикулярний, олігоартикулярний); 2) позасуглобові прояви (ураження очей, нирок та ін.); 3) ступінь активності процесу (на думку лікаря — візуальна аналогова шкала 100 мм): 0 — <10 мм, I — 10–35 мм, II — 35–65 мм, III — >65 мм; 4) наявність або відсутність РФ; 5) наявність або відсутність АНФ; 6) рентгенологічну стадію (I — IV за Штейнброкером); 7) функціональну здатність (I–IV за Штейнброкером); 8) ремісію.
Таблиця 23.14
Східноєвропейські критерії ЮРА
|
Клінічні ознаки |
артрит тривалістю 3 міс і більше; |
|
артрит другого суглоба, що виникає через 3 міс або пізніше після ураження першого; |
|
|
симетричне ураження дрібних суглобів; |
|
|
випіт в порожнині суглоба; |
|
|
контрактура суглоба; |
|
|
тендосиновіт або бурсит; |
|
|
м’язова атрофія (частіше регіональна); |
|
|
уранішня скутість; |
|
|
ревматоїдне ураження очей; |
|
|
ревматоїдні вузлики |
|
|
Рентгенологічні ознаки |
ОП, дрібнокістозна перебудова кісткової структури епіфізів; |
|
звуження суглобових щілин, кісткові ерозії, анкілоз суглобів; |
|
|
порушення росту кісток; |
|
|
ураження шийного відділу хребта |
|
|
Лабораторні ознаки |
позитивний РФ; |
|
позитивні дані біопсії синовіальної оболонки |
|
|
При виявленні 3 з 16 ознак при обов’язковій наявності артриту встановлюють діагноз істинного ЮРА, 4 ознак — визначеного ЮРА; 8 ознак — класичного ЮРА. Обов’язкова умова діагностики ЮРА — виключення ревматичних хвороб іншої нозологічної приналежності |
|
Ці східноєвропейські критерії ЮРА мають низьку специфічність, не дозволяють провести розмежування усередині групи, оскільки повна відповідність їм не виключає формування в катамнезі спондилоартритів. Діагностичні критерії ЮРА включають: 1) початок захворювання до 16-літнього віку; 2) ураження 1 або більше суглобів, що характеризується припухлістю/випотом або що має, як мінімум, 2 з наступних ознак: обмеження функції, болісність при пальпації, підвищення локальної температури; 3) тривалість суглобових змін не менше 6 тиж; 4) виключення усіх інших ревматичних хвороб. До переліку необхідних досліджень при ЮРА входять: 1) загальний аналіз крові (гіпохромія, лейкоцитоз з нейтрофільним паличкоядерним (20–30%) зрушенням, збільшення ШОЕ до 60–70 мм/год); 2) СРБ; 3) сироваткові імуноглобуліни (G — на ранньому етапі, А — у більш віддалені терміни); 4) РФ (у віці до 7 років відзначається вкрай рідко); 5) АНФ; 6) рентгенологічна картина. До неспецифічних лабораторних зрушень при ЮРА нележать: нейтрофільний лейкоцитоз (до 53•109/л при системному варіанті); гіпохромна анемія; збільшення ШОЕ; підвищення рівня гострофазових білків; виявлення у 20% пацієнтів РФ, у 40% — АНФ. РФ в сироватці крові проявляється через тривалий проміжок часу від початку захворювання. Для рентгенологічного дослідження суглобів характерними є ряд змін: набряк періартикулярних тканин, навколосуглобовий ОП, ерозії, анкілоз суглобів. Для розвитку ерозій і анкілозів суглобів потрібно чимало часу (рис. 23.1).
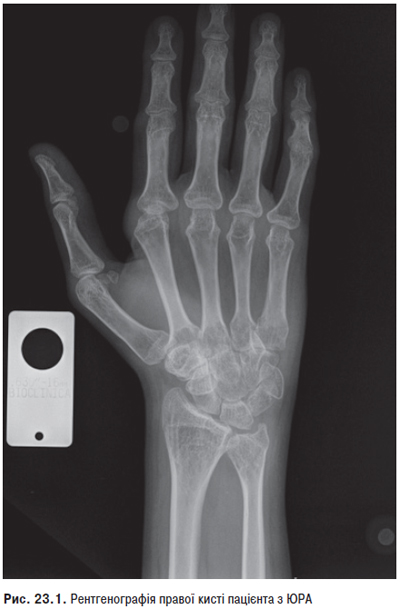
Диференціацію ЮРА проводять з СЧВ, серонегативними спондилоартритами, ДМ, ревматизмом.
Формулювання діагнозу ЮРА. Необхідно вказати варіант перебігу (системний, поліартикулярний, олігоартикулярний), ступінь активності (0, I, II, III), наявність або відсутність РФ, рентгенологічну стадію (за Штейнброкером), функціональний клас і ускладнення.
Лікування
Основні завдання терапії пацієнтів з ЮРА наступні: 1) стабілізація патологічного процесу; 2) запобігання його загостренню; 3) реабілітація функціональних порушень суглобів, ревмоортопедія. Перші 2 пункти включають призначення НПЗП, ГК, базисних препаратів. Основні принципи терапії (за Насонов Е.Л., 2006; 2011 (ред.); Никишина І.П., 2010) представлено в табл. 23.15.
Таблиця 23.15
Принципи лікування хворих на ЮРА
|
Симптоматична терапія |
||||
|
НПЗП |
Монотерапія |
На ранніх стадіях хвороби (6–12 тиж до верифікації діагнозу) |
||
|
При обмеженому ураженні суглобів без деструкції. З віку 0–2 років можливе призначення ібупрофену (30–40 мг/кг), індометацину 2–2,5 мг/кг в 3–4 прийоми |
||||
|
Поєднання з імуносупресорами |
У випадках високої активності процесу, поліорганного типу ураження, системного варіанта. При цьому дози НПЗП знижують в 2 рази |
|||
|
Селективні інгібітори ЦОГ-2 |
Німесулід |
Призначають дітям у віці старше 2 років (3–5 мг/кг в 2–3 прийоми) |
||
|
Мелоксикам |
Призначають дітям з 12 років (0,12–0,25 мг/кг в формі таблеток, свічок) |
|||
|
Неселективні |
Напроксен |
Напроксен призначають з 2 років (10–15 мг/кг, суспензія); диклофенак — з 6 років (таблетки і свічки 25 мг) |
||
|
Диклофенак |
||||
|
Ацетилсаліцилова кислота |
Її призначення недоцільне: є ризик розвитку синдрому Рейе |
|||
|
ГК |
Системне застосування |
Призначають при: а) виражених системних проявах; б) високій персистуючій активності; в) недостатній ефективності НПЗП, внутрішньосуглобового і в/в введення ГК. Застосовують преднізолон і метилпреднізолон (тріамцинолон, дексаметазон недостатньо ефективні та дають побічні реакції). ГК обов’язково поєднують з імуносупресорами або іншими видами лікування. Середня доза преднізолону становить 0,5–0,7 мг/кг/добу в 1–2 прийоми в ранішні години. При високій активності добову дозу розподіляють протягом дня. Пригнічувальну дозу застосовують не більше 1 міс. Після досягнення ефекту дозу поступово знижують до підтримувальної (0,1–0,2 мг/кг/добу), яку застосовують від декількох місяцев до декількох років |
||
|
Локальна терапія |
Показаннями до неї є активний артрит, тендосиновіт, ентезит з випотом, асиметричний артрит нижніх кінцівок у дітей раннього віку, створення умов для досягнення ефекту БПЗП. Застосовують бетаметазон, тріамцинолон, метилпреднізолон. У той самий суглоб ГК вводять не частіше 1 разу в 1–3 міс. Не рекомендується введення тріамцинолону в дрібні та середні суглоби, а також в суглоби несиновіального типу, оскільки можливий розвиток в них атрофії м’яких тканин. Введення ГК в кульшові суглоби може призвести до аваскулярного некрозу голівок стегнових кісток |
|||
|
Пульс-терапія |
Призначають при станах, загрозливих для життя пацієнта, резистентності до терапії. Пульс-терапія дає високий протизапальний ефект, пригнічує системні прояви, дозволяє подолати резистентність, але до стійкого терапевтичного ефекту, як правило, не приводить. Використовують в основному метилпреднізолон у дозі 15–20 мг/кг маси тіла дитини на одне введення. Одноразовий курс триває 3 дні, на другий день додають циклофосфамід 0,4 г/м2. Курси проводять кожні 1–3 міс протягом 1 року. При міні-пульс-терапії початкова максимальна доза становить 5–15 мг/кг щоденно протягом 3 днів. Потім дозу знижують на 62,5 мг 1 раз в 2–3 дні до повної відміни. Пульс-терапію метилпреднізолоном проводять таким чином: 3 дні по 250 мг, 2 дні по 187,5 мг, 2 дні по 125 мг, 2 дні по 93,75 мг, 2 дні по 62,5 мг з подальшим переходом на в/м введення метилпреднізолону: 2 дні по 60 мг, 2 дні по 40 мг, 2 дні по 20 мг, бетаметазону 7 мг 1 день. Схему пульс-терапії можна змінювати залежно від стану хворих |
|||
|
БПЗП |
||||
|
Метотрексат |
Як показали доказові дослідження, він ефективний і безпечний в дитячій ревматологічній практиці. Порівняно з іншими БПЗП його призначають у декілька разів частіше. Істотно не впливає на вираженість системних проявів. Ефективний при оліго- і поліартикулярних варіантах ЮРА в дозах 10–12 мг/м2/тиж. Початкову дозу 5–7,5 мг/м2/тиж підвищують до 12,5 мг 1 раз на тиждень під контролем аналізу крові і біохімічних досліджень. Ефект оцінюють через 2–3 міс. У разі недостатнього ефекту доза може бути збільшена до 15–20 мг/м2/тиж шляхом внутрішньом’язового або підшкірного введення. У вільні від прийому (введення) метотрексату дні необхідно приймати фолієву кислоту по 5 мг/тиж (1 мг/добу). При реакціях з боку ШКТ змінюють спосіб введення препарату (з орального на парентеральний), знижують дозу, дають симптоматичні засоби. Застосування метотрексату скасовують при 3-разовому підвищенні активності АлАТ і АсАТ (у 3–7% випадків). Такі побічні реакції, як алопеція, нодульоз, пневмоніт, синдром активації макрофагів, розвиваються рідко. Існує висока вірогідність загострення хвороби після відміни препарату, а також розвитку вторинної неефективності. Після досягнення ремісії його застосовують ще 2 роки, потім відміняють. Не доведено негативного впливу метотрексату на репродуктивне здоров’я пацієнтів, а також його онкогенності |
|||
|
Сульфасалазин |
Показаний пацієнтам з оліго- і поліартикулярним варіантами ураження в дозі 30–40 мг/кг/добу. Ефект настає через 1–2 міс. Лікування починають з дози 125–250 мг/добу, кожні 5–7 днів дозу підвищують на 125 мг протягом 3–4 тиж. Препарат протипоказаний при системному варіанті (неефективний і викликає ускладнення). Можливі поява диспептичних розладів, підвищення активності трансаміназ, цитопенія, спленомегалія, лихоманка, еритроцитурія. Таке серйозне ускладнення, як синдром Стівенса — Джонсона розвивається рідко. Як правило, після 3-місячного застосування сульфасалазину ускладнення вже не розвиваються |
|||
|
Циклоспорин |
Призначають в дозі 3,5–5,0 мг/кг/добу. Результат відзначають через 1–3 міс, його максимум — протягом 6–12 міс. Найбільш ефективний при системному варіанті, гальмує розвиток структурних змін у суглобах, купірує гострий коксит, увеїт. При асептичному некрозі голівок стегнових кісток чинить стимулюючий вплив на репарацію хряща і кістки. Особливо ефективна комбінація циклоспорину і метотрексату. У процесі лікування необхідно контролювати рівень артеріального тиску і креатиніну в крові. Циклоспорин (собливо форма для в/в введення) є засобом вибору при такому ускладненні ЮА, як синдром активації макрофагів |
|||
|
Гідроксихлорохін |
Вживані дози становлять 5–6 мг/кг/добу. Для лікування при ЮА вважається малоприйнятним через низьку ефективності і побічні реакції з боку очей |
|||
|
Пеніциламін |
Як пеніциламін (10 мг/кг/добу), так і солі золота (0,15–0,20 мг/кг/добу) для лікування при ЮА вважаються малоприйнятними оскільки їх ефективність не відрізняється від ефективності плацебо |
|||
|
Солі золота |
||||
|
Циклофосфамід, азатіоприн, хлорамбуцил |
Застосовують рідко, лікування супроводжується тяжкими побічними ефектами (інфекція, лейкопенія, неопластичні процеси, безпліддя та ін.) |
|||
|
Лефлуномід |
Застосовують (0,3–0,6 мг/кг/добу) у разі тяжкого, резистентного до базисних засобів, ЮА. У цілому досвід застосування лефлуноміду в педіатрії недостатній |
|||
|
Інфліксимаб |
Застосовують при тяжких формах ЮРА, резистентності до базисних засобів. Препарат виявляє виражену протизапальну дію. Схема введення: 0; 2; 6-й тиждень, потім кожні 8 тиж. Початкова доза становить 3 мг/кг. При недостатній ефективності дозу підвищують (може досягти 20 мг/кг на інфузію) або зменшують інтервал між введеннями до 4–5 тиж. Лікування поєднують з прийомом метотрексату (7,5–10 мг/м2/тиж). До початку лікування слід виключити туберкульоз (проводять внутрішньошкірні туберкулінові проби) та інші персистующі інфекції |
|||
|
Етанерцепт |
При лікуванні ЮА препарат вводять підшкірно 2 рази на тиждень в дозі 0,4 мг/кг |
|||
|
Адалімумаб |
Вводять підшкірно в дозі 40 мг (24 мг/м2) кожні 2 тиж дітям з масою тіла ≥30 кг |
|||
|
Ритуксимаб |
Показання до їх застосування при ЮА остаточно не вироблені. Абатацепт призначають при поліартикулярному ЮА дітям у віці старше 6 років в/в у дозі 10 мг/кг 1 раз на місяць |
|||
|
Абатацепт |
||||
|
Імуноглобулін (IgG) для в/в введення |
Поліартикулярний варіант |
З розрахунку 1,5–2,0 г/кг, вводять 2 рази на місяць протягом 2 міс, потім 1 раз на місяць протягом 6 міс. Після відміни препарату досягнутий ефект втрачається |
||
|
Системний варіант |
Не впливає на перебіг артриту, ефективний при системних проявах, знижує ризик розвитку інфекції. Доза становить 0,7–1,0 г/кг на курс |
|||
|
Комбінована імуносупресивна терапія |
|
|
Метотрексат 7,5–10 мг/м2/тиж + циклоспорин 4,4–4,5 мг/кг/добу |
Комбінована терапія дозволяє подолати резистентність до монотерапії БПЗП, загальмувати процес деструкції кісток |
|
Циклоспорин 4,4–4,5 мг/кг/добу + сульфасалазин 30–40 мг/кг/добу |
|
|
Метотрексат 7,5–10 мг/м2/тиж + сульфасалазин 30–40 мг/кг/добу |
|
|
Метотрексат 7,5–10 мг/м2/тиж + гідроксихлорохін 5–7 мг/кг/добу |
|
НПЗП призначають у вигляді монотерапії на ранній стадії хвороби (не більше 6–12 тиж до остаточної верифікації діагнозу), а також хворим з недеструктивним ураженням обмеженої кількості суглобів. Дози НПЗП, які застосовують в педіатрії, наведено в табл. 23.16.
Таблиця 23.16
Дози НПЗП, які застосовують у дітей
|
Вік, з якого можна застосовувати |
Препарат |
Доза, мг/кг/добу |
Максимальна доза (мг/добу) та частота прийому |
|
Без вікових обмежень |
Індометацин |
2 |
100 мг/добу в 2–4 прийоми |
|
Ібупрофен — суспензія |
35–40 |
800–1200 мг/добу в 2–4 прийоми |
|
|
З 1 року |
Напроксен — суспензія |
10–15 |
750 мг/добу в 2 прийоми |
|
З 2 років |
Німесулід — суспензія 10 мг/1 мл, таблетки 50 мг, що диспергуються |
3–5 |
200–250 мг/добу в 2–3 прийоми |
|
З 6 років |
Диклофенак — таблетки та свічки 25 мг |
2–3 |
100 мг/добу в 2–3 прийоми |
|
З 12 років |
Диклофенак — таблетки та свічки 50 мг, розчин для ін’єкцій |
2–3 |
100 мг/добу |
|
Німесулід — таблетки та саше 100 мг |
3–5 |
200–250 мг/добу в 2–3 прийоми |
|
|
Напроксен — таблетки 250 мг |
10–15 |
750 мг/добу в 2 прийоми |
|
|
Мелоксикам |
0,15–0,25 |
15 мг/добу в 1–2 прийоми |
|
|
Ацетилсаліцилова кислота |
75–90 |
3000 мг/добу в 3–4 прийоми |
|
|
З 14 років |
Піроксикам |
0,3–0,6 |
20 мг/добу в 1–2 прийоми |
У поєднанні з імуносупресорами їх призначають при поліартикулярному або системному варіанті ЮРА, високій активності хвороби. З метою запобігання побічним ефектам з боку ШКТ та нирок слід застосовувати селективні НПЗП (німесулід та мелоксикам у дітей у віці старше 2 років). У дітей віком старше 6 років використовують диклофенак, напроксен. При системному ЮА застосовують індометацин з урахуванням його негативних ефектів. Не рекомендується застосування ацетилсаліцилової кислоти (ризик розвитку синдрому Рейє). У хворих із загостренням системних проявів НПЗП можуть спровокувати синдром активації макрофагів та дисеміноване внутрішньосудинне згортання крові. ГК призначають усередину при виражених системних проявах, високій персистуючій активності, недостатньому ефекті від НПЗП, внутрішньосуглобовому та в/в введенні ГК. Добова доза становить 15–20 мг преднізолону (0,2–0,5 мг/кг/добу). Призначення ГК до 3 років призводить до зупинки росту. Необхідне поєднання ГК із БПЗП, після досягнення ремісії дозу ГК поступово знижують до підтримувальної з наступною відміною препарату. При дозі більше 15 мг/добу 1 раз на 3–4 дні дозу знижують на 1,25 мг; при 15–10 мг/добу 1 раз у 5–7 днів дозу знижують на 1,25 мг; при дозі 10–5 мг/добу зниження альтернуюче — кожний другий день дозу знижують на 1/8 таблетки; 5 мг/добу — продовжують кожний другий день дозу зменшувати на 1/8 таблетки до повної відміни. У разі розвитку синдрому відміни відзначають: біль у м’язах та суглобах, тремтіння, лихоманку, нудоту, блювання, депресію. За необхідності вводять в/в ГК. Дозу ГК знижують після купірування активності системних проявів та ексудативних змін в суглобах на тлі введення в/в імуноглобуліну, терапії імуносупресорами тривалістю не менше 1 міс. Лікування ГК супроводжують призначенням 500–1000 мг кальцію та 400 МО вітаміну D3. Необхідно пам’ятати, що тривалий прийом ГК навіть у низьких дозах спричиняє негативні наслідки. БПЗП призначають зразу після верифікації діагнозу (в перші 3–6 міс хвороби), відміняють у випадках клініко-лабораторної ремісії не менше 1–2 років. Необґрунтована відміна призводить до загострення хвороби та розвитку вторинної неефективності.
Слід зазначити, що практично в усі схеми комбінованої імуносупресивної терапії включають метотрексат. Патогенетично найбільш обґрунтованою вважається його комбінація з циклоспорином (табл. 23.17).
Таблиця 23.17
Показання до комбінованої імуносупресивної терапії ЮА
|
Комбінація |
Показання до застосування |
|
Метотрексат (10–25 мг/м2/тиж) + циклоспорин А (2,5–4 мг/кг) |
Рефрактерні варіанти системного ЮА, швидкопрогресуючий поліартикулярний варіант, поєднання поліартриту з увеїтом. Розвиток кокситу з асептичним некрозом кісток кульшового суглоба або без нього у хворих, які отримують метотрексат. Збереження високої активності запального процесу у хворих з системним ЮА, які одержують метотрексат або циклоспорин. Висока активність захворювання у пацієнтів з системним ЮА при позитивному клінічному ефекті лікування циклоспорином |
|
Метотрексат (10 мг/м2/тиж) + сульфасалазин (30–40 мг/кг/ добу) |
Високоактивний поліартикулярний варіант (особливо HLA-B27-асоційований), рефрактерні варіанти спондилоартритів з вираженим периферичним артритом і ентеритами. Прогресування ураження суглобів у хворих з олігоартритом з пізнім початком, які отримували сульфасалазин |
|
Метотрексат (10 мг/м2/тиж) + лефлуномід (0,3–0,5 мг/кг) |
Тяжкі рефрактерні до терапії варіанти ЮА при недостатній ефективності метотрексату |
|
Циклоспорин (4,4–4,5 мг/кг) + сульфасалазин (30–40 мг/кг/добу) |
Розвиток кокситу у хворих з олігоартритом з пізнім початком, які отримують сульфасалазин. Поява увеїту у хворих з оліго- або поліартикулярним варіантом ЮРА, що приймають сульфасалазин |
|
Метотрексат (7,5–10 мг/м2/тиж) + гідроксихлорохін (5–7 мг/кг/добу) |
Активність запального процесу, що зберігається у хворих з оліго- або поліартритом без ураження очей на тлі прийому метотрексату |
Особливості терапії при різних варіантах ЮА представлено також в табл. 23.18.
Таблиця 23.18
Особливості терапії при різних варіантах ЮА
|
Варіант ЮА |
Характер терапії |
|
Системний |
Проводять агресивну терапію, що включає НПЗП, ГК (системні і локальні), базисні препарати, імуноглобулін. Метотрексат призначають в дозах 12,5–15 мг/м2/тиж або в комбінації з циклоспорином. Не рекомендується застосування сульфасалазину через можливий розвиток побічних реакцій. Хворим з ознаками синдрому активації макрофагів призначають ГК (у тому числі в/в) і циклоспорин. На активність хвороби позитивно впливають блокатори ФНП-α, але системні прояви вони не купірують |
|
Поліартикулярний |
Потрібне раннє застосування БПЗП, зокрема метотрексату, зрідка призначають циклоспорин А, препарати золота. При їх неефективності призначають блокатори ФНП-α і лефлуномід |
|
Олігоартикулярний |
Ефективним є поєднання НПЗП з внутрішньосуглобовим введенням ГК. У разі рецидивування, відсутності відповіді на лікування, ураження очей призначають метотрексат. При його неефективності використовують циклоспорин А. Блокатори ФНП-α використовують при часто рецидивуючому артриті й увеїті |
|
HLA-B27-асоційований олігоартрит. Наявність ентезитів |
Ефективними є сульфасалазин і метотрексат. При високій активності процесу хороші результати спостерігаються у разі використання блокаторів ФНП-α |
Використовують також нефармакологічні (функціональне лікування, реабілітація) і хірургічні методи лікування (за відповідними показаннями), проводять навчання хворих.
Профілактика ОП у хворих на ЮА. З метою профілактики ОП на тлі лікування ГК в дозах ≥5 мг/добу обов’язковим є щоденний прийом препаратів кальцію (500–1000 мг) і вітаміну D3 (400 МО).
Критерії ефективності лікування ЮА включають зменшення частоти і вираженості рецидивів периферичного артриту, ентезиту, активності запалення (за лабораторними даними), поліпшення функціональної здатності. Для визначення ефективності лікування оцінюють наступні показники: 1) кількість «активних» суглобів (враховують 75 суглобів); 2) кількість суглобів з обмеженою функцією (враховують 75 суглобів); 3) ШОЕ і/або СРБ; 4) загальну оцінку активності хвороби, на думку лікаря (візуальна аналогова шкала); 5) оцінку загального самопочуття, на думку пацієнта або його батьків (візуальна аналогова шкала); 6) оцінку функціональної здатності (опитувальник «Childhood Healh Assesment Quesionnare»). Ефект лікування вважається помірно позитивним при 30% поліпшенні показників, хорошим — при 50%, дуже хорошим — при 70%.
Прогноз
Прогноз ЮА залежить від варіанта хвороби (табл. 23.19).
Таблиця 23.19
Прогноз ЮА залежно від варіанта хвороби
|
Поліартикулярний серонегативний варіант з раннім дебютом |
Можливий розвиток тяжкого деструктивного артриту з інвалідизацією |
|
|
Олігоартрит |
З раннім початком |
У 40% хворих розвивається деструктивний поліартрит. У пацієнтів з увеїтом можливий розвиток сліпоти |
|
З пізнім початком |
Можлива трансформація в АС |
|
|
Системний варіант |
У 40–50% дітей можливе настання тривалої ремісії. У ⅓ хворих відзначають безперервно рецидивуючий перебіг хвороби, у половини з них розвивається деструкція суглобів, а в дорослому віці виникає тяжка функціональна недостатність, амілоїдоз |
|
Найбільш серйозний прогноз відзначають при системному варіанті ЮА. Він важко піддається лікуванню, тривало зберігається активність, високою є інвалідизація з приводу ураження опорно-рухового апарату, можливі летальні випадки. Перебіг ювенільного спондилоартриту має доброякісний характер і відзначається рідкісною інвалідизацією.
Протокол медичної допомоги при ЮА, затверджений наказом МОЗ України від 22.10.2012 р. № 832 (витяг)
|
Положення протоколу |
Обґрунтування та необхідні дії |
|
А 2.1. Амбулаторний етап |
|
|
1. ЮА — група захворювань суглобового апарату, що мають хронічний прогресуючий перебіг. Хворі на ЮА потребують постійного прийому хворобо-модифікуючої терапії, яка дозволяє попередити первинну інвалідність та подовжити тривалість життя |
Обґрунтування. При наявності чітких симптомів синовіту принаймні одного суглоба, який не може бути пояснений іншим захворюванням, пацієнт повинен бути направлений до лікаря дитячого кардіоревматолога [Клінічна настанова (КН) 1.5, 2.1] [рівень доказовості А] Необхідні дії. Обстеження та спостереження лікарем дитячим кардіоревматологом |
|
2. Обстеження пацієнтів із ураженням суглобового апарату здійснюється амбулаторно та стаціонарно |
Обґрунтування. Діагноз ЮА, його форма встановлюється лікарем дитячим кардіоревматологом згідно з класифікаційними критеріями [КН 1] [рівень доказовості А] Необхідні дії. Забезпечення своєчасного встановлення діагнозу ЮА |
|
3. Досить часто при хронічному запальному процесі у суглобах спостерігається ураження очей — ревматоїдний увеїт |
Обґрунтування. Початок розвитку увеїту спостерігається незабаром після виникнення ураження суглобів, але увеїт може виникати як дебют ЮА [КН 2.5] [рівень доказовості А] Необхідні дії. Необхідно всіх хворих на ЮА направляти на консультацію до офтальмолога на предмет виключення чи встановлення діагнозу «ревматоїдний увеїт» якомога раніше та не пізніше ніж через 6 тиж від початку захворювання |
|
А 2.2. Госпіталізація |
|
|
Госпіталізація здійснюється у разі появи хвороби або ускладнень, обстеження та лікування яких потребує стаціонарного спостереження хворого |
Обґрунтування. Направлення на госпіталізацію здійснюється лікарем дитячим кардіоревматологом [КН 2] Необхідні дії. Хворі на ЮА госпіталізуються для обстеження та стаціонарного лікування |
|
А 2.3. Діагностика |
|
|
1. Встановлення діагнозу «ювенільний артрит» |
Обґрунтування. Для встановлення діагнозу «явний ЮА» за критеріями ILAR, 1997. ЮА може бути визначений, як артрит невстановленої причини з початком до 16-річного віку, тривалістю захворювання 6 і більше тижнів, при виключенні інших захворювань [таких як системний червоний вовчак, ревматична лихоманка, неоплазія, імунодефіцит та ін.] [КН 2.1] [рівень доказовості В] Необхідні дії. Обсяг діагностики:
Бажані: HLA B27; УЗД суглобів в режимі енергетичного допплера з метою виявлення та оцінки ступеня кровотоку в синовіальній оболонці |
|
2. Визначення клінічного варіанту ЮА |
Обґрунтування. На підставі клінічних проявів хвороби протягом перших 6 міс визначити клінічний варіант ЮА [КН 2.1] [рівень доказовості B] Необхідні дії. Алгоритм встановлення клінічного варіанту ЮА виконується відповідно до діагностичних критеріїв ЮА [Edmonton, 2001] [A.3.1] |
|
3. Визначення ступеня активності захворювання та стратифікація щодо наявності несприятливих прогностичних факторів |
Обґрунтування. Своєчасна оцінка ступеня активності та наявності факторів несприятливого перебігу [КН 2.2] [рівень доказовості A] Необхідні дії. Відповідно до результатів обстеження, алгоритму оцінки ступеня активності та наявності факторів несприятливого перебігу ЮА [A.3.2] провести аналіз стану пацієнта |
|
4. Виявлення ревматоїдного увеїту |
Обґрунтування. Своєчасна діагностика або виключення ревматоїдного увеїту [КН 2.4] [рівень доказовості B] Необхідні дії. Огляд окуліста повинен бути проведений як можна раніше |
|
5. Виділення терапевтичної групи пацієнтів з ЮА |
Обґрунтування. Для прийняття подальших терапевтичних рішень виділяється п’ять груп ЮА [КН 2.2] [рівень доказовості B] Необхідні дії. Для прийняття терапевтичних рішень виділяють 5 груп пацієнтів з ЮА:
|
|
6. Проведення скринінгу з метою виявлення туберкульозу |
Обґрунтування. Пацієнтам, яким показане лікування БА, необхідно проводити скринінг з метою виявлення туберкульозу будь-якої локалізації [КН 3.3] [рівень доказовості B] Необхідні дії. Отримання очищеної від білка проби Манту на туберкульоз рекомендовано до початку імунобіологічної терапії для всіх пацієнтів [рівень C]. Рекомендовано повторювати тестування приблизно 1 раз на рік всім пацієнтам, які продовжують отримувати імунобіологічну терапію [рівень D]. На початку лікування та протягом лікування рутинну перевірку на наявність туберкульозу слід проводити під час кожного візиту до лікаря [B 1–2], за необхідності в оглядовий процес залучати лікаря-фтизіатра. Рентгенографію ОГП проводити 1 раз на рік або за наявності показань |
|
А 2.4. Лікування |
|
|
1. Головним орієнтиром для вибору адекватної схеми лікування ЮА є не стільки форма захворювання, скільки прогноз перебігу та оцінка ймовірності інвалідизуючого результату захворювання. Якщо характер перебігу ЮА прогностично несприятливий та існує високий ризик інвалідизації, то терапія повинна бути обов’язково випереджаючою, агресивною, але за обов’язкової умови, що ризик, пов’язаний з лікуванням, істотно нижчий, ніж ризик прогресування хвороби |
Обґрунтування. Мета лікування — сповільнення структурних змін в суглобах, досягнення ремісії або мінімальної активності захворювання, попередження втрати працездатності та інвалідизації [КН 3.6, 3.7, рівень доказовості B] Необхідні дії. Лікування пацієнтів з ЮА виконується та коригується відповідно до результатів обстеження лікарем дитячим кардіоревматологом. Алгоритм лікування призначається відповідно до рекомендацій A.3.3. Дані динамічного спостереження за станом хворого вносяться до відповідної документації [карта стаціонарного хворого, амбулаторна картка] |
|
2. Як початкові препарати вибору для скорочення запалення та болю при лікуванні ЮА призначаються НПЗП |
Обґрунтування. Лікарі-педіатри, дитячі кардіоревматологи повинні призначити нестероїдні протизапальні лікарські засоби [НПЗП] як початкові ліки вибору для скорочення запалення та болю при лікуванні ЮА [КН 3.2.2, рівень доказовості B] Необхідні дії. Призначити тільки один НПЗП. При тривалому застосуванні НПЗП слід обирати найнижчу ефективну дозу. При застосуванні НПЗП доступна рідка форма для дітей, які не можуть ковтати таблетки. НПЗП можна призначати з метотрексату [МТ]. Знеболювальна дія очікується в перші години після прийому НПЗП. Протизапальний ефект НПЗП проявляється на 10–14-й день прийому препарату |
|
3. У разі відсутності протипоказань ХМПРП призначаються відразу після встановлення діагнозу ЮА за наявності факторів несприятливого прогнозу та середній/високій активності запального процесу |
Обґрунтування. Препаратом першої лінії серед синтетичних ХМПРП є МТ [КН 3] [рівень доказовості B] Необхідні дії. Пацієнтам з мінімальною активністю захворювання терапія МТ не завжди є необхідною. МТ повинен бути призначений пацієнтам з активним ЮА, які раніше не отримували інші синтетичні ХМПРП. МТ може призначатись в комбінації з іншими препаратами |
|
Прийом МТ може починатися в таблетованій формі в дозі 10 мг/м2/тиж, з подальшим її підвищенням залежно від ефективності на 5 мг кожні 2–4 тижні до 20 мг/м2/тиж. Для підвищення ефекту МТ можлива заміна на парентеральну форму. На фоні терапії МТ призначається фолієва кислота в дозі не менше 5 мг/тиж, залежно від дози МТ. У випадку протипоказань або непереносимості МТ призначається інший ХМПРП [сульфасалазин, циклоспорин, лефлуномід] із урахуванням віку, форми ЮА, активності запального процесу. При недостатній ефективності монотерапії МТ допускається його комбінація з іншим ХМПРП [сульфасалазин, циклоспорин] |
|
|
4. Додавання глюкокортикоїдів [ГК] до монотерапії ХМПРП або комбінації ХМПРП є раціональним в стартовій короткотривалій терапії за наявності показань |
Обґрунтування. Призначення ГК можливе у вигляді внутрішньосуглобових введень або/та системного використання [КН 3.2.3] [рівень доказовості B] Необхідні дії. Внутрішньосуглобово призначається тріамцинолон гексацетонід (за умови доступності), метилпреднізолон, бетаметазон, який може бути рекомендований як перша лінія лікування. Системне призначення ГК допускається хворим як швидкодіючий засіб у пацієнтів з високою активністю ЮА або у хворих із системним клінічним варіантом ЮА з органними проявами захворювання [увеїт, серозити та ін]. За необхідності проводиться пульс-терапія. Перевага надається метилпреднізолону. ГК системно призначається пацієнтам з поліартикулярною формою ЮА [РФ+] як доповнення до терапії ХМПРП до того часу, поки не буде спостерігатися клінічний ефект лікування. Тривале призначення системних ГК пов’язане із високим ризиком розвитку побічних ефектів, тому доза ГК при тривалому використанні не повинна перевищувати 0,2 мг/кг маси тіла за еквівалентом преднізолону |
|
5. Лікування пацієнтів з увеїтами, пов’язаними із ЮА, слід призначати послідовно та дифенційовано |
Обґрунтування. Лікування увеїту при ЮА спочатку повинно починатися з місцевих ГК. Близько 20% пацієнтів з ЮА та увеїтом не відповідають зовсім або мало відповідають на лікування місцевими ГК [КН 3.2.9] [рівень доказовості B] Необхідні дії. Лікування хворого на ЮА із явищами увеїту повинно призначатися консиліумом дитячого кардіоревматолога та офтальмолога. За наявності ураження суглобів без високої запальної активності та відсутності факторів ризику разом із місцевою терапією призначаються НПЗП. При високому ступені запальної активності, двобічному ураженні очей потрібне призначення ГК системно. При призначенні базисної терапії надається перевага МТ. Пацієнтам із обмеженою відповіддю або відсутністю відповіді проводиться заміна МТ на циклоспорин або комбінація базисних препаратів. При збереженні високої активності ЮА, торпідності перебігу увеїту рекомендований до призначення адалімумаб |
|
6. Рішення про додавання біологічних ХМПРП або перехід на інші синтетичні ХМПРП приймається, якщо мета терапії не була досягнута при застосуванні одного ХМПРП або їх комбінації. За наявності несприятливих прогностичних факторів слід розглянути застосування біологічного ХМПРП, а за відсутності несприятливих факторів розглянути можливість заміни синтетичного ХМПРП |
Обґрунтування. Блокатори фактору некрозу пухлини [ФНП] для лікування хворих на ЮА у дітей призначаються:
Блокатори ФНП [адалімумаб] для лікування ЮА у дітей повинні використовуватись в комбінації з МТ; якщо у пацієнта є непереносимість МТ або терапія МТ вважається недоцільною, адалімумаб може застосовуватись у вигляді монотерапії [КН 3] [рівень доказовості C]. Лікування блокаторами ФНП може бути продовжено більше 6 міс лише у разі досягнення ремісії або мінімальної активності. Впродовж тривалої терапії за умови відсутності адекватної ефективності блокаторів ФНП препарат відміняється [КН 3] [рівень доказовості B]. Заміна одного блокатора ФНП на інший може проводитись за умови розвитку побічних реакцій на попередній препарат або при втраті досягнутого ефекту, що потребує детального обґрунтування та згоди пацієнта [КН 3] [рівень доказовості B]. Блокатор рецепторів до ІЛ-6 для лікування хворих на ЮА призначається:
При рефрактерності хворих на ЮА до декількох синтетичних ХМПРП та біологічних ХМПРП рекомендовано призначення азатіоприну, циклоспорину A або циклофосфаміду [КН 3] [рівень доказовості B] |
|
Необхідні дії. Призначення імунобіологічних препаратів проводиться консиліумом організатора охорони здоров’я, лікарів-спеціалістів дитячих кардіоревматологів, наукових консультантів — спеціалістів з проблем дитячих ревматичних захворювань в умовах спеціалізованих лікувально-консультативних установ третього рівня надання медичної допомоги. Проведення лікування імунобіологічними препаратами, його контроль доцільно проводити в умовах лікувальних установ високоспеціалізованої (третинної) медичної допомоги, перевага надається установам, які мають досвід лікування імунобіологічними препаратами та централізації відповідних хворих. Під час прийому адалімумабу необхідно проводити моніторинг амінотрансфераз, білірубіну, нейтрофілів кожні 1–2 міс, наявності туберкульозної інфекції 1 раз на рік. Протягом лікування імунобіологічними препаратами рутинну перевірку на наявність туберкульозу слід проводити під час кожного візиту до лікаря [B1–2], за необхідності в оглядовий процес залучати лікаря-фтизіатра. Рентгенографію ОГП проводити 1 раз на рік або за наявності показань |
|
|
7. В окремих випадках показання до відкритої або артроскопічної синовектомії може бути розглянуто, якщо консервативна терапія неефективна |
Обґрунтування. Ортопедична корекція проводиться лікарями-ортопедами за направленням дитячого кардіоревматолога. Рекомендовано проведення відкритої або артроскопічної синовектомії, корекції стійких деформацій суглобів, якщо консервативна терапія неефективна в умовах спеціалізованого ортопедичного відділення [КН 3] [рівень доказовості C] Необхідні дії. Для виявлення доцільності та проведення артроскопічної процедури дитячий кардіоревматолог повинен направити хворого на ЮА на консультацію до ортопеда, а при показаннях до артроскопії хворого переводять в дитяче спеціалізоване ортопедичне відділення. Для виявлення доцільності проведення хірургічного втручання за наявності деформацій суглобів, стійких контрактур, анкілозів, асептичних некрозів кісткових структур, патологічних кіст лікар-педіатр, дитячий кардіоревматолог повинен направити хворого на ЮА до ортопеда. Об’єм оперативного втручання визначається лікарем-ортопедом. Готовність пацієнта до оперативного втручання вирішується сумісно дитячим кардіоревматологом та ортопедом. Шини, ортези повинні накладатися досвідченими лікарями ортопедами-ортезистами, які спеціалізуються на ураженнях рухового апарату |
|
8. Паралельно із медикаментозними методами лікування в умовах стаціонару хворі повинні отримувати психологічну допомогу в повсякденній соціальній реабілітації |
Обґрунтування. Для повсякденної соціальної адаптації необхідно роз’яснювати дитині можливості фізичного навантаження та активності [КН 3] [рівень доказовості C] Необхідні дії. Дитячий кардіоревматолог проводить бесіди із хворими та батьками хворих для пояснення об’єму фізичної активності у кожного хворого індивідуально |
|
9. Проведення профілактики та лікування зниження мінеральної щільності кісткової тканини [МЩК] |
Обґрунтування. Діти з ЮА мають низьку МЩК на ранній стадії захворювання незалежно від застосування ГК [КН 3] [рівень доказовості C] Необхідні дії. При довготривалому перебігу хвороби, довготривалому збереженні активності хвороби, тривалому прийомі ГК доцільне проведення денситометрії для визначення МЩК. В щоденний раціон хворих доцільне включення продуктів, збагачених кальцієм (твердий сир, молочні продукти тощо). Профілактичне лікування добавками кальцію, призначення вітаміну D пропонується всім пацієнтам зі зниженням МЩК та тим, які приймають ГК |
|
10. Паралельно із медикаментозними методами лікування в умовах стаціонару хворі повинні отримувати немедикаментозну терапію. |
Обґрунтування. В умовах стаціонару необхідно заохочувати пацієнтів із ЮА займатися регулярними фізичними вправами, які відповідають їх загальним можливостям та обмеженням, що накладаються їхньою хворобою [КН 3] [рівень доказовості C] Необхідні дії. Фізичні вправи рекомендується починати з гострого періоду залежно від ступеня запалення, кількості уражених суглобів та загальної оцінки активності захворювання. Регулярна фізична активність, яка відповідає загальним здатностям дітей та обмеженням, що накладаються розвитком ЮА, сприяє нормальному розвитку дитини та може протидіяти небажаному впливу захворювання на силу м’язів, витривалість та аеробну здатність. Комплекс рекомендованих вправ повинен бути впроваджений під контролем лікаря лікувальної фізкультури |
|
11. Фізіотерапевтичне лікування |
Обґрунтування. Лікар-кардіоревматолог повинен направити хворих для індивідуальної стратегії призначення фізіотерапії до лікаря фізіотерапевта [КН 3.7.5] [рівень доказовості C] Необхідні дії. Тепло [теплі/гарячі компреси, теплі ванни] та/або холод [льодяні масажі/холодні компреси] можуть бути рекомендовані для полегшення симптомів у дітей та підлітків із ЮА. Тепла ванна або душ уранці може зменшити ригідність та біль у м’язах. Застосування для масажу великого шматка льоду при гострому запаленні суглоба [наприклад заморозити воду у паперовій або пенополістирольній чашечці, а потім зрізати верх чашечки для розкриття поверхні льоду — легенько, круговими рухами, виконувати масаж льодом]. Тривалість масажу слід обмежувати 5 хвилинами для уникнення появи льодяного опіку. Для призначення індивідуального фізіотерапевтичного лікування хворий на ЮА консультується з лікарем-фізіотерапевтом. Лікар-фізіотерапевт призначає комплекс індивідуальних фізіотерапевтичних процедур (термотерапію, електро- чи ультразвукову терапію, масаж, лімфодренаж). Додаткові та альтернативні види фізіотерапії не повинні заміняти собою медикаментозні засоби та вправи, призначені для лікування ЮА |
|
A.2.5. Санаторно-курортне лікування та реабілітація |
|
|
I. Санаторно-курортне лікування показане:
|
Обґрунтування. Задачами відновного лікування на санаторно-курортному етапі являються:
Необхідні дії. Медичний відбір на санаторно-курортне лікування хворих на ЮА здійснює відбіркова санаторно-курортна комісія, до складу якої входять: дільничний педіатр (сімейний лікар), дитячий кардіоревматолог, завідуючий відділенням |
|
2. Протипоказами до санаторно-курортного лікування є:
|
Обґрунтування. Діти із системними проявами [системна форма], високим [II–III] ступенем активності, наявністю незворотних уражень суглобового апарату [анкілозування], втратою можливості до самообслуговування повинні проходити лікування в умовах стаціонару та/або амбулаторних умовах із залученням спеціалістів дитячих кардіоревматологів [джерела 10–16] [рівень доказовості C] Необхідні дії. Медичний відбір на санаторно-курортне лікування хворих на ЮА здійснює відбіркова санаторно-курортна комісія, до складу якої входять: дільничний педіатр (сімейний лікар), дитячий кардіоревматолог, завідуючий відділенням |
|
3. В період санаторно-курортного лікування хворі повинні продовжувати медикаментозну терапію, яку одержували на попередньому етапі лікування (стаціонар — поліклініка) |
Обґрунтування. Залежно від активності процесу, форми захворювання і характеру змін у суглобах призначають лікувальні комплекси: 1. ЮА, суглобова форма, проліферативні або проліферативно-фіборзні зміни, неактивна фаза захворювання (ремісія): грязьові аплікації на уражені суглоби. Т — 38–40 °C, тривалістю 10–12 хв, на курс 10 процедур. Влітку лікувальний комплекс доповнюється морськими купаннями при температурі води в морі не нижче 22–23 °C. З фізіотерапевтичних процедур для поліпшення периферичного кровообігу додатково до масажу можна призначати біорезонансну стимуляцію м’язів, що оточують уражені суглоби, лазеротерапію на суглоби (за наявності артралгій), фонофорез з гідрокортизоном на область суглобів для зменшення фіброзних проявів. 2. ЮА, суглобова форма, мінімальний ступінь активності, переважно з проліферативними змінами в суглобах: грязьові аплікації на уражені суглоби, Т — 40 °C, тривалістю 10 хв, через день, курс 10 процедур. З метою зниження активності процесу грязелікування чергується із застосуванням лазеротерапії на уражені суглоби, ДМХ або СМТ-терапії. 3. ЮА, суглобова форма, мінімальний ступінь активності, ексудативно-проліферативні зміни в суглобах: грязелікування проводять по щадній методиці — Т — 38 °C, в тому числі дітям молодшого шкільного віку тривалістю 8 хв, через 2 дні. Для зниження активності процесу і зменшення компонента на фоні продовження медикаментозної терапії призначається фізіотерапія — ДМХ, СМХ або ж лазеротерапія на більш «активні» суглоби. Підліткам з мінімальною активністю процесу грязелікування слід починати з температури 38 °C, поступово підвищуючи до 40 °C, від 10 до 15 хв, через день або 2 дні з перервою 1 день, курс лікування 10 процедур. У випадках ремісії захворювання, переважання проліферативно-фіброзних явищ в суглобах рекомендується призначати грязьові аплікації — Т 40–42 °C, 15–20 хв, через день або 2 дні з перервою 1 день. Курс лікування до 12 процедур. 4. ЮА, суглобова форма, помірна активність процесу: на фоні медикаментозної терапії застосовують бальнеолікування — хлоридні натрієві ванни мінералізації 20 г/л, Т — 36–37 °C, 10–12 хв, курс лікування 10 процедур, чередуючи з фізіотерапевтичними процедурами: СМТ, фонофорез грязі на область суглобів, ДМХ і СМХ-терапію. 5. ЮА, системна форма, помірна активність процесу: ці хворі продовжують одержувати в санаторії гормональну терапію. Разом із загальнозміцнюючою, медикаментозною і протизапальною терапією призначають фізіотерапевтичні процедури, які стимулюють гіпофізарно-наднирковозалозну систему, — ДМХ, СМТ-терапію, магнітолазерну терапію [джерела 10–16] [рівень доказовості C] Необхідні дії. Обов’язкові дослідження, які проводяться при вступі до санаторію: 1. Клінічний огляд з детальним описом анамнезу (скарг на біль в суглобах, ранкову скутість, оцінку ходи, деформації суглобів, гіпотрофії м’язів, біль при активних і пасивних рухах, проведення гоніометрії). 2. Лабораторні дослідження:
3. Для пролонгації ремісії хвороби, збереження функції опорно-рухового апарату, поліпшення якості життя рекомендується проводити своєчасні і повторні (1—2 рази) курси щорічного курортного лікування, при строгому дотриманні вимог спадкоємності на всіх етапах: стаціонар — поліклініка — курорт |
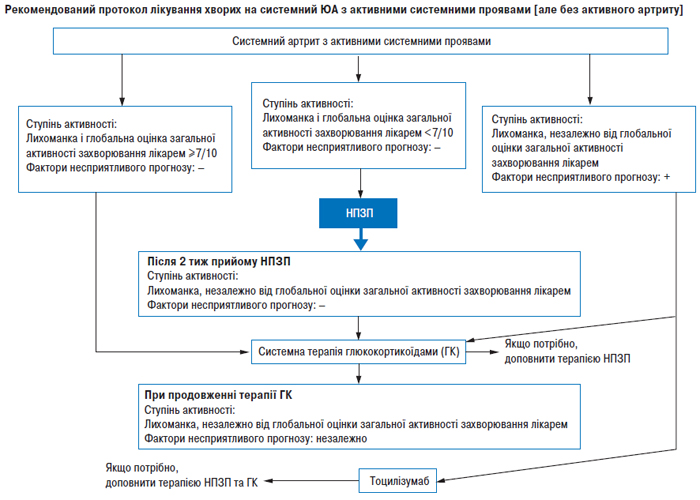
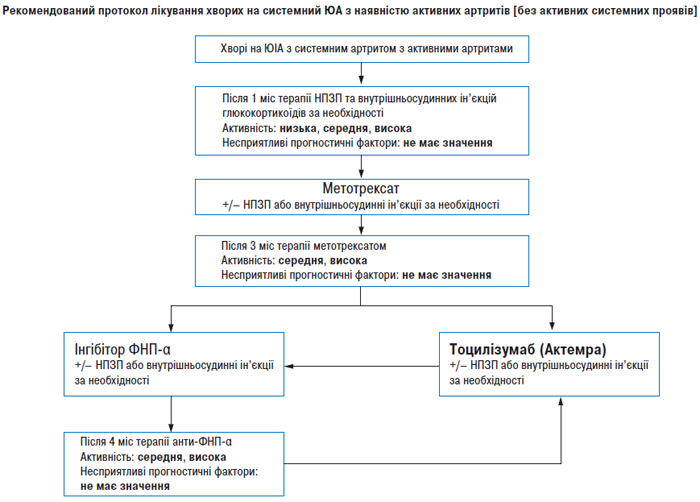
Глава 24. Спондилоартропатії
Анкілозуючий спондиліт
АС — хронічна системна запальна хвороба хребта і суглобів.
Переважно уражаються суглоби і зв’язки хребта із розвитком обмеження його рухливості. Залучаються внутрішні органи: серце, аорта, нирки.
Синоніми: ідеопатичний АС, хвороба Бехтєрева, хвороба Штрюмпеля — Марі — Бехтєрева.
Епідеміологія
Поширеність АС серед населення України становить 0,1% (усього близько 48 тис. хворих).
У країнах Європи зустрічається з частотою від 0,05 до 1,8%, в середньому — 0,1–0,2%.
Серед населення Африки негроїдної раси і японців зустрічається дуже рідко, а індійців Хайда в Британській Колумбії та індійців Піми в Аризоні — до 6%.
Чоловіки хворіють частіше — 3–9:1, частка жінок у структурі хвороби становить близько 15%.
Розвиток захворювання у близьких родичів відзначають у 2,4–4% випадків, що в 20 разів частіше, ніж в популяції.
Етіологія
Етіологія захворювання точно не встановлена, тому виділяють основні фактори розвитку АС: генетична зумовленість, імунопатологічні реакції, зовнішні тригерні фактори (особливо інфекційний).
Генетичну детермінованість пов’язують з носійством HLA-B27 (крім субтипів 06 та 09). Цей антиген у популяції виявляють у 7–10%, серед хворих на АС — 90–97%, а серед їх родичів — 30–50%.
Вважається, що в розвитку АС беруть участь ті мікроорганізми, у яких є загальні антигенні детермінанти з антигеном гістосумісності HLA-B27. Виявлено молекулярну подібність між HLA-B27 і антигенами, які знаходяться на поверхні клебсієл, ієрсіній, хламідій та інших грамнегативних мікроорганізмів. Інфікування організму такими мікробами за відсутності антигену, схожого на них, приводить до продукції захисних антитіл проти збудника, їх елімінацію та видужання. У осіб, які мають антиген, схожий на мікробні антигени, виробляються антитіла, які мають часткову аутоімунну активність. Комплекси антиген-антитіло, які утворюються в сплетінні Батсона (вертебральні венозні та лімфатичні сплетіння), сприяють виникненню запалення у крижово-клубових суглобах і суглобах хребта. Повторні інфікування можуть приводити до хронічного запалення.
Наявність гена HLA-B27 становить тільки 20% схильності до захворювання. Родичі хворих на АС, які мають HLA-B27, хворіють в 10 разів частіше, ніж носії HLA-B27, чиї родичі не хворіють. В останньому випадку ризик розвитку АС становить 1–2%.
Розвиток АС часто пов’язують із перенесеною сечостатевою або кишковою інфекцією. Поверхневі або капсульні антигени кишкових бактерій можуть бути тригерами хронічного запалення суглобів.
Порушення складу нормальної мікрофлори кишечнику може призводити до структурних змін в слизовій оболонці, внаслідок чого підвищується здатність до проникнення бактеріальних агентів до кровотоку. Ці зміни у частини хворих на АС проходять безсимптомно, їх виявляють при ректороманоскопії чи колоноскопії. Патологічні зміни слизової оболонки кишечнику і порушення його біоценозу супроводжуються недостатністю місцевої бар’єрної функції, пов’язаної зі станом загальної імунної реактивності організму.
У різних країнах чинними агентами АС можуть бути ті мікроорганізми, які є ендемічними у даному регіоні: в Іспанії — Salmonella typhi, в Англії — Klebsiella pneumoniae, в Скандинавських країнах — Yersinia enterocolitica.
Не виключається тригерна роль травм хребта чи кісток таза та вплив гормональних факторів.
Патоморфологія
Основні патоморфологічні прояви АС — ентезопатії, ентезити, остити та синовіти.
Ентезопатія — хондроїдна метаплазія, яка супроводжується подальшою осифікацією, оститом, прогресуючою деструкцією суглобових хрящів з ерозуванням субхондральних кісток, субхондральним остеосклерозом, анкілозуванням крижово-клубових і дрібних суглобів хребта. На пізніх стадіях такі зміни виявляють і в лобковому зрощенні.
Ентезити — запалення місць прикріплення до кістки сухожиль, зв’язок, фіброзної частини міжхребцевих дисків, капсул суглобів.
Характерні місця ураження ентезитів:
- з’єднання рукоятки з тілом груднини, ключиці;
- ділянка прикріплення зв’язок до остистих відростків;
- зв’язковий апарат міжхребцевих дисків;
- крижово-клубові суглоби;
- гребені клубових кісток;
- лобковий симфіз;
- вертлюг стегна;
- капсули і внутрішньокапсульні зв’язки великих синовіальних суглобів;
- надколінок;
- п’яткові кістки (ахіллодиніт, підошовний фасцит).
Синовіти морфологічно подібні до ревматоїдних.
Імунозапальний процес починається з ураження крижово-клубових з’єднань, потім приєднуються міжхребцеві, реберно-хребцеві суглоби, рідше периферичні, міжхребцеві диски, зв’язки хребця в місцях їх прикріплення до тіла хребця. Спочатку виникає інфільтрація лімфоцитами і макрофагами, потім розвивається активний фібропластичний процес з утворенням фіброзної рубцевої тканини, її кальцифікація й осифікація. Суглоби, які найчастіше уражаються при АС, мають загальне ембріональне походження: груднино-реберні, груднино-ключичні, міжхребцеві, крижово-клубові, лобковий симфіз, а також інші тканини, близькі за походженням, — судинна оболонка ока, інтима аорти.
Прискорення руйнування хрящів і розвиток анкілозів викликають рефлекторний спазм навколохребетних м’язів за рахунок посилення болю та розладів кровопостачання.
Розвиток анкілозів можливий і без попередніх ентезитів, хондроїдної метаплазії та синовітів.
Клінічна картина
Класифікацію АС наведено в табл. 24.1.
Таблиця 24.1
Класифікація АС (АРУ, 2004)
|
Форма |
Центральна (осьова) |
Ураження хребта і осьових суглобів (плечових, кульшових) без ураження периферичних суглобів |
|
Периферична форма |
Ураження периферичних суглобів окремо або в сполученні з ураженням хребта і осьових суглобів |
|
|
Вісцеральна форма |
Сполучення центральної або периферичної форми з ураженням внутрішніх органів (аортит, тощо) |
|
|
Перебіг |
Повільнопрогресуючий Повільнопрогресуючий з періодами загострення Швидкопрогресуючий |
За короткий термін приводить до нового анкілозу |
|
Клініко-рентгенологічні стадії |
І (початкова або рання) |
Помірне обмеження рухів у хребті, або в уражених суглобах; рентгенологічні зміни відсутні або виявляються нечіткість або нерівність поверхонь клубово-крижових суглобів, вогнища субхонрального остеосклерозу, розширення суглобових щілин |
|
ІІ (помірних уражень) |
Обмеження рухів у хребті чи периферичних суглобах, звуження щілини клубово-крижових суглобів, або їх часткове анкілозування, звуження міжхребцевих суглобових щілин, або ознаки анкілозу суглобів хребта |
|
|
ІІІ (пізня) |
Значне обмеження рухів у хребті або великих суглобах кінцівок внаслідок анкілозування, кістковий анкілоз клубово-крижових суглобів, міжхребцевих і реберно-хребцевих суглобів з наявністю осифікації зв’язкового апарату |
|
|
Ступінь активності |
0 (відсутня) |
Відсутність скутості та болю у хребті та суглобах кінцівок, ШОЕ до 20 мм/год, СРБ – |
|
І (мінімальна) |
Невелика скутість, біль у хребті та суглобах кінцівок зранку, ШОЕ до 20 мм/год, СРБ + |
|
|
ІІ (помірна) |
Постійний біль в хребті та суглобах кінцівок, ранкова скутість декілька годин, ШОЕ до 40 мм/год, СРБ ++ |
|
|
ІІІ (виражена) |
Постійний біль у хребті та суглобах кінцівок, скутість протягом всього дня, ексудативні зміни в суглобах, субфебрільна температура тіла, вісцеральні прояви, ШОЕ понад 40 мм/год, СРБ +++/++++ |
|
|
Ступінь функціональної недостатності |
І |
Зміна фізіологічних згинів та обмеження рухливості хребетного стовпа та суглобів кінцівок, самообслуговування збережено або незначно порушено |
|
ІІ |
Значне обмеження рухливості хребетного стовпа і суглобів кінцівок, внаслідок чого хворий повинен змінити професію, самообслуговування значно порушено |
|
|
ІІІ |
Анкілоз всіх відділів хребетного стовпа і кульшових суглобів, втрата працездатності, неможливість самообслуговування |
|
|
Рентгенологічні стадії сакроілеїту |
0 |
Норма |
|
І |
На тлі рівномірного ОП виявлені ділянки склерозу в субхондральному відділі, суглобова щілина нерівномірно розширена, суглобові поверхні втрачають чіткість (розмита суглобова щілина) |
|
|
ІІ |
Зростає субхондральний склероз, фрагментуються замикаючі пластинки, суглобові щілини нерівномірно звужені, окостеніння крижово-клубових зв’язок, картина «нитки перлів» |
|
|
ІІI |
Ерозії, значне звуження суглобової щілини, частковий анкілоз клубово-крижових суглобів, окостеніння зв’язкового апарату |
|
|
IV |
Анкілоз клубово-крижових суглобів |
Клінічна картина АС зазвичай розгортається у молодих осіб. Деякі хворі відзначають продромальні явища за кілька місяців чи навіть років до початку хвороби. Загальна слабкість, дратівливість, сонливість, парестезії, артралгії, міалгії, зменшення маси тіла — основні скарги хворих в продромальний період. Ірити, іридоцикліти, епісклерити можуть бути провісниками цього захворювання. Характерна особливість цих станів — резистентність до звичайних методів лікування.
Виділяють ранню та пізню стадію хвороби.
На ранній стадії (дебют) хворі скаржаться на біль та скутість у спині, що виникають та посилюються вночі. Ці відчуття зникають протягом дня та після фізичних навантажень. Деколи хворі вимушені серед ночі робити фізичні вправи. Типовими є біль в попереку та крижах. Біль у сідницях, розповсюджується по задніх поверхнях стегон і охоплює всю ногу, нагадує ішіас, який має «коливальний» характер, часто рецидивує. Хворі відчувають деяку скутість рухів в хребті.
Поступово процес поширюється вгору, виникає біль в грудній клітці, що має оперізувальний характер, розвивається міжреберна невралгія. Зменшується дихальна екскурсія грудної клітки. Біль посилюється спочатку при кашлі, чханні, а потім і в спокої.
Осанка хворого на ранній стадії залишається нормальною, хода не порушена, рухи в хребті порушуються при загостренні та вираженому больовому синдромі.
Частими є скарги на втомлюваність, слабкість, втрату апетиту, зменшення маси тіла, підвищення температури.
Виділяють декілька варіантів дебюту АС.
- Поступово наростаючий біль і скутість в нижній частині спини, сідницях або грудній клітці.
- Переважання скутості, міалгії, біль в місцях прикріплення зв’язок або «корінцевий» біль без неврологічних порушень.
- Початок з периферичного моно-, олігоартриту колінних суглобів.
- Початок (частіше у дітей) з ентезиту (ахіллодиніту, підошовного фасциту), болю в п’ятах при ходьбi.
- Переважання в дебюті двобічного ураження плечових і тазостегнових (рідше груднино-ключичних) суглобів.
- Розвиток в дебюті ентезопатії крижово-клубових суглобів, міжхребцевих дисків, груднино-реберних з’єднань, лобкового симфізу, ділянки прикріплення зв’язок до остистих відростків, гребеня клубової і вертлюга стегнової кістки, надколінка і п’яткових кісток.
Виділяють центральну та периферичну форму хвороби Бехтєрева.
Центральна форма АС має 2 типи: кіфозний та ригідний. Для першого типу характерний розвиток кіфозу грудного відділу та гіперлордозу шийного відділу хребта, для другого — зменшення чи зникнення поперекового лордозу та грудного кіфозу.
До патологічного процесу можуть бути залучені осьові суглоби — плечові та кульшові. Прогноз більш сприятливий при залученні тільки плечових суглобів. Артрит плечових суглобів не стійкий, виникає біль у плечовому поясі під час руху або при фізичному навантаженні, розвивається різка атрофія м’язів та обмеження рухів. Анкілоз цих суглобів розвивається рідко.
Кульшові суглоби можуть уражатися одночасно з хребтом або трохи пізніше. Поступово виникає біль у кульшовому суглобі з больовою іррадіацією в пахвинну зону, стегно, колінний суглоб. Розвиваються м’якотканинні контрактури, що разом із вираженим больовим синдромом значно обмежує ротаційні рухи, потім виникає анкілоз. Разом із анкілозами в кульшових суглобах розвиваються анкілози груднино-ключичних і надплечово-ключичних суглобів.
При периферичній формі АС уражаються колінні, гомілковостопні, ліктьові суглоби, а іноді й дрібні суглоби кистей і стоп (схоже на серонегативний варіант РА). Для периферичної форми типовим є підгострий дебют і рецидивуючий перебіг. У 10% пацієнтів уражаються скронево-нижньощелепні суглоби, що супроводжується утрудненням жування.
Вісцеральну форму АС діагностують при поєднанні центральної чи периферичної форми із ураженням внутрішніх органів: серця, аорти, нирок, судинної оболонки ока.
При ураженні серця може розвиватися аортит, який супроводжується аортальною регургітацією і перикардитом. Загалом, для цих уражень характерні незначні прояви і повільне прогресування кардіалгій та стенокардії, серцебиття, зниження толерантності до фізичних навантажень. Серцева недостатність розвивається дуже повільно. Описано випадки повної АВ-блокади з розвитком синдрому Морганьї — Едемса — Стокса.
Ураження легень відбувається опосередковано: залучення грудного відділу хребта приводить до обмеження дихальної екскурсії легень. Хворі стають схильними до респіраторних захворювань. Описано поодинокі випадки прогресуючого фіброзу верхівок легень. Легенева гіпертензія з часом може розвинутися у торакодіафрагмальну форму хронічного легеневого серця.
Нирки уражаються при високій активності запального процесу та швидкопрогресуючому перебігу хвороби. Застосування НПЗП приводить до розвитку анальгетичної нефропатії. Часто виявляється нефролітіаз. Тяжка ниркова недостатність розвивається на фоні амілоїдозу.
Ірит, іридоцикліт, увеїт — досить часті й ранні ознаки вісцеральної форми АС. У третини хворих на АС ці стани можуть бути «провісниками». Запалення судинної оболонки ока не піддаються традиційному лікуванню, рецидивують і мають хронічний перебіг. Позаскелетні прояви АС запам’ятовують за допомогою абревіатури із перших літер назви ураженних органів (Janson R.W., 1999) (табл. 24.2).
Таблиця 24.2
Позаскелетні прояви АС (за: Janson R.W., 1999)
|
А |
Aortic insufficiency, ascending aortitis |
Аортальна недостатність, висхідний аортит, порушення провідності, діастолічна дисфункція і перикардит (10% хворих) |
|
N |
Neurologic |
Неврологічні прояви внаслідок підвивиху в атланто-аксіальному з’єднанні, синдром «кінського хвоста» |
|
К |
Kidney |
Вторинний амілоїдоз нирок і хронічне запалення передміхурової залози |
|
S |
Spine |
Переломи в шийному відділі хребта, спінальний стеноз |
|
Р |
Pulmonary |
Фіброз верхніх часток легень, рестриктивні порушення |
|
О |
Ocular |
Передній увеїт (25–30% хворих) |
|
N |
Nephropathy |
Нефропатія (IgA) |
|
D |
Discitis or spondylodiscitis |
Дисцит або спондилодисцит (симптом Андерсена) |
Деякі автори виділяють септичний варіант хвороби Бехтєрева. Для нього характерні гострий початок з лихоманкою гектичного характеру, озноб, профузне підвищене потовиділення, ранній розвиток вісцеральних уражень (серця, аорти, нирок, очей). Пізніше, після лихоманок з’являються артрити та скутість у хребті.
Пізню стадію АС діагностують через 8–10 років від початку хвороби у чоловіків, а у жінок — через 15–20 років. Діагностику АС ускладнюють повільнопрогресуючий перебіг та мінімальна активність запального процесу.
Больові відчуття в попереково-крижовій ділянці зменшуються чи зникають зовсім, в місцях запалення розвивається анкілоз суглобів. Суттєве зменшення дихальної екскурсії до 1–2 см виникає за рахунок анкілозів реберно-хребцевих суглобів. Рестриктивний тип дихальної недостатності виникає не так часто, тому що компенсація розвивається за рахунок діафрагмального дихання. Грудна клітка фіксується в фазі часткового видиху. Задишка виникає після їди. Під час кашлю, чхання, різких рухів може виникати біль у міжлопатковій ділянці, грудній клітці. У шиї з’являється виражений біль при поворотах і нахилах, обсяг рухів обмежений майже до повної нерухомості. Прояви вертебробазилярної недостатності, радикулярний біль та дистрофічні зміни — часто виявляють у хворих на АС.
У жінок спостерігається менша вираженість клінічних проявів і більш повільний розвиток рентгенологічних змін. Тому діагностувати АС у жінок набагато важче, ніж у чоловіків. Повний анкілоз хребта розвивається рідко, частіше виявляють ізольований анкілоз шийного відділу хребта та ураження дистальних відділів кінцівок.
У підлітків АС характеризується тяжким перебігом із залученням кульшових суглобів. У чверті з них у пізній стадії виявляють дистальний артрит (табл. 24.3).
Таблиця 24.3
Методики клінічного обстеження хворих на АС
|
Рухливість шийного відділу хребта |
При максимальному нахилі голови між підборіддям та ручкою груднини відстань 0–2 см, а при максимальному відкиданні — 16–22 см |
|
Рухливість грудного відділу хребта |
Проба Отта: від VII шийного хребця вниз відмірюють 30 см, потім повторюють вимірювання під час максимального згинання у шийно-грудному відділі, у нормі збільшується відстань на 4–5 см |
|
Дихальна екскурсія грудної клітки |
На рівні IV ребра вимірюють межі на висоті вдиху та видиху, відстань 5–6 см. Симптом Богданова: однією рукою підтримуємо корпус пацієнта, а другою натискаємо на епігастральну ділянку (обмежуємо черевне дихання) під час вдиху зростає напруження м’язів шиї. Симптом нитки: на видиху зав’язують нитку навколо грудної клітини, яка повинна порватися підчас вдиху |
|
Рухливість поперекового відділу хребта |
Тест Томаєра: при максимальному нахилі вперед відстань між кінчиками середніх пальців та підлогою 0–10 см. Тест Шобера: від точки перехрестя хребта з лінією ромбу Міхаеліса відміряють вгору 10 см, відстань збільшується на 4–5 см при максимальному згинанні |
|
Симптом тазової компресії: хворий лежить на боці, лікар стискає таз. У хворих на АС стискання спричиняє біль в крижово-клубових суглобах |
|
|
Симптом Генслена: одна нога звисає з ліжка, а другу хворий піднімає до грудної клітки — при АС це супроводжується болем у крижово-клубових суглобах на боці ноги, що звисає |
|
|
Симптом Патрика: п’ята знаходиться на коліні другої ноги, якщо тиснути на зігнуте коліно з надаванням стегну положення згинання, відведення та зовнішньої ротації (FABER), виникає біль у крижово-клубових суглобах на протилежному боці |
|
|
Тести Кушелевського: I — надавлюють на верхні передні ості клубових кісток (хворий лежить на спині) II — надавлюють на крило клубової кістки (хворий лежить на боці) III — хворий лежить на спині, одночасно надавлюють на внутрішню поверхню зігнутого під кутом 90о і відведеного колінного суглоба та верхню передню ость протилежного крила клубової кістки. Поява болю у крижово-клубових суглобах при цих діях свідчить про запалення |
Діагностичні критерії
Римські критерії:
1. Біль у крижах — 3 міс, у спокої постійний.
2. Біль і скутість у грудній клітці.
3. Обмеження рухливості поперекового відділу хребта.
4. Обмеження екскурсії грудної клітки.
5. Ірит гострий або в анамнезі.
6. Двобічний сакроілеїт при рентгенологічному дослідженні.
За наявності двобічного сакроілеїту й одного з клінічних критеріїв або за наявності 4 з 5 критеріїв.
Нью-Йоркські критерії:
1. Обмеження рухливості поперекового відділу хребта у всіх площинах.
2. Наявність в цей час або в анамнезі болю в ділянці поперекового відділу хребта або попереково-крижового переходу.
3. Екскурсія грудної клітки <2,5 см на висоті IV межребер’я.
4. Рентгенологічні дані:
а) двобічний сакроілеїт III–IV стадії;
б) двобічний сакроілеїт III–IV стадії або двобічний сакроілеїт II стадії.
Достовірний АС за наявності:
- сакроілеїту III–IV стадії й одного з клінічних критеріїв;
- двобічного сакроілеїту II стадії або однобічного сакроілеїту III–IV стадії з критерієм 1 або двома критеріями 2 і 3.
Модифіковані Нью-Йоркські критерії:
1. Біль у крижах протягом 3 міс, вираженість якого зменшується при фізичних вправах; у спокої — постійний.
2. Обмеження рухливості поперекового відділу хребта в сагітальній та фронтальній площинах.
3. Зменшення екскурсії грудної клітки відносно норми, що відповідає вікові та статі.
4. Рентгенологічні дані: двобічний сакроілеїт II–IV стадії або однобічний сакроілеїт III–IV стадії.
Достовірний АС за наявності однобічного сакроілеїту III–IV стадії або двобічного сакроілеїту II–IV стадії й одного з клінічних критеріїв.
Диференційні ознаки запального та механічного болю (табл. 24.4).
Таблиця 24.4
Симптоми запального та механічного болю
|
Симптоми |
Характер болю |
|
| Запальний | Механічний | |
|
Час появи симптомів хвороби |
Переважно до 40 років |
Можливі у будь-якому віці |
|
Характерний початок |
Поступовий |
Гострий |
|
Тривалість симптомів |
3 міс та більше |
Меньше 4 тиж |
|
Тривалість ранкової скутості |
Більше 1 год |
Меньше 30 хв |
|
Наявність нічного болю |
+ |
– |
|
Ефект фізичних вправ |
Поліпшення |
Погіршення |
|
Покращання стану після відпочинку |
– |
+ |
|
Болісність у крижово-клубових суглобах |
Характерна |
Відсутній |
|
Обмеження рухів у хребті |
Рухи обмежені у всі боки |
Порушене згинання |
|
Стан дихальної екскурсії грудної клітки |
Обмежений |
Не порушений |
|
Наявність неврологічних порушень |
Спостерігаються рідко |
Можливі |
За критеріями ASAS запальний біль у спині:
- початок болю у віці до 40 років;
- поступовий початок;
- покращення після фізичного навантаження;
- немає покращення після відпочинку;
- нічний біль (з покращенням після пробудження).
4 ознаки з 5 свідчать про запальний характер болю.
Характерні клінічні особливості АС. У ⅔ хворих ураженню хребта передує запалення суглобів, яке має рецидивуючий характер і турбує впродовж 2–5 років. У ⅓ хворих спочатку уражається хребет. Рецидивуючий артрит має доброякісний перебіг. В уражених суглобах рентгенологічні ознаки не виявляються (деколи ОП). Можливі ревматоїдоподібні зміни суглобів кистей, які не супроводжуються вираженим запаленням і деструкцією. Через деякий час приєднується запалення крижово-клубових суглобів чи хребта.
У ряду хворих відзначають ураження очей, серця, аорти, нирок. До ураження очей відносять склерити, епісклерити, увеїти, ірити, іридоцикліти, хоріоідити, кератити, катаракту, склеромаляцію. Очні симптоми можуть на декілька років передувати ураженню хребта і суглобів. Із загостренням пов’язують виникнення іриту та іридоцикліту. Ураження очей можуть приводити до втрати зору, глаукоми.
Порок серця і перикардит виявляють у хворих з ураженням периферичних суглобів, аортити — при ураженні хребта, крижово-клубових і периферичних суглобів. Відносно часто виникає ниркова колька, а при біопсії нирок виявляють ознаки гломерулонефриту.
У жінок хвороба прогресує повільно, хребет деформується пізно, анкілоз крижово-клубових суглобів виявляють у 10% через 10 років. Порівняльно пізно виникають зміни в грудному відділі хребта.
Діагностувати АС у дітей досить важко. До перших симптомів належать болісність сухожиль і зв’язок, ірит, ураження аорти. Хвороба часто розпочинається із ураження колінних суглобів. Можливі ремісії від декількох місяців до декількох років. Загострення можуть бути спровоковані переохолодженням, фізичним перевантаженням, травмою, ангіною чи грипом. Періодично турбує біль у хребті. Активність запалення, за даними аналізів, низька. Рентгенологічних ознак не виявляють, а обмеження рухів у хребті та порушення постави виникають через 15–20 років.
Хвороба розпочинається з ураження великих суглобів, особливо тазостегнових. Тривалий коксит ізольований, без ознак загального запалення. Можуть бути уражені періартикулярні тканини. Функція хребта довгий час збережена, симптоми невиражені. У випадку стійкого артриту (більше 4 міс) виникає субхондральний ОП, кісткові кісти, рідко узурпація.
Морфологічні зміни на ранній стадії хвороби: розвиток імунного запалення, яке розпочинається в крижово-клубових суглобах, а потім залучаються міжхребцеві, реберно-хребцеві й периферичні суглоби. Запальна інфільтрація лімфоцитами і макрофагами замінюється утворенням сполучної тканини та осифікацією.
На пізній стадії виявляють синовіти, запалення місць прикріплення зв’язок, сухожиль, капсул суглобів, фіброзної частини міжхребцевих дисків (ентезопатії), кісток (оститів). Утворюються контрактури, анкілози (фіброзні й кісткові) осьового скелета, периферичних суглобів чи зв’язкового апарату хребта (рис. 24.1, 24.2).
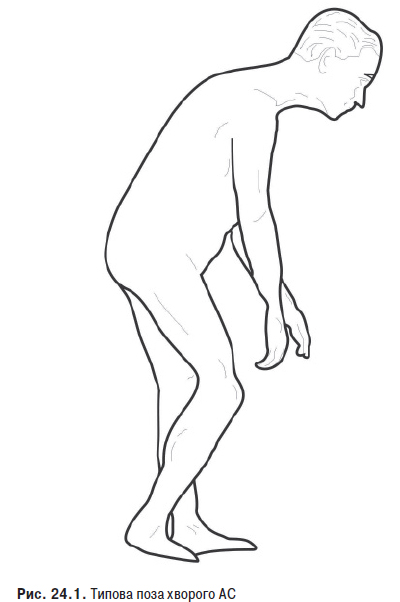
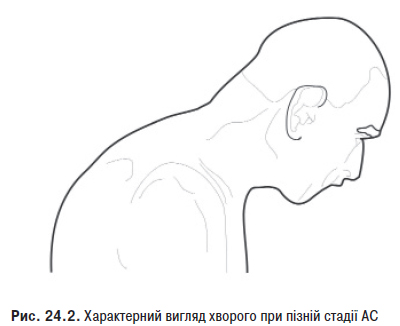
У 30–60% хворих виявляють безсимптомний мікроскопічний коліт з ураженням термінального відділу клубової та товстої кишки.
До розвитку порушень серцевої провідності приводить фіброз субаортальних відділів, можливе виникнення повної АВ-блокади.
У жінок первинні атаки АС змінюються довгими ремісіями (до 5–10 років). Ураження крижово-клубових суглобів однобічне, типовий «бамбуковий» хребет розвивається рідко. Більш часто спостерігаються периферичні артрити (дрібні суглоби кистей і стоп), вісцеральні ураження (аортит, вада серця, увеїт).
Перебіг АС у осіб похилого віку має свої особливості — слабкість зв’язкового апарату та ригідність хребта приводять до підвищеної чутливості до травм. Наслідком падіння може бути перелом шийного відділу хребта, тетрапарез і навіть смерть хворого.
Діагностика
У більшості пацієнтів при лабораторному обстеженні виявляють підвищені показники ШОЕ, вміст СРБ, IgA, реєструють HLA-B27 (у 90%), нормохромну анемію. При тяжкому перебігу хвороби відзначають підвищений вміст лужної фосфатази. За рахунок обмеження екскурсії легень знижується життєва ємність легенів, але гіповентиляція не розвивається. РФ та АНФ не виявляються.
Виявлення HLA-B27 не є достатнім критерієм діагностики АС. Його визначення допомагає діагностувати АС, якщо ще не сформувалися рентгенологічні ознаки сакроілеїту. Діагноз АС базується на даних анамнезу, результатах об’єктивного обстеження і наявності ознак сакроілеїту на рентгенограмах. Тестування на HLA-B27-антиген не обов’язкове.
Рентгенологічні зміни, характерні для анкілозуючого спондиліту
Ентезопатії («вусики») — характерні ознаки новоутвореної кістки виявляють в ділянці прикріплення зв’язок до остистих відростків хребців, гребенів клубових кісток, горбистих сідничних кісток, вертлюгів стегон, п’яткових кісток, надколінка, ключиці. У перелічених місцях утворюються вогнища деструкції.
Сакроілеїт — двобічний і симетричний на початкових стадіях, уражує нижні ⅔ суглобів. У результаті прогресування ерозивних змін настає «псевдорозширення» суглобової щілини з явищами остеосклерозу, а потім повне кісткове анкілозування. Стандартні рентгенограми на ранніх стадіях сакроілеїту не реєструють кісткових змін. Виявити на ранній стадії запальні зміни і набряк дозволяє МРТ. Для пацієнтів, яким протипоказано проводити МРТ (металічні імпланти чи водії ритму) або внаслідок проблем з обладнанням, можна застосовувати КТ. При КТ-дослідженні краще візуалізуються ерозії та кісткові зміни сакроілеальних зчленувань, однак неможливо виявити набряк кісткового мозку. Суттєвим недоліком методики є променеве навантаження на органи, в тому числі й гонади (рис. 24.3, 24.4).
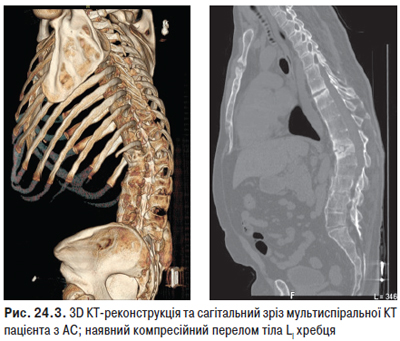
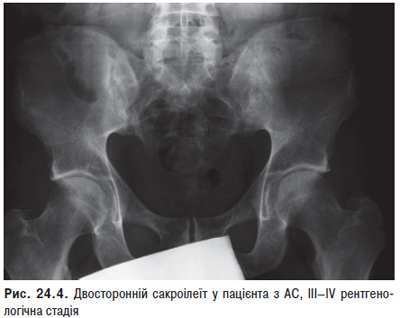
Ураження хребта — характерна поява симптому «блискучих кутів», потім — «квадратизації» тіл хребців. Ці зміни є наслідком запалення, тиску фіброзного кільця міжхребцевих дисків на кути тіл хребців. Формування міжхребцевих кісткових мостиків — синдесмофітів, анкілозування дуговідросткових суглобів і кальциноз зв’язок хребта приводять до повного зростання хребців, хребет набуває вигляду «бамбукової палиці» (табл. 24.5, рис. 24.5).
Таблиця 24.5
Рентгенологічні зміни, які виявляють на різних стадіях АС
|
Відділи |
Стадія |
Характерні рентгенологічні ознаки |
|
Крижово-клубові з’єднання |
Рання |
Розмитість контурів, псевдорозширення суглобової щілини через субхондральний ОП |
|
Пізня |
Субхондральні ерозії, звуження і повне зникнення (анкілоз) суглобової щілини |
|
|
Хребет |
Рання |
Ерозії в ділянці верхніх і нижніх передніх кутів тіл хребців. Зникнення нормальної увігнутості тіл хребців. Осифікація передньої поздовжньої зв’язки («квадратизація хребців») |
|
Пізня |
Осифікація зв’язок хребта починається між XII грудним і I поперековим хребцем. Утворення кісткових мостиків (синдесмофітів) спричиняє зміни за типом «бамбукової палиці» |
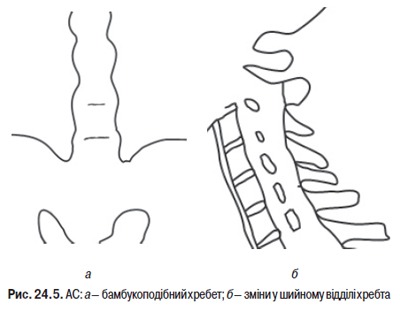
Рентгенологічні зміни в крижово-клубових суглобах виявляють при запальних спондилоартропатіях, подагрі, гіперпаратиреозі, хворобі Педжета, остеоартрозі, метастазах пухлин, конденсаційному остеїті (табл. 24.6).
Таблиця 24.6
Рентгенологічні стадії сакроілеїту
|
Стадія |
Зміни в суглобах |
|
I |
Є підозра на наявність змін |
|
II |
Відмічають ерозії та склероз |
|
III |
Виявляють ерозії, склероз і частковий анкілоз |
|
IV |
Є повний анкілоз |
Перелічені діагностичні критерії мають недолік — вони не дозволяють встановити діагноз АС на ранній стадії, оскільки достовірні рентгенологічні зміни проявляються через багато місяців після початку хвороби.
Визначення активності та ступеня тяжкості анкілозуючого спондиліту
Опитувальник для визначення індексу BASDAI (Bath Ankylosing Spondilitis Disease Activity Index) складається з 6 питань, на які пацієнт відповідає самостійно.
Для відповіді на кожне питання пропонується 10-сантиметрова візуальна аналогова шкала (ліва крайня точка відповідає відсутності даної ознаки, права крайня точка — крайньому ступеню вираженості ознаки; для останнього питання про термін скутості — 2 год і більше). Пацієнт повинен відповісти на кожне з наведених нижче питань, свою відповідь позначає рискою, яка перетинає 10-сантиметрову лінію в обраному ним місці.
1. Як би Ви розцінили рівень загальної слабкості (стомлюваності) за останній тиждень?
2. Як би Ви розцінили рівень болю в шиї, спині або кульшових суглобах за останній тиждень?
3. Як би Ви розцінили рівень болю (або ступінь припухлості) в суглобах (окрім шиї, спини або кульшових суглобів) за останній тиждень?
4. Як би Ви розцінили ступінь неприємних відчуттів, які виникають при дотику до будь-яких болісних ділянок або тиску на них (за останній тиждень)?
5. Як би Ви розцінили ступінь вираженості ранішньої скутості, яка виникає після прокидання (за останній тиждень)?
6. Як довго продовжується ранішня скутість, яка виникає після прокидання (за останній тиждень)?
Лікар за допомогою лінійки вимірює довжину відмічених відрізків ліній, вираховує суму та середню величину. Значення індексу BASDAI, яке перевищує 4, свідчить про високу активність захворювання.
Індекс BASFI (Bath Ankylosing Spondilitis Functional Index) складається з 10 питань, пов’язаних з оцінкою повсякденної активності, які відображені на аналогових шкалах.
Питання «Чи можете Ви…» (оцінюються за візуальною аналоговою шкалою за останній тиждень):
- Ліва крайня точка відповідає 0 мм — «без труднощів», права крайня точка відповідає 100 мм — «неможливо це зробити»
- Надіти шкарпетки або колготи без сторонньої допомоги
- Нагнутися вперед, щоб підняти ручку з підлоги без використання пристосувань
- Дотягнутися рукою до високо розташованої полиці без сторонньої допомоги
- Встати зі стільця без допомоги рук і без сторонньої допомоги
- Встати з підлоги із положення лежачи на спині без сторонньої допомоги
- Стояти без додаткової опори протягом 10 хв і не відчувати дискомфорту
- Піднятися наверх на 12–15 сходинок, не спиратися на перила або паличку (наступати однією ногою на кожну сходинку)
- Повернути голову і подивитися за спину, не повертаючи тулуб
- Займатися фізично активними видами діяльності (наприклад фізичними вправами, спортом, роботою в саду)
- Зберігати активність протягом усього дня (вдома або на роботі)
Лікар за допомогою лінійки вимірює довжину відмічених відрізків ліній, вираховує суму та середнє значення. Значення індексу BASFI, яке перевищує 4, свідчить про виражені функціональні порушення.
Приклад формулювання діагнозу: АС (хвороба Бехтєрева), центральна форма, активність І ступеня, двобічний сакроілеїт ІІІ ступеня, спондиліт поперекового відділу хребта ІІ ступеня, недостатня функціональність суглобів ІІ ступеня.
Лікування
Немедикаментозне лікування
Необхідно навчити пацієнта дотримуватися рухливого способу життя. Корисними для хворих будуть заняття плаванням, гра у волейбол, великий та малий теніс. Заняття ЛФК мають проводитися щоденно протягом 30–40 хв. Періодично контроль вправ повинен здійснювати інструктор з ЛФК. Особливу увагу необхідно надати організації сну: хворі повинні спати на твердому ліжку з низькою подушкою на ортопедичному матраці. Фізіотерапевтичне лікування проводять з метою запобігання розвитку контрактур, м’язових атрофій, обмежень рухів хребта. Використовують індуктотермію, діадинамічні струми, магніто- та лазеротерапію. У фазі ремісії ефективні масаж, бальнеотерапія та грязелікування. Санаторно-курортне лікування та фізіотерапія протипоказані при високій активності процесу і загостреннях.
Медикаментозне лікування
Нестероїдні протизапальні препарати
Найбільш ефективні на початку хвороби. Призначають в максимальній добовій дозі, декілька разів на день і ніч. Очікуваний ефект настає через 1–2 тиж. Пацієнти застосовують ці препарати тривалий час, замінюють іншими при втраті ефективності. Інгібітори ЦОГ-2 (еторикоксиб) призначають при високому ризику гастроінтестинальних ускладнень або непереносимості препаратів інших груп.
Еторикоксиб*
*В Україні зареєстрований як Аркоксія®
Фармакотерапевтична група. Високоселективний інгібітор ЦОГ-2. Код АТС M05BA04. Еторикоксиб є НПЗП — селективним інгібітором ЦОГ-2 у межах клінічного діапазону доз. У клінічних фармакологічних дослідженнях препарат дозозалежно інгібував ЦОГ-2 без інгібування ЦОГ-1 при застосуванні у дозах до 150 мг/добу. Еторикоксиб не інгібує синтез простагландинів шлунка та не впливає на функцію тромбоцитів.
Показання. Симптоматичне лікування ОА — 30 мг 1 раз на добу (у деяких пацієнтів з недостатнім послабленням симптомів підвищення дози до 60 мг може призводити до більш вираженого ефекту); РА — 90 мг 1 раз на добу; AС, болю і усунення запальної симптоматики при гострому подагричному артриті — в дозі 120 мг слід застосовувати протягом обмеженого періоду — максимально 8 днів. Нетривале лікування помірного та тяжкого гострого післяопераційного болю, пов’язаного зі стоматологічними та абдомінально-гінекологічними операціями.
Післяопераційний біль, пов’язаний зі стоматологічним втручаннями. Рекомендована доза — 90 мг 1 раз на добу.
Післяопераційний біль, пов’язаний з абдомінально-гінекологічними втручаннями. Рекомендована доза — 90 мг 1 раз на добу. Початкову дозу слід прийняти безпосередньо перед операцією. Застосування 120 мг/добу може забезпечити додаткову ефективність для зменшення більш тяжкого післяопераційного болю при ходьбі.
Рішення про призначення селективного інгібітору ЦОГ-2 має ґрунтуватися на оцінці всіх індивідуальних ризиків для пацієнта. Еторикоксиб застосовують перорально. Препарат можна приймати незалежно від їди. Початок ефекту препарату настає швидше при застосуванні без прийому їжі, що слід враховувати при необхідності швидкого полегшення симптомів.
Протипоказання:
- гіперчутливості до будь-якого компонента препарату;
- активна пептична виразка або шлунково-кишкова кровотеча;
- пацієнти, у яких виникав бронхоспазм, гострий риніт, назальні поліпи, ангіоневротичний набряк, кропив’янка або інші алергічні реакції після застосування ацетилсаліцилової кислоти або НПЗП, включаючи інгібітори ЦОГ-2;
- період вагітності та годування грудьми;
- тяжкі порушеннях функції печінки (альбумін сироватки крові <25 г/л або ≥10 балів за шкалою Чайлд-П’ю);
- нирковий кліренс креатиніну <30 мл/хв;
- діти та підлітки у віці до 16 років;
- запальні хвороби кишечнику;
- застійна серцева недостатність (NYHA ІІ–ІV);
- пацієнти з гіпертензією, у яких показники артеріального тиску постійно вищі за 140/90 мм рт. ст. та не контролюються адекватно;
- встановлена ішемічна хвороба серця, захворювання периферичних артерій та/або серцево-судинні захворювання.
Оскільки ризик виникнення порушень з боку серцево-судинної системи зростає при підвищенні дози та збільшенні тривалості експозиції селективних інгібіторів ЦОГ-2, слід проводити якомога коротші курси лікування при застосуванні найнижчих ефективних добових доз. Необхідність полегшення симптомів та ефективність терапії у пацієнтів слід періодично переглядати.
Побічні реакції:
- Часто — набряки/затримка рідини; запаморочення, головний біль; пальпітація; гіпертензія; гастроінтестинальні порушення (такі як абдомінальний біль, метеоризм, печія), діарея, диспепсія, відчуття дискомфорту в епігастральній ділянці, нудота; підвищення рівня АсАТ, АлАТ; екхімоз; астенія/слабкість, грипоподібні симптоми.
- Нечасто — гастроентерит, інфекції верхніх дихальних шляхів, інфекції сечовивідного тракту; анемія (переважно внаслідок шлунково-кишкової кровотечі), лейкопенія, тромбоцитопенія; послаблення або посилення апетиту, збільшення маси тіла; тривожність, депресія, погіршення розумової діяльності; дисгевзія, безсоння, парестезія/гіпестезія, сонливість; нечіткість зору, кон’юнктивіт; дзвін у вухах, вертиго; фібриляція передсердь, застійна серцева недостатність, неспецифічні зміни на ЕКГ, стенокардія, інфаркт міокарда; припливи крові, інсульт, транзиторні ішемічні порушення мозкового кровообігу; кашель, диспное, носова кровотеча; здуття живота, кислотний рефлюкс, зміна характеру перистальтики кишечнику, запор, сухість у роті, гастродуоденальні виразки, синдром подразненого кишечнику, езофагіт, виразка у ротовій порожнині, блювання, гастрит; спазми/судоми м’язів, скелетно-м’язовий біль/скутість; протеїнурія, підвищення рівня креатиніну в сироватці крові; набряк обличчя, свербіж, висип; біль у грудній клітці; підвищення рівня азоту сечовини крові, підвищення рівня КФК, гіперкаліємія, підвищення рівня сечової кислоти.
- Рідко — еритема; дуже рідко — кропив’янка, синдром Стівенса — Джонсона, токсичний епідермальний некроліз; зниження рівня натрію у крові.
- Дуже рідко — реакції гіперчутливості, включаючи ангіоневротичний набряк, анафілактичні/анафілактоїдні реакції, включаючи шок; сплутаність свідомості, галюцинації, безсоння; гіпертонічний криз; бронхоспазм; пептичні виразки, включаючи гастроінтестинальну перфорацію та кровотечу (частіше в осіб літнього віку); гепатит; порушення функції нирок, включаючи ниркову недостатність.
- Невідомо — тахікардія; панкреатит; жовтуха.
Глюкокортикоїди
Локальне застосування. Застосовують локально при лікуванні ентезопатій і резистентних синовітів периферичних суглобів. ГК (бетаметазон 0,25–2 мл внутрішньосуглобово або періартикулярно в ділянку ураженого суглоба не частіше 1 разу на 1–3 міс в той самий суглоб залежно від дози та розміру суглоба) вводять в порожнину суглобів при периферичному артриті, а також в ділянку крижово-клубових суглобів. Місцеве лікування проводиться при гострому передньому увеїті.
Бетаметазон (betamethasone)*
*В Україні зареєстрований як Дипроспан®
Склад: 1 мл суспензії містить 6,43 мг бетаметазону дипропіонату (еквівалентний 5 мг бетаметазону) та 2,63 мг бетаметазону натрію фосфату (еквівалентний 2 мг бетаметазону).
Препарат Дипроспан® є комбінацією розчинного і малорозчинного ефірів бетаметазону для внутрішньом’язових, внутрішньосуглобових, навколосуглобових, внутрішньосиновіальних та внутрішньошкірних ін’єкцій, а також для введення безпосередньо у вогнище ураження. Бетаметазон має високу глюкокортикостероїдну та незначну мінералокортикостероїдну активність. Бетаметазону натрію фосфат — легкорозчинний компонент, який швидко абсорбується з місця введення, що забезпечує швидкий початок терапевтичної дії. Бетаметазону дипропіонат — малорозчинний компонент, який повільно абсорбується з депо, що утворюється в місці ін’єкції, і зумовлює тривалу дію препарату.
Незначні розміри кристалів бетаметазону дипропіонату дозволяють використовувати голки невеликого діаметра (до 0,9 мм) для внутрішньошкірних введень та введення безпосередньо у вогнище ураження.
Форма випуску. Суспензія для ін’єкцій. Кортикостероїди для системного застосування. Код АТС H02A B01.
Показання до застосування. Захворювання м’язово-скелетної системи та м’яких тканин (у тому числі РА, ОА, бурсити, тендосиновіти, тендиніти, перитендиніти, АС, епікондиліт, радикуліт, кокцигодинія, ішіаз, люмбаго, кривошия, гангліозна кіста, екзостоз, фасцит, захворювання стоп).
Алергічні захворювання та стани (у тому числі бронхіальна астма, сінна лихоманка, алергічний бронхіт, сезонні або цілорічні риніти, медикаментозна алергія, сироваткова хвороба, реакції на укуси комах).
Дерматологічні стани (у тому числі атопічний дерматит, монетоподібна екзема, нейродерміти, контактний дерматит, виражений сонячний дерматит, кропив’янка, червоний плоский лишай, інсулінова ліподистрофія, гніздова алопеція, дискоїдний еритематозний вовчак, псоріаз, келоїдні рубці, звичайна пухирчатка, герпетичний дерматит, кістозні вугрі).
Колагенози (включаючи системний еритематозний вовчак, склеродермію, ДМ, ВП).
Пухлинні захворювання (паліативна терапія лейкозу та лімфом у дорослих; гострий лейкоз у дітей).
Інші захворювання та стани: адреногенітальний синдром, виразковий коліт, регіонарний ілеїт, синдром мальабсорбції, ураження стоп — бурсит на фоні твердої мозолі, шпори, тугорухливість великого пальця стопи; патологія, що потребує субкон’юнктивальних ін’єкцій препарату, патологічні зміни крові, які потребують проведення кортикостероїдної терапії, нефрит, нефротичний синдром.
Первинна недостатність кори надниркової залози (при обов’язковому одночасному введенні мінералокортикоїдів).
Побічна дія. Небажані явища, як і при застосуванні інших ГКС, зумовлені дозою та тривалістю прийому препарату. Ці реакції, як правило, зворотні, їх вираженість може бути зменшена шляхом зниження дози; цьому методу, порівняно з повною відміною препарату, зазвичай надають перевагу.
- Натріємія, підвищене виділення калію, гіпокаліємічний алкалоз, збільшення виведення кальцію, затримка рідини в тканинах;
- застійна серцева недостатність у хворих, схильних до цього захворювання; артеріальна гіпертензія;
- м’язова слабкість, міопатія, втрата м’язової маси, погіршення міастенічних симптомів при тяжкій псевдопаралітичній міастенії, ОП, асептичний некроз головок стегнової або плечової кістки, патологічні переломи довгих кісток, розриви сухожиль, нестабільність суглобів (після багаторазових введень);
- ерозивно-виразкові ураження ШКТ з можливою наступною перфорацією та кровотечею, панкреатит, метеоризм, виразки стравоходу;
- порушення загоєння ран, атрофія шкіри, стоншення та ламкість шкіри, петехії та екхімози, еритема обличчя, підвищене потовиділення, шкірні реакції, такі як дерматит, висипання, ангіоневротичний набряк;
- судоми, підвищення внутрішньочерепного тиску з набряком диска зорового нерва (зазвичай після завершення лікування), запаморочення, головний біль;
- порушення менструального циклу, розвиток кушингоїдної конституції, затримка внутрішньоутробного розвитку плода або росту дитини, порушення толерантності до глюкози, прояви латентного цукрового діабету, підвищення потреби у застосуванні ін’єкцій інсуліну чи пероральних антидіабетичних засобів;
- задня субкапсулярна катаракта, підвищення внутрішньоочного тиску, глаукома, екзофтальм;
- негативний баланс азоту (внаслідок катаболізму білка);
- ейфорія, зміна настрою, депресія (з вираженими психотичними реакціями), підвищена дратівливість, безсоння;
- анафілактична або надчутлива реакція на введення препарату, гіпотензивна або шокоподібна реакція;
- поодинокі випадки порушення зору, що супроводжують внутрішньоосередкову терапію в ділянці обличчя та голови, гіпер- або гіпопігментація, підшкірна та шкірна атрофія, стерильні абсцеси, припливи крові до обличчя після ін’єкції (внутрішньосуглобове введення) та нейрогенна артропатія.
Протипоказання:
- системні мікози;
- підвищена чутливості до бетаметазону або інших компонентів препарату, або до інших ГКС.
Системне застосування. Внутрішній прийом ГК не впливає на кістково-м’язові прояви АС. Системно, короткочасно призначають при лихоманці, увеїтах, загостреннях іридоциклітів, IgA-нефропатіях. Крім того, в число показань до системного застосування ГК у хворих АС входять:
- системні прояви (аортит, порок серця, АВ-блокади та ін.);
- висока активність запалення (клінічно і за данними маркерів запалення) протягом ≥3 міс;
- виражений периферичний артрит з порушеннями функції;
- коксит, резистентний до інших видів терапії.
Терапію ГК проводять короткими курсами (не довше 2–3 міс), доза преднізолону становить 15–20 мг/добу.
Пульс-терапія
Показання до проведення пульс-терапії у хворих на АС:
- висока запальна активність процесу;
- різкий больовий синдром і обмеження рухів у хребті;
- відсутність ефекту від НПЗП;
- стійкий торпедний перебіг хвороби;
- аортит.
У перелічених випадках пульс-терапію метилпреднізолоном проводять 2–3 раза в рік. В/в вводять 1000 мг метилпреднізолону упродовж 3 діб. Тривалість ефекту — близько 4 тиж.
Базисні протизапальні препарати
Сульфасалазин. Показаннями до його призначення є стійка висока активність АС із запаленням периферичних суглобів і ентезисів, недостатня ефективність НПЗП та симптоматичної терапії. Препарат достовірно усуває біль, скутість, але не впливає на функцію хребта. Під впливом сульфасалазину зменшується ШОЕ і знижується IgA, зменшуються прояви увеїту. Для оцінки ефективності застосовують в дозі 2–3 г/добу протягом 3–4 міс. За наявності ефекту використовують довгостроково у підтримувальній дозі 1,5 г/добу.
Метотрексат. Впливає на симптоми периферичного артриту, але не впливає на стан хребта.
Блокатори фактора некрозу пухлини
Рекомендації ASAS
Хворим, у яких достовірний діагноз виставлено за модифікованими нью-йоркськими критеріями, з центральною чи периферичною формою АС, які приймали як мінімум два НПЗП протягом 3 міс, сульфасалазин в дозі 2–3 г/добу, у яких проводили локальну терапію ГК (за показаннями) і зберігається висока активність АС (BASDAI > 4), а також прискорена ШОЕ, запалення за даними МРТ, рентгенологічне прогресування, — можливе призначення інгібіторів ФНП-α.
Під впливом цих препаратів достовірно зменшується вираженість ранішньої скутості, болю в спині, слабкості, периферичного артриту, знижуються гострофазові показники, покращується якість життя, уповільнюється прогресування змін в хребті, прояви увеїту, ентезитів.
Ефективність препаратів — блокаторів ФНП висока і досягає майже 80%. Деякі хворі відзначають покращання з наступного дня після введення препаратів, хоч очікуваний ефект настає на 6-му тижні лікування. Стійкий ефект спостерігається при довготривалому лікуванні (до 7 років спостережень). Під впливом лікування знижується частота рецидивів увеїту, зменшується вираженість ентезитів, артритів, уповільнюється прогресування ураження хребта та крижово-клубових суглобів.
У комплексну терапію АС включають також препарати системної ензимотерапії.
Для зняття м’язового спазму призначають центральні міорелаксанти, це дозволяє зменшити напруження м’язів і запобігає деформаціям хребта.
Хірургічне лікування
Показання до хірургічного лікування при АС представлені в табл. 24.7.
Таблиця 24.7
Показання до хірургічного лікування при АС
|
Характер операції |
Показания |
|
Протезування тазостегнових суглобів |
Сильний біль і обмеження рухів |
|
Остеотомія хребта |
Виражений кіфоз |
|
Протезування аортального клапана, встановлення кардіостимулятора |
Аортальна вада Повна АВ-блокада |
Ускладнення АС:
- переломи хребта (особливо шийного відділу) синдром «кінського хвоста»;
- прогресуючий фіброз верхівок легень;
- виникнення аортальної недостатності, АВ-блокад серця (у тому числі повна);
- амілоїдоз нирок;
- запалення передміхурової залози.
На жаль, повного виліковування досягнути неможливо.
Прогноз
У більшості випадків виражених функціональних порушень немає, працездатність збережена. Тривалість життя хворих зменшується при тяжкому варіанті хвороби, на що вказують постійно висока ШОЕ, анкілоз шийного відділу хребта, ураження тазостегнових суглобів, увеїт, фіброз легень.
Смертність хворих АС становить 5%. Головні причини смерті хворих — це травма хребта, серцева (аортальна недостатність), дихальна і ниркова (амілоїдоз) недостатність. У більшості пацієнтів працездатність збережена, незважаючи на прогресуючий характер хвороби. Тяжка інвалідність розвивається у 20% пацієнтів через 20–40 років хвороби.
Псоріатична артропатія
ПсА — асоційована з псоріазом хронічна запальна хвороба суглобів, хребта і ентезисів. Вона належить до групи серонегативних спондилоартропатій.
Код за МКХ-10
М07 Псоріатичні і ентеропатичні артропатії.
М07.0 Дистальна міжфалангова псоріатична артропатія (L40.5).
М07.1 Мутилюючий артрит (L40.5).
М07.2 Псоріатичний спондиліт (L40.5).
М07.3 Інші псоріатичні артропатії (L40.5).
Епідеміологія
ПсА розвивається у 7–39% хворих на псоріаз, які становлять 2–3% популяції. Захворюваність на ПсА становить 3,6–6,0 на 100 тис. населення, поширеність — 0,05–1%. Хвороба розвивається у віці 25–55 років однаково часто у чоловіків і жінок. Ураження суглобів у середньому виникає через 10 років після появи шкірних ознак псоріазу. У 75% випадків шкірні прояви передують суглобовим, в 10% вони починаються одночасно, в 15–20% — ураження суглобів випереджає (іноді на багато років) прояви псоріазу (Бурдейний А.П., 1997). Кореляції між характером, вираженістю, особливостями перебігу псоріазу і ПсА в більшості випадків немає.
Етіологія і патогенез
Етіологія ПсА не з’ясована. Вважається, що захворювання є наслідком взаємодії внутрішніх (генетичних, імунологічних) і зовнішніх чинників. Спостерігається спадкова схильність до розвитку псоріазу і ПсА більше ніж у 40% пацієнтів. Ідентифіковано 7 генів, відповідальних за розвиток псоріазу. При ПсА з підвищеною частотою виявляють гени головного комплексу гістосумісності HLA: B13, B17, B27, B38, DR4, DR7. При цьому HLA-B27 частіше визначається у пацієнтів з ПсА і рентгенологічними ознаками сакроілеїту, HLA-DR4 — при поліартикулярній, ерозивній формі ПсА. У розвитку псоріазу і ПсА важливу роль відіграють порушення Т-клітинного імунітету, високий рівень ФНП-α та інших прозапальних цитокінів (ІЛ-1, -6, -8), гранулоцитарно-макрофагального колонієстимулюючого фактора. Зокрема, такі клінічні прояви, як лихоманка, запалення ентезисів, деструктивні зміни в суглобах, ішемічний некроз, пов’язують з високою концентрацією ФНП-α. Існує прямий корелятивний зв’язок між рівнями ФНП-α, матриксної металопротеїнази 1-го типу, маркерів деградації хряща. У патогенезі ПсА беруть участь клітини CD8+/CD4+. Нині уточнюються причини посилення ремоделювання кісткової тканини при ПсА, а також механізми розвитку періоститів і анкілозів.
Класифікація
Виділяють 5 клінічних форм ПсА:
1. Асиметричний олігоартрит
2. Артрит дистальних міжфалангових суглобів
3. Симетричний ревматоїдоподібний синдром
4. Мутилюючий артрит
5. Псоріатичний спондиліт
Е. Фаучі та співавтори (2005) виділяють асиметричну, симетричну, аксіальну й інші форми ПсА. Їх характеристику наведено в табл. 24.8.
Таблиця 24.8
Форми ПсА (за: Фаучі Е. та співавт., 2005)
|
Форми ПсА |
Характеристика |
|
Асиметричний артрит (47%) |
Виникає через декілька років після появи псоріазу. Чоловіки і жінки хворіють однаково часто. Клінічній маніфестації артриту передує скутість. Характерні оніходистрофія, дактиліт (запалення проксимальних і дистальних міжфалангових суглобів), кульшові, колінні, гомілковостопні, а також скронево-нижньощелепні суглоби уражуються значно рідше. Залучаються суглоби-виключення: міжфалангові суглоби I пальця і проксимальний міжфаланговий суглоб V пальця кисті. Через тендовагініт уражені пальці набувають сосископодібного вигляду (це характерніше для пальців стоп). Шкіра над ураженими суглобами набуває багряно-синюшного забарвлення. Суглобовий біль виражений незначною мірою. Зрідка відзначають ізольований моноартрит променезап’ясткових суглобів з деструкцією. Між проявами оніходистрофії і перебігом артриту залежності немає. Деструкція суглобів виникає у ¼ хворих, ураження очей — у ⅓. Необхідно зазначити, що можливий не лише асиметричний моно-, олігоартрит, але й поліартрит, який переноситься більш тяжко |
|
Симетричний артрит (25%) |
Частіше (у 2 рази) виникає у жінок одночасно з псоріазом і за клінічним перебігом нагадує РА. На відміну від РА, деформація пальців хаотична з різною направленістю осей пальців. Мало характерні деформації за типом «бутон’єрки» і «шиї лебедя», але може розвинутися типова ліктьова девіація. Нерідко розвиваються згинальні контрактури, зумовлені фіброзом суглобових капсул. Спостерігається ранкова скутість, уражуються п’ястково-фалангові, дистальні і проксимальні міжфалангові суглоби, плеснофалангові, груднино-ключичні, великі суглоби нижніх кінцівок. У 25% визначається РФ, у 50% має місце деструктивний (мутилюючий) процес. Ураження очей відзначають рідко |
|
Аксіальна форма (23%) |
Частіше виникає через декілька років з моменту появи псоріазу. Ця форма може бути ізольованою (спондиліт/сакроілеїт), а також може поєднуватися з периферичним артритом. Відзначають біль у попереку і ранкову скутість, при периферичному ураженні можливий мутилюючий процес. Розвиваються ентезопатії, у тому числі уражується ахіллове сухожилля, і підошовний апоневроз, що супроводжується болем у п’яті. Оніходистрофія відзначається у багатьох пацієнтів, очі уражуються рідко. У 30% відзначається запалення кишечнику |
|
Інші форми ПсА |
ПсА з переважним ураженням дистальних міжфалангових суглобів: вважається найбільш типовим, але ізольовано зустрічається рідко. Часто поєднується з ураженням інших суглобів. Зрідка артрит міжфалангових суглобів поєднується з ураженням нігтів |
|
Мутилюючий (що спотворює) артрит: проявляється деструктивним артритом в основному пальців кистей і стоп. Остеоліз призводить до укорочення пальців, їх деформації («телескопічний палець») або деформації кисті («рука з лорнетом»). Функція кінцівки порушується через патологічну рухливість фаланг. Мутилюючий артрит розвивається у разі раннього початку хвороби, наявності вираженого шкірного синдрому (пустульозного псоріазу, еритродермії). У більшості випадків він поєднується з ураженням хребта |
|
|
Ювенільний ПсА |
|
|
Синдром SAPHO (Synovitis — артрит, Acne — вугри, Pustulosis — пустульозні висипання, Hyperostosis — гіперостоз, Osteomyelitis — остеомієліт) |
Класифікацію ПсА (Бадокин В.В., 1995) представлено в табл. 24.9.
Таблиця 24.9
Класифікація ПсА (за: Бадокин В.В., 1995)
|
1. Клінічна форма |
1. Тяжка 2. Звичайна 3. Злоякісна 4. ПсА у поєднанні із системними захворюваннями сполучної тканини, ревматизмом, хворобою Рейтера, подагрою |
|
|
2. Клініко-анатомічний варіант суглобового синдрому |
1. Дистальний 2. Моноолігоартритичний 3. Поліартритичний 4. Остеолітичний 5. Спондилоартритичний |
|
|
3. Системні прояви |
А. Без системних проявів |
|
|
Б. З системними проявами: трофічними порушеннями, генералізованою аміотрофією, поліаденією, кардитом, пороком серця, неспецифічним реактивним гепатитом, цирозом печінки, амілоїдозом внутрішніх органів, шкіри і суглобів, дифузним гломерулонефритом, ураженням очей, неспецифічним уретритом, поліневритом, синдромом Рейно та ін. |
||
|
4. Характер і стадія псоріазу |
А. Характер |
1. Вульгарний: вогнищевий, поширений 2. Ексудативний 3. Атиповий: пустульозний, еритродермічний, рупіоїдний (з особливо вираженим гіперкератозом) |
|
Б. Стадія |
1. Прогресування 2. Стаціонарна 3. Регресії |
|
|
В. |
Псоріаз нігтів |
|
|
5. Фаза і ступінь активності |
А. Активна |
1. Мінімальна 2. Помірна 3. Максимальна |
|
Б. Ремісія |
||
|
6. Рентгенологічна характеристика |
А. Периферичні та кореневі суглоби |
I. Навколосуглобовий ОП IIA. Те саме + звуження суглобової щілини, кістоподібні просвітлення кісткової тканини IIБ. Те саме + одиничні поверхневі узури III. Те саме + множинні узури IV. Те саме + кісткові анкілози |
|
Б. Крижово-клубові суглоби |
I. Нечіткість суглобової щілини, слабковиражений ОП II. Звуження або розширення суглобової щілини, субхондральний остеосклероз III. Те саме + часткове анкілозування IV. Те саме + повне анкілозування |
|
|
В. АС з: |
а) синдесмофітами або параспінальними осифікатами б) анкілозами міжхребцевих суглобів |
|
|
7.Функціональная здатність суглобів |
А. Збережена |
|
|
Б. Порушена |
II. Професійна здатність втрачена |
|
|
III. Втрачена здатність до самообслуговування |
||
Міжнародна класифікація ПсА досі не розроблена.
Клінічна картина
Як правило, ПсА починається поступово, зрідка — гостро. У сумнівних випадках потрібний ретельний пошук шкірних проявів, у тому числі огляд шкіри волосистої частини голови (рис. 24.6). Можливі варіанти дебюту ПсА наведено в табл. 24.10.
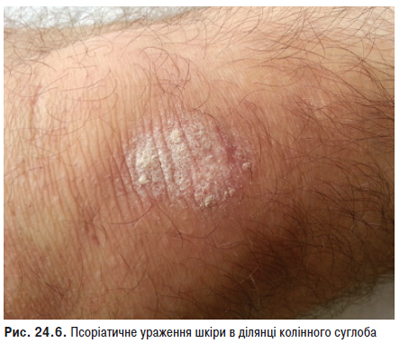
Таблиця 24.10
Можливі варіанти дебюту ПсА
|
Поступовий початок |
Артрит розвивається повільно. Можуть відзначатися продромальні явища — підвищена стомлюваність, порушення самопочуття, сну, субфебрилітет, зменшення маси тіла, міалгії, арталгії |
|
Початок з артралгій |
У 20% хворих тривало, іноді роками, відзначається суглобовий біль, які змінюється суглобовим синдромом. У деяких хворих суглобовий синдром так і не розвивається |
|
Початок з тендовагініту |
Тендовагініти згиначів пальців часто бувають першими симптомами ПсА |
|
Гострий початок |
Симптоми з’являються несподівано без явної причини і нагадують напад подагричного артриту або септичний артрит |
Характерним для ПсА є ураження дистальних міжфалангових суглобів. При цьому часто уражуються нігті. Клінічні прояви, властиві ПсА, описано в табл. 24.11.
Таблиця 24.11
Клінічні прояви, характерні для ПсА
|
Органи і системи |
Характерні прояви |
|
|
Очі |
Частіше розвивається кон’юнктивіт (у ⅓ хворих), рідше — увеїт |
|
|
Слизові оболонки |
Характерним є розвиток стоматиту, глоситу |
|
|
Лімфатична система |
Відзначається генералізована лімфаденопатія |
|
|
М’язова система |
Має місце прогресуюча атрофія м’язів |
|
|
Легені |
Зрідка розвивається легеневий фіброз |
|
|
Серцево-судинна система |
Можливий розвиток аортальної недостатності, міокардиту |
|
|
ШКТ |
Розвивається ураження печінки |
|
|
Нирки |
Виявляють ураження нирок |
|
|
Суглоби |
Асиметричний моно-, олігоартрит великих суглобів |
Поєднується з ураженням проксимальних і дистальних міжфалангових суглобів (1–2), дактилітом кистей або стоп (що підтверджує псоріатичне походження артриту) |
|
Симетричний поліартрит |
Уражуються дрібні суглоби кистей і стоп, променезап’ясткові, колінні і плечові суглоби. До процесу залучаються дистальні міжфалангові суглоби, розвиваються анкілози як дистальних, так і проксимальних міжфалангових суглобів. Симетричний артрит без перелічених уражень на тлі псоріазу за наявності дуже високих титрів РФ можна розцінювати як поєднання псоріазу і РА |
|
|
Мутилюючий артрит |
Характерний, але досить рідкісний (менше 5% випадків) варіант ПсА. Проявляється остеолізом кінцевих фаланг і голівок п’ясткових кісток. Наслідком остеолізу є «телескопічна» деформація пальців і кисті («рука з лорнетом») |
|
|
Спондиліт і сакроілеїт і/або артрит кульшових і плечових суглобів (псевдоанкілозуючий спондиліт) |
Спондиліт розвивається рідко — в 5% випадків. Поєднання з ураженням крижово-клубових або корінних суглобів нагадує АС. Але біль у спині відсутній (незважаючи на виражені рентгенологічні зміни) і сакроілеїт (є у ⅓ хворих) має асиметричний характер |
|
|
Ентезиси |
Ентезити розвиваються досить часто. Найчастіше уражується ахіллове сухожилля і розгиначі пальців |
|
При ПсА уражуються груднино-ключичні, акроміально-ключичні суглоби, відзначається синхондроз рукоятки грудини, м’язово-фасціальний біль, тендовагініти, ентезопатії. Часто відмічають ахіллобурсит, підп’ятковий бурсит, трохантерит. Ахіллобурсит може призвести до локального ОП кістки п’яти. Ураження очей, аорти і легень у вигляді легеневого фіброзу, аортальної недостатності та увеїту (іридоцикліту, епісклериту) розвиваються рідко, кон’юнктивіт — у ⅓ хворих. При будь-якій формі ПсА можливий розвиток спондиліту, ураження дистальних міжфалангових суглобів і також мутилюючого артриту. Остеоліз зустрічається при різних варіантах ПсА. Особливості, властиві ПсА:
- асиметричність ураження, у тому числі кісткового анкілозування;
- ураження дистальних міжфалангових суглобів;
- відсутність навколосуглобового ОП;
- чашоподібна деформація проксимальної частини фаланг (симптом «олівця в ковпачку»);
- остеоліз п’ясткових кісток, акроостеоліз;
- сакроілеїт (частіше однобічний).
Слід зазначити, що при ПсА можливе ураження будь-яких суглобів, у тому числі скронево-нижньощелепних. Найчастіше відзначають моно- або олігоартикулярне ураження: виникає артрит проксимальних і дистальних суглобів кистей, колінних суглобів з болем запального характеру, скутістю, вираженою в основному у ранкові години. Відносно рідко уражуються п’ястково- і плеснофалангові, плечові суглоби. ПсА характеризується стійким запальним процесом, ураженням усіх елементів суглоба і періартикулярних тканин. При гострому запаленні суглобів підвищується температура шкіри, змінюється її колір. Шкіра стає багряною з синюшним відтінком через наявність гіперемії і застою. Спостерігається гіперемія шкіри кінчиків пальців рук і стоп, дистрофічні зміни нігтьових фаланг. Шкіра пальців набуває рожевого блискучого вигляду, вона натягнута і гладка, як при склеродермії. При характеристиці ПсА застосовують терміни «пальці-сардельки» і «осьове ураження». «Пальці-сардельки» — це позначення набряклих (набряк дифузного характеру) пальців кистей або стоп внаслідок ураження міжфалангових суглобів, сухожильних піхв і м’яких тканин. Термін «осьове ураження» означає одночасне ураження дистального і проксимального міжфалангового і п’ястково-фалангового суглоба одного і того ж пальця. Термін «дактиліт» відображає характерний для ПсА артрит і тендосиновіт дистальних і проксимальних міжфалангових суглобів.
У пацієнтів з ПсА виникають кісткові і фіброзні зміни в ділянці кісток п’ят, що супроводжуються болем.
Відмітні особливості псоріатичного спондиліту. Перебіг псоріатичного спондиліту може бути безсимптомним, хворобу виявляють випадково при рентгенологічному обстеженні з іншого приводу. Ізольоване ураження осьового скелета може нічим не відрізнятися від хвороби Бехтєрева. Спочатку виникає біль запального характеру в поперековому відділі, потім послідовно уражується грудний, шийний відділ, реберно-хребцеві суглоби і формується «поза жебрака» (у таких випадках часто виявляють HLA-В27). Проте процес може початися в будь-якому відділі та мати однобічний характер. Сакроілеїт також може бути однобічним. Рентгенологічні зміни в хребті при ПсА можуть нагадувати «бамбукову палицю», але хребці не мають квадратної форми, оскільки паравертебральні осифікати не пов’язані з хребцями.
Особливості злоякісної форми ПсА:
- виявляють у чоловіків молодого віку (до 35 років) з пустульозним псоріазом, еритродермією;
- відмічають множинне ураження суглобів (генералізований поліартрит) з вираженим синовітом, больовим синдромом, фіброзними анкілозами, а також спондиліт;
- відзначають лихоманку гектичного типу, генералізовану лімфаденопатію, ураження слизових оболонок, ЦНС, внутрішніх органів;
- підвищується ШОЕ до 60 мм/год і більше;
- у крові відзначають зниження вмісту альбуміну, збільшення γ-глобулінів;
- розвивається виражена анемія;
- реєструють швидку втрата маси тіла, утворення трофічних виразок, пролежнів;
- посилене випадіння волосся;
- розвивається м’язова атрофія;
- характерним є розвиток міокардиту, збільшення розмірів серця, поява тахікардії, аритмій;
- виникає гепатит, гепатолієнальний синдром, амілоїдоз внутрішніх органів;
- внаслідок ураження зв’язково-м’язового апарату і остеолізу розвиваються підвивихи;
- відзначається долонна і підошовна кератодермія, баланіт.
Особливості перебігу ПсА у дітей:
- ПсА розвивається у віці 9–12 років, переважно у дівчаток (у 2 рази частіше);
- дебют ПсА у ⅓ дітей нагадує напад гострого подагричного артриту;
- у 50% ПсА передує псоріазу;
- у більшості випадків ПсА починається з моно-, олігоартриту суглобів кистей і надалі поширюється на інші суглоби;
- не супроводжується лімфаденопатією, гепатоспленомегалією, лихоманкою;
- явища остеолізу відзначають дуже рідко;
- перебіг сприятливий, стійка ремісія досягається у 20–50% дітей;
- хронічний поліартрит відзначається у 25% дітей, інвалідизація — у 10%.
У ⅓ хворих дітей діагностують моно- або олігоартрит (ураження 1–3 суглобів). Перебіг більш доброякісний, ніж у дорослих, проте не виключається і ураження внутрішніх органів.
Діагностика
Класифікаційні критерії ПсА за Vasey і Espinosa (1984):
1. Асиметричне ураження суглобів.
2. Ураження дистальних міжфалангових суглобів кистей і стоп.
3. Наявність симетричного ураження суглобів за відсутності РФ і підшкірних вузликів.
4. Виявлення при рентгенографії ерозивного ураження периферичних суглобів за наявності мінімальної остеопенії; поява ерозій і анкілозу дистальних міжфалангових суглобів.
5. Скутість і біль у спині протягом більше 4 тиж.
6. Рентгенологічне виявлення двобічного сакроілеїту II стадії або однобічного сакроілеїту III–IV стадії.
Ураження шкіри і нігтів, характерне для псоріазу, поєднується з однією з перелічених вище ознак.
Діагностичні критерії ПсА за Mathies:
1. Ураження дистальних міжфалангових суглобів кистей, зокрема великих пальців стоп. Суглоби болісні, припухлі, шкіра над ними синюшна або багряно-синюшна.
2. Одночасне ураження п’ястково-фалангового або плеснофалангового, проксимального і дистального міжфалангового суглобів одного і того ж пальця («палець-сосиска»).
3. Раннє ураження великого пальця стопи.
4. Талалгія (біль у п’ятах).
5. Рентгенологічні прояви: остеоліз з різноосьовими зміщеннями кісток, періостальні накладення, відсутність навколосуглобового ОП.
6. Рентгенологічні ознаки паравертебральної осифікації.
7. Клінічні або рентгенологічні ознаки сакроілеїту.
Представлено також діагностичні критерії Інституту ревматології РАМН (1989) (табл. 24.12).
Таблиця 24.12
Діагностичні критерії ПсА Інституту ревматології РАМН (1989)
|
№ з/п |
Критерії |
Бали |
|
1 |
Псоріатичні висипання на шкірі |
5 |
|
Псоріаз нігтьових пластинок |
2 |
|
|
Псоріаз у близьких родичів |
1 |
|
|
2 |
Артрит дистальних міжфалангових суглобів |
5 |
|
3 |
Артрит 3 суглобів одного пальця |
5 |
|
4 |
Асиметричний артрит |
2 |
|
5 |
Типові параартикулярні явища |
5 |
|
6 |
«Сосископодібна» дефігурація пальців стоп |
3 |
|
7 |
Різноспрямовані підвивихи суглобів пальців кистей |
4 |
|
8 |
Біль і ранкова скутість в хребті |
1 |
|
9 |
Остеоліз в ділянці суглобів |
5 |
|
10 |
Анкілоз дистальних міжфалангових (кисті, стопи) і плеснофалангових суглобів |
5 |
|
11 |
Рентгенологічні ознаки певного сакроілеїту |
2 |
|
12 |
Синдесмофіти або паравертебральні осифікати |
4 |
|
13 |
Серонегативність за РФ |
2 |
|
14 |
Зв’язок посилення шкірних проявів із загостренням суглобового синдрому або його появою |
4 |
|
Категорія діагнозу відповідно до суми балів |
||
|
ПсА класичний — 16 балів і більше ПсА визначений — 11–15 ПсА вірогідний — 8–10 ПсА відкидається — 7 балів і менше |
||
Примітки: за наявності серопозитивності за РФ, ревматоїдних вузликів, тофусів, тісного взаємозв’язку появи суглобового синдрому або його загострення з урогенітальною або кишковою інфекцією, відсутності псоріазу у момент обстеження або в анамнезі від загальної суми віднімають по 5 балів на кожну ознаку.
У табл. 24.13 наведено критерії ПсА CASPAR, 2006 (Насонов Є.Л., 2011 (ред.)).
Таблиця 24.13
Критерії ПсА (CASPAR, 2006)
|
1 |
Псоріаз:
|
Бали 2 1 1 |
|
2 |
Псоріатичная дистрофія нігтів:
|
1 |
|
3 |
Негативний РФ (окрім латекс-тесту) |
1 |
|
4 |
Дактиліт:
|
1 1 |
|
5 |
Рентгенологічні ознаки позасуглобової кісткової проліферації за типом крайових розростань (окрім остеофітів) на рентгенограмах кистей та ступнів |
1 |
Примітка: щоб відповідати критеріям CASPAR, пацієнти повинні мати ознаки запалення суглобів (артрит, спондиліт або ентезит) та 3 або більше балів із наведених 5 категорій.
Ювенільний ПсА входить до групи ювенільного спондилоартриту. Він, як і інші ювенільні артрити, може тривалий час проявлятися неспецифічною клінічною картиною, яку важко відрізнити від інших ювенільних спондилоартритів і ювенільного хронічного артриту. У таких випадках використовують класифікаційні критерії Ванкувера ювенільного ПсА (табл. 24.14).
Таблиця 24.14
Діагностичні критерії Ванкувера ювенільного ПсА (1989)
|
Певний ювенільний ПсА: |
|
Артрит і типовий псоріатичний висип, або |
|
Артрит і наявність хоча б 3 з наступних «малих» ознак: |
|
а) зміни нігтів (синдром «наперстка», оніхолізис); |
|
б) псоріаз у родичів 1-го або 2-го ступеня спорідненості; |
|
в) псоріазоподібний висип; |
|
г) дактиліт |
|
Вірогідний ювенільний ПсА: |
|
Артрит + хоч б 2 з «малих» ознак |
Зміни, що виявляють при лабораторних та інструментальних дослідженнях у хворих на ПсА, наведено в табл. 24.15.
Таблиця 24.15
Зміни, що виявляються при лабораторному та інструментальному обстеженні хворих на ПсА
|
Лабораторні дослідження |
|
|
Аналіз крові |
Характерним є підвищення ШОЕ, лейкоцитоз, еозинофілія, анемія (гіпохромна) |
|
Імунологічні дослідження |
У 15% пацієнтів з ПсА (а також при псоріазі без артриту) виявляють підвищення титрів РФ. Також підвищується рівень IgА |
|
Біохімічні дослідження |
Нерідко відзначають підвищення в крові рівня ліпідів і сечової кислоти. Клінічні прояви вторинної подагри спостерігаються рідко (у синовіальній рідині кристали сечової кислоти не виявляються). У крові також збільшується концентрація СРБ, фібриногену, серомукоїду, активність комплементу |
|
Інструментальні дослідження |
|
|
ЕхоКГ |
Зміни аналогічні тим, які реєструють при АС |
|
Рентгенографія суглобів |
Виявляється симетричність ураження (асиметричність кісткових анкілозів), відсутність навколосуглобового ОП, ураження дистальних міжфалангових суглобів, остеоліз, чашкоподібна деформація проксимальної частини фаланг («олівець у ковпачку»), однобічний або двобічний асиметричний сакроілеїт. Зміни хребта можуть іноді нагадувати «бамбукову палицю». У таких випадках необхідно виключити поєднання псоріазу і АС |
Рентгенологічні зміни, які відзначають в кістково-суглобовій системі при ПсА, описано в табл. 24.16.
Таблиця 24.16
Рентгенологічні зміни, які відзначають при ПсА в кістково-суглобовій системі
|
Суглоби кистей і стоп |
Виявляють субхондральний ОП, склероз, кісти, узурацію суглобових поверхонь (результат прориву кіст), анкілозування суглобів. Зазубрена кортикальна частина епіфізу плеснових кісток, остеоліз епіфізів |
|
Крижово-клубові суглоби |
Реєструють ознаки однобічного (у 50% пацієнтів) або двобічного (у ⅓ хворих) сакроілеїту. Поєднуються ділянки остеолізу і ОП (переважають склеротичні зміни), контури суглобових поверхонь нерівні. Клінічні ознаки цих уражень можуть бути відсутніми |
|
Хребет |
Уражується кісткова тканина і зв’язковий апарат, меншою мірою — хрящова тканина. Виявляють паравертебральне окостеніння, яке утворює місток над міжхребцевим простором. У міжхребцевих дисках виникають вогнища склерозу, форма дисків і хребців змінюється. Розвивається деструкція передньоверхніх і передньонижніх кутів тіл хребців. Змінюється висота міжхребцевих дисків, з’являється склерозування покривних пластинок і остеофіти. У поперековому відділі хребта виявляють масивні остеофіти або синдесмофіти, які часто розташовані з одного боку і на відстані від міжхребцевих дисків |
Найбільш характерні для ПсА рентгенологічні зміни:
- набряк м’яких тканин;
- звуження суглобової щілини;
- субхондральні кісти;
- підвивихи;
- кісткові анкілози пальців;
- симптом «ручки з ковпачком» (розширення основи дистальної фаланги з ерозією суглобової поверхні, входження в утворене заглиблення конусоподібної голівки середньої фаланги), тобто симптом «телескопічного пальця» (входження однієї кістки в іншу);
- розростання кісткової тканини навколо ерозій;
- остеоліз дистальних фаланг;
- однобічний сакроілеїт;
- окостеніння м’яких тканин навколо хребта (у тому числі в шийному відділі);
- асиметричні некрайові синдесмофіти.
Характер морфологічних змін в синовіальній оболонці і суглобових хрящах при ПсА. У біоптатах синовії виявляють ознаки васкуліту, крововиливи, вогнища мукоїдного набрякання, фіброзні зміни, дистрофію і некроз синовіоцитів. Хронічне запалення супроводжується ворсинчастою гіперплазією синовіальної оболонки, склерозуванням стінок судин, периваскулярною інфільтрацією лімфоцитами і плазматичними клітинами. У поверхневих і середніх шарах суглобового хряща зменшується кількість хондроцитів і глікозаміногліканів (табл. 24.17).
Таблиця 24.17
Морфологічні зміни, які виявляють в різних тканинах при ПсА
|
Синовіальна оболонка |
Відзначають проліферацію синовіоцитів, інфільтрація нейтрофілами (у I фазу хвороби) і лімфоцитами. В оболонці збільшується кількість судин, зменшується — макрофагів і експресія Е-селектину. Поступово збільшується вираженість фіброзу оболонки |
|
Хрящ |
Розвиваються ерозії хряща, поверх нього розростається синовіальна оболонка |
|
Шкіра |
У вогнищах переважають лімфоцити CD45RO |
|
Кістковий мозок |
Розвивається його фіброз |
Диференційний діагноз
Необхідно проводити диференціацію із хворобами, які супроводжуються подібними ураженнями шкіри, нігтів, суглобів (табл. 24.18).
Таблиця 24.18
Захворювання, які супроводжуються ураженнями шкіри нігтів, суглобів, подібними до ПсА
|
Ураження шкіри |
Необхідно диференціювати псоріаз зі сверблячими дерматозами, зокрема з себорейним дерматитом. Треба шукати бляшки на шкірі волосистої частини голови, в пупі, сідничних складках |
|
Ураження нігтів |
Слід відрізняти оніходистрофію від оніхомікозів. Для оніходистрофії є характерним симптом наперстка і оніхоліз |
|
Ураження суглобів |
Виявлення у хворого з псоріазом вузликів Гебердена і Бушара свідчить про наявність ОА, а кристалів урату натрію в синовіальній рідині — про подагру. Носійство HLA-B27, псоріаз, оніходистрофія, ентезопатії, асиметричний артрит, дактиліт при виявленні РФ — аргументи проти РА |
|
Дактиліт |
Необхідно враховувати, що він наявний і при синдромі Рейтера та поєднується з бленорейною кератодермією, кон’юнктивітом, уретритом |
У тих випадках, коли у хворих з псоріазом відсутнє ураження дистальних міжфалангових суглобів і є серопозитивність за РФ, слід думати про поєднання псоріазу і РА. Можливе поєднання псоріазу і АС. Його підозрюють в тих випадках, коли у HLA-В27-позитивних пацієнтів з псоріазом визначається швидке прогресування спондиліту, сакроілеїту, розвиток іриту, периферичного артриту. Розвиток у хворих на ПсА вторинної подагри реєструють рідко, хоча гіперурикемія — досить часте явище при ПсА.
Приклад формулювання діагнозу. ПсА, тяжка форма, поліартритний варіант, активність III ступеня, рентгенологічна стадія III, недостатність функції суглобів II ступеня, амілоїдоз нирок, хронічна ниркова недостатність III ступеня, поширений вульгарний псоріаз.
Лікування
Показання до госпіталізації в стаціонар:
- злоякісна форма з тяжкими системними проявами;
- висока активність патологічного процесу, яка зберігається протягом ≥3 міс;
- генералізоване ураження суглобів з ексудацією;
- швидке прогресування хвороби;
- відсутність ефекту від терапії НПЗП, а також базисними і протизапальними препаратами;
- виникнення побічних реакцій при прийомі базисних препаратів.
Сучасні принципи медикаментозної терапії ПсА наведено (за: Насонов Е.Л., 2006; 2011 (ред.)) в табл. 24.19.
Таблиця 24.19
Медикаментозна терапія ПсА
|
Групи препаратів |
Характер терапії |
||
|
НПЗП |
Призначають (диклофенак, індометацин, селективні НПЗП, еторикоксиб) при олігоартритах, ураженні дистальних міжфалангових суглобів. За відсутності ефекту протягом 2–3 тиж застосовують БПЗП. Необхідно враховувати, що при прийомі НПЗП можливе загострення шкірного псоріазу. НПЗП зменшують вираженість болю, скутість, кількість болісних та припухлих суглобів |
||
|
ГК |
Системне лікування |
10–15 мг/добу преднізолону призначають у разі високої активності процесу, наявності системних проявів, периферичного артриту з вираженою функціональною недостатністю, при неефективності НПЗП. При використанні ГК має місце ризик загострення псоріазу. Відсутні докази ефективності ГК, які базувалися б на результатах контрольованих досліджень. У режимі постійного прийому ГК не застосовують |
|
|
Локальна терапія |
Проводять при ентезопатії, моно- і олігоартриті, ураженні крижово-клубових з’єднань у поєднанні з НПЗП та БПЗП (бетаметазон 0,25–2 мл внутрішньосуглобово або періартикулярно в ділянку ураженого суглоба не частіше 1 разу на 1–3 міс в той самий суглоб залежно від дози та розміру суглоба) |
||
|
БПЗП |
Метотрексат |
Є препаратом вибору, показаний при: 1) високій активності патологічного процесу; 2) поєднанні артриту з поширеним псоріазом в прогресуючій стадії; 3) пустульозному і еритродермічному атиповому дерматозі. В плацебо-контрольованих дослідженнях підтверджена ефективність метотрексату у вигляді в/в пульс-терапії (1–3 мг/кг) та прийому усередину (7,5–15 мг/тиж). Метотрексат не гальмує розвиток рентгенологічних ознак деструкції суглобів як у режимі монотерапії, так і в складі комбінації метотрексат + циклоспорин. В цілому він є більш ефективним, ніж циклоспорин (3–5 мг/кг/добу). Перед початком терапії метотрексатом слід виконати тест на наявність вірусів гепатиту В та С |
|
|
Циклоспорин |
У дозі 3–5 мг/кг/добу позитивно впливає на клінічні прояви артриту та псоріазу, сповільнює прогресування рентгенологічних ознак деструкції суглобів. Викликає побічні реакції (нефротоксичність, артеріальна гіпертензія) і не має переваг перед метотрексатом. При лікуванні протягом 2 років слабо впливає на рентгенологічне прогресування |
||
|
Сульфасалазин |
Призначають в дозі 2 г/добу. Не впливає на стан ураженого хребта і прогресування артриту. Не гальмує розвиток рентгенологічних ознак деструкції суглобів. Помірний позитивний ефект відзначають при ураженні суглобів і шкіри. Високі дози препарату призводять до підвищення частоти побічних ефектів. Не має переваг перед циклоспорином та стандартною терапією (НПЗП + анальгетики + ГК) |
||
|
Солі золота |
Призначають при усіх варіантах ПсА, окрім ураження хребта і крижово-клубових з’єднань |
||
|
Амінохінолінові препарати |
При ПсА неефективні, можуть спровокувати загострення псоріазу, викликати ексфоліативний дерматит |
||
|
Азатіоприн і пеніциламін |
Призначають при неефективності перелічених вище препаратів |
||
|
Лефлуномід |
Офіційно рекомендований для лікування ПсА, ефективний при ураженні суглобів, сповільнює розвиток деструктивних змін. Ефективність підтверджена в міжнародному подвійному сліпому контрольованому дослідженні. Призначають усередину у дозі 100 мг/добу перші 3 дні, 20 мг/добу — в подальшому. Сповільнює рентгенологічне прогресування ПсА після 1 року лікування |
||
|
Інгібітори ФНП-α |
Призначають при постійно високій активності захворювання (кількість болісних суглобів — >3, припухлих — >3), гострому дактиліті, генералізованій ентезопатії, псоріатичному спондиліті. Ефективність підтверджена в багатоцентрових плацебо-контрольованих рандомізованих дослідженнях (IMPACT, IMPACT 2). Інфліксимаб в дозі 3–5 мг/кг вводять у вигляді монотерапії або в поєднанні з метотрексатом. Він ефективний відносно ураження шкіри і суглобів у випадках резистентності до інших базисних препаратів. Інфліксімаб затримує рентгенологічне прогресування, його добре переносять хворі. Адалімумаб (40 мг 1 раз на 2 тиж) також використовують у режимі монотерапії або в комбінації з метотрексатом. Він стримує рентгенологічне прогресування, покращує показники якості життя |
||
|
Ретиноїди |
На основні прояви ПсА чинять позитивний вплив (ацитретин), але їм властива тератогенна дія, гепатотоксичність, вони викликають сухість шкіри |
||
|
Лікування системних проявів ПсА |
|||
|
Пульс-терапія |
Рекомендується її проведення метотрексатом (100 мг) в комбінації з метилпреднізолоном (250 мг) |
||
Помітним терапевтичним ефектом володіють також хлорамбуцил, 6-меркаптопурин. Вони позитивно впливають на вираженість шкірного псоріазу.
Показання до системного застосування ГК при ПсА:
- злоякісний варіант перебігу хвороби;
- висока активність запального процесу (резистентність до НПЗП) протягом ≥3 міс;
- поширене ураження суглобів з наявністю вираженого ексудативного компонента;
- виражені вісцеральні ураження (кардіоміопатія, гепатит, дифузний гломерулонефрит, периферична невропатія);
- побічні ефекти метотрексату (прояви алергії, цитопенія, гепатопатія та ін.);
- пустульозний псоріаз, універсальна еритродермія.
ГК (преднізолон в дозі 30–40 мг/добу) застосовують в комбінації з метотрексатом (20–25 мг/тиж в/м або в/в) або циклоспорином. ГК достатньо ефективно усувають клінічну симптоматику, зумовлену запальним процесом, але меншою мірою впливають на шкірний синдром (пустульозний і еритродермічний варіанти). Тривалий (>3 міс) прийом ГК в дозі 5 мг/добу і більше вимагає призначення комбінованих препаратів кальцію (1000–1500 мг/добу) і вітаміну D3. Пульс-терапію метилпреднізолоном (без циклофосфаміду) можна застосовувати як доповнення до стандартної терапії при рецидивах і торпідному перебігу АС. У комплексне лікування при ПсА включають системну ензимотерапію.
Показаннями до локальної терапії ГК є:
- виражені прояви суглобового синдрому в обмеженій кількості уражених суглобів (при олігоартритичному варіанті);
- висока локальна активність, стійкий синовіт;
- дактиліт, талалгія, ентезити іншої локалізації;
- шкірний синдром.
Слід враховувати, що локальна терапія ГК може призвести до дестабілізації псоріазу, хоча це відзначають рідше, ніж при системному застосуванні. ГК сприяють переходу псоріазу в пустульозну або еритематозну форму.
Показання до призначення БПЗП хворим на ПсА. Їх призначають при:
- збереженні високої активності патологічного процесу протягом ≥3 міс;
- наявності ураження хребта і поширеного артриту;
- прогресуючій деструкції суглобів;
- поєднанні ураження хребта і/або суглобів з тяжкими формами псоріазу;
- злоякісній формі хвороби з багатьма системними ураженнями.
Поліпшення стану (у тому числі позитивна динаміка уражень шкіри) відзначається через 16 тиж лікування інгібіторами ФНП-α у більшості пацієнтів, які раніше отримували лікування БПЗП (дослідження IMPACT). Антагоністи ФНП-α виявляють позитивну дію як при монотерапії, так і при доповненні до терапії, що проводиться іншими БПЗП (метотрексатом). Вони гальмують рентгенологічне прогресування ПсА. В деяких випадках антагоністи ФНП-α можуть викликати появу вогнищ псоріатичного ураження шкіри або загострення вже наявного псоріазу. Лікування цими препаратами переривають у разі майбутньої хірургічної операції (у зв’язку з ризиком розвитку інфекції), при появі ознак вірусної або бактеріальної інфекції, розвитку застійної серцевої недостатності. Якщо на тлі лікування інгібіторами ФНП-α розвинулася інфекція, повторне призначення цих препаратів протипоказане. Лікування припиняють при вагітності. Найбільш тяжким ускладненням терапії інгібіторами ФНП-α є дисемінація туберкульозної інфекції.
Послідовність призначення лікарських препаратів при ПсА наведено в табл. 24.20.
Таблиця 24.20
Послідовність призначення лікарських препаратів при ПсА
|
ПсА |
НПЗП→БПЗП→ГК в/с→інгібітори ФНП-α |
|
Дактиліт |
НПЗП→ГК в/с, періартикулярно→інгібітори ФНП-α |
|
Ентезит |
НПЗП→ГК періартикулярно→інгібітори ФНП-α |
|
Псоріатичний спондилоартрит |
НПЗП→ГК в крижово-клубові з’єднання→пульс-терапія ГК→інгібітори ФНП-α |
|
Псоріаз шкіри та нігтів |
Стероїдні мазі→ПУВА-терапія→метотрексат (системно)→циклоспорин (системно)→інгібітор ФНП-α |
Інші лікувальні засоби. При лікуванні хворих на ПсА не застосовуються антималярійних препаратів, оскільки вони можуть спровокувати загострення псоріазу. При лікуванні шкірних проявів призначають похідні вітаміну А, мазі, що містять ГК (флуоцинолону ацетонід, тріамцинолону ацетонід). При злоякісному варіанті хвороби з системними проявами використовують гемосорбцію і плазмаферез. Проводять навчання хворих, призначають фізіотерапію, ЛФК, надають ортопедичну допомогу.
Ремісія захворювання виникає у 50% пацієнтів і в середньому може тривати близько 2 років. Критерії ремісії спондилоартритів (Насонов Є.Л., 2011 (ред.)) представлено в табл. 24.21.
Таблиця 24.21
Критерії ремісії спондилоартритів
|
Клінічні прояви |
Значення |
|
Стомлюваність |
Відсутня |
|
Біль в суглобах (включно із запальним болем у спині) |
Відсутній |
|
Тривалість уранішньої скутості (хв) |
≤15 |
|
Кількість болісних суглобів |
0 |
|
Кількість припухлих суглобів (дактиліт/ентезит) |
0 |
|
ШОЕ, мм/год: жінки чоловіки |
≤30 ≤20 |
Примітка: обов’язково мають бути присутніми 5 ознак із 6 протягом 2 послідовних місяців.
Показання до хірургічного лікування. При деструктивному ураженні колінних, кульшових, суглобів кистей та стоп відповідно проводять ендопротезування та реконструктивні операції. При стійких артритах колінних суглобів проводять артроскопічну або хірургічну синовектомію.
Прогноз
ПсА часто призводить до інвалідизації та скорочення тривалості життя. Деструкція суглобів і їх функціональна недостатність можуть розвинутися вже в 1-й рік хвороби. Амілоїдоз нирок і кардіоваскулярна патологія, що виникають, призводять до підвищення смертності (як чоловіків, так і жінок) порівняно з популяцією. До маркерів несприятливого прогнозу при ПсА відносять: 1) наявність тяжкого псоріазу; 2) початок захворювання у молодому віці; 3) поліартикулярний тип ураження; 4) носійство антигенів HLA-DR3 і -DR4. У цих випадках рекомендують проведення ранньої агресивної терапії.
Профілактика
Можлива тільки вторинна профілактика, яка полягає в: 1) проведенні адекватної терапії з використанням базисних засобів; 2) уповільненні прогресування патологічного процесу; 3) збереженні функції суглобів, проведенні реабілітації.
Реактивні артрити
РеА — запальні стерильні (негнійні) захворювання суглобів, які розвиваються після гострої кишкової або урогенітальної інфекції у генетично схильних осіб (носіїв HLA-B27) і належать до групи спондилоартритів. Якщо раніше вважалося, що РеА — це стерильне запальне захворювання, що виникає після певної інфекції в результаті імунних порушень, при цьому сам мікроорганізм і його антигени в порожнині суглоба не виявляються, то зараз у суглобових тканинах виявляють антигени мікробів (ієрсиній, сальмонел, хламідій) і мікроорганізми, здатні до розмноження на культурі тканин. Таким чином, межі між інфекційним і РеА розмиті, термін «реактивний артрит» використовують з обережністю.
У РеА і серонегативних спондилоартритів є спільні риси, які полягають у асоціації з носійством антигену HLA-B27, ураженні крижово-клубових з’єднань і хребта, очей, шкіри, слизових оболонок.
Код за МКХ-10
МО2 Реактивні артропатії
МО2.0 Артропатія, що супроводжує кишковий шунт
МО2.1 Постдизентерійна артропатія
МО2.2 Постімунізаційна артропатія
МО2.3 Хвороба Рейтера
МО2.8 Інші реактивні артропатії
МО2.9 Реактивна артропатія неуточнена
М03 Постінфекційні та реактивні артропатії при хворобах, класифікованих в інших рубриках
М03.2 Постінфекційна артропатія при ентериті, спричиненому Yersinia enterocolitica (А04.6)
Епідеміологія
Частота розвитку урогенітальних (індукованих Ch. trachomatis) артритів становить 4,6 випадку на 100 тис. населення, постентероколітичних — 5,0 на 100 населення (Насонов Е.Л., 2006 (ред.)). Хвороба розвивається переважно у віці 20–40 років, але РеА можуть виникати у дітей і літніх осіб. На ентерогенний РеА однаково часто хворіють чоловіки і жінки, на урогенітальний — в основному чоловіки (відношення чоловіків і жінок становить 6–25:1).
Етіологія і патогенез
Різні мікроби виявляють тригерну дію і викликають однотипну картину РеА. Однотипна артритогенність пов’язана з тим, що ліпополісахариди ієрсиній, сальмонел, хламідій здатні до перехресних реакцій, клітинні оболонки мають однотипні антигенні компоненти. У синовіальній рідині і тканині синовіальної оболонки хворих на РеА виявляють антигени до цих мікробів, а також рибосомальну РНК і ДНК хламідій. Здатність нуклеїнових кислот до реплікації не доведена. Антигени уреаплазми у суглобах не виявлені. У синовіальній оболонці та рідині виявляють здатні до поділу живі мікроорганізми. Їх персистенція та присутність антигенів в тканинах суглобів ініціює хронічний запальний процес. Участь інфекції у розвитку РеА підтверджується: 1) виявленням антитіл до хламідій та кишкових інфекцій; 2) наявністю асоціації між розвитком або загостренням суглобового синдрому з одного боку, інфекційними захворюваннями сечостатевого та кишкового тракту — з другого; 3) позитивним (не завжди) ефектом антибіотиків при лікуванні РеА.
У відповідь на тригерні агенти продукуються специфічні антитіла, які визначаються в сироватці крові і синовіальній рідині. Це робить можливим уточнення етіологічної природи артриту. У разі ієрсиніозного ентероколіту при хронічному перебігу артриту і у носіїв HLA-В27 високий титр антитіл не знижується тривалий час, що відображає збереження активної інфекції і стимуляцію лімфоїдної системи. Персистенція антигенів мікробів в синовіальній рідині і тканинах зумовлює тривале існування гуморальної імунної відповіді.
Генетичні чинники відіграють важливу роль в розвитку РеА. Це підтверджується їх тісною асоціацією з HLA-B27. При урогенних артритах HLA-B27 визначається в 80–90% випадків, рідше — при постентероколітичних. Патогенетична роль HLA-B27 остаточно не встановлена. Носійство HLA-B27 підвищує вірогідність урогенного РеА в 50 разів. Білок, що продукується геном, є рецептором для бактерій, а тому сприяє персистенції інфекції в організмі. Він має спільні антигенні детермінанти із пептидами мікробів, тканинами організму, наслідком чого є направленність імунної відповіді як проти інфекційного агента, так і проти власних тканин. Іншими факторами, що сприяють розвитку РеА, є неадекватна (генетично зумовлена) відповідь CD4+Т-клітин на інфекцію, особливості продукції цитокінів, недостатня елімінація мікробів та їх антигенів з порожнини суглоба, мікротравматизація. Окрім характеру збудника і генетичних чинників, в розвитку РеА (особливо постентероколітичних) важливу роль відіграє стан стінки кишечнику: в результаті запального процесу вона стає проникливою для мікроорганізмів та їх антигенів. Антигени надходять в кров, суглоби (безпосередньо або у складі імунних комплексів) і викликають запалення. При хламідійних артритах мікроби розносяться по організму фагоцитами крові.
Інфекція, з якою асоціюються РеА. РеА асоціюються з гострою кишковою і урогенітальною інфекцією (табл. 24.22).
Таблиця 24.22
Інфекція, з якою асоціюються РеА
|
Локалізація інфекції |
Збудники |
|
Гостра кишкова інфекція |
Yersinia enterocolitica (серотипи 3, 8, 9) Yersinia pseudotuberculosis Salmonella enteritidis Salmonella typhimurium Campylobacter jejuni Schigella Flexneri |
|
Гостра урогенітальна інфекція |
Chlamydia trachomatis (серотипы D, K) |
Урогенітальну інфекцію викликають серотипи D і K Chl. trachomatis. Інфекція передається статевим шляхом, перебіг безсимптомний у більшості (80%) жінок, часто не підлягає діагностуванню, що призводить до її поширення. У число збудників, що викликають так звані HLA-B27-незалежні РеА, входять: стрептококи, гонококи, бруцели, борелії, віруси (краснухи, епідемічного паротиту, вірусного гепатиту). До цієї групи також відносять артрити при носоглотковій інфекції (тонзилогенні, фарингогенні, включаючи РА). Збудники РеА представлено в табл. 24.23.
Таблиця 24.23
Збудники РеА
|
Найбільш часті збудники РеА |
|
|
Chlamydia trachomatis Yersinia enterocolitica (серотипи 3, 8, 9) Yersinia pseudotuberculosis Shigella Flexneri (серотип 2а) Shigella sonnei |
Salmonella enteritidis Salmonella typhimurium Campylobacter jejuni |
|
Рідкісні збудники РеА |
|
|
Borrelia burgdorferi Ureaplasma urealyticum Clostridium difficile Vibrio parahaemoliticum Chlamydia pneumoniae Стрептококи Ешерихії |
Клебсіела Стафілококи Аденовіруси Віруси грипу Менінгококи Вірус гепатиту В |
На сьогодні вважається, що етіологічну роль Clostridium difficile, Ureaplasma urealiticum, Mycoplasma hominis, Neisseria gonorrhoeae у розвитку РеА не доведено.
Асоціація між РеА і ВІЛ-інфекцією. Відомо, що у ВІЛ-інфікованих пацієнтів синдром Рейтера виявляють в 10 разів частіше, ніж в популяції, і перебіг його більш тяжкий.
Морфологічні зміни в суглобах при Реа. У гостру стадію в синовіальній оболонці виникають зміни, які нагадують інфекційний артрит: набряк, гіперемія, інфільтрація нейтрофільними лейкоцитами. У разі хронізації розвивається картина неспецифічного синовіту з інфільтрацією лімфоцитами і плазматичними клітинами. У синовіальній рідині переважають нейтрофільні лейкоцити, зустрічаються цитофагоцитуючі макрофаги (клітини Рейтера), які не мають специфічності. Загальна кількість клітин коливається від 10 000 до 50 000 в 1 мкл.
Клінічна картина
Клінічні прояви виникають на 1–4-му тижні хвороби. Слід враховувати, що у 10% пацієнтів інфекція може проходити в стертій або прихованій (безсимптомній) формі. Це стосується, передусім, урогенітального хламідіозу у жінок. До моменту розвитку суглобового синдрому прояви тригерної інфекції повністю купіруються. Виділяють наступні варіанти перебігу РеА: гострий (тривалість менше 6 міс), затяжний (тривалість від 6 до 12 міс), хронічний (більше 12 міс). У більшості випадків перебіг доброякісний з повним купіруванням артриту протягом 4–6 міс. У ½ пацієнтів виникають рецидиви. До чинників, які викликають рецидиви і хронізацію РеА, належать реінфекція, суперінфекція, активація персистуючої інфекції. Клінічні прояви РеА описано в табл. 24.24.
Таблиця 24.24
Клінічні прояви РеА
|
Симптоми |
Характеристика |
|
|
Загальні симптоми |
Слабкість, зниження апетиту, зменшення маси тіла, субфебрилітет (можлива лихоманка) |
|
|
Суглобовий синдром |
|
|
|
Ентезити |
Запалення в місцях прикріплення сухожиль і зв’язок до кісток. Частіше усього ентезити виникають в ділянці п’ят. Розвиваються тендовагініти окремих пальців стоп (рідко — кистей). Вони проявляються болем, набряклістю усього пальця (дактиліт, «палець-сосиска»), багряно-синюшним забарвленням шкіри (іноді) |
|
|
Ураження слизових оболонок |
Розвивається асептичне (неінфекційне) запалення слизових оболонок у вигляді:
Кон’юнктивіт може бути однобічним, короткочасним і навіть безсимптомним. Може розвинутися однобічний передній увеїт |
|
|
Ураження шкіри |
Характерним є розвиток кератодермії (keratoderma blennorragica) — гіперкератозу (що зливається або у вигляді папул і бляшок), який частіше за все локалізується в ділянці підошов, долонь. Проте окремі вогнища гіперкератозу можуть з’являтися на будь-якій частині тіла |
|
|
Ураження нігтів |
Розвивається жовте забарвлення нігтів на пальцях (частіше стоп), оніхолізис, інші прояви оніходистрофії |
|
|
Системні прояви |
Серцево-судинна система |
Аортит, недостатність аортального клапана, міокардит, перикардит, порушення АВ-провідності |
|
Легені |
Плеврит |
|
|
Нирки |
Гломерулонефрит |
|
|
Скелетні м’язи |
Міозит |
|
|
Периферична нервова система |
Поліневрит |
|
|
Лімфатична система |
Лімфаденопатія при урогенітальній інфекції |
|
Особливості суглобового синдрому при РеА наведені в табл. 24.25.
Таблиця 24.25
Особливості суглобового синдрому при РеА
|
Перші симптоми артриту з’являються в проміжок часу від 3 днів до 2 міс після кишкової або сечостатевої інфекції |
|
Перебіг артриту гострий, кількість уражених суглобів обмежена; у 85% хворих спостерігається моно- та олігоартрит |
|
Типовим є асиметричний характер ураження |
|
В усіх випадках має місце ураження суглобів нижніх кінцівок (за винятком кульшових) |
|
Спочатку уражуються колінні, гомілкові, плеснофалангові суглоби, пізніше приєднується ураження суглобів верхніх кінцівок та хребта |
|
У 50% випадків спостерігається ураження плеснофалангових суглобів великих пальців стоп; зрідка уражуються інші плеснофалангові суглоби та міжфалангові суглоби пальців стоп, суглоби заплесна |
|
Нерідко розвиваються дактиліти одного або декількох пальців стоп (частіше — перших) зі сосископодібною деформацією (її причиною є запалення періостальної кістки та періартикулярних структур) |
|
Внаслідок включення в запалення суглобів заплесна та зв’язок швидко формується плоскостопість («гонорейна стопа») |
|
У 20–25% хворих розвиваються ентезопатії (типова ознака серонегативних спондилоартропатій), які супроводжуються болем та припухлістю у місці уражених ентезисів |
|
Найбільш характерним варіантом ентезопатії є підошовний апоневрозит, ахіллобурсит, сосископодібна дефігурація пальців, трохантерит (біль в ділянці великих вертлюгів стегнової кістки при відведенні стегна) |
|
Проявом ентезопатії є також симфізит, синдром передньої грудної стінки (в запальний процес включаються груднино-реберні з’єднання) |
|
При хронічному перебігу (більше 12 міс) зменшується кількість уражених суглобів, збільшується вираженість сакроілеїту; ентезопатії стають стійкими та рефрактерними для лікування |
|
На початку хвороби ознаки ураження хребта відзначають у 50% хворих: має місце біль у нижній частині хребта, крижово-клубових з’єднаннях, обмеження рухів хребта, ранкова скутість та спазм паравертебральних м’язів |
|
Рентгенологічні зміни в осьовому скелеті відзначають тільки у 25% випадків |
|
Частота виявлення сакроілеїту прямо корелює з тривалістю хвороби та спостерігається у 35–45% хворих; характер ураження дво- (частіше) або однобічний |
|
Спондиліт розвивається у 10–15% випадків, рентгенологічно проявляється асиметричними синдесмофітами та параспінальними осифікатами (характерні також для ПсА) |
Спільні риси постентероколітичних і урогенітальних РеА наведено в табл. 24.26.
Таблиця 24.26
Спільні риси постентероколітичних і урогенітальних РеА
|
1. |
Розвиток хвороби у віці 20–40 років |
|
2. |
Початок артриту частіше гострий з яскравими місцевими ознаками запалення |
|
3. |
Моно- або олігоартикулярний тип ураження (переважно колінних і гомілковостопних суглобів), асиметричний артрит |
|
4. |
Сосископодібна дефігурація пальців стоп, артрит великого пальця стопи |
|
5. |
Періостит дрібних кісток стопи |
|
6. |
Часте залучення сухожильно-зв’язкового апарату: ахіллобурсит, підп’ятковий бурсит з болем і припухлістю в ділянці п’ят, утворенням пухких шпор (рентгенологічно) |
|
7. |
Однобічне ураження крижово-клубових з’єднань, розвиток локального спондиліту, паравертебральної асиметричної осифікації. Однобічний сакроілеїт розвивається у 22–23% хворих, при чому у більшості з них його виявляють тільки при рентгенологічному дослідженні (у ⅓ сакроілеїт супроводжується больовими відчуттями) |
|
8. |
Наявність позасуглобових проявів у вигляді ураження очей (кон’юнктивіт, передній увеїт), шкіри (кератодермія підошв, баланіт), слизових оболонок (виразки, ерозії в порожнині рота) |
|
9. |
Доброякісний перебіг суглобового синдрому: у більшості хворих через 6–7 тиж настає повний зворотний розвиток. Затяжний перебіг (більше 6 міс) артриту відзначають у 20–25% хворих. При хронізації процесу через 1–2 роки суглобові явища повністю зникають |
|
10. |
Відсутність РФ |
|
11. |
Наявність HLA-B27 |
Позасуглобові прояви РеА і частоту їх розвитку представлено в табл. 24.27.
Таблиця 24.27
Позасуглобові прояви РеА і частота їх розвитку
|
Органи і системи |
Характерні прояви |
|
Загальні симптоми |
Субфебрильна лихоманка, зменшення маси тіла |
|
Очі |
Кон’юнктивіт (у 60% хворих), передній увеїт |
|
Слизові оболонки |
Безболісні виразки ротової порожнини (25%) |
|
Шкіра |
Кератодермія (20%), ураження нігтів (10%), вузлувата еритема (Yersinia) |
|
Серце |
Перикардит, аортит (1%), аортальна недостатність, блокади серця (1%) |
|
ШКТ |
Інфекційний або асептичний ілеїт (коліт) |
|
Сечостатева система |
Кільцеподібний баланіт (30%), інфекційний або асептичний уретрит, простатит, цистит, сальпінгіт |
|
Інші |
Нейропатія, нефропатія, амілоїдоз нирок, тромбофлебіт |
Ураження хребта виявляють у 40% хворих. Сакроілеїт і спондиліт частіше розвиваються при хронічному перебігу, наявності HLA-В27. У жінок можливий атиповий перебіг РеА: може відзначатися ураження дрібних суглобів кистей, що вимагає виключення РА.
Діагностика
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих, наведено в табл. 24.28.
Таблиця 24.28
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих на РеА
|
Лабораторне обстеження |
||
|
Аналіз крові |
Виявляють збільшення в крові кількості лейкоцитів, тромбоцитів, підвищення показників ШОЕ, СРБ, розвиток анемії |
|
|
Імунологічні дослідження |
У 60–80% хворих виявляють HLA-B27. Спостерігається асоціація HLA-B27 з розвитком спондиліту, сакроілеїту, аортиту, увеїту. Відзначають підвищення в крові концентрації IgA. РФ і АНФ не визначаються. При тяжкому перебігу РеА можливе виявлення маркерів ВІЛ-інфекції |
|
|
Загальний аналіз сечі |
При уретритах виявляють піурію, при гломерулонефриті — протеїнурію, мікрогематурію |
|
|
Дослідження синовіальної рідини |
Проводять з метою виключення септичного і мікрокристалічного артриту. Виявляють велику кількість лейкоцитів (5000–10 000 в 1 мкл, переважно нейтрофілів), низьку в’язкість, пухкий муциновий згусток, збільшення вмісту білка, комплементу. Концентрація глюкози не знижується |
|
|
Інструментальні дослідження |
||
|
ЕКГ, ехоКГ |
Дозволяють виявити (виключити) наявність міокардиту, перикардиту, недостатності аортального клапана, порушення АВ-провідності |
|
|
Ренгенографія |
Затяжний і хронічний перебіг РеА |
Виявляють:
|
|
Хронічний перебіг |
Звуження суглобової щілини Розвиток ерозивних змін в дрібних суглобах стоп |
|
Зміни периферичних суглобів і зв’язок при РеА, аналогічні таким при АС.
Визначення «тригерного» мікроорганізму. З цією метою використовують мікробіологічні, імунологічні і молекулярно-біологічні методи (табл. 24.29).
Таблиця 24.29
Визначення «тригерного» мікроорганізму за допомогою мікробіологічних, імунологічних і молекулярно-біологічних методів
|
Мікробіологічні |
Посіви калу, зіскрібків епітелію з уретри і шийки матки на культуру клітин |
Посів зіскрібків з уретри проводять для виявлення попередньої урогенітальної інфекції Ch. trachomatis |
|
Імунологічні, молекулярно-біологічні |
Визначення антитіл до збудників, а також їх антигенів, фрагментів нуклеїнових кислот |
Отримані результати служать тільки непрямим доказом наявності в організмі відповідних збудників. У зв’язку зі значним поширенням інфекції Ch. trachomatis часто одержують хибнопозитивні результати серологічних досліджень. Це стосується і полімеразної ланцюгової реакції: з її допомогою виявляють Ch. trachomatis у здорових людей і при різних ревматичних хворобах. Для підвищення достовірності результатів досліджень рекомендують одночасно використовувати декілька тест-систем, зіставляти результати дослідження |
Найбільш доказовим лабораторним методом підтвердження тригерної інфекції є виділення Ch. trachomatis в культурі клітин, виявлення Yersinia enterocolitica, Yersinia pseudotuberculosis, Shigella Flexneri, Shigella sonnei, Campylobacter jejuni, Salmonella enteritidis при посіві калу. У разі негативних результатів використовують непрямі методи дослідження: для виявлення хламідіозу застосовують пряму імунофлуоресценцію, полімеразну ланцюгову реакцію, визначення антитіл в сироватці крові; для виявлення ентеробактерій — визначення рівня антитіл в сироватці.
Характерні рентгенологічні ознаки РеА. Ці ознаки характерні для усіх серонегативних спондилоартропатій і скорочено позначаються ABCDES (Williams D.C., 1999) (табл. 24.30).
Таблиця 24.30
Рентгенологічні ознаки серонегативних спондилоартропатій
|
А |
Ankylosis |
Анкілоз хребетного стовпа розвивається у 20% хворих, характерним є формування асиметричних синдесмофітів («ручка глека») |
|
В |
Bone reactivity |
Виявляють кісткові розростання в ділянці ентезисів — місцях прикріплення ахіллового сухожилля (рис. 24.7) і підошовної фасції, періостит |
|
Bone osteoporosis |
Відмічають ОП суглобових кісток як результат запального процесу в суглобі |
|
|
С |
Cartilage space |
Відзначають рівномірне звуження суглобової щілини. Кальциноз і утворення патологічного хряща не спостерігаються |
|
D |
Distribution |
Переважно уражуються суглоби нижніх кінцівок. Характерне ураження міжфалангового суглоба великого пальця стопи |
|
Е |
Erosions |
Часто утворюються ерозії в плеснофалангових суглобах і асиметричні ерозії в крижово-клубових з’єднаннях |
|
S |
Soft tissue swelling |
Відзначають набряк м’яких тканин, у тому числі дифузний набряк пальців стоп (дактиліт) |
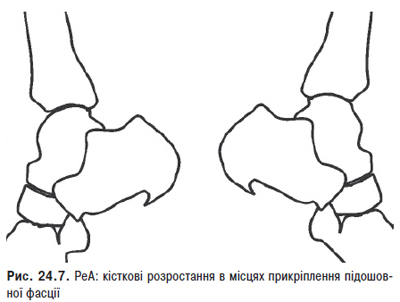
Характерні рентгенологічні особливості ураження опорно-рухового апарату при РеА:
- сполучення деструктивних змін із ремоделюванням кісткової тканини;
- наявність гетеротопних осифікацій;
- слабо виражені ерозії (частіше у плеснофалангових суглобах);
- асиметричність розташування ерозій, поєднання їх з проліферацією кісткової тканини;
- остеопенія мало виражена навіть у хворих зі стійким перебігом суглобового синдрому;
- наслідком ентезопатій є формування пухких шпор (кісткових розростань) п’яткових кісток та утворення ерозій;
- утворюються періостити на п’ясткових, плеснових (часто) кістках; характерним для РеА є розвиток періоститів діафізів основних фаланг пальців;
- періостальні нашарування виявляються на гребенях клубових кісток;
- деструкція та ремоделювання кістки виявляються у місці з’єднання лона та рукоятки грудини.
Відмітною особливістю рентгенологічної картини є те, що при РеА, як і при інших спондилоартритах, в зонах запалення (при ентезитах і ерозіях) діагностують остеосклероз, кісткову проліферацію, періостит, а не ОП, як при РА.
При діагностиці РеА необхідно враховувати наявність ряду чинників:
- попереднього уретриту або діареї, які треба підтвердити лабораторними методами;
- периферичного асиметричного моно-, олігоартриту суглобів нижніх кінцівок;
- болю в п’ятах, інших ознак ентезиту;
- дактиліту, шкірно-слизових проявів;
- носійства HLA-B27.
Загальноприйнятих діагностичних критеріїв РеА немає. Діагностичні критерії РеА Німецької ревматологічної асоціації (1995) представлено в табл. 24.31.
Таблиця 24.31
Діагностичні критерії РеА (Німецька ревматологічна асоціація, 1995)
|
1. |
Типове ураження суглобів (асиметричне, периферичне, олігоартикулярне, нижні кінцівки, особливо колінні і гомілковостопні) |
|
2. |
Типовий анамнез (діарея, уретрит) і/або клінічні прояви інфекції вхідних воріт |
|
3. |
Безпосереднє виявлення збудника у вхідних воротах (наприклад хламідій в зіскрібку з уретри) |
|
4. |
Виявлення специфічних антитіл, що аглютинують, з достовірним підвищенням титрів (наприклад у разі кишкових збудників) |
|
5. |
Наявність HLA-В27-антигену |
|
6. |
Виявлення субстрату збудника за допомогою полімеразної ланцюгової реакції або специфічних моноклональних антитіл |
|
Достовірний РеА: |
|
|
Встановлюється за наявності критеріїв 1 + 3, або 4, або 6. |
|
|
Вірогідний РеА: |
|
|
За наявності критеріїв 1 + 2 і/або + 5. |
|
|
Можливий РеА: |
|
|
Передбачається за наявності критерію 1. |
|
Класифікаційні критерії, прийняті на IV Міжнародній робочій нараді з діагностки РеА, наведено в табл. 24.32.
Таблиця 24.32
Класифікаційні критерії РеА (IV Міжнародна робоча нарада з діагностики РеА)
|
Великі критерії |
1) асиметричність суглобового ураження, залучення 1–4 суглобів і локалізація артриту на нижніх кінцівках (потрібна наявність 2 з 3 таких ознак); |
|
|
2) клінічно маніфестна інфекція кишкового або сечостатевого трактів (ентерит або уретрит за 1–3 дні — 6 тиж до розвитку захворювання) |
||
|
Малі критерії |
1) лабораторне підтвердження сечостатевої або кишкової інфекції (виявлення Chlamydia trachomatis у зіскрібку з уретри і каналу шийки матки або виявлення ентеробактерій в калі); |
|
|
2) виявлення інфекційного агента в синовіальній оболонці або рідині за допомогою полімеразної ланцюгової реакції |
||
|
Оцінка |
||
|
Визначений РеА |
Діагностують за наявності 2 великих і відповідних малих критеріїв |
|
|
Можливий РеА |
За наявності 2 великих критеріїв і 1 малого |
|
|
Лабораторні дослідження |
||
|
1. |
Для виявлення хламідійної інфекції застосовують метод (реакцію) прямої імунофлуоресценції (скринінговий метод). Його чутливість — 50–90%. |
|
|
2. |
Полімеразна ланцюгова реакція |
|
|
3. |
Серологічне дослідження з видоспецифічними антисироватками 3 класів імуноглобулінів |
|
|
4. |
Культуральний метод (найбільш специфічний) |
|
|
Оцінка |
||
|
Треба отримати позитивний результат в 2 будь-яких реакціях |
||
Слід враховувати зміни в інших аналізах: підвищений СРБ, лейкоцитоз, тромбоцитоз, помірна анемія, носійство HLA-B27, яке привертає увагу до локалізації запального процесу у хребті й асоційоване із системними проявами.
РеА диференціюють з інфекційними артритами, у тому числі хворобою Лайма, іншими спондилоартритами, серонегативним РА, подагрою та ін. Труднощі при діагностиці РеА виникають у разі безсимптомного перебігу хламідійного запалення урогенітальної системи. Треба пам’ятати, що у ВІЛ-інфікованих осіб перебіг РеА більш тяжкий.
Приклад формулювання діагнозу РеА. Реактивний артрит урогенітальний (хламідійний), затяжний перебіг, з переважним ураженням колінних і гомілковостопних суглобів, однобічний сакроілеїт, активність III ступеня, недостатність функції суглобів II ступеня. При формулюванні діагнозу РеА необхідно враховувати нижченаведену інформацію (табл. 24.33).
Таблиця 24.33
Дані, які необхідно враховувати при формулюванні діагнозу РеА
|
Форма |
Урогенітальна, постентероколітична |
|
|
Характер процесу |
Первинний, вторинний |
|
|
Варіант перебігу |
Гострий, затяжний, хронічний |
|
|
Клініко-морфологічна характеристика ураження |
Сечостатевих органів |
Уретрит, епідидиміт, простатит, баланопостит, цервіцит, ендометрит, сальпингіт |
|
Органа зору |
Кон’юнктивіт, гострий передній увеїт |
|
|
Опорно-рухового апарату |
Моно-, оліго-, поліартрит, сакроілеїт, спондиліт, ентезопатії |
|
|
Рентгенологічна характеристика |
Артриту (за Штейнброкером), сакроілеїту (за Келгреном або Дейлом), спондиліту (синдесмофіти, параспінальні осифікати, анкілоз міжхребцевих суглобів |
|
|
Ступінь активності та недостатність функції суглобів |
||
Особливості постентероколітичних РеА пов’язані з епідемічними спалахами і спорадичними випадками. За даними Інституту ревматології РАМН, при гострому ієрсиніозі артрити розвиваються в 20% випадків, шигельозі — в 1,5%, сальмонельозі — в 2–7,5%, кампілобактеріозі — менш ніж в 1%. 63% постентероколітичних РеА пов’язані з інфекцією Yersinia enterocolitica або Y. рseudotuberculosis, 5,4% — з Sh. Flexneri і менше 2% — з Salmonella tiphimurium. Тригерний агент не виявляється у 25–35% хворих. Існує прямий зв’язок між вираженістю суглобового синдрому і запальними змінами в стінці кишечнику, підтвердженими гістологічно. У хворих з постентероколітичними РеА відзначають зменшення вмісту в калі кишкової палички, біфідобактерій і збільшення водночас умовно-патогенної флори. Запальні зміни ініціюють розвиток РеА і сприяють його хронізації. Тому лікувальні заходи мають бути спрямовані на нормалізацію функції органів травлення. Характерні ознаки ентероартритів:
- розвиток артриту через 1–4 тиж після діареї;
- гострий характер суглобових змін (припухлість, почервоніння шкіри, підвищення локальної температури, різка болісність при рухах);
- уражуються великі й середні суглоби (переважно нижніх кінцівок) у вигляді оліго- і поліартриту;
- характерним є асиметричне ураження суглобів з розвитком бурситів, тендовагінітів;
- позитивні результати бактеріологічного дослідження, виявляються діагностичні титри антитіл до збудника;
- у хворих виявляють антиген гістосумісності HLA-B27 (у здорових зустрічається в 5–7%);
- у HLA-В27-позитивных хворих відзначають ураження очей (у вигляді кон’юнктивіту, іриту, кератиту, епісклериту), шкіри і слизових оболонок, сечостатевих (у вигляді уретриту, циститу, орхіту, баланіту) і внутрішніх органів у вигляді кардиту (при ієрсиніозі, сальмонельозі, шигельозі), плевриту (при шигельозі).
Ревматичні прояви при ієрсиніозі
Хвороба описана в 1962 р. Зустрічається як у домашніх тварин (кролі, собаки, коти, свині, корови, коні), так і у диких (миші, щурі). Від тварин збудник передається людині через овочі, фрукти, молочні продукти, воду. Іерсиніоз викликають мікроби Yersinia enterocolitica і Yersinia pseudotuberculosis, які входять в сімейство Enterobacteriaceae. Із 30 серотипів для людини є патогенними 3; 5; 8-й і 9-й. Ієрсинії зберігаються і розмножуються на овочах, коренеплодах, інших харчових продуктах, вони не стійкі до висихання і високої температури, високочутливі до дії звичайних дезінфектантів. Передача від хворої людини відбувається орально-фекальним шляхом. Мікроби, які здолали шлунковий бар’єр (соляну кислоту), проникають і розмножуються в лімфоїдному апараті дистального відділу клубової кишки, викликають запальний процесс — ілеїт, мезаденіт. Найчастіше захворювання виникає в липні — серпні.
Клінічна картина ієрсиніозу
Початок хвороби гострий або підгострий. Розвивається клінічна картина, що нагадує дизентерію, токсикоінфекцію: з’являється нудота, біль в животі, діарея, підвищення температури тіла до 38–39 °С, озноб, загальна слабкість. Діарея може бути з домішками крові та слизу. У частини хворих є симптоми подразнення верхніх дихальних шляхів, ангіна, що утрудняє своєчасне встановлення діагнозу. Може спостерігатися гепатит. У деяких хворих з’являється шкірний висип, що нагадує такий при краснусі й скарлатині. У подальшому на місці висипу залишається пігментація. За відсутності адекватного лікування хвороба може стати хронічною (ентерит, коліт, мезаденіт та ін.). У ⅓ хворих відзначають вузлувату еритему. Симптоми ураження м’язів і суглобів виявляють у 20% хворих, в основному при генералізованій тяжкій формі хвороби, хоча артрити зустрічаються і при стертих формах ієрсиніозу. При цьому хворі забувають про наявний епізод діареї. Артрит частіше виникає через 1–3 тиж після ентероколіту. Зазвичай артрит триває від 1 до 3 міс. У 30–50% пацієнтів з поліартритом відзначається кардит (міокардит, рідше — перикардит). Ураження серця частіше перебігає без істотних гемодинамічних порушень, повністю зворотне, хоча і може набути затяжного перебігу.
Переважно уражуються суглоби нижніх кінцівок з розвитком гострих запальних проявів. Може настати хронізація суглобового синдрому. Як і у хворих з іншими формами ентероартритів, спостерігається однобічне ураження крижово-клубових суглобів, кісток п’ят, наявність HLA-В27 в крові, що вимагає проведення диференційної діагностики з хворобою Бехтєрева (на ранній стадії). Нерідко до процесу залучається великий палець, акроміально-ключичний, груднино-ключичний суглоби, розвиваються тендоперіостити, тендосиновіти. Рентгенологічні зміни в суглобах не виявляються.
Діагностика ієрсиніозу
Необхідно враховувати дані анамнезу, наявність клінічних симптомів шлунково-кишкового захворювання з діареєю, гепатитом, міалгією, моно-, олігоартритом нижніх кінцівок, лейкоцитозом, антитілами в крові або збудником в різних середовищах. Основне значення має бактеріологічне дослідження інфікованого матеріалу. З метою посіву використовують кров, синовіальну рідину, біоптат тканин різних органів, сечу, фекалії. Зазвичай через 1 тиж від початку хвороби в крові виявляють антитіла до Y. enterocolitica. Реакцію вважають позитивною за наявності титру 1:200. При диференціації артритичного синдрому при ієрсинозі слід враховувати хворобу Бехтєрева (ранні етапи), при мігруючому артриті і ураженні серця — виключити ревматизм.
Ураження суглобів при дизентерії. Раніше артрити розвивалися у 10% хворих на дизентерію, зараз — у 0,05–0,07%, тобто, цей прояв стає рідкісним. Артрит може розвиватися на тлі гострої і хронічної діареї, викликаної частіше шигелами Флекснера, рідше — Зонне. Транзиторні артрити колінних, гомілковостопних і дрібних суглобів можуть спостерігатися вже в період розпалу дизентерії. У пізніші періоди реєструють клінічну симптоматику підгострого і навіть хронічного поліартриту. Гостре або підгостре запалення суглобів супроводжується вираженим болем, ексудативними проявами. У окремих хворих артрит розвивається поступово і має стійкіший перебіг (може тривати 1–2 роки). Як гострий, так і хронічний дизентерійний артрит повністю купіруються, не залишаючи жодних безповоротних змін в суглобах. У частини хворих розвивається атрофія м’язів, прилеглих до суглоба. Тільки у разі стійкого артриту при рентгенографії суглобів можна виявити ОП, при тривалому перебігу — звуження суглобової щілини. У деяких хворих розвивається сакроілеїт, запалення райдужки. Ці зміни, а також олігоартрит нижніх кінцівок, вимагають проведення диференційної діагностики з хворобою Бехтєрева. РеА діагностують легко, якщо виникає тріада або тетрада Рейтера. Достовірний діагноз встановлюють на підставі анамнезу, клінічних симпомів захворювання, виявлення бацил в калі і позитивних серологічних реакцій (проводиться реакція непрямої гемаглютинації, позитивний титр 1:200 і більше).
Суглобовий синдром при сальмонельозі. Сальмонельоз рідко ускладнюється артритами. У випадках сальмонельозу, викликаного S. typhimurium або enteritidis, артрити виникають через 1–2 тиж після початку проносу, супроводжуються болем і лихоманкою. Уражуються великі суглоби — колінні, гомілковостопні, рідше — променезап’ясткові. Поліартрит може розвиватися через 1–6 міс після діареї, він супроводжується лихоманкою, підвищенням ШОЕ, іноді — гіперлейкоцитозом, анемізацією. Синовіальна рідина стерильна, зміни суглобів повністю зворотні. Діагноз встановлюють на підставі анамнезу, клінічної картини хвороби, результатів посіву калу, серологічних досліджень (реакція аглютинації — титр 1:200 і більше, реакція непрямої аглютинації — титр такий самий).
Ураження суглобів при кампілобактеріозі. Кампілобактеріоз — інфекційне захворювання, яке характеризується ураженням ШКТ і має тенденцію до генералізації процесу. Збудником є Campylobacter jejuni — рухлива грамнегативна паличка, стійка в довкіллі та швидко гине при нагріванні. Джерелом збудника є велика рогата худоба, свині, вівці, свійська птиця, кролі, собаки, коти, гризуни, зайці, рідше — люди — носії збудника. Кампілобактерії виділяються в довкілля з випорожненнями тварин і людей. Чинниками передачі служать харчові продукти (м’ясо, молоко, овочі), забруднені збудником, які не були піддані термічній обробці, а також вода відкритих водоймищ. Бактерії, які здолали шлунковий бар’єр, досягають порожнистої, клубової й товстої кишки, де й локалізується запальний процес. Товста кишка залучається в більшості випадків, і патологічна картина нагадує дизентерію. Інкубаційний період становить 1–2 дні. Постійним симптомом є гостра діарея, яка зберігається декілька днів. Вираженість діареї може бути від легкої до тяжкої, запах калу смердючий, нерідко в калі виявляється кров. Хворі скаржаться на біль в животі, головний біль, нудоту, блювання, підвищення температури до 38–40 °С, постійну або рецидивуючу діарею. На тлі імунодефіциту, алкоголізму, патології серцево-судинної системи виникають ускладнення: пневмонія, артрит, міокардит, менінгіт, сепсис.
Суглобовий синдром при кампілобактеріозі нічим не відрізняється від суглобового синдрому при дизентерії, інших ентероартритах. При діагностиці кампілобактеріозу мають враховуватися наявність епідемічного спалаху, клінічна картина хвороби, виділення збудника з фекалій, сечі, крові, цереброспінальної рідини, результати серологічних досліджень (позитивна реакція аглютинації і пасивної гемаглютинації).
Особливості уроартритів — РеА, зумовлені сечостатевою інфекцією: у результаті розвитку запалення і підвищення проникності слизової оболонки сечостатевих шляхів антигени збудників потрапляють в кров, а потім в суглоби і інші органи. У синовіальній оболонці суглобів відкладаються як антигени, так і імунні комплекси. Уроартрити викликають передусім хламідії, і виникають вони частіше у чоловіків. Таким чином, розвитку артриту передує уретрит неспецифічного характеру. Набуті частіше статевим шляхом РеА розглядають як неповну хворобу Рейтера. Виділення з уретри часто мізерні, серозного характеру, хоча можуть бути і значними, гнійними. Хворі відчувають дискомфорт внизу живота, частішає сечовипускання. Отвір сечового каналу гіперемований. При несвоєчасному лікуванні виникає везикуліт, епідидиміт, запалення передміхурової залози, вагініт, бартолініт.
Артрит починається гостро або підгостро. Після короткочасного болю або різі в сечостатевому каналі виникає запалення в 1–2 суглобах нижніх кінцівок. Можливий і поступовий розвиток процесу при нормальній температурі тіла. Суглобовий синдром аналогічний такому при кишкових захворюваннях: уражуються суглоби нижніх кінцівок — колінні, гомілковостопні, пальців стоп з сосископодібною дефігурацією; процес асиметричний і олігоартикулярний; розвиваються бурсити (ахіллобурсит, підп’ятковий бурсит), періостити. Повний розвиток артриту відбувається за 1–6 міс, проте можливі рецидиви і хронічний перебіг із залученням великої кількості суглобів.
Лабораторні дослідження, що підтверджують діагноз уроартриту. У сироватці крові виявляють антитіла до збудників (хламідій). Необхідно дослідити матеріал з уретри, вивчити осад сечі для ідентифікації мікробів в уретрі й цервикальному каналі, якщо є симптоми урогенітального запалення.
ШОЕ у хворих може досягати 40–50 мм/год, хоча його збільшення не завжди чітко відображає ступінь запального процесу. У крові відзначають помірний лейкоцитоз, підвищений вміст серомукоїду, α- і γ-глобулінів, СРБ.
Синдром Рейтера
Синдром Рейтера (хвороба Рейтера, уретроокулосиновіальний синдром, синдром Фіссенже — Леруа) — форма РеА, що включає клінічну тріаду — уретрит, кон’юнктивіт і артрит. У 1981 р. йому дав визначення комітет ARA: «Епізод периферичного артриту тривалістю більше 1 міс, що зустрічається в асоціації з уретритом або цервіцитом». Проте сьогодні в синдром включають і постентероколітичні випадки. У літературі використовують замість терміну «синдром Рейтера» різні назви: урогенний РеА, індукований хламідіями РеА, уреаплазма-індукований РеА, SARA і BASE-синдром. Клінічна картина нагадує серонегативні спондилоартропатії. При посіві з синовіальної оболонки життєздатних мікроорганізмів не виявляють. Клінічна класифікація синдрому Рейтера представлена в табл. 24.34.
Таблиця 24.34
Клінічна класифікація синдрому Рейтера (АРУ, 2004)
|
Перебіг |
Гострий, підгострий, хронічно-рецидивуючий |
|
|
Ступінь активності |
0 (відсутній), I (низький), II (помірний), III (високий) |
|
|
Клініко-морфологічна характеристика |
Суглоби |
Асиметрично: поліартрит, рідше — оліго- або моноартрит |
|
Сечостатева система |
Уретрит; простатит; цистит; баланіт; ендоцервіцит; аднексит; пієлонефрит |
|
|
Орган зору |
Реактивний ірит; увеїт; кон’юнктивіт; епісклерит. |
|
|
М’язово-зв’язковий апарат |
Ентезопатія; тендиніт; тендовагініт; міалгія; біль в п’ятах; шпори п’ят; бурсит. |
|
|
Шкіра і слизові оболонки |
Бленорагічна кератодермія (pustulosis palmoplantaris) гіперкератоз; дистрофія нігтів і періоніхіальні зміни; псоріазоподібні зміни; ерозії слизових оболонок твердого піднебіння, щік, язика, губ; висипи у вигляді везикул на слизових оболонках з переходом до утворення виразок |
|
|
Серцево-судинна система |
Міокардит; ендокардит; недостатність аортального клапана (зустрічається рідко, як правило, латентна і малосимптомна) |
|
Примітка: при формулюванні діагнозу слід вказувати характер рентгенологічних змін, ступінь функціональної недостатності суглобів.
Виділяють два варіанти синдрому (табл. 24.35).
Таблиця 24.35
Варіанти синдрому Рейтера
|
Епідемічний варіант |
Виникає після ієрсиніозного, шигельозного та іншого коліту, який виявляють переважно в літньо-осінній період при несприятливій санітарно-епідеміологічній ситуації. При цьому варіанті може бути і урогенітальна інфекція (тобто може спостерігатися змішана інфекція) |
|
Спорадичний варіант (чи венеричний) |
Він пов’язаний з негонококовими уретритами, викликаними хламідіями, уреаплазмою |
Збудниками, що найбільш часто викликають розвиток синдрому Рейтера, є паразитуючі внутрішньоклітинно Chlamydia trachomatis і Chlamydia pneumoniae. Chlamydia trachomatis є найбільш поширеною інфекцією, що передається статевим шляхом (зустрічається в 2–6 разів частіше, ніж гонорея), і викликає у чоловіків 60% усіх негонококових уретритів. Для хламідійних уретритів характерний хронічний рецидивуючий перебіг. У жінок ця інфекція викликає розвиток хронічного запального процесу (у вигляді циститу, аднекситу, цервіциту, сальпінгіту) і трубного безпліддя. Синдром Рейтера зазвичай зустрічається у молодих осіб (до 40 років), хоча може розвинутися і в літньому віці. Виявляють хворобу і у дітей (шлях зараження залишається невідомим). Чоловіки хворіють в 20 разів частіше, ніж жінки, що важко пояснити.
Клінічна картина
Уретрит зазвичай починається через 7–28 днів після випадкового статевого контакту або ентероколіту. Прояви його не такі гострі, як при гонореї: виділення з уретри слизово-гнійного характеру незначні. Є неприємні відчуття в уретрі вранці при сечовипусканні. Уретрит може бути переднім або тотальним з простатитом. Больові відчуття і дизурія у багатьох випадках відсутні. Через 1–1,5 міс після сечостатевої інфекції або загострення хронічного вогнища розвивається артрит. Характерні риси артриту при синдромі Рейтера:
- асиметричність ураження;
- сосископодібна дефігурація пальців стоп;
- псевдоподагрична симптоматика (залучення суглобів великого пальця стопи);
- біль у п’ятах в результаті розвитку тендиніту, бурситів, запалення ентезисів, швидкого виникнення шпор;
- формування плоскої стопи в результаті ураження суглобів заплесна і зв’язкового апарату;
- розвиток сакроілеїту (частіше підтверджується рентгенологічно, клінічні прояви присутні у 20–30% хворих);
- ураження суглобів верхніх кінцівок у випадках гострого ентероколітичного початку, а також при хронічному прогресуючому перебігу хвороби.
Позасуглобові прояви синдрому Рейтера, причиною яких є хламідійна інфекція та імунопатологічні реакції:
- ураження очей у вигляді двобічного катарального слабовираженого, короткотривалого (1–2 дні) кон’юнктивіту, увеїту (може призвести до сліпоти). («Хворий не відчуває кон’юнктивіт, а лікар не бачить його»);
- наявність класичної тріади (уретрит-артрит-кон’юнктивіт) або тетради Рейтера (уретрит-артрит-кон’юнктивіт-ураження шкіри і слизових оболонок);
- ураження шкіри, нігтів. Розвивається кератодермія (частіше в початковий гострий період хвороби) долонь і підошов — поліморфні висипи, які важко відрізнити від пустульозного псоріазу. Можуть мати вигляд точкового, вогнищевого або дифузного кератозу. Гіперкератоз може виявлятися у вигляді твердих мозолеподібних утворень на тлі запалення, почервоніння і лущення шкіри;
- псоріазоподібні висипання можуть розташовуватися на шкірі чола, тулуба, кінцівок. Кератодермія часто поєднується з ураженням нігтів за типом оніходистрофії (підвищена ламкість, шорсткість, горбистість нігтів, зміна їх кольору), піднігтьового гіперкератозу, оніхолізису, пароніхії. На відміну від псоріазу при ремісії ці прояви зникають;
- частий розвиток баланіту або баланопоститу, еритеми слизових оболонок ротової порожнини, малоболісних ерозій, глоситу;
- пахвова лімфаденопатія;
- розвиток міокардиту, порушень серцевого ритму і провідності у ⅓ хворих;
- ураження нирок у вигляді нефриту, пієлонефриту, амілоїдозу;
- розвиток лихоманки до 39–40 °С при гострому початку захворювання (зазвичай спостерігається субфебрилітет).
До шкірних симптомів, відносно специфічних для синдрому Рейтера, відносять кільцеподібний баланіт і бленорагічну кератодермію, які переважно реєструють при урогенітальній формі і не вимагають спеціального лікування. Кільцеподібним баланітом (Сirculate balanitis) є безболісна виразка голівки статевого члена, а бленорагічна кератодермія — псоріазоподібні зміни на підошовній поверхні п’яти і в ділянці голівок плеснових кісток.
Ентезиси, що часто уражуються при хворобі Рейтера. Запалення місця з’єднання сухожилля з кісткою (ентезис) називається ентезитом. Зазвичай при хворобі Рейтера уражується ахіллове сухожилля і підошовна фасція, відзначається біль у п’яті та плеснових кістках (через ураження підошовної фасції), сосископодібна деформація пальців (дактиліт).
Прояви суглобового синдрому. Для нього характерна помірна вираженість запального процесу, наявність тривалої скутості. Ураження асиметричне за типом олігоартриту, частіше уражуються колінні, гомілковостопні і суглоби стоп. Артрит суглобів верхніх кінцівок (зап’ястка, пальців) розвивається відносно рідко. Можливий розвиток суглобових ерозій.
Характер перебігу синдрому Рейтера. Первинні випадки хвороби тривають 3–6 міс. За наявності позасуглобових проявів, ураження ділянки п’яти процес затягується до 12 міс. Реінфекція, персистенція інфекції призводить до частих рецидивів. Інтервали між рецидивами можуть досягати 5 і більше років. Хронічний перебіг олігоартриту (асиметричного, серонегативного), спондилоартриту відзначають у 20–30% хворих.
Особливості перебігу синдрому Рейтера у ВІЛ-інфікованих хворих. У них синдром Рейтера зустрічається в 0,5–3% випадків. Найчастіше він розвивається в період вже наявного тяжкого імунодефіциту, але може більше ніж на 2 роки випереджати клінічну маніфестацію або розвинутися на тлі початкових клінічних проявів СНІДУ. Кон’юнктивіт виникає рідко, частіше спостерігаються ентезопатії, підошовний фасцит, дактиліт, зміни нігтів і шкіри. Рідше виникають баланіт і стоматит. Виникнення тяжких артритів з ерозіями призводить до інвалідності.
Діагностика
Діагностичні критерії синдрому Рейтера (за: Willkens R.F. et al., 1981):
- серонегативна асиметрична артропатія (переважно суглобів нижніх кінцівок) + 1 або декілька наступних критеріїв:
- уретрит/цервіцит;
- дизентерія;
- запальні зміни з боку органа зору;
- ураження шкіри/слизових оболонок: баланіт, виразка слизової оболонки ротової порожнини і кератодермія.
Необхідно виключити наявність:
- АС;
- ПсА;
- інших ревматичних захворювань.
Дані лабораторних та інструментальних досліджень у хворих з синдромом Рейтера наведено в табл. 24.36.
Таблиця 24.36
Дані лабораторних та інструментальних досліджень у хворих з синдромом Рейтера
|
Клінічний аналіз крові |
Нейтрофільний лейкоцитоз, тромбоцитоз, анемія |
|
ШОЕ |
Підвищена |
|
Посіви крові на стерильність |
Результати негативні |
|
СРБ |
Концентрація в крові підвищена |
|
РФ |
Відсутній |
|
Антинуклеарні антитіла |
Відсутні |
|
Антитіла до ієрсиній і хламідій |
Визначаються |
|
Загальний аналіз сечі |
Піурія, бактеріурія |
|
Дослідження синовіальної рідини |
Лейкоцити 5000–10 000/мкл, при гострому процесі переважають нейтрофіли, хронічному — лімфоцити або моноцити. Вміст білка підвищений, глюкози — нормальний, в’язкість знижена. Можуть виявлятися клітини Рейтера — вакуолізовані макрофаги (містять лімфоцити, фрагменти ядер) |
|
ЕКГ |
Блокади серця |
|
ЕхоКГ |
Аортальна недостатність |
|
Рентгенографія: |
|
|
периферичних суглобів |
Артрит, ентезити |
|
хребта |
Спондиліт, ентезит |
|
кісток таза |
Сакроілеїт |
На початку хвороби відзначають збільшення ШОЕ, лейкоцитоз. Необхідно звертати увагу навіть на незначну лейкоцитурію, провести урологічне обстеження, зробити аналіз секрету передміхурової залози. Жінки мають бути обстежені гінекологом.
У 80–90% пацієнтів з синдромом Рейтера виявляють HLA-B27. Дослідження HLA-B27 корисне при стертій клінічній картині, відсутності даних про попередню інфекцію, а також позасуглобових проявів. У синовіальній рідині реєструють помірне збільшення загальної кількості клітин за рахунок нейтрофілів, появу цитофагоцитуючих макрофагів (клітин Рейтера), наявність хламідійних антигенів і антитіл до хламідій, високий рівень комплементу.
Зміни, що виявляються при рентгенологічному дослідженні кісток і суглобів хворих з синдромом Рейтера:
- «пухкі» розростання (шпори) кісток п’ят і їх ерозії;
- періостити кісток п’ят, фаланг пальців стоп;
- асиметричні ерозії плеснофалангових суглобів;
- однобічний малопрогресуючий сакроілеїт, при хронічному перебігу можливий двобічний процес і анкілозування крижово-клубових суглобів;
- асиметрична паравертебральна осифікація (вторинний спондилоартрит).
Синдесмофіти і ураження крижово-клубових суглобів при синдромі Рейтера і АС відрізняються. При АС відзначають симетричний характер розташування синдесмофітів і ураження крижово-клубових суглобів. При АС рентгенологічні ознаки сакроілеїту відзначаються в 100% випадків, при РеА — в 25%.
У процесі діагностики синдрому Рейтера слід враховувати низку чинників:
- молодий вік пацієнтів;
- розвиток тріади або тетради Рейтера після сечостатевої або кишкової інфекції;
- наявність гострого асиметричного артриту, переважно суглобів нижніх кінцівок з ураженням пальців стоп, ентезопатіями і бурситами п’ят;
- наявність запального процесу в сечостатевих органах, виявлення хламідій (у 80–90% хворих) в зіскрібках шийки матки або епітелію сечовипускального каналу.
Критерії певного і можливого хламідійного артриту. «Дійсним» хламідійним урогенним артритом вважають артрит при виявленні Chlamydia trachomatis в урогенітальній системі, виключенні інших ревматичних захворювань. Про «можливий» хламідійний артрит йдеться тоді, коли у хворого на серонегативний артрит є запальні зміни в урогенітальній системі, відсутні хламідії в зіскрібках сечовипускального каналу, каналу шийки матки, а в сироватці крові виявляються антитіла до хламідій в титрі 1:32 і більше.
Захворювання, з якими диференціюють синдром Рейтера. Синдром Рейтера диференціюють, передусім, з гонококовим артритом, псоріазом, синдромом Бехчета, ВІЛ-інфекцією. Гонококовий артрит частіше розвивається у жінок, проходить гостро з лихоманкою, ознобами, гонококи виявляють в сечостатевій системі, синовіальній рідині («безперечний» гонококовий артрит), крові. Спостерігається зворотний розвиток артриту під дією пеніциліну, відсутній антиген HLA-B27. Можливе поєднання псоріазу і синдрому Рейтера. Синдром Рейтера може розвиватися у ВІЛ-інфікованих. Саме захворювання не впливає на перебіг імунодефіциту, проте лікування імуносупресорами провокує клінічну картину СНІДУ. Одним з можливих показників наявності СНІДУ є поява висипання на долонях. При синдромі Бехчета виразкові зміни слизової оболонки рота, на відміну від синдрому Рейтера, рецидивують.
Лікування
Метою лікування РеА є усунення тригерної інфекції, досягнення стійкої ремісії (табл. 24.37).
Таблиця 24.37
Медикаментозне лікування при РеА
|
Характер терапії |
Умови, препарати |
Примітки |
|
Антимікробна терапія |
Виявлені вогнища «тригерної» інфекції |
Застосовують антибіотики відповідно до чутливості мікроорганізмів до повної ерадикації. Проводять мікробіологічний контроль |
|
Хламідійна інфекція підтверджена |
Необхідно обстежувати і лікувати не лише хворого, але і постійного статевого партнера |
|
|
Кишкова інфекція |
Антимікробна терапія, як правило, неефективна |
|
|
НПЗП |
Неселективні: диклофенак 100–150 мг/добу в 3 прийоми |
Призначають молодим особам, без факторів ризику розвитку НПЗП-гастропатій |
|
ЦОГ-2-селективні: целекоксиб 100–200 мг 2 рази на добу, мелоксикам 7,5–15 мг/добу, німесулід 100–200 мг 2 рази на добу, еторикоксиб 90 мг/добу |
Показані особам з ризиком розвитку НПЗП-гастропатій |
|
|
ГК |
Локальна терапія: в порожнину суглоба або періартикулярно вводять бетаметазон, метилпреднізолон, тріамцинолон або гідрокортизон. В крижово-клубові суглоби ГК вводять під контролем КТ (бетаметазон 0,25–2 мл внутрішньосуглобово або періартикулярно в ділянку ураженого суглоба не частіше 1 разу на 1–3 міс в той самий суглоб залежно від дози та розміру суглоба) |
Застосовують при синовіті, резистентному до НПЗП, непереносимості НПЗП за умови виключення інфекційного артриту |
|
Системна терапія ГК в низьких і середніх дозах (не більше 10 мг/добу преднізолону). |
Показана при важкому перебігу РеА з системними проявами і за наявності протипоказань до прийому цитостатиків і НПЗП (вагітність, період лактації, непереносимість) |
|
|
Локальна терапія при позасуглобових проявах |
При кон’юнктивіті застосовують краплі, що містять ГК. У разі іридоцикліту використовують інстиляції, субкон’юктивальні ін’єкції. Місцеву ГК-терапію призначають при ураженні інших слизових оболонок — стоматиті, баланіті, баланопоститі |
|
|
Сульфасалазин |
Призначають в дозі 2–3 г/добу протягом багатьох місяців (частіше в дозі 2 г/добу) |
Є препаратом першого ряду при затяжному і хронічному перебігу, відсутності ефекту від симптоматичної терапії протягом ≥3 міс. Добрі результати відзначають при лікуванні постентероколітичних РеА. Препарат зменшує вираженість ознак запалення в периферичних суглобах. При 6-місячній терапії позитивні результати спостерігаються у 62% випадків |
|
Метотрексат, азатіоприн, циклофосфамід |
Показання ті самі, що й для призначення сульфасалазину. Метотрексат призначають при неефективності сульфасалазину, починають з дози 7,5 мг і поступово підвищують до 15–20 мг/тиж |
|
|
D-пеніциламін, солі золота, гідроксихлорохін |
Малоефективні |
|
|
Інгібітори ФНП-α (інфліксимаб) |
Його застосування доцільне при тяжких формах РеА, резистентних до лікування іншими базисними засобами. Купіруються всі прояви хвороби (артрит, спондиліт, ентезити, дактиліти, гострий передній увеїт) |
Тривала (>3 міс) терапія ГК в дозі ≥7,5 мг/добу вимагає призначення комбінованих препаратів кальцію (1000–1500 мг/добу) і вітаміну D3 (які окрім кальцію і вітаміну D3 містять остеотропні мінерали).
Застосування пульс-терапії при РеА. Можливе використання пульс-терапії метилпреднізолоном (без циклофосфамыду) як доповнення до стандартної терапії при рецидивах і торпідному перебігу.
Антимікробні засоби, що призначаються при РеА. Мета їх призначення — ерадикація збудника при хламідійному уретриті з сечостатевої системи, що дозволяє знизити ризик хронізації і частоту рецидивів. З цією метою рекомендують (Насонова В.А., Насонов Е.Л., 2003 (ред.)) призначення всередину протягом 10–30 діб наступних антибіотиків: азитроміцину (1 г в 1-й день, потім по 0,5 г/добу), доксицикліну гідрохлориду (100 мг/добу), ломефлоксацину (0,4 г 1–2 рази на добу), офлоксацину (0,2 г 3 рази на добу), ципрофлоксацину (0,5 г 2–3 рази на добу). Окрім перелічених антибіотиків можуть бути застосовані всередину еритроміцин (1,5–2 г/добу), рокситроміцин (150 мг 2 рази на добу), кларитроміцин (250 мг 2 рази на добу), джозаміцин (750 мг 2 рази на добу), рифампіцин (0,9 г/ добу). Ефективними є тетрациклін (окситетрациклін) 1,5–2 г/добу, метациклін (0,6–1,2 г/добу). Пеніциліни і цефалоспорини неефективні через формування стійких форм хламідій. Антибактеріальну терапію урогенітального хламідіозу при РеА (Белов Б.С., 2001) описано в табл. 24.38.
Таблиця 24.38
Антибактеріальна терапія урогенітального хламідіозу при РеА
|
Антибіотики |
Добова доза |
Тривалість курсу (діб) |
|
Макроліди |
||
|
Спіраміцин |
9 млн од. у 3 прийоми |
28 |
|
Азитроміцин |
1,0 г у 1-й день, потім 0,5 г у 1 прийом |
29 |
|
Рокситроміцин |
0,3 г у 2 прийоми |
30 |
|
Кларитроміцин |
0,5 г у 2 прийоми |
30 |
|
Тетрацикліни |
||
|
Тетрациклін |
2,0 г у 4 прийоми |
30 |
|
Метациклін |
0,9 г у 3 прийоми |
30 |
|
Доксициклін |
0,3 г у 3 прийоми |
30 |
|
Миноциклін |
0,2 г у 2 прийоми |
30 |
|
Фторхінолони |
||
|
Офлоксацин |
0,6 г у 2 прийоми |
28 |
|
Ципрофлоксацин |
1,5 г у 2 прийоми |
28 |
|
Пефлоксацин |
0,8 г у 2 прийоми |
28 |
|
Ломефлоксацин |
0,4–0,8 г у 1 або 2 прийоми |
28 |
Про ефективність антибіотиків при лікуванні РеА свідчать наступні факти:
- своєчасне лікування уретриту попереджує розвиток артриту;
- вони зменшують тривалість суглобової атаки;
- можуть попередити рецидив у випадку загострення уретриту;
- незначною мірою впливають на перебіг хронічного урогенного артриту;
- антибіотики не впливають на тривалість та прогноз постентероколітичного артриту (виявляють швидку елімінацію збудника);
- тетрацикліни (доксициклін) пригнічують експресію матриксних металопротеїназ.
Одночасно урологом і гінекологом проводиться локальне лікування запального процесу в сечостатевих органах. З метою пригнічення запального процесу в суглобах призначають НПЗП. ГК застосовують у вигляді внутрішньосуглобових або навколосуглобових ін’єкцій. Усередину їх застосовують тільки при тяжких позасуглобових проявах (кардит, поліневрит та ін.). Гідроксихлорохін призначають при затяжних і хронічних формах артриту, проте ефективність його є низькою. Для відновлення функції опорно-рухового апарату призначають ЛФК, масаж, фізіотерапевтичні методи (фонофорез гідрокортизону, електрофорез хлориду літію, кальцію, аплікації озокериту, парафіну). Хворих із залишковими явищами направляють на санаторно-курортне лікування (сірчановодневі, радонові, нафталанні ванни, грязетерапія).
Особливості терапії хворих з синдромом Рейтера хламідійної етіології. Хламідійна інфекція тяжко піддається антибактеріальній терапії. Це пов’язано з особливостями розвитку цього мікроорганізму. Він має двофазний цикл розвитку, який полягає в чергуванні двох форм, — елементарних і ретикулярних тілець (табл. 24.39).
Таблиця 24.39
Цикл розвитку хламідійної інфекції
|
Елементарні тільця |
Розташовуються позаклітинно, метаболічно неактивні, стійкі до дії антибактеріальних засобів. Після поглинання їх клітинами (шляхом ендоцитозу) виявляються в ендосомах, де збільшуються в розмірах і перетворюються на ретикулярні тільця, що активно діляться |
|
Ретикулярні тільця |
Їх внутрішньоклітинні колонії можуть налічувати до 1000 мікроорганізмів. Ретикулярні тільця секретують білки, які застерігають їх злиття з лізосомами. Покидаючи клітини, вони інфікують інші і зазнають нового циклу розвитку |
Клітини, які залишили хламідії, можуть посилено проліферувати. Хламідії пригнічують експресію білків головного комплексу гістосумісності II класу, й інфіковані клітини не розпізнаються імунною системою. Стійкість до антибактеріальних препаратів визначається також низьким рівнем метаболізму часток. Нині є штами, стійкі до макролідів і багатьох антибіотиків. Антибіотики, до яких чутливі хламідії, представлені вище. Лікування пацієнтів з хворобою Рейтера проводять одним з препаратів групи макролідів або тетрациклінів, а фторхінолові препарати, у тому числі грепафлоксацин, клінафлоксацин, гатифлоксацин та ін.), застосовують як препарати 2-го ряду. Курс лікування триває, як ми вже зазначали, не менше 4 тиж з наступним щомісячним бактеріологічним контролем (мінімум трьохразовим). Застосування антибіотиків при хламідійному уретриті попереджає розвиток артриту. Водночас дані про вплив їх на суглобовий синдром суперечливі. При усуненні урогенітальної інфекції не розвивається хронізація процесу, рідше настають рецидиви. При урогенному артриті хламідійна інфекція більшою мірою стійка до антибіотиків, ніж при уретриті, неускладненому суглобовим синдромом. Неускладнений урогенітальний хламідіоз можна усувати 7–10 днів, але такі курси недопустимі при розвитку хламідіозного РеА. При виявленні трихомонад перед курсом антибіотиків призначають протягом 7–10 днів метронідазол по 0,5 г 2–3 рази на день. Доказів підвищення ефективності лікування хворих на хламідійний РеА при тривалому застосуванні (до 3 міс) антибіотиків немає. Вагітним жінкам протипоказане призначення доксицикліну і офлоксацину. Високоефективні схеми антибактеріальної терапії досі не розроблені, тому через 3 тиж після завершення лікування рекомендується повторне тестування. Якщо його результати позитивні або відновилася урогенітальна симптоматика, потрібний повторний курс лікування зі зміною антибіотика.
Лікування артриту включає призначення НПЗП, ГК (внутрішньосуглобово, в синовіальні сумки при бурситах, в болісні зони при ентезопатіях), препаратів системної ензимотерапії. При агресивному перебігу суглобового синдрому призначають метотрексат 10–25 мг/тиж протягом 1,5–4 міс. Ефективним може бути азатіоприн, зовсім неефективні препарати золота.
Особливості лікування постентероколітичних артритів. Призначають антибактеріальні препарати, незважаючи на те, що патологічний процес в кишечнику може бути завершений, а бактеріологічне дослідження дає негативні результати. Застосовують тетрациклін, до якого чутливі усі ентеробактерії. Курс має тривати не менше 2 тиж. Проте, як правило, антибактеріальна терапія при постентероколітичних артритах неефективна. При ієрсиніозі, крім тетрацикліну, застосовують хлорамфенікол (2 г/добу), офлоксацин (0,2 г 3 рази на добу), ципрофлоксацин (0,5 г 2 рази на добу), гентаміцин (160–240 мг/добу); при шигельозі — хлорамфенікол 2 г/добу, ко-тримоксазол 2 таблетки 2 рази на добу, нітрофуранові похідні (фуралтадон, фуразидин та ін.) по 0,1–0,15 г 4 рази на добу; при сальмонельозі — хлорамфенікол 2 г/добу. Тривалість курсу така сама (до 2 тиж). У разі дисбіозу призначають біфідобактерії біфідум 5–15 доз щоденно протягом 1,5–2 міс. При лікуванні артриту застосовують НПЗП, ГК для введення внутрішньосуглобово, в синовіальні сумки, в місця прикріплення сухожиль, а також усередину при системних проявах. При РеА НПЗП призначають в повних дозах. Вибір препаратів проводять залежно від індивідуальної переносимості і чутливості. Контрольовані дослідження ефективності ГК при РеА не проводилися. При хронізації артриту призначають сульфасалазин в дозі 1,5–2 г/добу протягом 6–12 міс, метотрексат. Результати плацебо-контрольованих досліджень ефективності сульфасалазину носять суперечливий характер. Не проводилися контрольовані дослідження ефективності при РеА метотрексату, азатіоприну, солей золота. Після стихання запального синдрому призначають ЛФК, масаж, фізіотерапевтичні процедури. Санаторно-курортне лікування включає застосування сірчановодневих, радонових, нафталанних ванн, грязелікування.
Профілактичні заходи при синдромі Рейтера включають:
- дотримання заходів санітарії і гігієни статевого життя;
- обмеження статевих контактів;
- використання презервативів;
- своєчасне лікування запальних захворювань сечостатевої сфери;
- обстеження статевих партнерів хворих з синдромом Рейтера і їх лікування.
Принципи диспансеризації хворих на РеА. Рекомендується через 1–3 міс здійснити перший клініко-лабораторний контроль, надалі його повторюють кожні 6 міс протягом 3 років. Вважають за необхідне після цього проводити щорічні обстеження (загальний стан, тест-контроль активності запалення, стан імунної системи, виключення сечостатевої інфекції). Велике значення має проведення профілактики ре- і суперінфекцій.
Прогноз
У більшості хворих він є сприятливим, зазвичай перша атака артриту триває 2–3 міс (рідше до 1 року). Повторне інфікування супроводжується рецидивами. Хронізацію РеА виявляють у 20–50% пацієнтів, передусім за наявності в дебюті тріади Рейтера. Порушення працездатності спостерігається у 15% хворих. Несприятливий прогноз реєструють у пацієнтів з ураженням серця і амілоїдозом нирок. До факторів ризику несприятливого прогнозу і хронізації хвороби належать: чоловіча стать, вік до 16 років, носійство HLA-B27, висока лабораторна активність упродовж 3 міс, урогенна форма захворювання, наявність позасуглобових проявів, низька ефективність НПЗП, олігоартрит, сосископодібна дефігурація пальців стоп, артрит кульшових суглобів, обмеження рухливості хребта. Ієрсиніозний РеА рецидивує в 5% випадків, сальмонельозний — в 25%, індукований хламідіями — в 68%.
HLA-B27-незалежні реактивні артропатії
Артрити, що виникають після носоглоткової інфекції (так звані ринофарингеальні артрити). Початку артриту передує гостра інфекція (частіше стрептококова) верхніх дихальних шляхів — гостре респіраторне захворювання, гостра ангіна, фарингіт, бронхіт. У середньому через 10–15 днів після початку інфекції виникає гостре або підгостре запалення суглобів: припухлість, гіперемія, підвищення місцевої температури, обмеження рухів через біль і запалення. Вираженість цієї симптоматики збільшується протягом 2–3 днів. У разі попереднього або супутнього прийому антибіотиків, протизапальних і антигістамінних засобів, перебіг суглобового синдрому може бути дуже млявим. Припухлість суглобів (передусім колінних) зумовлена випотом в порожнину суглобів, періартикулярні тканини не уражуються. При пальпації суглобів відзначають болісність, місцеву гіпертермію. У більшості пацієнтів (71%) запалюються декілька суглобів, розвивається поліартрит. Найчастіше уражуються гомілковостопні суглоби (у 75%), трохи рідше (у 58–60%) — колінні. Середня тривалість суглобового синдрому, що розвинувся при носоглотковій інфекції, становить 1–2 міс, у деяких хворих спостерігається затяжний перебіг (≥6 міс). Після повторної гострої інфекції, переохолодження у частини хворих виникають рецидиви артриту. У разі хронізації суглобового процесу слід виключити РА. При значному випоті в порожнину колінних суглобів виникає необхідність диференціації з інтермітуючим гідрартрозом. У цілому перебіг його доброякісний з повним відновленням функції.
При лабораторному обстеженні відзначають збільшення ШОЕ до 25–30 мм/год (рідко до 40–45 мм/год), вмісту в крові серомукоїду, гаптоглобіну, СРБ, α2-, γ-глобулінів та імуноглобулінів (рідко). У більшості хворих (62%) титр антистрептолізину-О перевищує 1:500. Діагностичні критерії артриту, що виникає після носоглоткової інфекції:
- виникнення артриту після гострого респіраторного захворювання, ангіни, фарингіту, трахеобронхіту;
- швидке (протягом 2–3 днів) збільшення вираженості ознак запалення в суглобах;
- переважне ураження у вигляді поліартриту із залученням в більшості випадків гомілковостопних і колінних суглобів;
- відсутність летючості суглобового ураження;
- доброякісний характер перебігу артриту з повним зворотним розвитком запального процесу;
- відсутність у цих хворих антигену гістосумісності HLA-B27.
Лікування
Метою лікування є санація вогнища інфекції і пригнічення активного суглобового процесу. Застосовують антибіотики ряду пеніцилінів, макроліди (еритроміцин, кларитроміцин, рокситроміцин, спіраміцин, азитроміцин). Для пригнічення активності запального процесу в суглобах використовують НПЗП в загальноприйнятих лікувальних дозах. При затяжному перебігу можливе місцеве (внутрішньосуглобове і навколосуглобове) застосування ГК (бетаметазон, тріамцинолону ацетонiд), а також призначення усередину амінохінолінових препаратів (гідроксихлорохін). Після купірування запалення застосовують ЛФК, масаж, фізіотерапевтичні методи (фонофорез гідрокортизону, електрофорез кальцію, хлориду літію, аплікації парафіну, озокериту). Хворих із залишковими явищами артриту направляють на санаторно-курортне лікування з використанням сірчановодневих, радонових, нафталанних ванн, грязетерапії.
Постімунізаційні артрити
Ці артрити найчастіше є одним з проявів медикаментозної хвороби і виникають в результаті потрапляння в організм антигенів мікробів під час вакцинації. Нерідко суглобовий синдром поєднується з алергічними проявами — кропив’янкою, набряком Квінке, бронхоспастичним синдромом, підвищенням температури тіла. Артрит виникає відразу після введення (особливо повторного) антигену, рідше — через 2–4 тиж. Гостре або підгостре запалення суглобів розвивається за типом моно- або олігоартриту. У великих суглобах (часто уражуються ліктьові суглоби) розвиваються припухлість, скутість, обмеження руху, з’являється ексудат. У синовіальній рідині виявляють у великій кількості лейкоцити (переважно нейтрофіли). У більшості випадків прояви повністю зворотні (явища можуть тривати від декількох днів до 1–2 міс). У 4% випадків артриту спостерігається важкий перебіг з сильним болем, ексудатом і навіть з деструкцією суглобових поверхонь великих суглобів.
Діагностика
Зазвичай при рентгенологічному дослідженні суглобів у хворих з постімунізаційним артритом зміни в суглобах не виявляють, за винятком застосування вакцини проти віспи, коли спостерігається первинне ураження кісток вірусом (розвивається інфекційний артрит), їх деструкція. Процес переходить на синовіальну оболонку, суглобові тканини. При встановленні діагнозу постімунізаційного артриту передусім слід враховувати факт попереднього введення вакцини, загальну картину медикаментозної хвороби, швидку зворотність суглобового синдрому.
Лікування
Призначають ГК, антигістамінні, препарати кальцію, НПЗП.
Артрит у хворих з обхідним шунтуванням кишок. Артрит розвивається у 20–80% хворих, яким проведено обхідне шунтування кишечнику з приводу пухлини, ожиріння тяжкого ступеня, а також у хворих з резекцією ≥3 м тонкої кишки з наступним накладенням анастомозу з товстою кишкою. Основою патогенезу артриту є надлишкове збільшення кількості мікробів в сліпій петлі кишечнику з наступною антигенною стимуляцією, формуванням імунних комплексів, відкладенням їх в суглобах і шкірі. До характерних особливостей артриту при кишковому обхідному шунтуванні належать: 1) поліартикулярний, асиметричний, мігруючий характер ураження; 2) залучення суглобів як верхніх, так і нижніх кінцівок; 3) можливість рецидивування артриту у 25% хворих; 4) частий (у 80% хворих) одночасний розвиток шкірних уражень у вигляді плямистопапульозного і везикулопустульозного висипу; 5) відсутність у більшості випадків рентгенологічних змін.
Клінічну симптоматику артриту полегшує призначення усередину антибіотиків, НПЗП. Повне усунення артриту настає після ліквідації сліпої петлі кишечнику.
Серонегативні спондилоартрити при виразковому коліті та хворобі Крона
Хронічні запальні захворювання кишечнику (ЗЗК) (виразковий коліт (неспецифічний виразковий коліт), хвороба Крона) належать до серонегативних спондилоартритів.
Виразковий коліт
Виразковий коліт (ВК, неспецифічний виразковий коліт) — хронічне прогресуюче запальне захворювання з виразково-деструктивними змінами слизової оболонки ободової і прямої кишки, що характеризується ускладненнями (звуження просвіту кишки, перфорація стінки кишки, кровотечі, сепсис). У основі ВК лежить хронічне рецидивуюче виразково-некротичне запалення слизової оболонки товстої кишки неспецифічного характеру. У 4% хворих виявляють ураження дистального відділу клубової кишки.
Код за МКХ-10
М07.5 Артропатія при виразковому коліті (К51.-).
Епідеміологія
На частку ВК припадає від 4,9 до 10,9 випадку на 10 тис. стаціонарних хворих. Частота ВК становить 40–80 випадків на 100 тис. населення. Він частіше зустрічається у віці 20–50 років, серед хворих переважають жінки.
Етіологія і патогенез
Етіологія залишається невідомою. У організм з довкілля попадають антигени, що викликають ревматичні захворювання. Вони потрапляють через шкіру, слизову оболонку дихальних і сечостатевих шляхів, ШКТ. Однією з функцій кишечнику (окрім переробки і всмоктування поживних речовин) є усунення потенційно небезпечних антигенів, що надходять з їжею (площа всмоктувальної поверхні кишечнику досягає 1000 м2). Деструкція антигенів, що надходять з їжею, відбувається під впливом соляної кислоти, пепсину, жовчних кислот, ферментів підшлункової залози і тонкого кишечнику, мікроорганізмів — товстого. Порушення продукції перелічених вище інгредієнтів веде до розвитку дисбіозу і вимагає проведення замісної терапії.
Проникненню антигенів через ШКТ перешкоджає лімфоїдна тканина, що становить близько 25% маси слизової оболонки. Вона включає пейєрові бляшки, власну пластинку слизової оболонки, внутрішньоепітеліальні Т-клітини. Запальний процес, що виникає в кишечнику, супроводжується порушенням цілісності слизової оболонки, підвищенням проникності кишкової стінки. У результаті з просвіту кишечнику в кров проникають нежиттєздатні бактеріальні антигени (у 1 г калових мас товстого кишечнику знаходиться близько 1012 мікробів). Мікробні антигени, що потрапляють в кров, надалі відкладаються в синовіальній оболонці суглобів, викликаючи місцеву запальну реакцію. Крім того, антигени викликають системну імунну відповідь, результатом якої є відкладення імунних комплексів в синовіальній оболонці суглобів, шкірі, інших органах.
Класифікація
Виділяють легку, середньотяжку, тяжку і блискавичну форми хвороби. Гостра форма (до 10% випадків) триває до 6 міс, надалі можливі тяжкі ускладнення, які загрожують життю (летальність становить 30–100%), або хвороба набуває хронічний перебіг. «Блискавичні» форми нерідко закінчуються смертю в перші дні хвороби. Хронічні рецидивуючі форми (до 70%) характеризуються циклічністю: є фази загострення, згасаючого загострення, ремісії. Можлива хронічна безперервна форма. За поширеністю процесу виділяють коліт тотальний, а також сегментарний — дистальний (проктит, проктосигмоїдит), лівобічний, правобічний. Можливе як поверхневе ураження, так і глибоке (виразки, псевдополіпи, склероз стінки кишки).
Клінічна картина
Захворювання може починатися з неприємних відчуттів при дефекації, потім з’являються домішки крові у випорожненнях. Повне розгортання клінічної картини спостерігають через 1–2 міс. Іноді захворювання розпочинається із запорів, болю в животі, болю в суглобах, субфебрильної температури. Можливий дизентерієподібний початок: пронос з кров’ю, інтоксикація, лихоманка, розгорнута картина хвороби формується вже через 3–5 днів. У вираженій стадії головним симптомом є кровотечі. Частота випорожнень коливається від 3–4 до 20 разів на добу при тяжкій формі. Вони частішають після їди і вранці після пробудження. Біль в животі — другорядна ознака, яка спостерігається у ⅔ хворих. Біль переймоподібний, локалізується в лівій клубовій ділянці, гіпогастрії, посилюється перед випорожненням, зменшується після них, у деяких пацієнтів його вираженість збільшується через 0,5–1,5 год після їди. Досить часто відзначають тенезми з болем в прямій кишці і неповним випорожненням. Негативно позначається на загальному стані: виникають слабкість, зниження працездатності, головний біль, безсоння, астенізація, зменшення маси тіла, порушується білковий, водно-сольовий обмін, вітамінний баланс, розвивається гіпохромна анемія.
Ускладнення. Виникають ускладнення, як місцевого, так і системного характеру (табл. 24.40).
Таблиця 24.40
Ускладнення ВК
|
Місцеві ускладнення |
Спостерігаються аноректальні ускладнення (анальні тріщини, періанальні абсцеси), запальні поліпи товстої кишки, дисбіоз, перфорація товстої кишки, «токсична кишка». Розвиваються звуження і укорочення товстої кишки, при цьому втрачається зв’язок між болем в животі і прийомом їжі, хворі можуть не відчувати позиву до дефекації і рідкий кал виливається в ліжко |
|
Позакишкові прояви |
До них відносять шкірні зміни — піодермію, фурункульоз, трофічні виразки на гомілках, вузлувату еритему. При тяжких формах відзначають ураження слизових оболонок порожнини рота (афтозний стоматит, глосит, гінгівіти), ірити, іридоцикліти, епісклерити. Серед позакишкових проявів на другому місці знаходиться ураження суглобів — артрит, поліартрит, спондиліт, сакроілеїт, васкуліт, поліневропатія, міокардіодистрофія. Можливий розвиток склерозуючого холангіту, жовчнокам’яної хвороби, холестазу, гепатиту, жирової дистрофії печінки, інтерстиціального панкреатиту з зовнішньосекреторною недостатністю |
Високим є ризик розвитку раку товстої кишки. При ЗЗК (ВК, хворобі Крона) відзначають схильність до тромбозів, тромбоемболій (у тому числі артерій головного та спинного мозку). Розиваються інсульти, судинна мієлопатія, тромбоз венозних синусів. Іноді розвивається гостра або хронічна запальна демієлінізуюча полірадикулоневропатія, множинна мононевропатія, краніальна невропатія, плечова плексопатія, міастенія, ДМ.
Ревматичні прояви виразкового коліту
Для цього захворювання характерні: 1) мігруючий артрит великих суглобів (у 15%); 2) сакроілеїт і спондилоартрит (частіше за наявності HLA-B27), які розвиваються на тлі імунних порушень: дисбалансу між Т-хелперами і Т-супресорами, підвищеного титру антитіл, ЦІК, зниження продукції ІЛ-2 і відповіді лімфоцитів на нього. Сакроілеїт, спондиліт можуть випереджати запалення в кишечнику, що утрудняє своєчасне встановлення діагнозу. Особливості периферичного артриту при ВК:
- гострий початок;
- олігоартикулярний мігруючий характер;
- асиметричність ураження;
- синовіальна рідина запального характеру і містить до 50 000 лейкоцитів (переважно нейтрофілів) в 1 мкл;
- результати посіву синовіальної рідини негативні, кристали не виявляються;
- явища артриту купіруються через 1–2 міс, деформації і рентгенологічні зміни не розвиваються.
Слід зазначити, що перелічені особливості характерні і для суглобового синдрому на тлі хвороби Крона. Частоту ураження суглобів при ВК і хворобі Крона надано в табл. 24.41 (West S., 1999).
Таблиця 24.41
Частота ураження суглобів при ВК і хворобі Крона
|
Суглоби |
ВК, % |
Хвороба Крона, % |
|
Плечовий |
20 |
20 |
|
Ліктьовий |
30 |
10 |
|
Зап’ясний |
15 |
15 |
|
Міжп’ясткові і фалангові |
25 |
10 (міжп’ясткові) |
|
Кульшовий |
80 |
– |
|
Колінний |
70 |
80 |
|
Гомілковостопний |
50 |
40 |
|
Плеснові і пальців стоп |
10 |
– |
З вищенаведених даних видно, що переважно уражуються суглоби нижніх кінцівок (колінні та гомілковостопні). Дрібні суглоби уражуються при ВК частіше, ніж при хворобі Крона, хоча в цілому частота ураження цих суглобів невисока. Периферичний артрит супроводжують позакишкові прояви: гангренозна піодермія (12%); афтозний стоматит (менше 10%); передній увеїт (5–15%); вузлувата еритема (9%). Артрит при ВК і хворобі Крона розвивається при ураженні товстого кишечнику. У 60–70% випадків він виникає при загостренні процесу в кишечнику, проте, як ми вже зазначили, може випереджати симптоми ВК, хвороби Крона. Особливості артриту та спондиліту у хворих із ЗЗК наведено у табл. 24.42.
Таблиця 24.42
Особливості артриту та спондиліту у хворих із ЗЗК
|
Ураження суглобів |
|
Розвиток артриту пов’язаний з ентеропатією, високою активністю хвороби (артрит є системним проявом ЗЗК) |
|
Спостерігається кореляція між загостренням ентеропатії та артриту |
|
Іноді артрит може виникати раніше (на місяці та роки), ніж розів’ється клінічна картина ентеропатії |
|
Асиметричний та мігруючий характер ураження, обмеженість кількості уражених суглобів (до 5) |
|
Переважне ураження суглобів нижніх кінцівок |
|
Чергування загострень (тривають до 2–3 міс) та ремісій |
|
Розвиток ентезопатій — сосископодібних пальців (дактилітів), ахіллодинії, талалгії |
|
Можливий розвиток ерозій в п’ястково-фалангових та плеснофалангових суглобах |
|
Розвиток зрідка локального ОП, періоститів, гіпертрофічної остеоартропатії (при хворобі Крона) |
|
У хворих з норицями (хвороба Крона) може розвинутися септичний артрит |
|
Ураження хребта |
|
При ЗЗК спондиліт як єдине ураження опорно-рухового апарату буває рідко, частіше він поєднується із олігоартикулярним периферичним суглобовим синдромом |
|
Аксіальне ураження часто передує розвитку ентеропатії |
|
Клінічна картина сакроілеїту та спондилоартриту при ЗЗК не відрізняється від такої при АС |
|
Відзначають біль в хребті запального характеру, обмеження рухів у поперековому відділі хребта та екскурсії грудної клітки, ранкову ригідність |
|
Спондиліт супроводжується ураженням периферичних суглобів (у тому числі дрібних суглобів кистей та стоп) та дактилітами (чого не спострігається при АС) |
|
Ураження аксіального скелета у жінок із ЗЗК відзначають у більш молодому віці, ніж при АС |
Виділяють 2 типи артритів при ЗЗК (табл. 24.43).
Таблиця 24.43
Типи артритів при ЗЗК
|
Типи артритів |
Характеристика |
|
Олігоартикулярний |
Суглобовий синдром передує клінічним проявам ЗЗК, асиметрично уражуються до 5 суглобів. Загострення артриту асоціюються з загостренням ентеропатії. Перебіг артриту гострий, ефективність ГК висока з повним купіруванням проявів запалення. Відзначають інші системні прояви (вузлувата еритема, увеїт та ін.) |
|
Поліартикулярний |
Процес двобічний, симетричний, характер перебігу первинно-хронічний. Уражуються багато суглобів і найбільш часто — п’ястково-фалангові. Загострення артриту, розвиток синовіту не збігаються із загостренням ентеропатії та іншими системними проявами |
Слід зазначити, що спондиліт при ЗЗК відповідає класифікаційним критеріям спондилоартропатій, розроблених Європейською групою з вивчення серонегативних спондилоартритів (ESSG), а також діагностичним критеріям ідіопатичного АС.
Особливості проявів сакроілеїту і спондилоартриту. Сакроілеїт розвивається у 4–18% хворих на ВК, спондиліт — у 3–6%. Перебіг їх малосимптомний. Характерне поступове посилення болю і скутості в крижово-клубовій ділянці, грудному відділі хребта як при рухах, так і в спокої. Сакроілеїт двобічний, можливе анкілозування. Рентгенологічно він виявляється раніше спондиліту. Спондиліт проявляється (окрім болю і скутості) обмеженням при згинанні, зменшенням дихальної екскурсії грудної клітки. Процес може закінчитися знерухомленням хребта в результаті осифікації міжхребцевих зв’язок, зрощення тіл хребців. Спондиліт супроводжується розвитком іридоцикліту (у 53–75% хворих виявляють HLA-B27), а периферичний артрит ускладнюється кератокон’юнктивітом.
Інші ревматичні прояви ВК. До них належать вузлувата еритема (відзначають у 9% пацієнтів) і гангренозна піодермія (близько 12%), можливий розвиток васкулітів, артеріальних і венозних тромбозів. У основі гангренозної піодермії лежить деструктивно-проліферативний васкуліт з абсцедуванням, мікротромбозом венул, капілярів, лімфогістіоцитарною інфільтрацією. Множинні папульозно-пустульозні висипання, які з’являються переважно на шкірі нижніх кінцівок, схильні до утворення абсцесів і виразок. Ці зміни розвиваються на тлі пригнічення реакцій гіперчутливості уповільненого типу, порушення хемотаксису, здатності до фагоцитозу. Гангренозна піодермія зберігається і після хірургічного лікування ВК. Терапія ГК у високих дозах може призвести до розвитку некрозу скелетних м’язів, тяжкого поліміозиту, а застосування сульфаніламідів — до розвитку вовчакоподібного синдрому.
Поєднання ВК з іншими хворобами. Описано поєднання ВК з саркоїдозом (Крикунов В.П., Головизнин М.В., 1997). При цьому разом з характерним запальним процесом в кишечнику відзначають характерні гранулематозні утворення в легенях, грудних лімфатичних вузлах, печінці. Легеневі прояви саркоїдозу (задишка, кашель, кровохаркання) можуть випереджати клінічні симптоми ВК. Як і ВК, саркоїдоз може супроводжуватися розвитком іридоцикліту і вузлуватої еритеми.
Труднощі діагностики ревматичних проявів ВК. Труднощі діагностики ревматичних проявів ВК полягають в тому, що сакроілеїт, спондиліт, гангренозна піодермія можуть випереджати розвиток запального процесу в кишечнику. Прогресування цих ускладнень не залежить від перебігу процесу в кишечнику (загострення і ремісії). За наявності ревматичних проявів диференціацію проводять з хворобами Бехчета, Кавасакі, артеріїтом Такаясу, системними васкулітами (табл. 24.44).
Таблиця 24.44
Відмітні ознаки синдромів Бехчета, Кавасакі, Такаясу
|
Хвороби |
Відмітні ознаки |
|
Бехчета |
Можливий розвиток виразок в товстій кишці, проте вони з’являються на тлі нормальної (не запаленої) слизової оболонки, і одночасно виникають виразки на статевих органах |
|
Кавасакі |
Окрім лімфаденопатії, папульозних висипань на шкірі і слизових оболонках, розвивається запальне ураження вінцевих артерій (коронарит). Водночас ВК супроводжується розвитком міокардиту, іноді тяжкого з кардіомегалією, серцевою недостатністю |
|
Артеріїт Такаясу |
Може супроводжуватися стенозом верхньобрижової артерії, проявами ішемічного коліту. Колоноскопія, рентгенологічне дослідження дозволяють диференціювати ці хвороби. У тяжких випадках проводять ангіографію брижових артерій |
Слід враховувати, що на фоні ВК може розвинутися ВП.
Лікування виразкового коліту, його ревматичних проявів
Основу базисної терапії ВК становлять препарати месалазину, системні і локальні ГК, імуносупресори. Мінімальна доза месалазину при загостренні ВК становить 4 г/добу. Вищі дози (6–10 г/добу) сприяють швидкій ліквідації клінічних симптомів хвороби, досягненню ремісії (клінічної і ендоскопічної). Тривалість лікування при загостренні визначається швидкістю зменшення клінічних проявів і запального процесу. Мінімальна підтримувальна доза месалазину становить 2 г/добу. Безперервне лікування триває протягом 2 і більше років. Монотерапія месалазином застосовується при легкій і середньотяжкій формах ВК. За відсутності ефекту, а також при тяжких формах хвороби використовують засоби другої лінії — преднізолон і метилпреднізолон. Їх призначають усередину в дозі 1 мг/кг/добу до отримання ефекту (4–6 тиж), потім дозу знижують по 10 мг кожні 10 днів до 30 мг/добу. Надалі темп зниження становить 2,5 мг кожні 7–10 днів, підтримувальну дозу приймають щодня або 10 мг через день. Курс лікування триває 6–9 міс, відміняють преднізолон (метилпреднізолон) при добовій дозі, що не перевищує 5 мг. За неможливості вживання усередину і важкій формі ГК (дексаметазон, бетаметазон, гідрокортизон) вводять парентерально. При легкій і середній тяжкості ВК призначають будесонід у вигляді капсул і піни для ректального введення. Доза препарату при термінальному ілеїті і правобічному коліті становить 9 мг/добу усередину (по 3 мг 3 рази), при ураженні поперечної і низхідної кишки її збільшують до 18 мг/добу. Ректальну піну (2–4 мг/добу) призначають при дистальному і лівобічному коліті. Будесонід застосовують до 8 тиж з подальшим зниженням дози протягом 2 тиж. При неефективності препаратів першої і другої лінії призначають азатіоприн (2–2,5 мг/кг/добу) або циклоспорин А (4 мг/кг/добу). У тяжких випадках лікування починають з в/в введення циклоспорину протягом 2 тиж з подальшим переходом на пероральний прийом азатіоприну. У зв’язку з відстроченим ефектом цитостатиків їх поєднують з ГК.
Лікування периферичного артриту включає призначення сульфасалазину, метотрексату, циклоспорину А. Лікування сакроілеїту і спондиліту проводять так само, як і при хворобі Бехтєрєва (вони мають незалежний від запалення в кишечнику перебіг). У разі розвитку системних васкулітів призначаються ГК, імуносупресори у високих дозах. Тривалий прийом ГК (більше 7,5 мг/добу преднізолону протягом 3 міс і більше) вимагає проведення профілактики ОП — призначення кальцію (1000–1500 мг/добу) і вітаміну D3 (800 МО/добу).
Суттєве значення мають лікування дисбіозу, корекція білкового обміну, водно-електролітного балансу, анемії, призначення ентеросорбентів, препаратів, що нормалізують перистальтику, ферментів підшлункової залози, вітамінів, седативних і психотропних засобів.
Хвороба Крона
Хвороба Крона (термінальний, регіонарний ілеїт, гранулематозний ентероколіт) є гранулематозним запаленням травного тракту невідомої етіології з переважною локалізацією в термінальному відділі клубової кишки. До поширених системних проявів хвороби Крона належать артрити і спондиліти.
Код за МКХ-10
М07.4 Артропатія при хворобі Крона (регіонарному ентериті) (К50.-)
Епідеміологія
На хворобу Крона хворіють 2–75 осіб на 100 тис. населення. Середній вік хворих становить 24–26 років, чоловіки хворіють в 2 рази частіше, ніж жінки. При хворобі Крона частіше уражується термінальний відділ клубової кишки, але до патологічного процесу може залучатися будь-який відділ ШКТ. Ураження товстої кишки діагностують в 30% випадків, множинну локалізацію — в 40%.
Етіологія і патогенез
Етіологія хвороби не встановлена. Спостерігається дефіцит Т-супресорів, посилена продукція лімфокінів, розвиток запалення. Запальний процес починається в підслизовому шарі: утворюються гранульоми, які містять епітеліоїдні і гігантські клітини Лангханса, розвивається гіперплазія лімфоїдних фолікулів, пейєрових бляшок. Потім інфільтрація лімфоцитами і плазматичними клітинами поширюється на м’язовий і субсерозний шари, що призводить до звуження просвіту кишки. Слизова оболонка залишається інтактною, вона піднята над гранулематозними інфільтратами (вигляд «бруківки»). Можливе ураження лімфоїдних вузликів з утворенням щілиноподібно-лінійних виразок, які проникають в підслизовий, м’язовий шар, утворення свищів, абсцесів, стриктур. Процес носить сегментарний характер: спостерігається чергування ділянок нормальної і патологічної тканини (протяжність уражених сегментів становить від 3–4 см до 1 м).
Клінічна картина
Загострення хвороби Крона може нагадувати гострий апендицит: процес локалізується в термінальному відділі клубової кишки, підвищується температура тіла, з’являється озноб, біль в правій клубовій ділянці, нудота, блювання. Розвивається метеоризм, пронос зі слизом, іноді з домішками крові. Може пальпуватися інфільтрат, який часто розцінюють як апендикулярний. При тонкокишковій локалізації процесу спостерігаються метеоризм, біль в животі, не пов’язані з характером і кількістю їжі, бурчання і переливання в ньому, діарея, субфебрильна температура. Відзначають випорожнення до 5–6 разів на добу зі слизом, ректальні кровотечі з розвитком анемії. Розвивається синдром мальабсорбції, зменшується маса тіла, приєднується слабкість. Може виникнути стеноз кишки з розвитком тонкокишкової непрохідності. Часто спостерігаються позакишкові прояви: вузлувата еритема, увеїти, іридоцикліти, моноартрити, афтозний стоматит, гіперкератоз, оперізувальний лишай, псоріаз, фурункульоз та ін.
При ураженні товстої кишки клінічна картина розвивається повільно. Характерним є біль в животі невизначеного характеру, діарея, зменшення маси тіла, лихоманка. Кал напівоформлений, періодично водянистий з частотою 4–12 разів на добу. Посилюються дисбіоз, токсичні прояви. Розвиваються стенози товстої кишки з ознаками непрохідності, ректовагінальні, кишково-міхурні, товстотонкокишкові свищі й ураження періанальної ділянки (тріщини, лінійні виразки). Позакишкові прояви такі самі, як при ураженні тонкої кишки.
Ревматичні прояви у пацієнтів з хворобою Крона. Найбільш поширеними варіантами ревматичних проявів є: периферичний артрит, спондиліт, сакроілеїт, ураження очей, дифузний некротизуючий гломерулонефрит, гранулематозне ураження різних органів і систем («метастатична хвороба Крона»). Артрит, який діагностують у пацієнтів з хворобу Крона, має ряд характерних проявів:
- уражуються суглоби нижніх кінцівок;
- суглобова атака зазвичай пов’язана із загостренням запального процесу в кишечнику;
- на початку уражується один суглоб: колінний або гомілковостопний;
- потім протягом декількох днів симетрично залучаються інші (колінні, гомілковостопні, плечові, ліктьові);
- не характерне ураження дрібних суглобів (відзначають рідко);
- окрім болю з’являється ранкова скутість, припухлість запалених суглобів, локальна гіперемія шкіри;
- артрит не супроводжується стійкими деформаціями і контрактурами;
- тривалість загострення становить від 1–2 тиж до 1 міс і більше;
- артрит великих суглобів супроводжується розвитком вузлуватої еритеми;
- вузлики локалізуються під шкірою на передній поверхні гомілок, ліктьових згинах, передпліччях;
- при гістологічному дослідженні виявляють: гостру запальну реакцію сполучної тканини, васкуліт судин шкіри, вогнища фібриноїдного некрозу, лімфогістіоцитарну інфільтрацію;
- замість артриту можуть проявлятися мігруючі артралгії.
Прояви спондиліту і сакроілеїту при хворобі Крона відзначають у 15–20% пацієнтів, вони нічим не відрізняються від таких при ВК:
- прояви спондиліту і сакроілеїту нагадують АС;
- ураження суглобів хребта частіше відзначають у чоловіків (3:1);
- хворі скаржаться на біль в спині, тривалу скутість, особливо вночі і після пробудження;
- при рухах, виконанні фізичних вправ скутість зменшується;
- порушуються рухливість хребта, дихальна екскурсія грудної клітки;
- при об’єктивному обстеженні виявляють болісність крижово-клубових суглобів;
- початок спондиліту, сакроілеїту може випереджати, збігатися і відставати від початку запалення в кишечнику;
- існує взаємозв’язок процесу з HLA-B27 (у носіїв ризик виникнення спондиліту, сакроілеїту в 7–10 разів вищий);
- частота носійства HLA-B27 при сакроілеїті/спондиліті у хворих на ВК становить 70%, хворобі Крона — 55%;
- рентгенологічні ознаки спондиліту і сакроілеїту такі самі, як і при АС.
Інші ревматичні прояви ВК і хвороби Крона:
- тендиніт ахіллового сухожилля, підошовний фасцит;
- синдром Марі — Бамбергера (гіпертрофічна остеоатропатія), пальці — «барабанні палички»;
- ОП внаслідок застосування ГК;
- інфекційний артрит кульшового суглоба, абсцес у ділянці поперекового м’яза в результаті утворення нориць у пацієнтів з хворобою Крона;
- васкуліт;
- амілоїдоз.
Ураження очей у вигляді кон’юнктивіту і епісклериту розвивається у 50% хворих з поліартритом. Причиною їх розвитку є відкладення ЦІК в стінці судин з наступним розвитком запалення. Ці ускладнення з’являються при загостренні хвороби, їх вираженість зменшується при ремісії або успішному лікуванні.
Таке ускладнення, як дифузний некротизуючий гломерулонефрит, є рідкісним і проявляється протеїнурією, гематурією, нефротичним синдромом. У сироватці крові виявляють підвищений рівень ЦІК, титру РФ, в біоптатах — відкладення IgM і G, компонентів комплементу (в базальній мембрані гломерул).
Для так званої метастатичної хвороби Крона є характерним гранулематозне ураження різних органів і систем: гранулематозний міозит, гранульоми кісток і суглобів. Вони супроводжуються значним больовим синдромом, обмеженням рухів, деструктивними змінами в кістках.
З інших проявів у пацієнтів з хворобою Крона можливий розвиток періоститу у вигляді синдрому Марі — Бамбергера і пальців — «барабанних паличок».
Діагностика ревматичних проявів хвороби Крона
При гістологічному дослідженні біоптатів виявляють: неспецифічний синовіт; гіпертрофію ворсинок синовіальної мембрани, набряк її; лімфогістіоцитарну інфільтрацію; гранулематозний васкуліт судин синовіальної мембрани. У синовіальній рідині виявляють: підвищений вміст білка, нейтрофільний лейкоцитоз. При рентгенографії реєструють прогресуючий ОП, відсутність узурацій, при тривалому перебігу можливий розвиток періоститів, одиничних ерозій.
Перебіг артриту не відрізняється від постентероколітичного артриту при ієрсиніозі, сальмонельозі, шигельозі, кампілобактеріозі. У випадку ураження дрібних суглобів (спостерігається рідко) слід виключити РА. У такому разі відсутні стійкі суглобові зміни, кісткова деструкція, в крові й синовіальній рідині не визначається РФ. Проте можливе поєднання РА з хворобою Крона з розвитком ерозивного артриту, деформації суглобів.
Ендоскопічна картина при хворобі Крона не відрізняється від такої при саркоїдозі. Проте при саркоїдозі гранульоми розташовуються в слизовій оболонці, а при хворобі Крона запальний процес захоплює усі шари кишкової стінки. Для саркоїдозу не характерні виразкові й норицеві ураження кишкової стінки.
Можливе поєднання хвороб Крона та Бехчета. При цьому разом з афтозним стоматитом, передньозаднім увеїтом і виразкуванням геніталій відзначають гранулематозне запалення товстої кишки і термінальних відділів тонкої. Сульфасалазин ліквідує запалення в кишечнику, проте не впливає на прояви хвороби Бехчета.
Лікування хвороби Крона і її ревматичних проявів
У період загострення рекомендується механічно і хімічно щадна дієта з достатнім вмістом білка, вітамінів, мінеральних речовин (дієта № 4, потім 4б, 4в). Використовують елементні дієти, парентеральне харчування. Лікування хвороби Крона є аналогічним терапії ВК: призначають препарати месалазину, ГК (в тому числі будесонід), імуносупресори (азатіоприн, 6-меркаптопурин, метотрексат). Мінімальна терапевтична доза месалазину має перевищувати 4,5 г/добу, мінімальна підтримувальна доза становить 2,5 г/добу. Безперервне лікування триває декілька років. Найбільш ефективним є призначення азатіоприну (2–2,5 мг/кг/добу) протягом 3–4 років з подальшим зниженням дози протягом 1–2 років. Меркаптопурин (1,5 мг/кг/добу) призначають при толерантності до азатіоприну. Метотрексат (25 мг/тиж) призначають протягом 12 міс при непереносимості азатіоприну і меркаптопурину. Інфліксимаб показаний при рефрактерності до усіх видів терапії і норицевих формах хвороби Крона. При тяжких формах хвороби Крона інфліксимаб вводять (інфузійно) одноразово в/в в дозі 5 мг/кг, при норицевих формах 3 рази вводять в тій самій дозі з інтервалом 2 і 4 тиж (0-, 2-, 6-й тиждень). За наявності гіпертермії і нориць застосовують ципрофлоксацин і метронідазол у вигляді коротких курсів. У 15–20% випадків стабілізувати перебіг хвороби не вдається. Хворобу Крона вилікувати неможливо як за допомогою медикаментозної терапії, так і хірургічних методів. Абсолютні показання до хірургічного втручання включають перфорацію, кишкову непрохідність, токсичний мегаколон, абсцеси, кровотечі, важку дисплазію, рак товстої кишки. До відносних показань належать відсутність ефекту від консервативної терапії, затримка фізичного розвитку дітей і підлітків, нориці, дисплазія низького ступеня.
При артриті призначають НПЗП в ін’єкціях (диклофенак натрію) і усередину, ГК внутрішньосуглобово. ГК застосовують також при вузлуватій еритемі (бетаметазон 0,25–2 мл). Ефективність лікарських засобів при лікуванні ревматичних проявів у хворих на ЗЗК наведена в табл. 24.45.
Таблиця 24.45
Ефективність лікарських засобів при лікуванні ревматичних проявів ЗЗК
|
НПЗП |
Препарати першої лінії. Вони можуть бути причиною загострення симптомів ентеропатії, виразкового процесу |
|
|
ГК |
Системно |
При системному застосуванні в дозах не менше 30–40 мг/добу швидко пригнічують активність артриту та спондиліту. Їх не призначають за наявності нориць у пацієнтів із хворобою Крона (можливий розвиток бактеріємії) |
|
Локально |
В суглоби вводять у випадку обмеженої кількості уражених суглобів |
|
|
Сульфасалазин |
Дозволяє контролювати запальний процес в периферичних суглобах, але не впливає на стан хребта. Ефективний при ВК, коліті внаслідок хвороби Крона. Терапевтична доза становить до 3,0 г/добу, при неефективності її підвищують до 6–8 г/добу. Для профілактики рецидивів призначають сульфасалазин у дозі 1,5 г/добу |
|
|
Азатіоприн, метотрексат |
Призначають при неефективності сульфасалазину та розвитку залежності від ГК |
|
|
Інфліксимаб |
Призначають у стандартному режимі (3–5 мг/кг на 0; 2; 6-й тижні, а потім кожні 6–8 тиж). Пригнічує запальний процес в кишечнику, контролює прояви артриту, спондиліту, сповільнює деструктивні зміни в осьовому скелеті, сприяє зворотному розвитку ускладнень (нориці, інфільтрати черевної порожнини, періанальні тріщини), системних проявів ЗЗК (гострого переднього увеїту, хронічного холангіту, гангренозної піодермії) |
|
|
Солі золота, похідні хіноліну, D-пеніциламін |
Неефективні |
|
Хірургічне втручання купірує усі ревматичні прояви, за винятком спондиліту і сакроілеїту, проте хвороба Крона рецидивує.
Профілактика ОП у пацієнтів з хворобою Крона. Проведення профілактики потрібне усім, у кого планується тривала (більше 3 міс) терапія ГК в дозі ≥5 мг/добу. Призначають комбіновані препарати кальцію (1000–1500 мг/добу), вітаміну D3 (800 МО/добу) і остеотропних мінералів. Їх також призначають на тлі лікування ОП стронцію ранелатом, алендроновою кислотою, кальцитоніном та ін.
Глава 25. Артрити, пов’язані з інфекцією
Ураження опорно-рухового апарату при інфекційних захворюваннях
Код за МКХ-10
М00–М03 Інфекційні артропатії.
М01 Пряме інфікування суглоба при інфекційних та паразитарних хворобах, класифікованих в інших рубриках.
Етіологія інфекційних артритів
Розрізняють артрити: 1) бактеріальні (стафілококові, гонококові, бруцельозні, спирохетозно-сифілітичні, мікобактеріальні, туберкульозні та ін.); 2) вірусні (при краснусі, епідемічному паротиті, вітряній віспі та ін.); 3) грибкові; 4) паразитарні. Інфекційні артрити бувають гострими і хронічними, гнійними і специфічними.
Патогенез інфекційних артритів, їх ознаки
Інфікування суглобів відбувається при гематогенному поширенні мікробів з вогнища інфекції, потраплянні їх в суглоб при травмі, використанні медичних інструментів. Мікроорганізми, що проникли в порожнину суглоба, інтенсивно розмножуються в синовіальній оболонці й їх можна виявити в синовіальній рідині. Розвитку інфекційного процесу в суглобі сприяє наявність імунодефіциту, резистентності до антибіотиків, масивна інфекція (в антибактеріальному захисті важливе значення мають фагоцитарна система і система комплементу, функція яких в цих випадках порушується). До ознак інфекційних артритів належать: 1) зв’язок з певною інфекцією; 2) позитивні серологічні реакції; 3) ураження суглобів у вигляді моно- й олігоартриту; 4) симетричний характер ураження; 5) хороший ефект від антибактеріальної терапії.
Бактеріальні (гнійні) артрити
Код за МКХ-10
М00 Піогенний артрит.
М00.9 Інфекційний артрит неуточненої етіології.
Т84.5. Інфекція та запальні реакції, зумовлені ендопротезуванням.
Епідеміологія
Захворюваність на бактеріальний артрит становить 2–10 на 100 тис. населення, серед хворих на РА — 30–40 на 100 тис. Інфекція протезованих суглобів виникає у 0,5–2,0% всіх випадків протезування.
Етіологія і патогенез гострого септичного артриту
Він може бути викликаний стафілококами, стрептококами, грамнегативною флорою (ешерихіями, протеєм, клебсієлою та ін.). Гнійний процес поширюється на суглоб з розташованого поблизу гнійного вогнища в кістці (остеомієліт) або в м’яких тканинах — м’язах, сухожильних піхвах, слизових сумках тощо. Джерелом інфекції, яка гематогенно потрапляє в суглоб, можуть бути фурункул, карбункул, ангіна, абсцес м’яких тканин, інфекційні захворювання — скарлатина, тиф, пневмонія. Гематогенним шляхом розвиваються стрептококові артрити. Шляхом безпосереднього поширення виникають стафілококові артрити. При вогнепальних ранах розвивається змішана інфекція піогенними коками і анаеробами. Гнійний процес може розвиватися у вигляді емпієми суглоба або капсульної флегмони. У першому випадку процес обмежується синовіальною оболонкою, в другому — гній поширюється на суглобову капсулу і періартикулярні тканини.
Клініка гострого бактеріального артриту
Він зазвичай розвивається гостро, проявляється припухлістю, гіперемією шкіри і гіпертермією, інтенсивним болем. Одночасно відзначаються гектична лихоманка, озноб, підвищене потовиділення. У аналізах крові виявляють виражений лейкоцитоз, зрушення формули вліво, підвищення ШОЕ. Частіше уражуються суглоби, які несуть значне навантаження: колінні, гомілковостопні, кульшові, плечові, ліктьові та ін. Дрібні суглоби та ступні уражуються рідко.
Діагностика
При підозрі на інфекційний артрит слід негайно зробити пункцію суглоба з лікувальною й діагностичною метою. Зміни, які виявляють при обстеженні хворих з бактеріальним артритом, наведені у табл. 25.1.
Таблиця 25.1
Зміни, які виявляють при лабораторному та інструментальному обстеженні хворих на інфекційний артрит
|
Лабораторні дослідження |
|
|
Аналіз крові |
Лейкоцитоз із зрушенням формули вліво, виражене підвищення ШОЕ |
|
Синовіальна рідина |
Сірувато-жовта або кров’яниста, мутна, з аморфним осадом, кількість лейкоцитів (переважно нейтрофілів) перевищує 50 тис./мм3 (нерідко 100 тис./мм3). Вміст глюкози низький (<50% її концентрації у сироватці крові) |
|
Бактеріологічне дослідження |
Синовіальну рідину сіють на живильне середовище для ана- та аеробних збудників. У разі інфекції протезованого суглоба досліджують біоптат кісткової тканини, взятої біля з’єднання цементу з протезом |
|
Інструментальні дослідження |
|
|
Рентгенографія |
Дозволяє виключити остеомієліт, визначити наявність ОП, звуження суглобової щілини, крайових ерозій |
|
Радіоізотопне сканування (технецій, галій, індій) |
Дозволяє виявити зміни, характерні для бактеріального артриту, на ранніх стадіях хвороби (коли рентгенологічно зміни ще не виявляють) |
|
КТ |
Метод інформативний при ураженні крижово-клубових та груднино-ключичних з’єднань |
|
Магнітно-резонансне дослідження |
На ранніх стадіях виявляє набряк м’яких тканин, випіт у порожнині суглоба, остеомієліт |
У синовіальній рідині виявляють велику кількість лейкоцитів (до 50 тис. в 1 мкл) з переважанням нейтрофілів. Рідина каламутна, в’язкість її знижена, муциновий згусток рихлий. Необхідно провести мікроскопію осаду, використовуючи забарвлення мазків за Грамом, а також посів рідини. У разі застосування антибіотиків до пункції результати посіву можуть бути негативними. Для встановлення можливого вогнища інфекції здійснюють посіви з носоглотки, мокроти, сечі, калу. У процесі діагностики слід враховувати ряд чинників, наведених в табл. 25.2.
Таблиця 25.2
Чинники, які слід враховувати при діагностиці бактеріального артриту
|
Гострий початок хвороби |
|
Тяжкий загальний стан хворого з гектичною лихоманкою, ознобами |
|
Зміни в крові та синовіальній рідині |
|
Виявлення мікроорганізмів в синовіальній оболонці або синовіальній рідині |
|
Запалення, що швидко прогресує |
|
Результати рентгенологічного дослідження |
Зміни в суглобах, що виявляють при рентгенологічному дослідженні, зазвичай включають: 1) епіфізарний ОП; 2) звуження суглобової щілини; 3) деструктивні зміни хряща і кісток суглоба при пізній діагностиці.
Лікування
При гострому бактеріальному артриті слід дотримуватися такої лікувальної тактики:
- іммобілізація кінцівки на термін 7–10 днів;
- своєчасний початок ЛФК;
- щоденний дренаж інфікованої синовіальної рідини;
- призначення антибіотиків з урахуванням чутливості мікрофлори і введення їх парентерально (в/в, в/м);
- видалення наявних некротичних мас хірургічним шляхом;
- використання для оцінки ефективності терапії визначення кількості лейкоцитів, фарбування за Грамом і посіву синовіальної рідини.
Антибіотики, ефективні при бактеріальних (септичних) артритах
Сучасні антибіотики добре проникають в порожнину суглобів при в/в та в/м введенні в організм, тому в суглоби їх не вводять. У першу добу захворювання здійснюють емпіричну терапію антибіотиками, в подальшому — відповідно до чутливості висіяних збудників. Лікування, яке призначають при врахуванні результатів фарбування мазків за Грамом, представлене в табл. 25.3.
Таблиця 25.3
Лікування, яке призначають при врахуванні результатів фарбування за Грамом
|
Мікроорганізми не визначаються |
Цефепім (4 г/добу, 2 введення), ампіцилін + сульбактам (6–12 г/добу, 4 введення), іміпенем + циластатин (2 г/добу, 4 введення) |
|
Грампозитивні коки (гроноподібні колонії) |
Цефазолін (6 г/добу, 3 введення), оксацилін (8–12 г/добу, 4–6 введень), ванкоміцин |
|
Грампозитивні коки у вигляді ланцюжків (можливо, стрептококи) |
Ампіцилін (8 г/добу, 4 введення) |
|
Грамнегативні палички |
Цефтріаксон (2–4 г/добу, 2 введення), ципрофлоксацин (800 мг/добу, 2 введення), цефотаксим (6 г/добу, 3 введення) |
До видалення ексудату з суглоба, усунення лихоманки і болю антибіотики вводять в/в або в/м. Тривалість курсу становить 4–6 тиж, можливе підвищення середніх добових доз антибіотиків в 1–1,5 раза. Після поліпшення стану пацієнтів переходять на прийом препаратів перорально ще 2–4 тиж. Якщо зберігається підвищення ШОЕ, лікування слід продовжити. Зміну антибіотиків проводять за відсутності позитивної динамики через 2 доби. У схеми лікування включають антибіотики (Ананьєва Л.П., 2003), наведені в табл. 25.4.
При виявленні грамнегативної флори (Esherichia coli, Proteus, Klebsiella, Pseudomonas), порушеннях імунної системи ефективним є азтреонам (група монобактамів). При септицемії його вводять в/в або в/м 2 г 2–3 рази на добу, у тяжких випадках — 2 г 3–4 рази. При септицемії, що викликана грамнегативною флорою, тяжких інфекціях кісток і суглобів високоефективними є аміноглікозиди і, зокрема, амікацин, який вводять в/в або в/м із розрахунку 15 мг/кг/добу 2 рівними дозами. Тривалість в/в інфузії становить 30–90 хв, болюсне введення триває не менше 7 хв. Як емпіричну терапію найчастіше використовують комбінацію аміноглікозидів і цефалоспоринів, в критичних випадках — іміпенем (0,5–1 г в/в 3–4 рази на добу). При інфікуванні протезованного суглоба антибіотики призначають на основі мікробіологічного дослідження кісткового біоптату. А терапію проводять не менше 6 тиж у вигляді наступних схем: оксацилін + рифампіцин; ванкоміцин + рифампіцин; цефепім + ципрофлоксацин або цефтазидим + ципрофлоксацин (див. табл. 25.4).
Таблиця 25.4
Антибіотики, які застосовують при септичному артриті
|
Характер інфікування |
Антибіотики |
|
Емпірична антимікробна терапія |
Амоксицилін/клавуланат, ампіцилін/сульбактам, кліндаміцин, лінкоміцин, цефуроксим |
|
Негоспітальна грампозитивна інфекція |
Офлоксацин, цефазолін, ципрофлоксацин, оксацилін + гентаміцин, цефазолін + гентаміцин, цефазолін + нетилміцин, спіраміцин |
|
Госпітальна грамнегативна інфекція |
Ванкоміцин, ванкоміцин + ципрофлоксацин, ванкоміцин + гентаміцин, ципрофлоксацин + фузидієва кислота |
|
Грамнегативна інфекція |
Цефтазидим, цефтріаксон, цефотаксим, лінкоміцин, кліндаміцин, цефалексин |
|
Бактеріальний артрит на тлі післяопераційного остеомієліту |
Іміпенем, меропенем |
|
Інфіковані протезовані суглоби |
Цефазолін + гентаміцин, лінкоміцин + гентаміцин, кліндаміцин + гентаміцин, ванкоміцин + гентаміцин |
Симптоматична терапія включає призначення анальгетиків і НПЗП. Надзвичайно важливим є дренування суглобової порожнини, яке здійснюють шляхом аспірації через голку, відкритого або артроскопічного дренування.
Показання до відкритого хірургічного дренування суглобів
Відкрите хірургічне дренування здійснюють за наявності:
- інфікованого кульшового, плечового (можливо) суглобів, груднино-ключичного з’єднання;
- артриту, який розвинувся внаслідок потрапляння в суглобову порожнину чужорідного тіла;
- неефективної аспірації вмісту суглоба, неможливості видалення гною через голку;
- протезованих суглобів;
- супутнього остеомієліту, у тому числі остеомієліту хребців у випадку стиснення спинного мозку.
При інфекціях протезованих суглобів (кульшових, колінних) проводять одномоментну артропластику з видаленням інфікованих тканин, протезних компонентів, встановленням нового протезу з наступним лікуванням антибіотиками протягом 3–6 міс. Прогноз при бактеріальному артриті може бути досить серйозним (летальність відмічають в 10–15% випадків). Незворотна втрата функцій суглоба розвивається у 25–50% хворих.
Менінгіт
Код за МКХ-10
М01.0 Менінгококовий артрит (А39.8).
М03.0 Постменінгококовий артрит (А39.8).
Епідеміологія менінгококового артриту
Частіше він розвивається у дітей з важким перебігом епідемічного цереброспінального менінгіту, викликаного Neisseria meningitis. Цей менінгіт часто супроводжується септицемією і може ускладнитися гострим бактеріальним артритом, який, як правило, виникає через 1–4 дні з моменту появи менінгіту. Інфікування суглобів відбувається на тлі масивної бактеріємії.
Клінічні особливості артриту
Частіше розвивається артрит з ураженням 2 і більше суглобів. Моно- або олігоартриту може передувати мігруючий поліартрит, який супроводжується припухлістю, гіперемією і болем в суглобах. Залучення до запального процесу навколосуглобових м’яких тканин нехарактерне. Проте, можливі внутрішньосуглобові і періартикулярні крововиливи. Іноді відмічають геморагічно-некротичний шкірний висип. Порушення згортальних властивостей крові пов’язане з розпадом менінгококів, появою в крові бактеріальних і лізосомальних ферментів, активацією системи комплементу. При своєчасно розпочатому лікуванні симптоми поліартриту швидко купіруються без залишкових проявів. При запізненні з терапією в суглобах виникає гнійне запалення. Високі дози антибіотиків купірують прояви гнійного артриту разом з симптомами менінгіту.
Труднощі діагностики менінгококового артриту полягають в тому, що клініка артриту може розвинутися до появи ознак менінгіту, а також в період реконвалесценції. Крім того, артрит може бути єдиним проявом менінгококової інфекції.
Існує точка зору, що артрит, який виникає у хворих з менінгітом, не завжди має інфекційний характер: після бактеріального артриту у пацієнтів може розвиватися імунний (реактивний) оліго- або поліартрит.
Інфекційний ендокардит
Код за МКХ-10
М03.6 Артропатія при інфекційному ендокардиті (I33.0).
Характер артриту у хворих з інфекційним ендокардитом
Його перебіг аналогічний артритам гонококової й менінгококової етіології. Гнійний моно- або олігоартрит поєднується з інфекційним ураженням сухожиль та їх піхв (ахілобурситом, тендосиновітом в ділянці кистей і стоп). Можливий розвиток остеомієліту. В деяких випадках артрит може бути єдиним проявом ІЕ. З самого початку ендокардиту виявляють поліартралгії, міалгії. Також артрити можуть носити реактивний характер, супроводжуватися стерильним серозним випотом в суглоби і крововиливами в них.
Хвороба укусу пацюків (rat bite fever)
Код за МКХ-10
А25 Гарячка від укусу пацюків.
А25.1 Стрептобацильоз.
Етіологія і патогенез захворювання
При укусі пацюка людина інфікується грамнегативною бактерією Streptobacillus moniliformis, що міститься в його глотці (у 50% здорових особин). Можливе зараження через продукти, воду, молоко, забруднені фекаліями пацюків (хаверхільська лихоманка). Ознаки захворювання виникають в середньому через 2–10 діб після укусу. У перші 2–5 діб хвороби відзначаються лихоманка неправильного типу, головний біль, нудота, блювання, поліартралгії, міалгія, збільшення периферичних лімфатичних вузлів, виникає петехіальний, пустульозний, макулопапульозний висип на шкірі кінцівок, долонях. У ділянці укусу розвивається запалення. У цей період розвивається артрит і, таким чином, він поєднується зі шкірними й іншими проявами хвороби. Може розвинутися інтерстиціальна пневмонія, ендокардит, міокардит, перикардит, панкреатит, простатит, абсцеси різних органів, у тому числі, печінки і селезінки. Ендокардит виникає при вже змінених раніше клапанах.
Особливості артриту
Початок артриту носить гострий або підгострий характер, уражується один або декілька суглобів. Артрит не завжди буває гнійним. Уражені суглоби, як правило, віддалені від місця укусу. Частіше уражуються великі суглоби кінцівок, груднино-ключичні, дрібні суглоби кистей і стоп.
Діагностика захворювання
Збудник (Streptobacillus moniliformis) може бути виділений шляхом посіву з крові і синовіальної рідини. Він погано виявляється при мікроскопії, для його розмноження в культурі потрібні певні умови.
При лікуванні хвороби укусу пацюка, що ускладнився гострим бактеріальним артритом, призначають пеніцилін у дозі 18 млн ОД/добу протягом 10–42 днів (Ивашкин В.Т., Султанов В.К., 2005). Для підвищення ефективності лікування проти L-форм збудника додатково призначають стрептоміцин. Показано видалення гною з уражених суглобів, їх іммобілізація.
Еризипелоїд
Код за МКХ-10
А26 Еризипелоїд.
А26.7 Еризипелоїдна септицемія.
Епідеміологія і патогенез еризипелоїду (рожа свиней, повзуча еритема)
Це гостре інфекційне захворювання належить до професійних хвороб: його виявляють у м’ясників (різники), робітників м’ясобоєнь, кухарів, ветеринарів, робочих, що обробляють рибу. Воно викликається грампозитивною бактерією Erysipelothrix rhusiopathie. Джерелом, резервуаром інфекції часто є свині. Збудник проникає через пошкоджену шкіру і через 1–7 днів розвивається запалення, яке надалі захоплює увесь палець, може поширитися на кисть, зап’ясток і передпліччя. Розвиваються лімфангоїт, регіонарний лімфаденіт, підвищується температура тіла. Можливий розвиток септичної форми еризипелоїду з появою вогнищ інфекції в суглобах, ендокарді, міокарді, легенях, ЦНС без утворення гнійників, абсцесів. У 1/3 хворих відмічають шкірний висип.
Особливості бактеріального артриту
Бактеріальний артрит міжфалангового суглоба нерідко виникає в ділянці еритеми. Він супроводжується різким болем, локальною гіперемією і гіпертермією. Можливий розвиток бактеріального артриту віддалених великих суглобів (колінних) без шкірних проявів еризипелоїду. У таких випадках з синовіальної рідини висівається збудник. Артрит триває декілька тижнів і може супроводжуватися деформацією суглобів. У деяких випадках артрит може набувати хронічного рецидивуючого перебігу. Деструкція суглобів відмічається рідко і носить помірний характер. У процесі діагностики враховують характерну клінічну картину, результати посіву синовіальної рідини і біоптатів шкіри.
Лікування
Комплексна терапія включає призначення пеніциліну по 200 тис. ОД кожних 4 год протягом 7 днів або тетрацикліну по 0,5 г 4 рази протягом 7 днів (Ивашкин В.Т., Султанов В.К., 2005).
Чинга-артрит
Етіологія
Це професійне захворювання характерне для мисливців, які полюють на морських зайців, нерпу, тюленів. Вважається, що збудником хвороби є різновид грампозитивного диплококу, якого виявляють у внутрішніх органах звірів, їх шкірі, в салі. У палець людини збудник проникає через пошкоджену шкіру. Він розмножується по ходу судин, викликає лімфоїдну запальну інфільтрацію. Гнійне запалення і некроз, як правило, не виникають. Розвивається запалення шкіри, фасції, м’язів, кісток з їх деструкцією.
Клінічні особливості
Характерним є розвиток інфекційного моноартриту пальців рук зі схильністю до хронічного перебігу і деформації. Через 1–2 тиж після зараження виникає сильний пульсуючий біль, який локалізується в одному з міжфалангових або п’ястково-фалангових суглобів. Уражений суглоб припухає, шкіра над ним гіперемована, ущільнена, напружена, рухи дуже болючі. Іноді приєднується вторинна гнійна інфекція, виникають лімфангоїт, лихоманка, в периферичній крові відзначається нейтрофільний лейкоцитоз (зазвичай зміни в периферичній крові відсутні). Інтенсивність запального процесу підвищується протягом наступних 2–4 тиж, він захоплює періартикулярні тканини, увесь палець, іноді — кисть, передпліччя. Збільшення регіонарних лімфатичних вузлів не виявляють, загальна інтоксикація виражена помірно, температура тіла нормальна. Перебіг артриту тривалий — до 6 міс, можливі рецидиви, деструкція кісткової тканини, утворення деформацій, контрактури. У деяких випадках виникає надмірна рухливість суглоба. Повне одужання зазвичай настає через 2–4 міс.
При рентгенологічному дослідженні суглобів виявляють звуження суглобової щілини, локальний ОП епіфізів фаланг, їх деструкцію, патологічні підвивихи. На пізніх стадіях розвивається анкілоз. Збудник захворювання може бути виділений з уражених тканин.
Лікування
Антибіотики та сульфаніламідні препарати неефективні (Ивашкин В.Т., Султанов В.К., 2005). У разі приєднання вторинної гнійної інфекції призначають пеніцилін або тетрациклін. Проводять блокади ураженого суглоба, при затяжному перебігу артриту — хірургічну обробку суглоба, а також артротомію у разі виражених деструктивних змін.
Черевний тиф, паратифи
Код за МКХ-10
А01 Черевний тиф та паратиф.
М01.3 Артрит при черевному тифі або паратифозній гарячці (А01).
Характер кістково-суглобових уражень
Найчастіше вони виникають наприкінці захворювання або через декілька місяців після одужання, при цьому уражується поперековий відділ хребта. При тифо-паратифозних захворюваннях можуть відмічати артрити й артралгії (тифозний псевдоревматизм), явища артриту іноді домінують (артротиф). Дрібні суглоби до патологічного процесу, як правило, не залучаються. Тифозний і паратифозний процеси уражують міжхребцеві диски (у них відмічають глибокі деструктивні зміни), періартикулярні зв’язки — настає окостеніння lig. longitudinale anterius.
Клінічні прояви спондиліту
Несподівано з’являється сильний біль в попереку, який іррадіює в таз, ділянку сідниць, стегна. Будь-яка зміна пози, рух посилюють біль, різко обмежують рухливість хребта, збільшують скутість. При рентгенографії хребта виявляють значне звуження або зникнення міжхребцевих просторів. Надалі з’являються крайові виступи — кісткові містки між хребцями. Розвиваються кісткові розростання симетрично на бічних поверхнях тіл хребців, біля основи поперечних відростків. Деструкція хребців, сплющення призводять до кіфозу, сколіозу. Передня поздовжня зв’язка костеніє (осифікується) і формує блок, кістковий анкілоз з 2–3 хребців (подібні зміни відмічають при синдромі Форестьє). Після осифікації вираженість болю зменшується або зникає зовсім.
Гонорея
Код за МКХ-10
А54.4. Гонококова інфекція кістково-м’язової системи.
М01.3 Гонококовий артрит (А54.4), бурсит (М73.0), остеомієліт (М90.2), синовіт (М68.0), тендосиновіт (М68.0).
М68.0 Синовіт або тендосиновіт при гонореї (А54.4).
М73.0 Гонококовий бурсит (А54.4).
Епідеміологія гонококових артритів
На думку американських фахівців, гонококові артрити складають половину бактеріальних артритів дорослих, а в цілому бактеріальні (інфекційні) артрити діляться на 2 групи: гонококові і негонококові. Поширеною є точка зору, що сексуально активні пацієнти з гострим моноартритом некристалічної або іншої невідомої етіології повинні отримувати превентивну протигонококову терапію. Гонококовий артрит частіше відмічають у молодих жінок у зв’язку зі стертою у них клінічною сиптоматикою гонореї. Гонорейний артрит розвивається як при гострій, так і хронічній гонореї і є результатом метастатичного (гематогенного) або імунного (реактивного) ураження. Первинне вогнище інфекції локалізується в урогенітальній сфері, звідки гонокок гематогенним шляхом потрапляє в порожнину суглоба. Слід враховувати, що при дисемінованій гонококовій інфекції (гонококовому сепсисі) можуть розвиватися інфекційний (гонококовий) ендокардит, міокардит, перикардит, менінгіт, ураження печінки, нирок, шкіри.
Клініка гонококового артриту
Розрізняють: гострий серозний; гострий гнійний; підгострий серофібринозний артрит; гонорейний поліартрит.
Гострий серозний гонококовий артрит характеризується рядом особливостей (табл. 25.5).
Таблиця 25.5
Характерні особливості гострого серозного гонококового артриту
|
Гострий початок |
|
Різкий біль в ураженому суглобі |
|
Висока температура тіла, озноби |
|
Високий лейкоцитоз, підвищення ШОЕ |
|
Ураження колінних, гомілковостопних або декількох великих суглобів |
|
Залучення до процесу періартикулярних тканин (слизові оболонки сумок, зв’язки, сухожилля або їх піхви) |
|
Швидкий розвиток атрофії м’язів у ділянці ураженого суглоба і контрактури |
|
Часте виникнення ахілобурситу, болісного припухання в місці прикріплення ахілового сухожилля до горба п’яти |
|
Поява вузликових папул на червоній основі, везикул і пустул з гнійним або геморагічним вмістом на шкірі в ділянці дистальних відділів кінцівок, поблизу уражених суглобів, на спині |
Гострий гнійний гонококовий артрит. Для нього характерні:
- виражене опухання і гіперемія суглоба;
- сильний біль;
- лихоманка;
- тяжкий загальний стан;
- виражений лейкоцитоз, різке підвищення ШОЕ;
- випіт гнійного характеру, в якому виявляють гонококи;
- швидкий розвиток атрофії прилеглих м’язів;
- розвиток некрозу тканин, лізис хряща з наступним розвитком анкілозу.
Підгострий серофібринозний гонококовий артрит. До його проявів належать :
- затяжний, іноді хронічний перебіг;
- розвиток ерозій хряща, суглобових поверхонь кісток;
- виникнення проліферативних явищ в м’яких тканинах, стійкої деформації й обмеження рухів у суглобі;
- розвиток при затяжному перебігу «шпор» кісток п’ят, які викликають біль при ходьбі і натисканні на п’яту.
Гонорейний поліартрит. Гонорейний поліартрит — це імунна (реактивна) форма з ураженням 2–4 великих суглобів, нестійкими ексудативними явищами в них. Він закінчується повним одужанням.
Характер перебігу гонорейного артриту. Після введення в клінічну практику антибіотиків (пеніцилінотерапії) гонорейні артрити (передусім гнійні) стали виявляти значно рідше з легшим перебігом. Вони закінчуються повним зворотним розвитком суглобових проявів через 1–3 тиж після рано розпочатої пеніцилінотерапії.
Діагностика гонорейного артриту. У ранній період захворювання з крові можуть бути виділені гонококи. Виявляють підвищення ШОЕ, лейкоцитоз. При дослідженні синовіальної рідини відмічають цитоз з переважанням нейтрофілів. У 50% хворих в ній можна виявити гонокок. На рентгенограмі при гострому гнійному артриті виявляють деструкцію хряща і кістки, при інших формах — тільки епіфізарний ОП. Діагноз «гонорейний артрит» верифікується виділенням збудника з крові або виявленням його в мазках і посівах з урогенітального тракту, в синовіальній рідині. Допоміжне діагностичне значення має реакція зв’язування комплементу з гонококовим антигеном (Борде — Жангу). Може використовуватися метод полімеразної ланцюгової реакції (ПЛР). У сироватці крові виявляють протигонококові IgА, IgG, IgМ.
Критерії безсумнівності і вірогідності діагнозу гонорейного артриту наведені в табл. 25.6.
Таблиця 25.6
Критерії безсумнівності і вірогідності діагнозу гонорейного артриту
|
Критерії безсумнівності діагнозу гонорейного артриту |
|
Виявлення гонококу в синовіальній рідині й урогенітальному тракті (гонококи в мазках) Артрит вважається гонорейним, якщо після нового статевого контакту з’являється лихоманка з ознобом (гонококовий сепсис), характерні висипання на шкірі, запалення суглоба (зазвичай колінного) |
|
Критерії можливого діагнозу гонорейного артриту |
|
Виявлення протигонококових антитіл в сироватці крові Виявлення гонококу у вогнищах, розташованих на відстані Негайний ефект пеніциліну зі зменшенням вираженості місцевого запалення, болю і поверненням суглоба до нормального вигляду за 1–2 тиж, повним одужанням через 1–2 міс Гонококи виявляють тільки в сечостатевих шляхах, а пеніцилінотерапія дає швидкий ефект (протягом 1–3 днів) |
Диференціація гонорейного артриту. Передусім, гонорейний артрит доводиться диференціювати з хворобою Рейтера, для якої характерні наступні фактори:
- носійство антигена гістосумісності HLA B27;
- хвороба уражує переважно чоловіків;
- запальні прояви в суглобах виражені меншою мірою;
- синовіальна рідина асептична;
- у матеріалі з уретри визначають хламідійну інфекцію;
- артрит поєднується з кон’юнктивітом, баланітом, ураженням слизових оболонок рота, кератодермією підошов і долонь;
- у крові виявляють підвищений титр антитіл до хламідій;
- відмічена резистентність до лікування пеніциліном.
Лікування хворих на гонорейний артрит представлено в табл. 25.7.
Таблиця 25.7
Характер терапії хворих на гонорейний артрит
|
Призначають ліжковий режим |
|
Проводять тимчасову іммобілізацію суглоба |
|
Виконують пункції суглоба з видаленням ексудату |
|
Призначають пеніцилін по 6—10 млн ОД/добу протягом 7–14 днів Швидкий регрес усіх клінічних проявів через 2–3 дні є додатковим діагностичним критерієм гонококового артриту |
|
Надалі переходять на ампіцилін перорально протягом ще 10 днів |
|
Хороший ефект дають оксацилін (0,5 г 4 рази на добу в/в або в/м), метицилін (1 г 4–6 разів на добу) |
|
При непереносимості препаратів пеніцилінового ряду призначають еритроміцин (1,5–2 г/добу), тетрациклін (1,5–2 г в 4 прийоми), цефазолін (0,5–1 г 3–4 рази на добу в/м) |
|
Одночасно з антибіотиками призначають НПЗП, хоча антибіотики дають швидкий ефект і не завжди є необхідність в НПЗП |
|
Після затихання запальних явищ призначають масаж, ЛФК, фізіотерапевтичні процедури |
При гонококовому артриті спочатку протягом 7–10 діб призначають цефтріаксон (1–2 г/добу в/в в 1 введення), цефотаксим (3 г/добу в 3 введення), в подальшому протягом 14–28 діб per os ципрофлоксацин (1000 мг/добу в 2 прийоми), офлоксацин (800 мг/добу в 2 прийоми). При лікуванні гонореї, викликаної бета-лактамазопродукуючими штамами гонококу, застосовують нетилміцин, спектиноміцин (тробіцин), амоксицилін з клавулановою кислотою, цефтріаксон, цефотаксим та ін. При змішаній гонорейно-хламідійній інфекції застосовують тетрацикліни (доксициклін), макроліди (азитроміцин), фторхінолони, рифампіцин.
Сифіліс
Код за МКХ-10
А50-А53 Сифіліс.
А51.4 Інший вторинний сифіліс.
М90.1 Вторинний сифілітичний періостит (А51.4).
А52.7 Інші прояви пізнього сифілісу (синовіт, тендосиновіт).
М90.2 Сифіліс кісток.
М73.1 Сифілітичний бурсит (А52.7).
М63.0 Міозит при сифілісі (А51.4, А52.7).
М03.1 Постінфекційна артропатія при сифілісі. Суглоби Клаттона (А59.5).
Особливості ураження опорно-рухового апарату в різні періоди сифілісу. Інкубаційний період (від моменту зараження до появи твердого шанкру) в середньому триває 20–40 днів. Протягом хвороби у нелікованих хворих виділяють 3 періоди (табл. 25.8).
Таблиця 25.8
Періоди сифілісу
|
Період хвороби |
Характеристика |
|
Первинний |
Триває від появи твердого шанкру до першого генералізованого висипання на шкірі. Його тривалість становить 6–8 тиж |
|
Вторинний |
Тривалість вторинного періоду включає час від першого генералізованого висипання до появи третинних сифілідів — горбків і гум. Основний прояв цього періоду — висип на шкірі і слизових оболонках, зумовлений проникненням в них трепонем. Висип великий, поліморфний: розеоли, папули, пустули. Він тримається декілька днів, потім зникає на невизначений час і з’являється знову. Окрім висипу, до вторинних сифілідів належать алопеція і лімфаденопатія |
|
Третинний |
Третинний період починається на 3–4-й рік захворювання і триває до кінця життя. Сьогодні відмічається рідко. Сифіліди представлені 2 елементами: 1) горбками синюшного кольору величиною з вишневу кісточку, розташованими в товщі шкіри; 2) гумами — безболісними вузлами в глибині підшкірної клітковини розміром до волоського горіха. У міру зростання вони набувають синюшно-червоного забарвлення, безболісно покриваються виразками і гояться, залишаючи «зірчасті» рубці |
Ураження опорно-рухового апарату відмічають у вторинний і третинний періоди. Артрити виявляють рідше, ніж ураження кісток і окістя (остеоперіостити). У вторинний період до специфічного процесу можуть залучатися усі органи і системи. Основне значення має ураження кісток і суглобів, ЦНС і деяких внутрішніх органів (гепатит з жовтяницею, міокардит, нефрит, гастрит, нейросифіліс). Ураження кісток (дифузні періостити, нічний біль в кістках) визначають у 5% хворих. Найчастіше уражуються кістки черепа і великогомілкові. Ураження кісток зазвичай має перебіг у вигляді поліартритичного синовіту (гідрартрозу). При спробі до руху з’являється біль, а під час руху він зникає. У вторинний період хвороби можливий розвиток сифілітичного менінгіту (він може бути асимптомним), гідроцефалії, ураження судин (менінговаскулярного сифілісу), зрідка — невритів, поліневритів, невралгії. У третинний період серед гумозних уражень виявляють сифіліди окістя, кісток і суглобів. Частіше уражуються кістки гомілок, передпліччя, черепа, колінні, ліктьові та гомілковостопні суглоби.
Ураження кісток при сифілісі. Розвивається в ранній вторинний період, а іноді навіть в продромальний період, і проявляється кістковим болем, який посилюється в нічний час (dolores osteocopi nocturni). Будь-яких змін з боку кісток при об’єктивному дослідженні не виявляють. Найчастіше біль відчувають в довгих трубчастих кістках нижніх кінцівок. Дещо рідше у вторинний період на кістках черепа, великогомілковій кістці виникають періостити й остеоперіостити — невеликі припухлості щільної консистенції, болісні при пальпації і такі, що супроводжуються сильним нічним болем. Надалі запальні інфільтрати розсмоктуються або осифікуються (рідше).
Ураження суглобів при сифілісі. Частіше розвивається сифіліс суглоба, значно рідше — гострий сифілітичний поліартрит. Цей варіант ураження відмічають на початкових стадіях сифілісу, його називають раннім сифілітичним артритом. Використовують також терміни «сифілітична артропатія», «поліартритичний синовіт з гідрартрозом». У більшості випадків хворих турбують симптоми артралгії, артрит характеризується припуханням суглобів і наявністю ексудату. Ураження суглобів кистей рук і стоп нехарактерне.
Поліартрит розвивається у вторинний період сифілісу і носить транзиторний характер. Частіше уражуються колінні, плечові, променезап’ясткові, рідше ліктьові і гомілковостопні суглоби. Можлива симетричність ураження, а також залучення до процесу суглобів шийного відділу хребта. В основному конфігурація суглобів не змінена, в ⅓ випадків відмічають деформацію, пов’язану з накопиченням ексудату в порожнині суглоба. У таких випадках суглоб набуває веретеноподібної форми за рахунок випинання заворотів. Атрофія м’язів в ділянці уражених суглобів виражена слабо, почервоніння шкіри не виявлено. Уражені суглоби малоболючі, у тому числі й при їх пальпації. Функція суглобів збережена, хворий продовжує вести звичайний спосіб життя. Поліартрит супроводжується нічним болем в кістках (частіше — гомілки), що зменшується до ранку. У цих випадках можна виявити (при пальпації, рентгенологічному дослідженні) періостити. Під впливом низьких доз протисифілітичних препаратів посилюється нічний біль, що має діагностичне значення. В деяких випадках сифілітичний синовіт має перебіг за типом підгострого поліартриту (псевдоревматизму Фурньє) і клінічно нагадує ревматичні, ревматоїдні і туберкульозні поліартрити. Прояви гострого і підгострого сифілітичного поліартриту швидко зникають під дією специфічної терапії.
Форми ураження суглобів, що відмічають в третинний період. Ураження суглобів (головним чином колінних і ліктьових) в третинний період виявляють у вигляді 2 форм. При першій формі перебіг процесу має вигляд гідрартрозу з прогресуючим кулястим здуттям суглоба (так званий суглоб Клаттона), викликаним ексудатом, специфічною інфільтрацією капсули, синовітом. Незважаючи на значну вираженість змін, рухи в суглобах безболісні, а функція їх не порушена.
При другій формі (остеоартрит) в епіфізі кістки утворюються обмежені гумозні інфільтрати. Вони ведуть до руйнування кісткової тканини, хряща, появи ексудату. Суглоб поступово деформується, проте рухи в ньому зберігаються, пацієнти майже не відчувають болю. Найчастіше уражується колінний, ліктьовий і променезап’ястковий суглоби. У подібних випадках про сифілітичну природу ураження свідчать:
- поступовий розвиток ураження;
- масивні випоти в порожнину суглобів;
- відсутність гострих запальних явищ (дефігурація без ознак запалення або «біла пухлина»);
- відсутність вираженого порушення функції суглоба;
- задовільний загальний стан хворих.
При рентгенографії виявляють округлі гумозні дефекти в епіфізах.
Критерії діагностики сифілітичного ураження опорно-рухового апарату
Діагностика сифілітичного ураження суглобів ґрунтується на:
- даних анамнезу;
- наявності вторинних і третинних сифілідів;
- вираженій дефігурації суглобів за рахунок масивного випоту і потовщення періартикулярних тканин;
- збереженні функції суглоба, незважаючи на значні зміни;
- виявленні блідих трепонем у папулах, пустулах, везикулах;
- наявності позитивної реакції Вассермана, імунофлуоресценції, реакції іммобілізації блідих трепонем. Дві останні реакції є специфічними для сифілісу і позитивні незалежно від періоду хвороби.
Реакція Вассермана може бути негативною в третинний період (у ⅓ хворих) і призводити до діагностичних помилок.
Лікування хворих із сифілітичним ураженням суглобів
Лікування проводиться дерматовенерологом в умовах спеціалізованого стаціонару. Використовуються антибіотики пеніцилінового ряду, за необхідності призначають НПЗП. У разі непереносимості препаратів пеніциліну використовують макроліди і тетрациклін.
Туберкульоз
Код за МКХ-10
М90.0 Туберкульоз кісток та суглобів (А18.0).
В90.2 Віддалені наслідки туберкульозу кісток та суглобів.
М49.0 Туберкульоз хребта.
М68.0 Синовіт або тендосиновіт при туберкульозі (А18.0).
М01.1 Туберкульозний артрит (А18.0).
Актуальність проблеми кістково-суглобового туберкульозу
Сьогодні в країні відзначається загрозлива ситуація в результаті епідемії туберкульозу. Інфікованість туберкульозом сягає 80% у великих містах. Майже у кожної людини віком старше 25 років може виникнути туберкульозна інфекція через наявність в організмі персистуючих форм мікобактерій. Ураження кісток і суглобів відмічають у 1–3% хворих на туберкульоз. Кістково-суглобовий туберкульоз однаковою мірою уражує периферичні суглоби і хребет. Частіше він виникає у віці 40–50 років, набагато пізніше за процес в легенях, лімфатичних вузлах, і пов’язаний з реактивацією інфекції в первинному комплексі або поширенням з лімфатичних вузлів. До груп ризику щодо захворюваності на туберкульоз кісток і суглобів відносяться особи: 1) що зловживають алкоголем; 2) що вживають наркотики; 3) літнього і старечого віку; 4) ВІЛ-інфіковані; 5) пацієнти з різними імунодефіцитними станами.
Клініка туберкульозу суглобів
Класичною формою туберкульозу суглобів є моноартрит. У 80% випадків туберкульозу суглобів уражуються кульшовий, колінний, плечовий суглоби. Дуже рідко залучаються гомілковостопні і ліктьові суглоби. Розрізняють три форми суглобового туберкульозу : 1) остеоартрит; 2) синовіт первинно-кісткового походження; 3) синовіт первинно-синовіального походження (так званий ревматизм Понсе). Наслідки туберкульозного артриту включають деструкцію, деформацію, зміну архітектури суглоба та анкілоз.
Туберкульозний остеоартрит. Остеоартрит складає 80–90% суглобового туберкульозу. Розвивається в результаті занесення мікобактерій в кістковий мозок, епіфізарні відділи кісток. Розрізняють три стадії процесу (табл. 25.9).
Таблиця 25.9
Стадії туберкульозного остеоартриту
|
Стадії |
Характеристика |
|
Передартритична |
У епіфізах формується вогнище, так званий первинний остит, який тривалий час може бути «німим». У цю стадію в синовії можуть розвиватися туберкульозні горбки в результаті проникнення в синовіальну тканину мікобактерій по кровоносних і лімфатичних шляхах. І лише при виникненні загальної інфекції, травмах, операціях, несприятливих умовах праці і побуту, зниженні загальної реактивності організму виникає нетривалий реактивний синовіт, який рідко розцінюється як специфічний. У першу стадію хворих турбують припухлість суглоба, помірний біль у ньому. Зрідка вже в цей період відмічають ознаки загальної інтоксикації: слабкість, субфебрилітет, схуднення |
|
Артритична |
Процес з епіфізарної кістки переходить на суглоб і періартикулярні тканини. Припухлість суглоба стає стійкою, розвивається артралгія, підвищується локальна температура шкіри без її гіперемії. Загальний стан погіршується, з’являються ознаки туберкульозної інтоксикації: субфебрильна температура тіла, підвищене потовиділення, слабкість, зменшення маси тіла. При пальпації суглоба — локальний біль. Ексудат у суглобі може бути великим, супроводжуватися значним набряком періартикулярних тканин. Розвивається значна м’язова атрофія. У подальшому виникають «сирнистий» розпад кісткового вогнища, прорив його в порожнину суглоба, ще більше обсіменіння синовіальної оболонки, капсули горбками. Уся порожнина суглоба проростає фунгозними масами, які зазнають «сирнистого» розпаду. Утворюється абсцес, він проривається через шкіру: з’являються нориці з гнійними виділеннями. Хрящ розплавляється, відбувається деструкція суглобових відділів кісток, їх укорочення, зміщення. Інтоксикація підвищується, збільшується ШОЕ. Рентгенологічно визначаються ділянки кістково-хрящової деструкції в епіфізах кісток |
|
Постартритична |
У цій останній стадії виявляється повне руйнування суглоба («руїни суглоба»). Функція суглоба повністю втрачена |
У передартритичну стадію на тлі нормальної будови кісткової тканини або дифузного ОП рентгенологічно можна виявити вогнищеву перебудову рисунка кісткових трабекул, а потім — обмежену кісткову порожнину. У цій порожнині іноді міститься секвестр. При прориві кісткового вогнища в порожнину суглоба виявляють руйнування суглобових кінців кісток, їх зміщення, підвивихи. У подальшому розвиваються ознаки вторинного ОА.
Туберкульозний синовіт первинно-кісткового походження. Це рідкісна форма туберкульозу суглобів. Головною ознакою цієї форми є локалізація процесу переважно в синовіальній оболонці (частіше колінного суглоба). Кісткові вогнища мають невеликі розміри, виявляють рідко і складно (важче, ніж при кокситах). Перебіг синовіту дуже стійкий, оскільки підтримується мікобактеріями, наявними в порожнині суглоба. Розвивається серозний випіт в порожнину суглоба. Рентгенологічне дослідження виявляє лише ОП і кісткові узури в місцях прикріплення синовіальної оболонки. При морфологічному дослідженні визначають лімфоїдну інфільтрацію синовіальної оболонки, невелику кількість туберкульозних горбків. «Сирнистий» розпад не відбувається, що є найважливішою відмінністю від остеоартриту.
Туберкульозний синовіт первинно-синовіального походження (ревматизм Понсе). Згідно з даними американських авторів, його відмічають частіше, ніж діагностують. Має місце специфічний синовіт, перебіг якого нагадує поліартрит, — уражуються декілька суглобів. Переважно уражуються дрібні суглоби кистей, запалення стійке, без деформації (настає повний зворотний розвиток суглобових явищ). Відмічається у хворих з вісцеральним туберкульозом — уповільненим, прихованим туберкульозним процесом в лімфатичних вузлах, легенях. Імунні комплекси відкладаються переважно в синовіальній оболонці. Розвиток поліартриту співпадає з гострою інфекцією (ангіна, грип) або виникає після неї. У цих умовах настає активізація латентної туберкульозної інфекції. При рентгенологічному дослідженні будь-яких характерних змін не виявляють. До відмітних ознак ревматизму Понсе відносять:
- туберкульоз в анамнезі або в теперішній час;
- стійкий артрит без деформацій;
- асиметричність ураження;
- відсутність ерозійних змін на рентгенограмах;
- переважний вміст в синовіальній рідині мононуклеарів;
- відсутність підшкірних ревматоїдних вузликів, а також РФ;
- позитивні проби Манту і Пірке;
- ефективність протитуберкульозної терапії.
Туберкульозний коксит. При кокситі переважає первинно-кісткова форма ураження, тобто туберкульозний остеоартрит. Характерними ознаками туберкульозного кокситу є:
- ранній і постійний біль в паху, який іррадіює в стегно;
- поступове збільшення вираженості болю, поява кульгавості;
- наявність в положенні на спині згинальної контрактури і тенденції до відведення стегна;
- наявність припухлості на зовнішній поверхні стегна (утворюється «натічник» або «холодний абсцес»);
- при рентгенографії визначаються вогнища деструкції у вертлюжній западині, голівці або шийці стегна;
- у центрі вогнищ деструкції видно секвестри (так звані шматочки цукру, що тануть).
Туберкульозний гоніт. Він, як правило, починається з артралгії. Потім приєднується водянка суглоба. В одних випадках водянка розвивається поступово (при проростанні специфічних гранульом в синовіальну тканину), в інших — швидко, несподівано (при прориві навколосуглобового вогнища в порожнину суглоба). При рентгенологічному дослідженні виявляють деструктивні зміни в ділянці виростків великогомілкової кістки.
Туберкульоз плечового суглоба. Частіше хворіють чоловіки. Типовим проявом туберкульозу цієї локалізації є «суха костоїда» (caries sicca). Виявляють обмеження рухів в плечі, приведення його до тулуба. Сам суглоб зазвичай не опухає, розвивається виражена атрофія навколишніх м’язів. На рентгенограмі суглоба на латеральній частині голівки плечової кістки виявляють округлі і «бухтоподібні» дефекти без секвестрів.
Частота туберкульозу ліктьового суглоба становить до 5% випадків. Основні скарги — біль, набряклість, обмеження рухів. Переважно інфекцією первинно уражається ліктовий відросток чи плечова кістка, рідше — синовіальна оболонка. На початкових стадіях захворювання в патологічний процес залучається лише кісткова тканина, на пізніх — власне суглоб, що може викликати деструкцію та патологічні переломи (рис. 25.1).
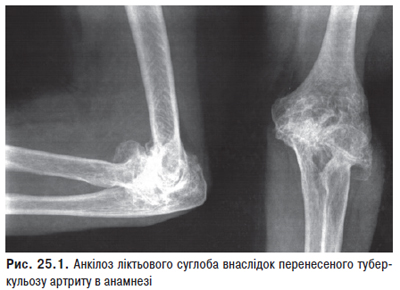
Туберкульоз хребта (специфічний спондиліт). Його характерні ознаки наведені в табл. 25.10.
Таблиця 25.10
Характерні ознаки туберкульозу хребта
|
Частіше відмічають у дітей і молодих людей віком 20–30 років |
|
Скарги на біль у спині і обмеження рухливості хребта через біль |
|
Як правило, уражується грудний або поперековий відділ хребта |
|
Характерним є ураження 2 суміжних хребців |
|
Диск між хребцями руйнується, при пальпації цього місця визначається біль |
|
Можна виявити випинання одного з остистих відростків |
|
У тілі хребців утворюються невеликі некротичні вогнища, які виявляють тільки на томограмі (у тому числі комп’ютерній) |
|
При звичайній рентгенографії виявляють ураження тільки зовнішнього шару хребця |
|
Початковою рентгенологічною ознакою є звуження міжхребцевої щілини, деструкція звернених одна до одної поверхонь тіл хребців, дефекти кісткової структури (рис. 25.2) |
|
У пізній період виявляється сплющення (колапс) передніх відділів тіл хребців (клиноподібні хребці) |
|
По обох боках хребта виявляються веретеноподібні тіні (натічні абсцеси) |
|
У результаті зменшення висоти міжхребцевих дисків збільшується грудний кіфоз і формується горб (хвороба Потта) |
Діагностика кісткового туберкульозу
Діагностика кісткового туберкульозу є досить складною. Успіхи в боротьбі з туберкульозом явно перебільшені. Тому правильний діагноз найчастіше лікарі встановлюють при процесі, що далеко зайшов. Навіть у високорозвинених країнах більше 50% випадків позалегеневого туберкульозу не розпізнаються за життя. Своєчасній діагностиці сприяє врахування ознак, наведених у табл. 25.11.
Таблиця 25.11
Основні диференційно-діагностичні ознаки кісткового туберкульозу
|
Анамнестичні дані (перенесений туберкульоз легень або інших органів; активний процес на даний час; контакт з туберкульозним хворим) |
|
Наявність субфебрилітету, підвищеного потовиділення, астенізації |
|
Неодноразові кровохаркання у минулому |
|
Моноартикулярний тип ураження суглобів (кульшового або колінного) |
|
Наявність типових рентгенологічних змін |
|
Виявлення мікобактерій туберкульозу у синовіальній рідині |
|
Позитивна проба Манту (не суворо специфічна) |
|
Ефективність терапії ex juvantibus туберкулостатичними препаратами |
|
Позитивні результати бактеріологічних досліджень, які включають посіви крові і синовіальної рідини |
Диференціація туберкульозного артриту
В артритичній стадії його доводиться диференціювати з РА. На користь РА свідчать: 1) одночасне ураження 3 і більше суглобів (це нехарактерно для туберкульозного процесу); 2) залучення до процесу нових суглобів; 3) відсутність мікобактерій в суглобовому вмісті; 4) наявність РФ. Туберкульозний коксартрит і гонартрит доводиться диференціювати також з ОА. Цьому допомагають рентгенологічні дослідження, а також врахування того факту, що реактивний синовіт не досягає такого ступеня розвитку, як при туберкульозі. Крім того, він добре піддається лікуванню НПЗП.
Лікування хворих з туберкульозом кісток і суглобів
Туберкульозний артрит піддається тривалій туберкулостатичній терапії, яка проводиться в спеціалізованих установах. Схеми лікування складені на основі терапії легеневих форм туберкульозу. Курси лікування тривають 1–2 роки. Вони включають ізоніазид (до 300 мг/добу (5 мг/кг маси тіла) усередину), рифампіцин (до 600 мг/добу усередину (10 мг/кг), піразинамід до 2 г (15–30 мг/кг)) щодня. Через 2 міс припиняють прийом піразинаміду. Якщо раніше хворий вже отримував протитуберкульозну терапію або має місце резистентність, в схему додатково включають етамбутол (15 мг/кг маси тіла щоденно).
Бруцельоз
Код за МКХ-10
А23 Бруцельоз.
М49.1 Бруцельозний спондиліт (А23.-).
Етіологія бруцельозу
Найбільшу небезпеку для людини становлять бруцели «козиного типу» (збудники так званої мальтійської лихоманки — Br. Militensis), великої рогатої худоби (Br. Abortus bovis) і свиней (Br. Abortus suis). Чинниками передачі служать м’ясо, сире молоко, шерсть, сеча. Бруцели проникають в організм людини через шкіру або слизові оболонки. Потрапляючи в шлунок, більшість бруцел гинуть під дією шлункового соку, деяка частина потрапляє у тонку кишку. З неї із током лімфи бруцели потрапляють в мезентеріальні та заочеревинні лімфовузли, де активно розмножуються. Інкубаційний період триває від 7 до 30 днів. Наприкінці інкубаційного періоду відбувається прорив бруцел в кровообіг з розвитком бактеріємії. Вони осідають в органах, багатих ретикуло-ендотеліальною тканиною (кістковий мозок, селезінка, печінка, периферичні лімфовузли), де відбувається їх подальше розмноження з повторним надходженням в кров. В уражених органах утворюються специфічні бруцельозні гранульоми. Захворювання починається з лихоманки, яка до 6–8-го дня сягає 39–40 °С та вище. Вона зберігається 25–40 днів і нерідко має хвилеподібний характер. Бруцельоз має схильність до хронічного перебігу. Окрім тривалої лихоманки, він характеризується ураженням опорно-рухової, нервової, серцево-судинної, сечостатевої та інших систем.
Клініка бруцельозу
Залежно від характеру перебігу хвороби виділяють наступні форми бруцельозу: гостра (тривалість перебігу до 3 міс); підгостра (до 6 міс); хронічна (більше 6 міс); резидуальні форми (клініка наслідків). При гострій і підгострій формі відмічають підвищення температури тіла, озноб, що завершуються профузним потовиділенням. Збільшуються периферичні лімфовузли, вони не спаяні з навколишніми тканинами. У ділянці сухожиль і м’язів утворюються щільні, болісні вузлики — фіброзити і целюліти. У тяжких випадках можуть розвинутися міокардит, ендокардит, перикардит. Виникають катари верхніх дихальних шляхів, бронхіти, бронхопневмонії, бронхоаденіти. Печінка і селезінка збільшені, болісні при пальпації. Ураження ЦНС проявляється головним болем, дратівливістю, емоційною нестійкістю, у тяжких випадках — менінгізмом, менінгітом.
Опорно-руховий апарат при гострому бруцельозі уражується лише у частини хворих. Біль у суглобах короткотривалий, він зникає у міру зменшення вираженості ознак інтоксикації. Рецидиви виникають через 1–2 міс і пізніше. Деяка частина хворих одужують через 1–2 роки. Проте нерідко процес стає хронічним. Найчастіше при хронічній формі уражується опорно-руховий апарат (виділяють хронічну кістково-суглобову форму бруцельозу). Розвивається нейробруцельоз: радикуліти, плексити, міжреберні невралгії, розлади чутливості, парези, неврити, менінгіти, менінгоенцефаліт. Уражується сечостатева система (орхіти, епідидиміти, ендометрити, викидні). Виділяють також легеневу і гепатолієнальну форми. Бруцельозний артрит розвивається у вигляді 2 форм: інфекційного і РеА.
Особливості інфекційної форми бруцельозного артриту
Після проникнення бруцел в порожнину суглоба відмічають швидке прогресування процесу з грубими руйнуваннями кісткової тканини, розвитком анкілозу суглоба. Синовіальна рідина має серозно-гнійний характер, містить бруцели. Сьогодні цю форму виявляють рідко.
Прояви, характерні для реактивної форми
Ураження суглобів відмічають на тлі генералізованої лімфаденопатії, збільшення печінки, селезінки. Зазвичай уражується декілька суглобів, запалення носить доброякісний характер і при своєчасному призначенні специфічної терапії функція суглобів відновлюється повністю. Синовіальна рідина ідентична такій при алергічному артриті. Часто виявляють двобічне ураження крижово-клубових суглобів. Характерна поява біля суглобів запальних вузлових інфільтратів з наступним розвитком фіброзних утворень. Для бруцельозу характерне ураження позасуглобових м’яких тканин з розвитком періартритів, бурситів, тендовагінітів, фасцитів, фіброзитів, міозитів (вони виникають на тлі високої температури тіла). Часто розвивається ахілодинія. Ураження хребта, що супроводжується осифікацією бокових зв’язок, переважно відмічають у чоловіків (70%).
Діагностика бруцельозного артриту, спондиліту
При реактивній формі бруцельозного артриту рентгенологічні зміни в суглобах не виявляють. При інфекційній формі (інфікуванні суглоба) руйнування кісткової тканини відмічають рідко, частіше в ліктьових і колінних суглобах виявляють локальний ОП, звуження суглобової щілини, крайові узурації, склеротичні зміни. Для бруцельозного спондилоартриту характерне ураження декількох хребців з наступними проявами:
- крайові узури;
- зазублена поверхня тіл хребців;
- асиметричні, невеликих розмірів крайові остеофіти;
- зменшення висоти міжхребцевих дисків;
- бічне зміщення хребця при деструкції з одного боку;
- осифікація бокових зв’язок (рис. 25.3).
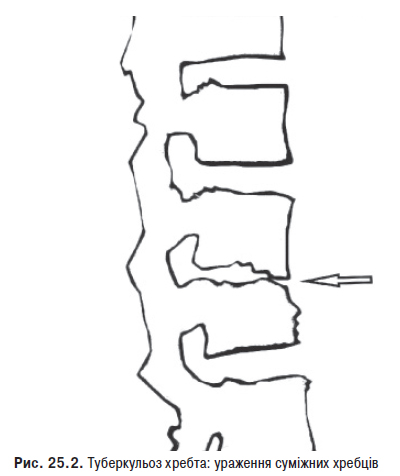
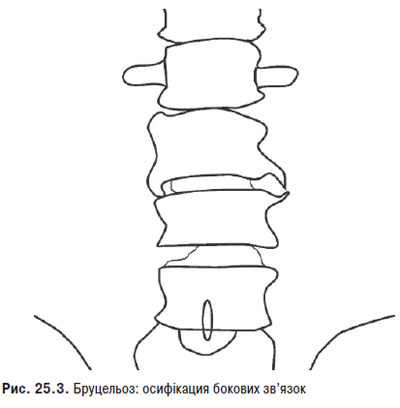
Лабораторні зміни, характерні для бруцельозу
При бруцельозі відмічають: підвищення ШОЕ до 25–35 мм/год; лейкопенію з відносним лімфоцитозом (40–80%).
Через 9–12 днів стає позитивною реакція аглютинації Райта (діагностичним вважається титр не менше 1:200) і пластинчата реакція аглютинації Хедльсона з цілісною сироваткою і концентрованим антигеном.
Позитивні реакції зв’язування комплементу, пасивної гемаглютинації, шкірна проба Бюрне (результати враховуються через 9–12 днів), опсонінова активність (визначають кількість опсонінів у сироватці крові).
При бактеріологічному дослідженні крові, кісткового мозку, сечі, лімфатичних вузлів виявляють бруцели (відповідь при посіві можна отримати тільки через 25–30 днів, оскільки бруцели ростуть повільно).
Критерії, що враховують при встановленні діагнозу «бруцельоз» представлені в табл. 25.12.
Таблиця 25.12
Критерії, які слід враховувати при діагностиці бруцельозу
|
Місце проживання пацієнта, споживання сирого молока |
|
Перебування за 1–4 тиж до хвороби в місцевості з епізоотією бруцельозу |
|
Професія, що припускає контакт з худобою (ветеринарні лікарі, зоотехніки, доярки та ін.) |
|
Підвищення температури тіла, озноб, біль у м’язах |
|
Невідповідність між високою температурою тіла і відносно задовільним станом хворих |
|
Поєднання високої лихоманки, ознобу, що супроводжуються підвищеним потовиділенням, із збільшенням усіх периферичних лімфовузлів, селезінки, печінки |
|
Наявність моно- й олігоартриту |
|
Поєднання моно-, олігоартриту, що супроводжується ураженням навколосуглобових м’яких тканин, з ознобом, підвищеним потовиділенням |
|
Запальні зміни в суглобах за відсутності змін на рентгенограмі |
|
Наявність тріади, нехарактерної для будь-якого іншого запального захворювання суглобів : 1) збільшення розмірів селезінки і печінки; 2) збільшення периферичних лімфовузлів; 3) гранулоцитопенія з відносним лімфоцитозом понад 40% |
|
Ураження 2–3 хребців із деструкцією поверхні тіл та осифікацією бокових міжхребцевих зв’язок |
|
Виявлення бруцел в синовіальній рідині, крові, харкотинні, кістковому мозку, сечі, лімфатичних вузлах |
|
Позитивні реакції Райта, Хедльсона, зв’язування комплементу, опсонінової активності |
Захворювання, з якими диференціюють бруцельозний артрит
Іноді доводиться диференціювати його з гонококовим артритом (виникає після урогенної інфекції, гонококи виявляють у сечостатевих органах, позитивна реакція Борде — Жангу), артритом при виразковому коліті (є клініка виразкового коліту, переважно уражуться суглоби нижніх кінцівок, перебіг артриту доброякісний). Бруцельозний спондиліт диференціюють з хворобою Бехтєрева, туберкульозним спондилітом. При хворобі Бехтєрева завжди є клініка двобічного сакроілеїту (біль в крижах, сідницях, стегнах), при бруцельозі сакроілеїт — рентгенологічна знахідка. При диференціації з туберкульозом слід враховувати, що при ньому процес локалізується в IX–X хребцях, а бруцельозний спондиліт уражує III–IV поперекові хребці.
Лікування бруцельозного артриту
Лікування гострих і підгострих форм бруцельозу починають з антибіотиків. Ефективними при бруцельозі є левоміцетин (по 0,5 г кожні 4 год/добу до нормалізації температури тіла, потім 0,5 г через кожні 6 год), тетрациклін (2 г/добу), доксициклін (0,2 г/добу), стрептоміцин (в/м 0,5 г в 2 рази на добу), рифампіцин (0,9 г/добу). Тривалість лікування становить 14 і більше днів. Другий і наступні курси лікування проводять через інтервали 10–14 днів. При хронічному бруцельозі антибіотики застосовують лише у разі загострення. Використовують лікувальну бруцельозну вакцину, протибруцельозний гамма-глобулін.
При артритах і періартритах призначають НПЗП з анальгезивною дією (фенілбутазон, ацетилсаліцилова кислота, індометацин, диклофенак, ібупрофен та ін.). Ефективним є внутрішньосуглобове і періартикулярне введення гідрокортизону, бетаметазону, тріамцинолону. Використовується фізіотерапія (індуктотермія, діадинамічний струм, діатермія, УВЧ), ЛФК, масаж, радонові, сірко-водневі ванни, грязелікування.
Профілактика бруцельозу
З метою профілактики хвороби застосовується жива вакцина (імунітет зберігається 1–2 роки).
Лайм-бореліоз
Актуальність проблеми хвороби Лайма для України
Хвороба Лайма — одна з найчастіших природновогнищевих інфекцій: захворюваність в різних країнах становить від 10 до 50 і більше випадків на 100 тис. населення. У Росії антитіла до збудника виявляють у 13–25% жителів ендемічних районів. В Україні до 2000 р. хвороба Лайма офіційно не реєструвалася. У тому ж році в країні зареєстровано 58 випадків хвороби, більшість з яких — у Києві (22 випадки хвороби Лайма, причому 8 з них заразилися в межах міста), Львівській і Дніпропетровській областях. Окремі випадки захворювання зареєстровані в Донецькій, Закарпатській, Запорізькій, Івано-Франківській, Київській, Миколаївській, Рівненській, Харківській і Чернівецькій областях.
Код за МКХ-10
А69.2. Хвороба Лайма.
М01.2. Артрит при хворобі Лайма.
Етіологія й епідеміологія хвороби
Збудник належить до грамнегативних джгутикових бактерій (спірохет) — борелій комплексу Borrelia burgdorferi sensu lato, які відрізняються генотипічною (описано більше 10 генотипів) і фенотипічною різноманітністю. Борелії, виділені в різних географічних зонах, розрізняються за морфологією і складом білків (це характерно для Євразії). Генетичні види відрізняються нуклеотидною послідовністю ДНК. Відмінності геномів визначають антигенну структуру борелій, а отже і клінічну симптоматику. У одному кліщі можна виявити борелії, що належать до двох різних генотипів. Можливе одночасне зараження бореліозом і вірусом кліщового енцефаліту. Циркуляцію в природних вогнищах забезпечують переносники борелій — кліщі роду Ixodes: тайговий кліщ Ixodes persulcatus і лісовий кліщ Ixodes ricinus. Природні вогнища в Європі можуть бути пов’язані одночасно з двома цими переносниками. Природна зараженість кліщів сягає 20–60%, при цьому показники нижчі у лісового кліща. Збудник інокулюється при укусі кліща з його слиною, сприйнятливість населення висока. Захворювання характеризується весняно-літньою сезонністю: у вогнищах з тайговим кліщем зараження відбувається навесні і в першу половину літа, лісовий кліщ активний навесні і наприкінці літа — на початку осені.
Життєвий цикл кліщів триває 2–3 роки. Дорослі кліщі присмоктують до великих лісових тварин восени і навесні й живляться їх кров’ю. Потім жіночі особини відкладають яйця на землі, де через декілька тижнів з них з’являються незрілі особини кліщів — ларви, які живляться кров’ю дрібних тварин (переважно мишей) навесні і влітку, потім гібернують до весни. Навесні вони перетворюються на німфу (наступну стадію розвитку) і живляться кров’ю мишей протягом літа, а восени перетворюються на дорослих особин. При живленні кров’ю дрібних тварин ларви та німфи інфікуються бореліями. Потім вони або дорослі кліщі, у свою чергу, інфікують різних тварин, людей. Бореліозом можуть захворіти кішки, собаки, коні, велика рогата худоба. Вони також можуть принести кліщів в житло людини.
Географія розподілу генотипів збудників
У Європі поширені B. Burgdorferi sensu stricto, B. Garinii і B. Afzeluii, причому в одному кліщі можна виявити два генотипи, що веде до плюриінфекції. У США є єдиний генотип — B. Burgdorferi sensu stricto. Від генотипу залежить клінічна симптоматика: так, B. Afzelii викликає ураження шкіри, B. Burgdorferi — суглобів, B. Garinii — нервової системи. Генотипові й антигенні відмінності збудників визначають особливості патогенезу і клініки захворювання. У різних генотипів різна чутливість до антибіотиків, тому, очевидно, для різних регіонів України необхідні свої схеми лікування. Труднощі серодіагностики хвороби Лайма обумовлені існуванням різних генотипів збудника, характерних для кожного регіону. У США для серодіагностики можна використовувати один генотип (B. Burgdorferi), для Європи — 3 генотипи.
Вікові групи, що потерпають від хвороби Лайма
Хворобу виявляють у пацієнтів усіх вікових груп незалежно від статі. У США лайм-артрит частіше розвивається у дітей, у дорослих перебіг інфекції субклінічний. Загалом частіше хвороба уражує осіб зрілого та літнього віку, у них відзначають хронізацію процесу.
Патогенез хвороби Лайма
Борелії в організмі можуть поширюватися гематогенно, лімфогенно, периневрально, що веде до поліорганних уражень. Після проникнення борелій в шкіру активізуються Т-клітини, В-лімфоцити, починається продукція специфічних антитіл. Активація В-лімфоцитів супроводжується підвищенням рівня сироваткового IgM, появою кріопреципітатів, антинуклеарних антитіл, РФ, антитіл до кардіоліпіну, ЦІК. Борелії персистують в тканинах дуже тривалий час і при їх активізації виникають рецидиви захворювання. Наявні в тканинах збудники й їх антигени ініціюють клінічні прояви, підтримують хронічне запалення, стимулюють аутоімунні реакції (виробляються антитіла до різних тканин). У флагелярного антигена борелій і клітин нервової системи, синовіальної оболонки, серцевого м’яза є спільні антигенні детермінанти. Цим пояснюється розвиток хронічного артриту, нейробореліозу (борелії проникають в ЦНС, де відмічається продукція антитіл класів IgG і IgM), кардиту. Персистенція збудника в організмі супроводжується високим титром антитіл в крові, який може зберігатися протягом декількох років. Комплекси антиген — антитіло відкладаються в судинах, тканинах. У пошкоджених тканинах виявляють периваскулярні інфільтрати, що складаються з лімфоцитів, плазматичних клітин, макрофагів. Борелії визначаються периваскулярно, в тканинах, усередині клітин.
Клініка хвороби Лайма
В перебігу хвороби Лайма виділяють ранній і пізній періоди (Насонова В.А., Насонов Е.Л., 2003 (ред.); Насонов Е.Л., 2010 (ред.)). Ранній включає інкубаційний період, локальну стадію мігруючої еритеми та стадію дисемінації, триває від 1–3 до 6 міс. Пізній період починається через 6–12 міс після виникнення первинних симптомів і проявляється синдромами — шкірним, неврологічним, офтальмологічним, ревматологічним, кардіологічним та ін.
Стадії хвороби Лайма
Перебіг хвороби має стадійний характер. Виділяють (Насонова В.А., Насонов Е.Л., 2003 (ред.)) наступні стадії: локальна, диссемінації і персистенції інфекції (табл. 25.13).
Таблиця 25.13
Характеристика стадій хвороби Лайма
|
Ранній період |
Інкубаційний період |
Він триває від декількох діб до 1 міс (у середньому 10–12 діб) |
||
|
Локальна стадія мігруючої еритеми |
У місці присмоктування кліща виникає локальне шкірне запалення — кліщова мігруюча еритема (КМЕ). Її відмічають на ранній стадії ураження усіма генотипами борелій. У 20–30% пацієнтів КМЕ відсутня, у 50–80% відзначають грипоподібний синдром |
|||
|
Стадія дисемінації |
Ураження шкіри |
Вторинні еритеми |
Є маркерами дисемінації, локалізуються в місцях, віддалених від місця присмоктування кліща, морфологічно нагадують КМЕ |
|
|
Доброякісна лімфоцитома, або лімфаденоз |
Найчастіше локалізується на мочці вуха або в ділянці соска молочної залози |
|||
|
Ураження суглобів |
Доброякісний рецидивуючий артрит |
|||
|
Хронічний артрит |
||||
|
Ураження нервової системи |
ЦНС |
Менінгіт, енцефаліт, мієліт, енцефалопатія |
||
|
Периферична нервова система |
Неврити черепномозкових нервів, радикулопатії, плексити (плечовий, попереково-крижовий), периферична нейропатія, множинні мононеврити, моторна нейропатія |
|||
|
Ураження внутрішніх органів |
Серце |
Ураження розвивається через 3–12 тиж від початку хвороби. Проявляється АВ-блокадами, порушеннями серцевого ритму, серцевою недостатністю (рідко) |
||
|
Печінка |
Безжовтяничний гепатит (рідко) |
|||
|
Пізній період |
Хронічний перебіг |
Моносиндромне ураження |
Нервова система |
Полінейропатії, енцефалопатії, енцефаломієлополірадикулоневрит (рідко) |
|
Шкіра |
Прогресуюча атрофія або хронічний атрофічний акродерматит |
|||
|
Суглоби |
Стійке запалення великих суглобів (переважно колінних), що триває більше 1 року |
|||
|
Полісиндромне ураження |
Поєднуються ураження шкіри, нервової системи і суглобів |
|||
Клінічні прояви I стадії хвороби
В середньому через 1–3 тиж після укусу кліща у 60–80% хворих розвивається кліщова мігруюча еритема (КМЕ), що є маркером хвороби Лайма. Одночасно з нею або дещо пізніше відмічають збільшення регіонарних лімфатичних вузлів, лихоманку (до 38–39 °С), симптоми інтоксикації: головний біль, відчуття втоми, іноді нудота, блювання, біль у м’язах, радикулоалгія, артралгія. Симптоми інтоксикації помірно виражені й зберігаються протягом 3–7 діб.
Характеристика КМЕ. Починається з невеликої еритемної плями на місці присмоктування кліща. Пляма швидко збільшується, може набути ціанотичного відтінку, периферичні ділянки її утворюють яскравий червоний валик неправильної форми з фестончастими краями. Діаметр еритеми перевищує 5–10 см, вона найчастіше виявляється в пахвовій ділянці, на животі, шиї — місцях, де любить присмоктуватися кліщ. Кліщова еритема в середньому зберігається 4–10 тиж і безслідно зникає.
Характеристика II стадії хвороби Лайма
Вона називається ранньою дисемінованою інфекцією, виникає в результаті гематогенного поширення збудника і характеризується широким спектром клінічних проявів, розвитком різних синдромів: шкірного, неврологічного, ревматологічного, кардіологічного, очного та ін. Ці синдроми проявляються в різних поєднаннях і розвиваються в різний час з моменту виникнення хвороби. Маркером стадії дисемінації служить вторинна еритема, яка з’являється у віддалених від місця укусу місцях і є результатом гематогенного поширення збудника. Характерним шкірним проявом є лімфоцитома (доброякісна лімфоцитома, лімфаденома шкіри), що є пухлиноподібним ущільненням, яке може локалізуватися в мочці вуха, в ділянці соска молочної залози, які стають набряклими, набувають яскравого малинового кольору (іноді фіолетового) і злегка болючі та щільні на дотик. Лімфоцитома може поєднуватися з іншими симптомами хвороби Лайма. Для цієї стадії характерні такі неспецифічні прояви, як нодозна еритема, періорбітальний набряк, геморагічна пурпура, уртикарний васкуліт, панікуліт, долонний висип (капілярит). З боку опорно-рухового апарату в II стадії відмічають артралгії, осалгії, короткі атаки зворотного артриту, тендиніти, бурсити, міалгії, міозит. У II стадії хвороби Лайма можливий розвиток ряду проявів з боку нервової системи: менінгіт; множинні мононеврити; енцефаліт; мієліт; черепномозкові неврити; судомний синдром; неврит лицьового нерва; церебральна атаксія; рухові або чутливі радикулоневрити.
У цій стадії можуть відмічати ураження інших органів і систем: лімфатичної системи (генералізована лімфаденопатія, спленомегалія); очей (кон’юнктивіт, ірит, хоріоїдит, панофтальміт, геморагії в сітківці); респіраторної системи (фарингіт); серця (АВ-блокада, міокардит, перикардит); шлунково-кишкового тракту (гепатит); сечостатевої системи (протеїнурія, мікрогематурія, орхіт).
У процесі дисемінації борелії уражують печінка (у 15–19% хворих). Ознаки гепатиту виражені слабко: відзначаються диспептичні прояви, відчуття важкості в правому підребер’ї, помірне збільшення печінки, підвищення функціональних проб (рівня білірубіну, активності АлАТ, АсАТ).
При морфологічному дослідженні виявляють ознаки запалення, виражені різною мірою: від помірної лімфоцитарної інфільтрації до ушкодження клітин (як при гострому вірусному гепатиті). У тканині печінки виявляють борелії. Загалом перебіг гепатиту сприятливий. Хороший терапевтичний ефект дає тетрациклін (доксициклін 200 мг/добу, міноциклін 200 мг/добу протягом 14–21 днів), клафоран (4 г/добу — 14 днів), пеніцилін.
Інфікованість бореліями є чинником ризику приєднання вірусного гепатиту (у 30% пацієнтів виявляють його маркери), в таких випадках підвищена активність трансаміназ зберігається декілька місяців.
Неврологічні прояви у пацієнтів із хворобою Лайма відзначаються на всіх стадіях (табл. 25.14).
Таблиця 25.14
Неврологічні прояви, що відмічають при хворобі Лайма
|
I стадія |
Мігруючий біль у різних відділах хребта і по ходу нервових стовбурів |
|
II стадія |
Ураження ЦНС (менінгіт, енцефаліт, мієліт, енцефалопатія), периферичної нервової системи (черепно-мозкові неврити, периферична нейропатія, мононеврити, радикулопатії, плечовий і попереково-крижовий плексити, прояви, що нагадують синдром Гійєна — Барре, моторна нейропатія та ін.). Ці зміни можуть відмічати окремо або в різних поєднаннях. Хворі скаржаться на чутливі і рухові порушення. Грудний радикуліт проявляється болем, відчуттям стиснення, гіпо- і гіпестезією. Характерним ураженням є парез лицьових нервів, який може бути одно- або двобічним і не супроводжується розвитком повного паралічу м’язів обличчя (відновлення починається через 2–3 тиж). За наявності менінгіту (частіше розвивається у дітей) відзначають головний біль, нудоту, блювання, світлобоязнь, біль при рухах очних яблук. У спинномозковій рідині виявляють лімфоцитарний цитоз, підвищений вміст білка, глюкози, IgG і IgM. Енцефаліт проявляється порушеннями сну, концентрації уваги, пам’яті, підвищеною збудливістю. При КТ у мозку виявляються зворотні вогнищеві зміни |
|
III стадія |
Хронічний поліневрит, енцефаломієліт, атаксія, розлади пам’яті, деменція |
Основним типом ураження периферичної нервової системи при нейробореліозі є множинна мононейропатія. При краніальній нейропатії уражуються лицьовий, окоруховий, трійчастий, зоровий, слуховий нерви. Уражується речовина ЦНС у вигляді гострого або підгострого енцефаліту (сонливість, порушення пам’яті, уваги, парези, атаксія, хорея, напади). Іноді розвивається гострий або підгострий поперечний мієліт з пара- або тетрапарезом, тазовими порушеннями. ІІІ стадія характеризується прогресуючим енцефаломієлітом. Він розвивається через декілька місяців або років після інфікування. Зрідка виникають паркінсонізм, енцефалопатія. Внаслідок ураження периферичної нервової системи розвивається дистальна сенсомоторна полінейропатія з парестезіями в кистях та стопах, множинна мононейропатія.
Особливості суглобового синдрому. На думку дослідників, хвороба Лайма є моделлю хронічного артриту інфекційної природи. Ураження суглобів, синовіальної оболонки настає в результаті гематогенного поширення збудника, потрапляння його в суглоб в перші дні або тижні захворювання. Протягом подальших декількох місяців розвивається специфічна імунна (клітинна і гуморальна) відповідь. У руйнуванні хряща і кістки активно беруть участь цитокіни і, зокрема, ІЛ-1. У крові виявляються ЦІК, кріопреципітати, РФ, антинуклеарні антитіла, антитіла до кардіоліпіну, тканин. Як вже відмічено, існують спільні антигенні детермінанти у флагелярного антигена борелій і синовіальної оболонки. Особливостями суглобового синдрому вважають:
- залучення (переважно) великих суглобів — колінних, кульшових, плечових;
- моноолігоартикулярний тип ураження;
- слабку вираженість синовіту;
- запалення періартикулярних тканин, якщо суглоб знаходиться поблизу місця присмоктування кліща;
- відсутність ураження хребта, сакроілеїту;
- ураження сухожильно-зв’язкового апарату у вигляді тендинітів, тендосиновітів, бурситів, ентезопатій, фіброзитів;
- ураження м’язів у вигляді міозитів, міалгій;
- поєднання суглобових проявів з неврологічними;
- підвищена частота виявлення HLA-DR2, -DR4.
У пацієнтів із хворобою Лайма відмічають 2 варіанти ураження суглобів: доброякісний рецидивуючий артрит і хронічний артрит (табл. 25.15).
Таблиця 25.15
Варіанти ураження суглобів при хворобі Лайма
|
Доброякісний рецидивуючий артрит |
Розвивається після періоду артралгій в терміни від декількох днів до декількох місяців з моменту виникнення мігруючої еритеми. Частіше розвивається асиметричний моноолігоартрит із залученням колінних суглобів (ураження дрібних суглобів нехарактерне). Суглобові атаки тривають від одного до декількох тижнів, рецидивують в проміжки від декількох тижнів до декількох місяців. Такий варіант артриту (за типом реактивного) може тривати до 5 років |
|
Хронічний артрит |
У 10% хворих (носіїв HLA-DR2 і -DR4) артрит набуває хронічного характеру і супроводжується утворенням панусу, ерозією хряща, розвитком лігаментитів, бурситів, ентезопатій. На пізньому етапі виявляють ОП, зменшення товщини і втрату хряща, кортикальні і крайові узури, осифікацію меніска і періартикулярних тканин, прояви вторинного ОА (субхондральний склероз, кісти, остеофіти). Відзначається серопозитивність (IgM-фактор) при використанні імуноферментного методу і серонегативність в латекс-тесті. У міру збільшення тривалості артриту в синовіальній рідині збільшується кількість ЦІК |
В основі розвитку синовіту при хворобі Лайма лежать імунопатологічні аутоімунні реакції. У синовіальній оболонці виявляють характерні морфологічні зміни:
- гіперплазія синовіальних клітин;
- неспецифічна гіпертрофія ворсин;
- лімфоплазмоклітинна інфільтрація;
- утворення фолікулів, відкладення фібрину;
- ураження мікроциркуляторного русла за типом облітеруючого ендартеріїту,
- борелії усередині судин синовіальної мембрани і периваскулярно; невелику кількість спірохет виявляють і в синовіальній рідині.
Борелії можуть персистувати в синовіальній оболонці протягом багатьох років і бути тригером хронічної лімфоплазмоклітинної імунної відповіді. У перихондральній частині синовіальної оболонки формується синовіальний панус — судинна і фіброзна тканина, яка вростає в хрящ і руйнує його.
Зміни м’язової системи. На ранній стадії хвороби Лайма відмічають набряклість м’язів, на пізніших стадіях — інтерстиціальний, вогнищевий вузликовий міозит. Клінічно міозит проявляється слабкістю та міалгією. Електроміографія істотно не змінюється, рівень КФК нормальний.
Ураження серця. Ознаки ураження серця виникають в період від 3 до 12 тиж від початку захворювання у 8–10% хворих. Розвиваються транзиторні АВ-блокади різного ступеня. При повній блокаді може бути необхідна імплантація водія ритму. Можливе порушення внутрішньошлуночкової провідності, часто розвиваються тахіаритмії (суправентрикулярні, шлуночкові). Лайм-кардит (міокардит, перикардит) рідко призводить до вираженої серцевої недостатності. Проте він може персистувати і викликати розвиток дилатаційної кардіоміопатії.
Прояви офтальмобореліозу. До них відносять неспецифічний фолікулярний кон’юнктивіт; кератит, нейротрофічний кератит; ураження склоподібного тіла; передній увеїт; неврит зорового нерва; нейроретиніт.
Зміни, що відмічають у III стадії хвороби Лайма
Ця стадія розвивається у нелікованих хворих і пов’язана з персистенцією збудника. Вона характеризується ураженням шкіри (хронічний атрофічний дерматит, вогнищеві склеродермоподібні зміни), опорно-рухового апарату (хронічний артрит, ентезопатії, періостит, міозит), нервової системи (хронічний поліневрит, енцефаломієліт, атаксія, розлади пам’яті, деменція), очей (кератит). Хронічний енцефаломієліт клінічно нагадує розсіяний склероз, може проявлятися порушеннями сну, мови, поведінки. Розвиток полінейропатії супроводжується розладами чутливості, дистальними парезами, радикулярним болем.
До пізніх проявів хвороби Лайма відноситься хронічний атрофічний акродерматит, який відмічають частіше у жінок літнього віку, хоча може розвинутися і у молодих хворих обох статей. Процес локалізується на розгинальних поверхнях кінцівок (коліна, лікті, тил кисті, підошви). Спочатку там з’являються інфільтративно-запальні прояви — плями, які супроводжуються інфільтратами, набряком шкіри і мають тенденцію до периферичного зростання і злиття. Запалення розвивається протягом декількох місяців або років. Шкіра на цьому місці поступово атрофується (нагадує цигарковий папір), через неї видно гіперпігментовані вени і сухожилля. Можливий розвиток склеротичного процесу, що нагадує склеродермію.
Особливості перебігу хвороби Лайма у дітей. У пацієнтів цієї категорії відмічають ряд особливостей клініки і перебігу хвороби (табл. 25.16).
Таблиця 25.16
Деякі особливості перебігу хвороби Лайма у дітей
|
Найчастішим місцем присмоктування кліща є голова (треба оглядати завушну ділянку, волосисту частину) |
|
Мігруюча еритема супроводжується грипоподібним синдромом і це є основою для призначення антибіотиків |
|
Значно частіше, ніж у дорослих, розвивається артрит |
|
Першим проявом хвороби може бути тільки артрит (коли не помічається укус кліща, а шкірна еритема не розвивається) |
|
У деяких нелікованих дітей через 10–12 років розвивається енцефалопатія, яка проявляється головним болем, загальною слабкістю, зниженням уваги, порушенням пам’яті, відставанням у навчанні |
|
Серопозитивність виявляють у 2 рази частіше, ніж у дорослих |
Діагностика хвороби Лайма
З метою діагностики хвороби Лайма виконують ЕКГ, ехоКГ, УЗД печінки, суглобів, гістологічне вивчення біоптатів шкіри, синовіальної оболонки, біохімічні дослідження. В Інституті ревматології РАМН використовують клініко-серологічний підхід до діагностики цієї патології (Ананьева Л.П., 1997; Насонов Е.Л., 2010 (ред.)). Діагноз «хвороба Лайма» вважається достовірним в наступних випадках:
1. У пацієнта розвивається типова кліщова мігруюча еритема. Діагноз кліщової мігруючої еритеми має бути встановлений лікарем.
2. За відсутності типової КМЕ діагноз встановлюється у разі розвитку у хворого одного або поєднання декількох типових синдромів при підтвердженні інфекції лабораторно:
- ураження нервової системи у вигляді серозного менінгіту (при виключенні кліщового вірусного енцефаліту), менінгорадикуліту (синдром Баннварта), периферичного невриту, невритів черепних нервів;
- ураження суглобів за типом моно- або олігоартриту рецидивуючого або хронічного характеру;
- ураження серця: порушення провідності за типом АВ-блокади II або III ступеня або міоперикардит;
- одинична лімфоцитома мочки вуха або соска молочної залози;
- хронічний атрофічний акродерматит.
Уніфіковані клінічні і серологічні критерії діагнозу хвороби Лайма не розроблені.
Лабораторна верифікація хвороби Лайма
Методи лабораторної верифікації бореліозної інфекції наведені в табл. 25.17.
Таблиця 25.17
Методи лабораторної верифікації хвороби Лайма
|
1. |
Метод непрямої імунофлуоресценції (основний), що виявляє антитіла до збудника (для Центрального і Північно-Західного регіонів Росії межею норми є титр 1:40, діагностичний 1:80 і вище або 4-разове наростання титру в процесі спостереження) |
|
2. |
Метод ензиммічених антитіл |
|
3. |
Метод «вестерн-блотинг» (застосовується при близьких до норми результатах і для виключення псевдопозитивних результатів). Ці два методи виявляють реактивність до 12 і більше антигенів борелій |
|
4. |
Посіви на спеціальні культуральні середовища біоптатів шкіри, синовіальної оболонки, біологічних рідин (крові, синовіальної і спинно-мозкової рідини) |
|
5. |
Забарвлення тканин (шкіри) сріблом для виявлення борелій в їх зрізах (використовується при КМЕ) |
|
6. |
Виявлення в досліджуваному матеріалі специфічних фрагментів ДНК борелій за допомогою високочутливого і високоспецифічного методу ПЛР |
У Європі серодіагностика хвороби Лайма ускладнена тим, що інфекція може викликатися різними серогрупами збудника. При визначенні антитіл до борелій отримують псевдопозитивні і псевдонегативні результати. При оцінці отриманих результатів серологічного дослідження рекомендується враховувати:
- клінічні дані;
- при рано розпочатому етіотропному лікуванні серологічна реакція може бути негативною;
- у Європі імунна відповідь хворих на борелії виражена слабко;
- серологічне дослідження на обох континентах не стандартизовано і лабораторії дають різні результати;
- у хворих, що перенесли інфекцію, роками може зберігатися високий титр антитіл до борелій без будь-яких клінічних проявів;
- розвиток будь-якого іншого артриту у пацієнта, що має безсимптомну інфекцію, позитивний серологічний тест на хворобу Лайма призведе до помилкової діагностики лайм-артриту.
Для ідентифікації збудника хвороби використовують як прямі (виявлення борелій в біоптатах за допомогою світлової та електронної мікроскопії; проведення ПЛР; культивування на поживних середовищах), так і непрямі методи (виявлення антибореліозних антитіл; проведення тесту специфічної стимуляції лейкоцитів). При трактуванні результатів серологічних тестів рекомендується враховувати наступні факти:
- частота виявлення низького титру антибореліозних антитіл в перші тижні після інфікування становить 10–20%;
- при дисемінації титр антитіл, що перевищує пороговий, визначається у 1/3 пацієнтів;
- високі показники серологічних тестів відмічають у нелікованих хворих з хронічним перебігом хвороби;
- у цілому факт підвищення титрів антитіл до борелій має допоміжне значення.
При проведенні серологічних досліджень слід дотримуватися ряду рекомендацій: зіставляти результати з анамнезом, клінікою і даними інших лабораторних досліджень; застосовувати дослідження парних сироваток; при невизначеній симптоматиці, титрах антитіл, не нижчих за порогові, використовувати імуноблот.
Диференційно-діагностичні ознаки Лайм-артриту
Вони наведені в табл. 25.18.
Таблиця 25.18
Диференційно-діагностичні ознаки Лайм-артриту
|
Артрит виникає в середньому через 0,5 року від початку хвороби |
|
Характерними варіантами прояву артриту є: 1) артралгії; 2) один епізод артриту; 3) рецидивуючий артрит; 4) хронічний артрит |
|
У більшості випадків ураження має моно- і олігоартикулярний характер |
|
Уражуються великі суглоби (колінні, кульшові, плечові) |
|
Вираженість синовіту слабка або помірна |
|
Залучаються періартикулярні тканини, розвиваються тендиніти, тендовагініти, бурсити, набряк м’язів |
|
Ураження хребта і розвиток сакроілеїту не характерні |
|
При УЗД виявляють ознаки наявності ексудату в суглобі і залучення до патологічного процесу періартикулярних тканин |
|
У 60–80% хворих виявляють антитіла до борелій |
|
У 77% хворих визначають антигени HLA-DR4 та/або -DR2 |
|
HLA-B27 визначається, як у популяції |
За наявності системних (поліорганних) проявів перебіг хвороби Лайма може під маскуватися проявами РеА, ревматизму, РА, ЮА, серонегативних спондилоартритів, СЧВ. Хронічний рецидивуючий перебіг артриту вимагає включення хвороби Лайма в диференційний діагноз хронічного артриту, насамперед в ендемічних регіонах.
Хворобу Лайма доводиться диференціювати з рядом неврологічних і психічних захворювань:
- діабетична й ідіопатична периферична нейропатія;
- цервікальна й люмбальна радикулопатія різної етіології;
- пухлини мозку і мозочка;
- розсіяний склероз, бічний аміотрофічний склероз;
- ураженням нервової системи при антифосфоліпідному синдромі;
- судомні розлади;
- синдром хронічної втоми, депресивні стани, шизофренія.
Існує трансплацентарна передача збудника і вона супроводжується внутрішньоутробною інфекцією, вадами розвитку плода, його загибеллю.
З бореліозною інфекцією пов’язують розвиток ряду синдромів. До них відносять вогнищеву склеродермію, склерозуючий ліхен, еозинофільний фасцит, прогресуючу геміатрофію обличчя.
Основні принципи терапії хвороби Лайма, Лайм-артриту
Вони наведені (Ананьєва Л.П., 2003; Насонов Е.Л., 2010 (ред.)) в табл. 25.19.
Таблиця 25.19
Основні принципи терапії хвороби Лайма
|
Стадія захворювання |
Вік хворих |
Препарати |
Примітки |
|
Препарати вибору |
|||
|
I. Рання стадія захворювання (неускладнена мігруюча еритема та/або легкі прояви дисемінації — одиничні вторинні еритеми, кардит з АВ-блокадою I стадії, стерта неврологічна симптоматика) |
Дорослі усередину протягом 14–21 дня |
Амоксицилін 0,5–1,0 г 3 рази на добу |
Тривалість лікування становить 14–21 день (іноді до 1 міс і більше). Рекомендується враховувати, що чим довше зберігаються прояви хвороби до лікування, тим більше часу необхідно для їх купірування і тим вище вірогідність неповного одужання. Доксициклін протипоказаний дітям віком до 8 років, вагітним і жінкам, що годують грудьми. Контрольовані дослідження свідчать, що усі вищеперелічені препарати викликають приблизно однаковий ефект |
|
Амоксицилін/клавуланат 0,375 г 3 рази на добу |
|||
|
Доксициклін 0,1 г 2 рази на добу |
|||
|
Діти віком старше 8 років усередину протягом 14–21 дня |
Амоксицилін/клавуланат 0,375 г 3 рази на добу |
||
|
Доксициклін 1–2 мг/кг/добу в 2 прийоми (не більше 100 мг на прийом) |
|||
|
Діти молодше 8 років усередину протягом 14–21 дня |
Амоксицилін 50 мг/кг/добу в 3 прийоми (не більше 500 мг на прийом) |
||
|
Амоксицилін/клавуланат 20–40 мг/кг/добу (з розрахунку на амоксицилін) в 3 прийоми |
|||
|
Альтернативні схеми лікування |
|||
|
Дорослим усередину |
Цефуроксим аксетил по 0,5 г 2 рази на добу |
||
|
Дітям усередину |
Цефуроксим аксетил 30 мг/кг/добу в 2 прийоми |
||
|
При непереносимості вищеперелічених засобів |
|||
|
Дорослим усередину протягом 7–10 діб |
Азитроміцин 500 мг/добу |
||
|
Дорослим усередину протягом 14–21 доби |
Кларитроміцин 500 мг 2 рази на добу |
||
|
Еритроміцин 500 мг 4 рази на добу |
|||
|
Дітям усередину протягом 7–10 діб |
Азитроміцин 10 мг/кг/добу |
||
|
Дітям усередину протягом 14–21 доби |
Кларитроміцин 7,5 мг/кг/добу в 2 прийоми |
||
|
Еритроміцин 12,5 мг/кг/добу |
|||
|
II. Дисеміновані і пізні форми (у тому числі хронічний нейробореліоз, атрофічний акродерматит, хронічний лайм-артрит) |
Препарати вибору (схеми лікування) |
||
|
В/в протягом 14–28 діб |
Цефтріаксон по 1 г 1 раз на добу |
Цефалоспорини III покоління на ранній стадії не мають переваг перед наведеними вище схемами. Цефтріаксону віддають перевагу при середньо-тяжкому і тяжкому перебігу бореліозу, ураженні нервової системи, АВ-блокадах II–III стадії. Дозу пеніциліну підвищують до 16 млн ОД/добу при менінгоенцефалітах |
|
|
Цефуроксим по 2 г 2–3 рази на добу |
|||
|
Альтернативні препарати (схеми лікування) : |
|||
|
В/в або в/м протягом 14 діб |
Бензилпеніциліну натрієва сіль 18–24 млн ОД/добу, розділені на 6 введень |
||
|
Бензилпеніциліну натрієва сіль по 500 тис. — 1 млн ОД 8 разів на добу (з інтервалом 3 год) |
|||
|
III. Лайм-артрит без супутніх неврологічних проявів |
Усередину протягом 28–30 діб |
Амоксицилін 0,5–1,0 г 3 рази на добу |
|
|
Амоксицилін/клавуланат 0,375 г 3 рази на добу |
|||
|
Доксициклін 0,1 г 2 рази на добу |
|||
|
IV. Виражений лайм-артрит із залученням періаперіартикулярних тканин |
1-ша схема лікування в/в протягом 14 діб |
Цефтріаксон по 1 г 1 раз на добу |
Доксициклін протипоказаний дітям віком до 8 років, вагітним і жінкам, які годують грудьми |
|
Цефуроксим по 2 г 2–3 рази на добу |
|||
|
Потім усередину 14 діб |
Амоксицилін 0,5–1,0 г 3 рази на добу |
||
|
Амоксицилін/клавуланат 0,375 г 3 рази на добу |
|||
|
Доксициклін 0,1 г 2 рази на добу |
|||
|
2-га схема лікування в/в протягом 14–28 діб |
Доксициклін 200–400 мг/добу в 2 введення |
||
|
V. Лайм-артрит з неврологічними проявами |
В/в або в/м протягом 14 діб |
Бензилпеніциліну натрієва сіль 18–24 млн ОД/добу, розділені на 6 введень |
Ефект антимікробної терапії розвивається повільно. За відсутності поліпшення проводять повторні курси через 2–3 міс, які також можуть бути малоефективними |
|
Бензилпеніциліну натрієва сіль 500 тис. — 1 млн ОД 8 разів на добу з інтервалом 3 год |
|||
|
В/в протягом 14–28 діб |
Цефтріаксон по 1 г 1 раз на добу |
||
|
Цефуроксим по 2 г 2–3 рази на добу |
|||
|
VI. Рецидиви лайм-артриту |
Усередину протягом 28 діб |
Амоксицилін 0,5–1,0 г 3 рази на добу |
НПЗП і ГК додають за відсутності ефекту від антибіотиків. Доксициклін протипоказаний дітям до 8 років, вагітним і жінкам, які годують грудьми. Після 3 курсів неефективної антибіотикотерапії рекомендують артроскопічну синовектомію |
|
Амоксицилін/клавуланат 0,375 г 3 рази на добу |
|||
|
Доксициклін 0,1 г 2 рази на добу |
|||
|
В/в протягом 14–28 діб |
Цефтріаксон по 1 г 1 раз на добу |
||
|
Додатково |
НПЗП |
||
|
ГК внутрішньосуглобово |
|||
|
При гіперчутливості до пеніцилінів (цефалоспоринів) |
|||
|
Усередину протягом 14 діб |
Кларитроміцин по 0,5 г 2 рази на добу |
||
|
В/м протягом 14 діб |
Левоміцетин сукцинат по 1,5 г 3 рази на добу |
||
Найбільше поширені схеми антибактеріальної терапії хвороби Лайма, які грунтуються на рекомендаціях експертів Американської асоціації інфекційних хвороб (табл. 25.20).
Таблиця 25.20
Антибактеріальна терапія хвороби Лайма
|
Рання стадія |
||
|
Неускладнена еритема або дисемінована хвороба Лайма без неврологічних проявів |
Per os доксициклін (100 мг 2 рази на добу), амоксицилін (500 мг 3 рази на добу) або цефуроксим (500 мг 2 рази на добу) протягом 14–21 днів |
Доксициклін не призначають дітям до 8 років, вагітним, у період годування грудьми |
|
Азитроміцин (500 мг/добу, 7–10 днів), кларитроміцин (500 мг 2 рази на добу, 14–21 день), еритроміцин (2 г/добу в 4 прийоми, 14–21 день) |
Ці антибіотики менш ефективні, їх не застосовують в якості 1-ї лінії. Призначають при протипоказаннях та непереносимості інших антибіотиків |
|
|
Цефалоспорини III покоління |
Не рекомендовані в якості 1-ї лінії терапії, так як за ефективністю не перевершують попередні схеми для прийому per os |
|
|
Ураження нервової системи (менінгіт, радикулоневрит), АВ-блокада II–III стадії |
Цефтріаксон (2 г в/в або в/м 1раз на добу) 10–28 днів |
Препарат вибору |
|
Цефотаксим (2 г в/в або в/м 3 рази на добу), бензилпеніцилін (пеніцилін G натрієва сіль) (14–24 млн ОД в/в в 6 прийомів) 14–28 днів |
Препарати 2-ї лінії |
|
|
Пізня стадія |
||
|
Лайм-артрит |
Per os доксициклін (100 мг 2 рази на добу), амоксицилін (0,5 г 3 рази на добу), цефуроксим (0,5 г 2 рази на добу) протягом 28 днів |
Повторний курс (для повного купірування симптомів артриту) проводять не раніше ніж через 3 міс, краще застосовувати цефалоспорини III покоління. У разі поєднання артриту з ураженням нервової системи доцільне парентеральне лікування бета-лактамними антибіотиками. У 7–12% хворих не виявлено позитивної відповіді на антибіотикотерпію |
|
Нейробореліоз |
Цефтріаксон 2 г в/в або в/м, 14–28 днів |
Препарат вибору |
|
Цефотаксим 2 г 3 рази на добу, бензилпеніцилін 18–24 млн ОД в/в в 6 прийомів, 14–28 днів |
Альтернативні засоби |
|
У зв’язку з тим, що різні генотипи борелій мають різну чутливість до антибіотиків, існує необхідність розробити схеми лікування для кожного регіону України. Особливості лікування хвороби Лайма антибіотиками наведені в табл. 25.21.
Таблиця 25.21
Особливості лікування хвороби Лайма антибіотиками
|
Лікування слід починати максимально рано (при виявленні КМЕ) і проводити до повного усунення симптомів |
|
При лікуванні, початому в перші 10 днів хвороби, настає одужання |
|
Лікування антибіотиками виправдане на будь-якій стадії хвороби (незалежно від її давності) |
|
Терапія має бути індивідуалізована: залежати від клінічної форми, характеру і тяжкості перебігу |
|
Слід враховувати, що в період лікування антибіотиками (особливо пеніцилінами) можливе короткочасне збільшення вираженості симптомів хвороби і інтоксикації (розвиток реакції Яриша — Герксгеймера) |
|
Показаннями до в/в застосування антибіотиків є неврологічна симптоматика, розвиток кардиту |
|
Якщо після курсу антибіотиків усередину розвиваються неврологічні прояви, потрібне призначення антибіотиків в/в |
|
Відповідь на лікування може розвиватися повільно і тоді протягом найближчих місяців вимагається повторний курс лікування до повного зникнення симптомів |
|
Якщо протягом 3 міс лікування позитивного ефекту не відмічено, потрібний ще один курс із застосуванням цефтріаксону (препарату вибору) |
|
За відсутності відповіді на лікування антибіотиками у хворих з хронічним артритом колінних суглобів рекомендується лікування протизапальними препаратами і внутрішньосуглобовим введенням ГК |
|
При неефективності і цієї терапії протягом 6–12 міс рекомендують артроскопічну синовектомію |
У хворих з КМЕ кращі результати отримують при призначенні доксицикліну 0,2 г/добу усередину протягом 14 днів, а також при використанні цефуроксиму 1 г/добу протягом 10 діб. Дещо гірші результати відмічають при застосуванні пеніциліну 2 млн ОД в/м 10–14 днів, тетрацикліну 1,2 г/добу протягом 14 днів. Оптимальні результати отримують при призначенні антибіотиків в перші 5 днів хвороби з тривалістю терапії не менше 14 днів. При неефективності наведеної вище терапії не раніше, ніж через 3 міс призначають пеніцилін 20 млн ОД/добу в/в з 4–6-годинними інтервалами протягом 10–14 діб або цефтріаксон 2 г/добу в/в одноразово 14 діб. Результативність лікування оцінюють за динамікою клінічних симптомів (мікробіологічний критерій недоступний). У зв’язку з тим, що пізні симптоми хвороби можуть розвинутися після лікування КМЕ, ефективність лікування слід оцінювати не раніше 6–12 міс від початку хвороби Лайма. Тетрацикліни при хронічному запаленні в суглобах інактивують металопротеїназу в синовіальній тканині; пригнічують активність фосфоліпази А2; знижують окиснювальну активацію латентної колагенази; інгібують функціональну активність нейтрофілів (хемотаксис, фагоцитоз, утворення реактивних форм кисню).
У 50% пацієнтів, що отримували антибактеріальне лікування з приводу мігруючої еритеми, розвивається «постлаймівський» синдром: протягом 0,5–3 років відмічають головний та м’язово-скелетний біль, постійну втому, зниження пам’яті, порушення уваги, вегетативних функцій та ін.
Профілактика хвороби Лайма
Вакцинацію в Україні не застосовують. Рекомендують використання репелентів, носіння одягу, що закриває тіло, огляди тіла і своєчасне видалення кліщів. Антибіотики призначають при виявленні борелій у переносника. Превентивну терапію (доксициклін по 0,1 г 1–2 рази на добу протягом 3–5 діб) при їх виявленні починають не пізніше перших 3 діб після присмоктування кліща (Ананьєва Л.П., 2003). З профілактичною метою протягом перших 5 діб після присмоктування кліща автор рекомендує прийом усередину протягом 5 діб азитроміцину (1,0 г в 1-шу добу і 0,5 г в наступні) або амоксициліну/клавуланату (0,375 г 3 рази на добу), або одноразове в/м введення бензатинбензилпеніциліну 2,4 млн ОД. Контрольне обстеження рекомендується через 1–3 міс.
Диспансеризація пацієнтів із хворобою Лайма
Після перенесеної гострої стадії хворих слід спостерігати протягом 1 року. За відсутності клінічної симптоматики клініко-серологічне обстеження проводиться не менше 1 разу в 6 міс. Якщо симптоми зберігаються або поновлюються, тактика ведення визначається індивідуально. Спостереження здійснюється інфекціоністом або терапевтом. Якщо розвивається ураження суглобів, нервової системи, шкіри залучаються відповідні фахівці.
Прогноз при хворобі Лайма
Своєчасна та адекватна антибактеріальна терапія попереджує прогресування хвороби, розвиток пізніх стадій. У 10% пацієнтів навіть своєчасне та адекватне лікування може бути неефективним. Непрацездатність відмічають в 10% випадків ураження суглобів, хвороба набуває рецидивуючого або хронічного перебігу.
Вірусні артрити
Код за МКХ-10
В06.8 Краснуха з іншими ускладненнями: артрит (М01.4).
В15–В19 Вірусний гепатит.
М03.2 Постінфекційна артропатія при вірусному гепатиті (В15–В19).
В02 Оперізувальний лишай (герпес зостер).
В02.8 Оперізувальний лишай з іншими ускладненнями.
В20–В24 Хвороба, зумовлена вірусом імунодефіциту людини.
В26. Епідемічний паротит.
В26.8 Епідемічний паротит з іншими ускладненнями: артрит (М01.5).
В34.0 Аденовірусні інфекції, неуточнені.
Етіологія вірусних артритів
Вони розвиваються під час деяких вірусних інфекцій: грип, кір, краснуха, інфекційний гепатит, вітряна віспа, епідемічний паротит. Часто їх викликають парвовіруси В19, альфа-віруси, ентеровіруси, аденовіруси, віруси герпесу.
Парвовірусна інфекція. Інфекція, викликана парвовірусами В19, є досить поширеною і передається повітряно-краплинним шляхом. Перебіг може бути субклінічним, викликаючи в той же час розвиток ревматичних синдромів. Часто хворіють діти: підвищені титри антитіл IgG виявляють у 60% молодих людей віком до 20 років. Інкубаційний період захворювання становить 5–14 днів, потім з’являється еритематозний і папульозний висип, що зудить. Він спочатку локалізується на щоках і нагадує бешихове запалення або почервоніння після ляпаса. Надалі подібна еритема з’являється на плечах, стегнах. Тривалість захворювання становить від 1–2 тиж до 3 міс.
Парвовірусна В19-інфекція асоціюється з наступними ревматичними синдромами:
- транзиторна артралгія (у 20% інфікованих пацієнтів);
- ревматоїдоподібний синдром;
- системні некротизуючі васкуліти (ВП, гранулематоз Вегенера, хвороба Кавасакі, Шенляйна — Геноха);
- хвороба Стілла у дорослих;
- хвороба Кикучі.
Особливості суглобового синдрому при парвовірусній В19 інфекції наведені в табл. 25.22.
Таблиця 25.22
Особливості суглобового синдрому при парвовірусній В19 інфекції
|
Артрит розвивається одночасно з шкірною еритемою або випереджає висип на 7 і більше днів |
|
Суглобовому синдрому передують продромальні розлади у вигляді нездужання, м’язового і головного болю, шлунково-кишкових розладів |
|
Рідко розвивається у дітей (близько 5%); у 50% з них розвивається синовіт, у інших — тільки артралгія; іноді клініка артриту нагадує ЮРА |
|
Тяжчий і стійкіший артрит розвивається у дорослих, при цьому жінки хворіють в 3 рази частіше |
|
Характерним є симетричне двобічне ураження суглобів кистей і стоп, променезап’ясткових, ліктьових, колінних, що супроводжується розвитком набряклості, скутості в суглобах, болю |
|
Ознаки артриту зберігаються близько 10 днів, проте можливий перебіг його більше 2 років з періодами ремісії і загострення |
|
Розвитку деформації суглобів не відмічено |
|
У 20% пацієнтів виникає лихоманка |
|
Одночасно з суглобовим синдромом виявляють ураження слизових оболонок у вигляді темно-червоних плям |
|
За клінікою артрит нагадує РА, проте відсутні клінічні і рентгенологічні ознаки прогресування артриту |
|
У 65% хворих з суглобовим синдромом, що триває більше 2 міс, виявляють носійство HLA-DR4 (що асоціюється з РА) |
Критерії діагнозу парвовірусної В19-інфекції. Про цю інфекцію свідчить поява в сироватці крові антитіл IgM (після гострої інфекції антитіла присутні в крові протягом 2–3 міс). Також виявляють антитіла до ДНК, АНФ, РФ.
Краснуха. Інкубаційний період при цьому захворюванні триває 14–18 днів. Потім з’являються лихоманка, головний, м’язовий біль, біль у горлі, генералізована лімфаденопатія і висип. Повне одужання настає через 7–10 днів.
Для суглобового синдрому, що розвивається у хворих із краснухою, характерний ряд проявів (табл. 22.23).
Таблиця 25.23
Характерні прояви суглобового синдрому у хворих із краснухою
|
Рідко розвивається у дітей; серед дорослих частіше уражує жінок |
|
Частіше артрит розвивається одночасно з висипом, але може випереджати його на декілька днів |
|
Уражуються зап’ястково-п’ясткові, проксимальні і дистальні міжфалангові суглоби, рідше — колінні, променезап’ясткові і ліктьові |
|
Характерне асиметричне двобічне ревматоїдоподібне ураження; зрідка розвивається моноартрит |
|
Артрит супроводжується ранковою скутістю, набряком і почервонінням суглобів, болем, розвитком іноді тендосиновітів, синдрому зап’ясткового каналу |
|
Рентгенологічні зміни в суглобах відсутні |
|
Синовіальна рідина запального типу |
|
В аналізах крові відзначаються лейкопенія, відносний лімфоцитоз, збільшення ШОЕ і титру РФ IgM |
|
Біль у колінних суглобах, парестезії в кистях можуть бути пов’язані з радикулоневритом |
Тяжкий поліартрит також може розвинутися через 2–10 тиж після вакцинації живою вакциною проти краснухи.
Вірусні гепатити. У продромальний період вірусних гепатитів В і С відмічають цілий ряд симптомів: загальне нездужання, лихоманка, головний біль, анорексія, нудота, біль у животі, макулопапульозний, петехіальний шкірний висип, кропив’янка, ангіоневротичний набряк, артралгія. Артрит, як правило, розвивається в цей період, він 2-бічний, симетричний. До процесу залучаються проксимальні міжфалангові суглоби, рідше — колінні, плечові, кульшові, ліктьові, гомілковостопні, а також хребет. Хворих турбують скутість, біль при рухах, різка болючість при пальпації, відзначається гіпертермія шкіри. При лабораторному обстеженні хворих на вірусний гепатит із суглобовим синдромом виявляють лейкопенію, підвищення ШОЕ, активності АлАТ, АсАТ. Може бути підвищений титр АНФ, виявлений РФ. Білірубін в крові підвищується після завершення артриту. При рентгенологічному дослідженні змін у суглобах не виявляють. Синовіальна рідина запального типу. Прояви артриту зменшуються після появи жовтяниці.
Інфекція Herpes zoster. Вірус вітряної віспи уражує задні вузли нервових стовбурів. Це супроводжується болем в ділянці відповідних нервових закінчень, появою висипу у вигляді везикул ідентичної локалізації. Частіше хворіють особи віком старше 50 років, однаковою мірою чоловіки і жінки. Розвивається артрит дрібних суглобів кистей, альгодистрофічний синдром. Артрит дрібних суглобів кистей і променезап’ясткових суглобів з’являється одночасно з ураженням шкіри. Він проявляється набряком суглобів, болем, може привести до розвитку контрактури і обмеження рухливості. Захворювання може ускладнитися моторним паралічом, що нагадує полімієліт, і менінгоенцефалітом. Артрит в таких випадках локалізується на боці ураження.
Епідемічний паротит. Епідемічний паротит належить до частих вірусних інфекцій. Інкубаційний період становить 18–21 день. Переважно хворіють діти. Чоловіки хворіють в 7 разів частіше, ніж жінки. Характерним проявом хвороби є збільшення і болючість привушних залоз. Можливий розвиток ускладнень — артриту, панкреатиту, орхіту, кардиту, менінгіту, енцефаліту. Для проявів суглобового синдрому у хворих з епідемічним паротитом характерні наступні особливості:
- артрит розвивається у 1 на 200 хворих (0,5%) через декілька днів після початку хвороби, а частіше через 4 тиж;
- має гострий, асиметричний, мігруючий характер;
- переважно уражуються колінні, гомілковостопні суглоби, рідше — плечові, ліктьові, променезап’ясткові, п’ястково-фалангові, проксимальні міжфалангові;
- відмічають виражену скутість, біль при рухах, ексудат в порожнині суглоба, шкірну еритему, локальну гіпертермію.
При диференціації слід пам’ятати про синдром Шегрена, при якому також відмічають рецидивуюче збільшення привушних залоз.
Аденовірусна інфекція. Інфекція проявляється катаральними явищами з боку верхніх дихальних шляхів, лихоманкою, нездужанням, міалгіями. Виникає гострий поліартрит з переважним ураженням колінних суглобів. Етіологічний діагноз встановлюють ретроспективно за високим титром антитіл до вірусу.
ВІЛ-інфекція. ВІЛ, що належить до сімейства ретровірусів, ушкоджує клітини, що продукують рецепторний білок глікопротеїн СD4, який є маркером Т-лімфоцитів СD4+. Ці лімфоцити відіграють центральну роль у розвитку імунної відповіді. У результаті зараження виникає інфекційне захворювання — синдром набутого імунодефіциту (СНІД) людини.
До факторів ризику розвитку ВІЛ-інфекції відносять наркоманію, гомосексуальні і бісексуальні контакти, безладне статеве життя, переливання крові, гемодіаліз, роботу в медичних установах.
Основні групи клінічних симптомів, що виділяють при ВІЛ-інфекції, наведені в табл. 25.24.
Таблиця 25.24
Групи ВІЛ-інфікованних пацієнтів, які виділяють за характером клінічних симптомів
|
Латентний (безсимптомний) перебіг від декількох днів до декількох років |
|
Мононуклеазоподібна гостра інфекція: після зараження протягом перших тижнів з’являються лихоманка, макулярний і папульозний висип, міалгії, фарингіт, артралгії, шлунково-кишкова симптоматика і неврологічні розлади |
|
Генералізована персистуюча лімфаденопатія (синдром лімфаденопатії) зі збільшенням лімфатичних вузлів більше 1 см в діаметрі в 2 і більше ділянках тіла протягом 3 і більше місяців за відсутності інших причин |
|
СНІД із вторинними інфекційними хворобами, пухлинами, неврологічними проявами (деменція, мієлопатія, периферична нейропатія) |
До неспецифічних симптомів недіагностованої ВІЛ-інфекції на різних стадіях належать втома, депресія, схуднення, діарея, ураження шкіри, синдром фіброміалгії, лімфаденопатія, гепатоспленомегалія. У 20–70% хворих із ВІЛ-інфекцією відмічають ревматичні синдроми, які можуть виходити на перший план і сприйматися як самостійне захворювання. Суглобові і кісткові синдроми, що розвиваються при ВІЛ-інфекції, представлені в табл. 25.25.
Таблиця 25.25
Суглобові і кісткові синдроми, що розвиваються при ВІЛ-інфекції
|
Артралгії |
Хвороба Рейтера |
|
РеА |
ПсА |
|
Гострий симетричний артрит |
Остеомієліт |
|
Болючий суглобовий синдром |
Аваскулярний некроз |
|
ВІЛ-артрит |
Альгодистрофія |
|
Септичний артрит (піогенний) |
Кісткова лімфома |
Артрити розвиваються у 30% ВІЛ-інфікованих осіб (табл. 25.26).
Таблиця 25.26
Характеристика ВІЛ-асоційованих ревматичних розладів
|
Характер розладу |
Клінічна характеристика |
|
|
Артралгія |
Найчастіше виявляють у колінних, плечових та ліктьових суглобах. Вважають, що її причиною є транзиторна ішемія кісток. Артралгія з’являється в гостру фазу ВІЛ-інфекції (на тлі фарингіту, виразок в ротовій порожнині, міалгії, лихоманки, загальної слабкості) |
|
|
Артрит |
Виникає як на ранніх стадіях ВІЛ-інфекції, так і на тлі розгорнутої клінічної картини СНІДу. Може з’явитися за декілька місяців до появи перших симптомів ВІЛ-інфекції у вигляді гострого або підгострого моно-, олігоартриту із залученням суглобів нижніх кінцівок. Можливий розвиток ревматоїдоподібного симетричного поліартриту дрібних суглобів кистей та стоп. Артрит може набути затяжного та хронічного перебігу. ВІЛ-асоційований артрит супроводжується сильним болем, порушенням функції суглобів (колінних, гомілкових, плечових, ліктьових) без наявності ознак запалення в синовіальній рідині |
|
|
Септичний артрит |
СНІД може супроводжуватися розвитком септицемії, гострого бактеріального артриту. Септичні ускладнення у вигляді септичного артриту, тендосиновіту і бурситу виникають у 1% ВІЛ-інфікованих. Їх розвитку сприяють в/в введення наркотиків, наявність гемофілії. Виникненню артриту може передувати герпетична інфекція. Причиною розвитку септичного артриту можуть бути піогенні мікроорганізми, Mycobacterium haemophilum, M. kansasi, M. avium, M. terrae, M. fortuitum, грибкова інфекція (гістоплазмоз, криптококоз, споротрихоз, бластомікоз) |
|
|
РеА |
Синдром Рейтера відмічають у 0,4–10% ВІЛ-інфікованих осіб, він може розвинутися за декілька років до встановлення діагнозу інфекції. Але найчастіше його відмічають при тяжкому імунодефіциті. Перебіг синдрому хронічний з рецидивами, ремісіями. Можливий ерозивний процес в суглобах з інвалідизацією хворих. HLA-B27 виявляють з такою ж частотою, як взагалі серед пацієнтів з цим синдромом |
|
|
ПсА |
Псоріаз виявляють у 1,5–2% ВІЛ-інфікованих, перебіг його, як правило, тяжкий, має місце резистентність до загальноприйнятої терапії |
|
|
Подагра |
Захворюваність на подагру та частота гіперурикемії вищі, ніж у загальній популяції |
|
|
Васкуліти |
У ВІЛ-інфікованих виявляють гранулематоз Вегенера, вузликовий поліартеріїт, синдром Черджа — Стросс, ізольований васкуліт ЦНС, гіперергічний васкуліт, пурпуру Шенляйна — Геноха, аневризми великих артерій. Васкуліти стають причиною інсультів |
|
|
Кріоглобулінемія II, III типу |
Розвивається при поєднанні ВІЛ-інфекції та вірусного гепатиту С. У таких випадках клінічної ремісії кріоглобулінемії досягають шляхом використання інтерферону та рибавірину |
|
|
Хвороба Бехчета |
Відмічають у 15% ВІЛ-інфікованих пацієнтів, характеризується типовими проявами (виразки ротової порожнини, геніталій, ураження шкіри, очей) при негативному симптомі патергії |
|
|
Синдром дифузної лімфоцитарної інфільтрації |
Діагностичні критерії цього синдрому включають: 1) ВІЛ-серопозитивність; 2) двобічне збільшення привушних залоз або сухість у роті, які зберігаються більше 6 міс; 3) гістологічно: лімфоцитарна інфільтрація слинної або слізної залози за відсутності гранулематозних та неопластичних уражень, або результат сцинтиграфії слинних залоз. Виявляється через декілька років після сероконверсії ВІЛ, проявляється ксерофтальмією, ксеростомією, збільшенням привушних залоз, інфільтрацією внутрішніх органів CD8+-лімфоцитами, ураженням інтерстиціальної тканини легень, м’язів, розвитком асептичного менінгіту, паралічів черепних нервів, інтерстиціального нефриту, ниркової недостатності |
|
|
Поліміозит |
Відмічається на всіх етапах перебігу СНІДу. Клінічна картина така, як і при ідіопатичній формі. У біопсійному матеріалі виявляють ознаки мікроваскуліту, міозиту |
|
|
Піоміозит |
Виникає при низькому рівні Т-хелперів, при цьому в 90% висівається Staphylococcus aureus. Відмічають біль у м’язах, припухлість, почервоніння, підвищення температури тіла |
|
|
Ураження кісток |
Остеонекроз |
Частота випадків остеонекрозу (переважно стегнової кістки, в тому числі двобічний) у ВІЛ-інфікованих сягає 4% на рік. Можливий безсимптомний перебіг остеонекрозу з тяжкими наслідками |
|
Остеопенія |
Є наслідком імунної активації, дії прозапальних цитокінів, підвищує ризик переломів кісток. Застосування бісфосфонатів супроводжується підвищенням МЩКТ |
|
|
Остеомаляція |
Її розвиток — наслідок застосування тенофовіру (проявляє нефротоксичний ефект). Прояви остеомаляції регресують після відміни препарату, призначення фосфатів |
|
|
Остеомієліт |
Відмічають при низькому рівні CD4+-клітин, поєднується із септичним артритом, викликаним S. аureus, полімікробною інфекцією |
|
Переважно уражуються великі суглоби нижніх кінцівок, плечові (всього 2–4 суглоби). Можливий (у 10%) так званий болючий суглобовий синдром з дуже сильним болем у великих суглобах верхніх і нижніх кінцівок. Спондилоартропатії, що включають РеА, неповний і повний синдром Рейтера, відмічають у 20% ВІЛ-інфікованих пацієнтів (у 10 разів частіше, ніж у популяції). ВІЛ-інфікування призводить до тяжкого перебігу синдрому Рейтера і хвороби Лайма. Для синдрому Рейтера у ВІЛ-інфікованих характерний ряд особливостей:
- швидке прогресування;
- виражені суглобові прояви: крайові ерозії, згинальні контрактури;
- тяжка ентезопатія (веде до інвалідизації);
- виражені позасуглобові прояви (уретрит, цервіцит, кон’юнктивіт, виразковий процес у роті);
- аденопатія, субфебрилітет, схуднення, діарея;
- наявність HLA-B27 у 75% хворих;
- синовіальна рідина запального характеру.
Для СНІД-стопи характерне гостре болісне ураження у вигляді дактиліту, тендиніту, ентезопатії, ахілодинії. Одночасно виявляють гострий болючий олігоартрит з переважним ураженням суглобів нижніх кінцівок (колінних і гомілковостопних). Ці прояви тривають від 1 тиж до декількох місяців і часто призводять до інвалідизації. ПсА у ВІЛ-інфікованих виявляють у 3 рази частіше, ніж в популяції. Його особливості наступні:
- псоріаз розвивається на фоні вже наявного артриту;
- початок артриту гострий;
- прояви псоріазу тяжкі, відзначається виражений себорейний синдром;
- суглобові і шкірні прояви швидко прогресують;
- відмічена асоціація між тяжкістю ураження шкіри, нігтів і суглобів.
Аваскулярний некроз у ВІЛ-інфікованих найчастіше розвивається в колінних суглобах. До м’язових синдромів, що розвиваються у хворих з ВІЛ-інфекцією, відносять міалгію, медикаментозну міопатію, кахектичну міопатію, фіброміалгію, піоміозит, поліміозит/дерматоміозит. Для поліміозиту характерні типова проксимальна м’язова слабкість, м’язовий біль, схуднення, підвищена концентрація в крові КФК, ЛДГ, альдолази, ознаки запалення в біоптатах.
До імунних синдромів у ВІЛ-інфікованих належать:
- імунна тромбоцитопенія;
- Кумбс-позитивна гемолітична анемія;
- вовчакоподібний синдром;
- дифузний інфільтративний лімфоцитарний синдром (що нагадує синдром Шегрена).
Вовчаковоподібні прояви, що відмічають у ВІЛ-інфікованих, наведені в табл. 25.27 (за Calabrese L.Y., 1989).
Таблиця 25.27
Вовчакоподібні прояви у ВІЛ-інфікованих
|
1. |
Загальні: лихоманка, нездужання, схуднення |
|
2. |
Дерматологічні: «метелик», пов’язаний із себорейним дерматитом, алопеція, шкірний васкуліт, афтозні виразки |
|
3. |
Неврологічні: психоз, судоми, периферична нейропатія |
|
4. |
М’язово-скелетні: артралгія (артрит, міалгія), міозит |
|
5. |
Ниркові: протеїнурія/гематурія, ниркова недостатність |
|
6. |
Лімфаденопатія |
|
7. |
Гематологічні: лейкопенія/лімфопенія, імунна тромбоцитопенія, Кумбс-позитивна гемолітична анемія |
|
8. |
Імунологічні: АНФ, АФЛ, гіпергаммаглобулінемія, ЦІК, активація клітинного імунітету (неоптерин та ін.), підвищення концентрації кислотолабільного α-інтерферону |
До проявів дифузного інфільтративного лімфоцитарного синдрому відносяться сухий кератокон’юнктивіт, ксеростомія, збільшення привушних залоз, генералізована лімфаденопатія, лімфоцитарна інфільтрація різних органів, підвищення рівня Т-лімфоцитів СD8+ в крові. Цей синдром відрізняється від синдрому Шегрена (табл. 25.28).
Таблиця 25.28
Відмінності дифузного інфільтративного лімфоцитарного синдрому від синдрому Шегрена
|
Розвивається переважно у чоловіків (віком до 40 років з лімфаденопатією) |
|
Частота ксерофтальмії нижча |
|
Ксеростомія помірніша |
|
Розвивається лімфоцитарне ураження внутрішніх органів: лімфоцитарний гепатит, гастрит, нефрит, пневмонія (інтерстиціальна), асептичний менінгіт |
|
Слинні залози інфільтруються СD8+, а не Т-лімфоцитами СD4+ |
|
Відмічена асоціація з HLA-DR5 (при синдромі Шегрена з HLA-B8 і DR3) |
У ВІЛ-інфікованих пацієнтів виявляють васкуліт з ураженням артерій усіх калібрів: дрібного, середнього, великого. Найчастішим проявом васкуліту є нейропатія з порушеннями чутливості і рухів. Можливий розвиток системного некротизуючого васкуліту (ВП, синдрому Черджа — Стросс, «перехресного» поліангіїту), лейкоцитокластичного васкуліту (васкуліту гіперчутливості, пурпури Шенляйна — Геноха), первинного ангіїту ЦНС. Ураження серця у хворих з ВІЛ-інфекцією проявляється міокардитом, дилатаційною кардіоміопатією.
До хвороб, несумісних зі СНІДом, відносять саркоїдоз, СЧВ, РА. Якщо ВІЛ-інфекція виникає у хворих на СЧВ і РА, активність імунопатологічного процесу знижується. СЧВ на тлі нелікованого СНІДу переходить у стадію ремісії, але може знову виникнути на фоні антиретровірусної терапії. У хворих на СЧВ можуть бути помилково позитивними тести на антитіла до ВІЛ. На фоні імуносупресивної терапії СВЧ ВІЛ-інфекція швидко прогресує. Перебіг РА на фоні інфікування ВІЛ переходить у ремісію.
Лікування суглобових проявів у ВІЛ-інфікованних має ряд особливостей:
- протипоказані метотрексат, цитотоксичні препарати (призводять до генералізації ВІЛ-інфекції);
- використовують НПЗП (високоефективним є фенілбутазон); при міопатичних проявах окрім НПЗП призначають короткі курси (6–8 тиж) преднізолону (20–30 мг/добу);
- при ВІЛ-асоційованому синдромі Рейтера ефективний циклоспорин А (1–2 мг/кг/добу);
- при дифузному інфільтративному лімфоцитарному синдромі використовують комбінацію азидотимідину з α-інтерфероном і преднізолон (40–60 мг/добу).
Слід враховувати, що застосування метотрексату у ВІЛ-інфікованих може спровокувати розвиток саркоми Капоші, а інгібіторів ФНП-α — полімікробні інфекції. Антиретровірусна терапія в поєднанні з ГК, в/в Ig та циклофосфамідом дають позитивний ефект при розвитку ускладнень васкуліту. Перебіг хвороби Бехчета поліпшується під впливом антиретровірусної терапії. Антиретровірусна терапія дає позитивний ефект при синдромі дифузної лімфоцитарної інфільтрації. Преднізолон при цьому синдромі використовують у високих дозах (1 мг/кг/добу), але ефект його тимчасовий. Імуносупресивна терапія (преднізолон 30–60 мг/добу, в тому числі в комбінації з зидовудином) купірує у ВІЛ-інфікованих прояви поліміозиту.
Саме лікування СНІДу може стати причиною виникнення ускладнень ревматичного характеру (табл. 25.29).
Таблиця 25.29
Ускладнення ревматичного характеру, які виникають внаслідок терапії СНІДу
|
Органи ураження |
Препарати, які викликають ураження |
|
Суглоби |
Причиною артралгії, моно- й олігоартриту, тендосиновіту, адгезивного капсуліту, контрактури Дюп’юїтрена може бути застосування індинавіру, ритонавіру |
|
М’язи |
Міопатію індукує зидовудин. Вона розвивається через 6 міс після початку терапії, стан хворих поліпшується після зниження дози, припинення прийому препарату. Гіперчутливість до абакавіру, використання статинів (особливо поєднання ловастатину, симвастатину з антиретровірусними препаратами) викликає рабдоміоліз (та гостру ниркову недостатність) |
|
Метаболічні процеси |
Основним захворюванням та застосуванням ставудину та диданозину може бути зумовлений високий рівень сечової кислоти |
Грибкові артрити
Код за МКХ-10
В35–В39 Мікози.
М01.6 Артрит при мікозах (В35–В49).
М01.8 Артрит при інших інфекційних і паразитарних хворобах, класифікованих в інших рубриках.
В37 Кандидоз.
В37.8 Кандидоз інших локалізацій.
В38.8 Інші форми кокцидіоїдозу.
В39 Гістоплазмоз.
В40 Бластомікоз.
В40.8 Інші форми бластомікозу.
В42.8 Інші форми споротрихозу.
В44 Аспергільоз.
В44.8 Інші форми аспергільозу.
В45.3 Криптококоз кісток.
М63.2 Міозит при мікозі (В35–В49).
Етіологія грибкових артритів
Викликати артрит можуть Histoplasma capsulatum, Cryptococcus neoformans, Coccidioides immitis (в усіх трьох поширення аерогенне), Blastomices dermatitidis (аерогенне поширення й інокуляція), Maduromycoses (інфікування босих стоп), Candida species (ендогенне поширення), Aspergillus fumigatus (вдихання часток), Sporothrix schenckii (інокуляція гриба при подряпинах, скалках). До розповсюджених грибкових захворювань відносяться актиномікоз, кандидомікоз, споротрихоз та ін. Інші поширені в певних районах світу.
Патогенез і характер ураження кістково-суглобової системи
Грибковий гнійний артрит розвивається внаслідок поширення інфекції з найближчого вогнища (ураженої шкіри, нігтів, вогнища остеомієліту) або інокуляції при травмах, або ендогенного поширення. При цьому розвивається моноартрит (можливе ураження 2 суглобів), перебіг якого в’ялий і діагностика затягується на місяці і роки. Перебіг артриту характеризується болем, почервонінням шкіри, гнійним випотом. Гострий перебіг артриту нехарактерний, виняток становлять артрити, викликані грибами Candida і Blastomyces. Артрити мають стійкий перебіг, і за зовнішніми і рентгенологічними даними вони схожі на туберкульозні (їх часто плутають).
Найбільш частим грибковим ураженням кістково-суглобової системи є остеомієліт. При грибковій інфекції кістково-суглобовий апарат уражується, як правило, в період генералізації. Первинні вогнища виникають в кістках черепа, нижньої щелепи, хребцях, епіфізах і діафізах трубчастих кісток. У них розвивається остеомієліт: деструктивні і періостальні процеси з утворенням гнійників, нориць в навколишніх тканинах. Суглоби уражуються повторно із сусідніх вогнищ.
Діагностика грибкових артритів. Основою для діагнозу «грибковий артрит» є наступні критерії:
- типове ураження шкіри поблизу суглоба;
- ураження нігтів;
- виявлення гриба в зскрібку шкіри або гнійних виділеннях;
- виявлення гриба в синовіальній рідині.
Встановленню діагнозу сприяють шкірні тести з грибковими антигенами, специфічні серореакції, виявлення міцелію у виділеннях із нориць, гнійному вмісті порожнини. Зміни, що виявляють при рентгенологічному дослідженні, включають: вогнища кісткової деструкції в епіметафізах; звуження суглобової щілини; нерівність суглобових поверхонь.
Лікування грибкових артритів. Найефективнішим препаратом проти більшості видів гриба є амфотерицин В (0,25–1 мг/кг/добу). Тривалість терапії залежить від виду збудника, ступеня тяжкості хвороби, переносимості препарату, наявності побічних ефектів. Середня тривалість курсу лікування становить 6–12 тиж, загальна доза препарату — 1–3 г. Використовують також 5-фторцитозин в комбінації з амфотерицином В, кетоконазол, флуконазол. При резистентності до препаратів проводиться хірургічна санація вогнищ інфекції.
Глава 26. Гостра ревматична лихоманка та хронічна ревматична хвороба серця
ГРЛ — системне запальне захворювання сполучної тканини з переважним ураженням серця, що є ускладненням А-стрептококового фарингіту (ангіни) у зв’язку з розвитком аутоімунної відповіді на епітопи стрептококу та перехресної реактивності зі схожими епітопами тканин людини (у шкірі, суглобах, серці і мозку).
Коди за МКХ-10 представлені в табл. 26.1, 26.22.
Таблиця 26.1
Коди за МКХ-10 нозологічних одиниць групи ГРЛ
|
Найменування нозологічної одиниці за МКХ-10 |
Код МКХ-10 |
Примітка |
|
ГРЛ без залучення серця |
I00 |
Код використовується для діагнозу у випадках без ураження серця і його клапанів, а також головного мозку (хореї) |
|
Гострий ревматичний перикардит |
I01.0 |
Виключені: інші (неревматичні) перикардити — I01—I02 |
|
Гострий ревматичний ендокардит |
I01.1 |
У тому числі з ревматичним артритом |
|
Гострий ревматичний міокардит |
I01.2 |
У тому числі з ревматичним артритом |
|
Інші гострі ревматичні хвороби серця |
I01.8 |
У тому числі з ревматичним артритом, але без хореї. При ревматичному неуточненому гострому ураженні серця — I01.9 |
|
Ревматична хорея, хорея Сиденхема |
I02.0, I02.9 |
I02.0 — з будь-яким гострим, підгострим, зворотним ураженням серця і його клапанів; I02.9 — без ураження серця |
Таблиця 26.2
Коди за МКХ-10 нозологічних одиниць групи ХРХС
|
Найменування нозологічної одиниці за МКХ-10 |
Код МКХ-10 |
Примітка |
|
Мітральний стеноз |
I05.0 |
При гострому або зворотному вальвуліті — I01.1. При неуточненій за патогенезом (ревматизм) і характером (виду) мітральній ваді серця — I05.9. При комбінованому ураженні (стеноз і недостатність) — коди групи I05. При поєднаному ураженні декількох клапанів — коди групи I08. Якщо не уточнено, які клапани уражені, — I09.1 |
|
Недостатність мітрального клапана |
I05.1 |
|
|
Комбінована вада серця (стеноз з недостатністю) |
I05.2 |
|
|
Аортальний стеноз |
I06.0 |
При гострому або зворотному вальвуліті — I01.1, при неуточненій за патогенезом (ревматизм) і характером (виду) аортальній ваді серця — I06.9 |
|
Недостатність аортального клапана |
I06.1 |
|
|
Аортальна комбінована вада (стеноз із недостатністю) |
I06.2 |
|
|
Трикуспідальний стеноз |
I07.0 |
При гострому або зворотному вальвуліті — I01.1, при неуточненій за патогенезом (ревматизм) і характером (вид) трикуспідальній ваді — I07.9 |
|
Трикуспідальна недостатність |
I07.1 |
|
|
Трикуспідальна комбінована вада (стеноз з недостатністю) |
I07.2 |
|
|
Інші уточнені ревматичні хвороби серця, ревматична хвороба легеневого клапана |
I09.8 |
|
|
Поєднання ураження мітрального й аортального клапанів |
I08.0 |
При гострому або зворотному вальвуліті — I01.1, при неуточненій за патогенезом і характером (вид) поєднаній ваді серця — I08.9 |
|
Поєднання ураження мітрального і тристулкового клапанів |
I08.1 |
|
|
Поєднання ураження аортального і тристулкового клапанів |
I08.2 |
|
|
Поєднання ураження мітрального, аортального і тристулкового клапанів |
I08.3 |
|
|
Ревматичний міокардит |
I09.0 |
Для гострого або підгострого міокардиту або фази його загострення — I01.2 |
|
Хронічний ревматичний перикардит |
I09.2 |
Для гострого або підгострого перикардиту або фази його загострення — I01.0 |
|
Ревматичні хвороби серця неуточнені |
I09.9 |
Для гострого або підгострого панкардиту або фази його загострення, або для неуточненого ревматичного ураження серця — I01.9 |
МКХ-10 також включає М12.0 Хронічна постревматична артропатія (Жакку). Використання терміну «ревматизм» не рекомендується, виключені терміни «в’ялий» та «безперервно-рецидивуючий» перебіг.
Епідеміологія гострої ревматичної лихоманки
В основному вона розвивається у схильних осіб молодого віку (7–15 років). Захворюваність ГРЛ в Україні становить 4,4 на 100 тис. населення. Рівень захворюваності не залежить від статі.
Етіологія гострої ревматичної лихоманки
До чинників, що викликають розвиток ревматичної лихоманки при А-стрептококовій інфекції, відносять молекулярну мімікрію; порушення клітинного і гуморального імунітету; наявність генетичної схильності; носійство В-лімфоцитарного алоантигену D8/17. Частота «сімейного» ревматизму в 6 разів вища, ніж у популяції. ГРЛ асоціюється з носійством HLA-A2, -B5, -B35, -DRS, -DR7.
Патогенез
Основою патогенезу ГРЛ є аутоімунні реакції в організмі у відповідь на надходження антигенів β-гемолітичного стрептококу, а також перехресна реактивність зі схожими аутоантигенами різних тканин людини (цей феномен дістав назву молекулярної мімікрії). При цьому захворюванні уражується сполучна тканина переважно в серцево-судинній системі.
А-стрептококи типу М3, М5 і М18 містять епітопи, що спричиняють імунізувальний ефект і викликають розвиток патологічного процесу. Стрептолізин О і S, гіалуронідаза, стрептокіназа, протеази, дезоксирибонуклеаза, М-протеїн, пептидоглікан та інші речовини, що виробляються стрептококами, пошкоджують лізосомальні мембрани, основну речовину сполучної тканини (табл. 26.3).
Таблиця 26.3
Ферменти, які продукують А-стрептококи
|
Гіалуронідаза |
Викликає деполімеризацію глюкуронової кислоти |
|
Гіалуронова кислота, капсули |
Гальмує фагоцитарну активність нейтрофілів |
|
Стрептолізин О |
Пошкоджує мембрани лізосом, викликає запальні реакції, чинить кардіотоксичну дію |
|
Стрептолізин S |
Також пошкоджує мембрани лізосом, в експерименті викликає артрит |
|
Стрептокіназа |
Активує кінінову систему |
|
М-протеїн |
Чинник вірулентності, індукує аутоімунітет |
Токсини стрептококів викликають розвиток запалення в сполучній тканині. З’являються антитіла до антигенних компонентів сполучної тканини — структурних глікопротеїдів, протеогліканів, АФЛ. Утворюються імунні комплекси, що збільшують вираженість запалення. Порушується функція тканинних базофілів, в результаті їх посиленої дегрануляції в сполучну тканину і русло крові виділяються медіатори запалення — гістамін, серотонін, брадикініни. У патогенезі ГРЛ активну роль відіграють моноцити/макрофаги, лімфоцити і цитокіни, що виділяються ними. Збільшується кількість В-лімфоцитів, зменшується — Т-лімфоцитів (процентно і абсолютно). Імунний запальний процес призводить до дезорганізації сполучної тканини. Дезорганізація сполучної тканини відбувається у декілька етапів (табл. 26.4).
Таблиця 26.4
Стадії запального процесу в сполучній тканині при ревматичній лихоманці
|
Стадії запального процесу |
Характеристика патологічного процесу |
|
Мукоїдне набухання |
Деполімеризується основна речовина сполучної тканини, в ній накопичуються мукополісахариди (в основному кислі). Сполучна тканина розволокнюється. Патологічний процес має зворотний характер |
|
Фібриноїдний некроз |
Розпад колагену, дезорганізація волокон, їх набрякання, відкладення фібриноїду. Процес незворотний |
|
Формування специфічних ревматичних гранульом |
Гранульоми формуються навколо вогнищ фібриноїдного некрозу і представлені великими базофільними гістіоцитами, базофілами, моноцитами, лімфоцитами, плазматичними клітинами |
|
Стадія склерозу |
Склерозуються гранульоми |
Одночасно з гранульомами в тканині відмічають прояви неспецифічного запалення: набряк, інфільтрація лімфоцитами, нейтрофілами, еозинофілами, просочення тканин фібрином і білками, що містяться в плазмі крові. З ураженням клапанів серця при першій атаці ГРЛ асоціюється високий рівень ІЛ-1α та неоптерину. Утворюються антитіла до L-форм стрептококу, антикардіальні антитіла, АФЛ (АКЛ-IgG у 54% пацієнтів). З ЕКГ-змінами (АВ-дисоціацією, блокадами) корелює рівень ЦІК, у формуванні яких бере участь АСЛ-О. Високий вміст в крові АКЛ-IgG, як і неоптерину, є чинником ризику уражень клапанів. При лабораторних обстеженнях також виявляють лейкоцитоз, зрушення лейкоцитарної формули вліво, підвищення ШОЕ, рівня α2— і γ-глобулінів, СРБ, серомукоїду, гаптоглобіну, фібриногену, АсАТ, імуноглобулінів. Персистенція стрептококу, реінфекція зумовлюють прогресування і загострення ревматичного процесу.
Ревматичні гранульоми. Вони виникають в серці, розташовуються в периваскулярній сполучній тканині або в інтерстиції міокарда з переважною локалізацією в лівому шлуночку, перегородці, сосочкових м’язах, ендокарді, зовнішній оболонці судин. В останні роки гранульоми при патологоанатомічному дослідженні виявляють все рідше. Ревматична гранульома складається з базофільних клітин (великих, неправильної форми) гістіоцитарного походження; гігантських багатоядерних клітин; лімфоїдних, плазматичних і гладких клітин. Цикл формування і рубцювання гранульоми займає в середньому 3–4 міс.
Прояви неспецифічної запальної реакції при ревмокардиті. Вона характеризується набряком міжм’язової сполучної тканини, виходом із судин фібрину, інфільтрацією клітинними елементами і, передусім, поліморфно-ядерними лейкоцитами, лімфоцитами. Неспецифічне запалення прямо корелює з активністю патологічного процесу і вираженістю кардиту. Неспецифічні реакції включають зміни судин мікроциркуляторного русла в усіх органах, залучення до патологічного процесу серозних оболонок із розвитком серозного, серозно-фібринозного запалення. Дезорганізація сполучної тканини, ексудативне запалення, васкуліт відмічають і в суглобових тканинах.
Найбільш характерна локалізація патологічного процесу при ревмокардиті включає мітральний, аортальний, тристулковий клапани. Результатом наявного вальвуліту є кардіосклероз.
Зміни, що лежать в основі ураження нервової системи при ГРЛ. У їх основі лежить залучення до ревматичного процесу судин мозку. Зміни клітин смугастого тіла, субталамічних ядер великого мозку і мозочка є причиною виникнення малої хореї.
Причина виникнення при ГРЛ ураження шкіри і підшкірної клітковини. Причиною їх розвитку є васкуліт, вогнищева запальна реакція.
Класифікація
Класифікації ГРЛ представлені нижче (табл. 26.5 і 26.6).
Таблиця 26.5
Класифікація ГРЛ (Насонова В.А. и соавт., 1999)
|
I. Клінічні форми |
ГРЛ, повторна ревматична лихоманка |
|
|
II. Клінічні прояви |
А. Основні |
Кардит, артрит, хорея, кільцеподібна еритема, ревматичні вузлики |
|
Б. Додаткові |
Лихоманка, артралгія, абдомінальний синдром, серозити |
|
|
III. Результати |
А. Одужання |
|
|
Б. ХРХС |
Без вад серця З вадами серця |
|
|
IV. Недостатність кровообігу |
А. За класифікацією М.Д. Стражеска та В.Х. Василенка |
Стадії: 0, I, IIA, IIБ, III |
|
Б. За класифікацією Нью-Йоркської кардіологічної асоціації (NYHA) |
Функціональні класи: 0, I, II, III, IV |
|
Таблиця 26.6
Клінічна класифікація ГРЛ (АРУ)
|
Клінічні прояви |
Наслідки |
Серцева недостатність |
||
| Основні | Додаткові | Стадія | ФК | |
|
Кардит Артрит Хорея Кільцеподібна еритема Ревматичні вузлики |
Лихоманка Артралгії Абдомінальний синдром Серозити |
Без вад серця Вади серця Одужання |
I II A II Б III |
I II III IV |
Примітки: термін «ревматизм» замінений на термін «ГРЛ»; серцева недостатність вказується відповідно до критеріїв класифікації Стражеска — Василенка з вказівкою функціональних класів (ФК) за Нью-Йоркською класифікацією.
Клініка гострої ревматичної лихоманки
Патологічні зміни, які виявляють у пацієнтів з ГРЛ при обстеженні, наведені в табл. 26.7.
Таблиця 26.7
Дані, які отримують при фізикальному та інструментальному обстеженні хворих на ГРЛ
|
Орган, система |
Характер ураження |
Клінічні прояви |
|
Шкіра |
Кільцеподібна еритема |
Висипання бліднорожевого кольору і кільцеподібної форми локалізуються на тулубі і проксимальних відділах кінцівок, відмічають у 4–17% пацієнтів. Локалізація на обличчі нехарактерна. Висипання не піднімаються над поверхнею шкіри, не сверблять. Кільцеподібна еритема з’являється на висоті ГРЛ і досить швидко зникає, сліди на шкірі після неї не залишаються. Необхідно враховувати, що кільцеподібну еритему виявляють і при інших захворюваннях. Елементи кільцеподібної еритеми можуть зливатися на плечах, тулубі, обличі, шиї, ногах |
|
Ревматичні вузлики |
Відзначаються рідко — у 1–3% хворих. Вони невеликого розміру, локалізуються під шкірою у фасціях, апоневрозах, суглобових сумках, місцях прикріплення сухожиль в ділянці колінних, ліктьових суглобів, потиличній кістці. Ревматичні вузлики є щільними, з безболісними округлими утвореннями в діаметрі від декількох міліметрів до 2 см. Вони з’являються непомітно для хворих і безслідно зникають через 1–2 міс |
|
|
Серце |
Систолічний шум |
Причиною його появи є мітральна регургітація. Інтенсивність шуму залежить від стадії захворювання. Він пов’язаний з I тоном, займає велику частину систоли, добре вислуховується на верхівці серця, проводиться в ліву пахвову ділянку. Від зміни положення тулуба і фази дихання інтенсивність шуму не залежить |
|
Мезодіастолічний шум |
Він низькочастотний, з’являється у пацієнтів з гострим кардитом з мітральною регургітацією. Йде за III тоном або зливається з ним, добре вислуховується на верхівці серця в положенні на лівому боці при затримці дихання на видиху |
|
|
Протодіастолічний шум |
Є віддзеркаленням аортальної регургітації, починається відразу після II тону, характер його високочастотний, дуючий, згасаючий. Добре прослуховується вздовж лівого краю груднини після глибокого видиху при нахилі тулуба вперед. Як правило, поєднується із шумом систоли |
|
|
Перикардит |
Нині відмічають рідко, перебіг сприятливий. Проявляється шумом тертя перикарду, зниженням вольтажу усіх зубців ЕКГ, конкордантною елевацією сегмента ST, зміною зубця Т, ехоКГ, рентгенологічними ознаками |
|
|
Легені |
Пневмонія |
Ревматичні пневмонії частіше локалізуються в нижніх відділах легень, супроводжуються ексудативним плевритом, лихоманкою, лейкоцитозом, збільшенням ШОЕ. Внаслідок ураження судин малого кола кровообігу виникають ціаноз, задишка. Аускультація малоінформативна, рентгенологічно реєструється розширення корення, посилення легеневого малюнка і його тяжистість. Можливий розвиток таких ускладнень, як кровохаркання, кровотеча, рецидивуючий набряк легень, спонтанний пневмоторакс, хронічне легеневе серце |
|
Плеврит |
Відноситься до частих ознак ревматичної лихоманки: відзначається у 50% хворих дітей і у 15% дорослих. Часто має ексудативний характер, двобічний, з невеликою кількістю випоту. Адгезивний процес розвивається рідко, перебіг доброякісний. У плевральній рідині переважають лімфоцити або нейтрофіли |
|
|
ШКТ |
Абдомінальний синдром |
Біль у животі виникає в результаті ураження мезентеріальних судин. У дітей можливий розвиток перитоніту, картини гострого живота. Біль у животі зазвичай має дифузний характер (може локалізуватися переважно в правому підребер’ї), симптом Щоткіна — Блюмберга позитивний, напруження м’язів передньої черевної стінки виражене незначною мірою. Лейкоцитоз може досягати 30–40•109/л, підвищується температура тіла. Абдомінальні прояви виникають після ангіни або скарлатини у пацієнтів з ознаками ревматизму (поліартрит, кардит, вада серця, плеврит) і добре купіруються антиревматичною терапією. У 50% хворих порушується функціональний стан печінки (внаслідок ревмоваскуліту печінки). Лікування ГРЛ може бути причиною загострення хронічних захворювань органів травлення, розвитку НПЗП-гастропатії, токсичного (в тому числі холестатичного) гепатиту |
|
Нирки |
Можливий розвиток гломерулонефриту, застійної нирки (при тяжкій серцевій недостатності) |
|
|
Ендокринна система |
Описаний розвиток ревматичного тиреоїдиту, недостатності надниркової залози |
|
|
Нервова система |
До ревматичних уражень нервової системи відносять менінгіт, арахноїдит, енцефаліт, можливий розвиток гострого порушення мозкового кровообігу, психозів. Однією з основних ознак ревматичної лихоманки є хорея Сиденгама — наслідок енцефаліту з переважним ураженням стріопалідарної системи. Хорея переважно відмічається у дітей віком 6–14 років. Такі ознаки малої хореї, як гіперкінези, порушення координації, м’язова гіпотонія, судинна дистонія, психоемоційні порушення, виявляють у 6–30% хворих на ГРЛ. Хорея може бути (у 5–7% пацієнтів) єдиним проявом ГРЛ (з кардитом або артритом вона поєднується рідко). Відзначаються хаотичні, одно- або двобічні насильницькі рухи різних м’язових груп. Мова стає невиразною, виникають труднощі під час прийому їжі, змінюється почерк. Діти стають пасивними, замкнутими, дратівливими. Гіперкінези зникають під час сну, посилюються при хвилюванні. Ускладнено виконання координаційних проб. У деяких хворих може відзначатися виражена м’язова гіпотонія. Часто хорея супроводжується проявами вегетативної дистонії. В останні роки вираженість клінічних проявів хореї зменшилася. Вони зникають у більшості дітей через 1–2 міс від початку лікування (хорея може зникати і мимоволі) |
|
|
Суглоби |
Ураження суглобів відзначається у ⅔ дітей з першою атакою ГРЛ і ½ — з другою. Починається гостро, супроводжується лихоманкою, припухлістю суглобів, болем у них, обмеженням рухів, почервонінням шкіри і підвищенням локальної температури тіла. В останні роки суглобовий синдром носить характер моно- або олігоартриту з ураженням колінних, гомілковостопних, променезап’ясткових, ліктьових суглобів. Характерна летючість запалення, суглоби уражуються неодночасно, досить часто симетрично. НПЗП добре купірують прояви суглобового синдрому, деформації суглобів не виникає. Артрит часто поєднується з кардитом, в деяких випадках можливий його затяжний перебіг. У 10–15% випадків відмічають тільки поліартралгії (симптоми запалення відсутні) |
|
|
Орган зору |
Можуть розвинутися ірит, іридоцикліт |
|
|
Кров |
Відзначаються нейтрофільний лейкоцитоз, помірна анемія. Можливе збільшення вмісту плазматичних клітин в кістковому мозку |
|
До клінічних особливостей ревматичного артриту відносяться:
- виникнення артриту після перенесеної гострої стрептококової інфекції;
- переважний розвиток при першій атаці ревматичної лихоманки;
- ураження в основному великих суглобів мігруючого характеру;
- гострий і підгострий характер запалення;
- висока ефективність НПЗП.
В окремих випадках у хворих на ГРЛ має місце хронізація суглобового синдрому, синовіт може зберігатися протягом декількох років. У деяких хворих можливий розвиток ревматоїдоподібної деформації дрібних суглобів без ерозивного процесу (артропатія Жакку). У теперішній час ревматологи відмічають як редукцію суглобового синдрому, так і його пролонгацію.
Частота розвитку при ГРЛ ревмокардиту, його характеристика. Ревмокардит відмічають у 70–85% дітей з першою атакою ГРЛ. Характер симптоматики визначається переважним ураженням однієї з оболонок серця — ендокарда, міокарда, епікарда. Порушується загальний стан хворих, частота серцевих скорочень (ЧСС), розширюються межі серця (переважно вліво), послаблюються тони серця. З’являється систолічний шум, виявляють додаткові III і IV тони. ЕКГ реєструє дисфункцію синусно-передсердного вузла у вигляді тахікардії, брадикардії, дихальної аритмії, міграції водія ритму, виникає АВ-дисоціація, АВ-блокада. Знижується скорочувальна функція і тонус міокарда. При УЗД серця виявляються ознаки ураження клапанів. У 1–2% хворих при первинному ревмокардиті з’являються клінічні ознаки перикардиту. Рентгенологічне дослідження дещо частіше виявляє адгезивний плевроперикардит. Сьогодні розвиток серцевої недостатності I–II ст. (за М.Д. Стражеском і В.Х. Василенком) відмічають рідко. На тлі лікування у більшості дітей нормалізується ЧСС, знижується інтенсивність шумів і зменшуються розміри серця, відновлюється звучність тонів, зникають ознаки серцевої недостатності. У 20–25% дітей після первинного ревмокардиту формується вада серця. Частота її формування залежить від ступеня тяжкості перенесеного ревмокардиту (в останні роки вона зменшилася в 2,5 раза). Найчастіше формуються мітральна недостатність, рідше — недостатність аортального клапана, мітрально-аортальна вада і мітральний стеноз. У 7–10% дітей після перенесеного первинного ревмокардиту залишається пролапс мітрального клапану. Нині вади серця формуються повільніше, виражені нерізко, компенсація зберігається багато років. У ряді випадків вади формуються під час другої атаки ГРЛ, що підтверджується результатами інструментальних досліджень.
Особливості первинного ревмокардиту включають:
- наявність хронологічного зв’язку з перенесеною стрептококовою інфекцією верхніх дихальних шляхів;
- проміжок часу між закінченням інфекції і першими проявами ревматичної лихоманки становить близько 2–4 тиж;
- виникнення захворювання переважає в дитячому і юнацькому віці;
- гострий і підгострий початок хвороби;
- дебют захворювання — з поліартриту або поліартралгії;
- відсутність кардіальних скарг або виявлення їх при ретельному цілеспрямованому пошуку;
- відносно часте виявлення панкардиту;
- значна мінливість симптомів запального ураження серця;
- наявність кореляції між клінічними проявами і лабораторними показниками активності запалення.
Частота розвитку клінічних симптомів ревмокардиту у дітей з ГРЛ представлена в табл. 26.8 (за: Насонова В.А., Кузьміна Н.Н., 1997).
Таблиця 26.8
Частота розвитку клінічних симптомів ревмокардиту у дітей з ГРЛ
|
Симптоми |
Частота |
Симптоми |
Частота |
|
Неприємні відчуття в ділянці серця |
4–6 |
Нормальна ЧСС |
30–40 |
|
Підвищена стомлюваність |
12–15 |
Розширення меж серця |
80–85 |
|
Порушення загального стану і підвищення температури тіла |
60–70 |
Поява додаткового III тону |
45–75 |
|
Тахікардія |
30–40 |
Поява IV тону |
15–25 |
|
Брадикардія |
20–30 |
Труднощі при визначенні ревматичного походження кардиту. Їх наявність пояснюється:
- слабкою вираженістю системних проявів;
- маломаніфестними проявами первинного ревмокардиту;
- низькими (часто) титрами антистрептококових антитіл;
- відносно рідкісним висіванням стрептококу із зіву;
- відсутністю даних, що підтверджують стрептококову етіологію попередньої носоглоткової інфекції.
Частота ураження внутрішніх органів при ГРЛ. Нині ураження внутрішніх органів відмічають рідко. На початку атаки ГРЛ у 5–7% дітей виявляють абдомінальний синдром — біль різної локалізації й інтенсивності. Вкрай рідко в період першої атаки відмічають ураження легень (у вигляді васкуліту, плевриту, ревматичної пневмонії) і нирок (гломерулонефрит). Їх розвиток можливий і при повторній атаці ГРЛ.
Чинники, що впливають на характер перебігу ГРЛ. Одним із визначальних чинників є вік пацієнтів (табл. 26.9).
Таблиця 26.9
Особливості перебігу ГРЛ залежно від віку дітей
|
Дитячий вік. Школярі середнього і старшого віку |
Початок хвороби гострий. Через 2–3 тиж після перенесеної ангіни підвищується температура тіла до 38–39 °С, з’являються біль у великих суглобах (частіше колінних), в ділянці серця, серцебиття, задишка (розвивається кардит). Такі прояви відзначають у 50% дітей, у інших — моносиндромний перебіг (артрит, кардит або хорея) |
|
Підлітки і молоді люди |
Початок хвороби поступовий. Після зменшення вираженості клініки ангіни з’являються субфебрильна температура тіла, біль у великих суглобах, ознаки кардиту, що розвивається |
Особливості клінічного перебігу ревматичного процесу в різних вікових групах (за: Насонова В.А., Кузьміна Н.Н., 1997) наведені також в табл. 26.10.
Таблиця 26.10
Особливості клінічного перебігу ГРЛ у різних вікових групах
|
Вік |
Особливості перебігу |
|
Підлітковий вік (15–17 років) |
Нерідко до патологічного процесу залучаються дрібні суглоби кистей і стоп |
|
У ⅓ хворих відзначають малосимптомний перебіг ГРЛ із слабкою вираженістю запалення |
|
|
Ураження клапанного апарату виявляють у 60% хворих |
|
|
Виражені ознаки ураження серця при слабкій вираженості суб’єктивних симптомів |
|
|
Вади серця формуються у ⅓ підлітків, що перенесли першу атаку ГРЛ |
|
|
Темп формування вад серця швидший, ніж у дітей |
|
|
Відзначається висока питома вага ізольованих аортальних і комбінованих мітрально-аортальних вад серця |
|
|
У цьому віці відбувається трансформація недостатності мітрального клапана в поєднану ваду |
|
|
Часто (25–30%) розвивається церебральна патологія (хорея, нервово-психічні порушення) і повторні атаки ГРЛ (10–15%) |
|
|
У підлітків зі сформованими вадами серця нерідко розвиваються порушення ритму, серцева недостатність |
|
|
Юнацький вік (18–21 рік) |
Нерідко залучаються дрібні суглоби кистей і стоп, груднино-ключичні, крижово-клубові зчленування |
|
Ревмокардит розвивається в абсолютної більшості хворих, у ⅓ уражується клапанний апарат, у 10% — перикард |
|
|
Максимальний ступінь активності процесу відзначається у 90% хворих (перебіг атаки частіше гострий і підгострий) |
|
|
Вада серця формується у 20% пацієнтів, при цьому переважає недостатність мітрального клапана (є у 50%) |
|
|
У 27% хлопчиків формується пролапс мітрального клапана |
|
|
Дорослі |
Ревмокардит відмічають у 90% з першою атакою ГРЛ і у 100% при повторній |
|
Після однієї атаки вада серця формується в 39–45% випадків |
|
|
Частота ураження суглобів при першій атаці становить 70–75%, при подальших атаках вона знижується |
|
|
До патологічного процесу нерідко залучаються крижово-клубові зчленування |
|
|
У осіб літнього і старечого віку первинні ревматичні атаки не відзначаються, але можливі рецидиви хвороби, що проявляються слабо вираженими вісцеропатіями |
У разі ревматичної лихоманки, перебіг якої супроводжується вираженим поліартритом і шкірними проявами, сьогодні рідко розвивається значне ураження серця.
Ревматична лихоманка перебігає гостро: вираженість симптомів швидко збільшується і регресує, в середньому розвиток клінічних проявів становить 2–3 міс. Середня тривалість атаки може затягнутися до 3–6 міс (затяжний перебіг). Клінічні прояви хвороби можуть бути маломаніфестними (латентний перебіг) і про перенесений ревмокардит можна іноді судити тільки ретроспективно за виявленими резидуальними змінами перенесеного ревмокардиту або існуючою клапанною вадою серця.
ХРХС. Ревматичні вади серця (РВС). До особливостей ХРХС відносяться розвиток крайового фіброзу стулок серцевих клапанів (як результат запалення), формування вади серця — недостатності та/або стенозу. Повторні атаки (рецидиви) ГРЛ провокуються інфекцією, викликаною β-гемолітичним стрептококом. У таких випадках переважно розвивається кардит. Існує 2 концепції формування вад серця у хворих на ГРЛ:
- вада серця наслідок перенесеного вальвуліту, що розвивається протягом 1–2 років після атаки ГРЛ;
- формування вади серця починається через декілька років після атаки ГРЛ.
Встановлено, що через декілька років після першої атаки ГРЛ фібробласти клапанів можуть здійснювати експресію DR4 і поверхневих білків. Це зумовлює розвиток хронічного вальвуліту за відсутності збудника хвороби. Ці дані дозволяють припустити можливість латентного перебігу ревматичного процесу. Повторні атаки ревматичної лихоманки збільшують вираженість вад.
Класифікації ХРХС та набутих вад серця наведені в табл. 26.11, 26.12.
Таблиця 26.11
Класифікація ХРХС (АРУ)
|
Активність процесу |
Клінічні прояви |
Стадії набутих вад |
Серцева недостатність |
|
|
|
Стадія | ФК | ||
|
Неактивна |
Вади серця |
I |
I |
I |
|
Активна: |
II |
IIA |
II |
|
|
I (мінімальна) |
III |
IIБ |
III |
|
|
II (помірна) |
IV |
III |
IV |
|
|
V |
– |
– |
||
Таблиця 26.12
Класифікація набутих вад серця (затверджена VI Національним конгресом кардіологів України, 2000 р.)
|
Класифікація набутих вад серця |
Коментарі |
||||
|
Етіологія |
|||||
|
Ревматична Неревматична |
У цьому виданні розглянуто тільки ревматичні вади |
||||
|
Локалізація |
|||||
|
Мітральна вада Аортальна вада Вада тристулкового клапана Вада клапана легеневої артерії |
Незважаючи на рідкісність набутих вад клапана легеневої артерії, вони відображені в МКХ-10 і включені в запропоновану класифікацію |
||||
|
Характер ураження |
|||||
|
Ізольований |
«Чистий» стеноз або недостатність одного клапана |
||||
|
Комбінований:
|
За наявності стенозу і недостатності одного з клапанів |
||||
|
Поєднаний |
При ураженні декількох клапанів |
||||
|
Стадія |
|||||
|
I, II, III, IV, V |
Нижче описані для кожної вади |
||||
|
Стадії набутих вад серця |
|||||
|
Стадії |
Вади серця |
||||
|
Мітральний стеноз |
Мітральна недостатність |
Аортальний стеноз |
Аортальна недостатність |
||
|
I |
Компенсації |
Компенсації |
Повної компенсації |
Повної компенсації |
|
|
II |
Легеневого застою |
Субкомпенсації |
Прихованої серцевої недостатності |
Прихованої серцевої недостатності |
|
|
III |
Правошлуночкової недостатності |
Правошлуночкової декомпенсації |
Відносної коронарної недостатності |
Субкомпенсації |
|
|
IV |
Дистрофічна |
Дистрофічна |
Вираженої лівошлуночкової недостатності |
Декомпенсації |
|
|
V |
Термінальна |
||||
Стадії вад серця за А.Н. Бакулєвим та Є.А. Дамір з уточненнями та доповненнями (Книшов Г.В., Бендет Я.А., 1997) представлено в табл. 26.13. За даними інституту серцево-судинної хірургії ім. акад. М.М. Амосова, ізольоване ураження мітрального клапана становить 67,2% випадків, клапана аорти — 9,6%, поєднане ураження аортального та мітрального клапанів — 14,4%. Чистий та превалюючий стеноз мітрального клапана відмічають у 62,4% хворих з набутими вадами серця, мітральну недостатність — у 34,3%, ураження аортального клапана — у 11%. Найчастіше при ревматичному процесі уражується мітральний клапан.
Таблиця 26.13
Характеристика вад серця
|
Мітральний стеноз (ревматичний I05/0) |
||
|
I стадія (компенсації) |
Клініка |
Характеризується повною компенсацією кровообігу, працездатність не обмежена. Скарги відсутні |
|
ЕхоКГ |
Площина мітрального отвору більша за 2 см2. Підвищення тиску в лівому передсерді до 10–15 мм рт. ст. (норма 3–5 мм рт. ст.) |
|
|
ЕКГ |
Ознаки перевантаження лівого передсердя (Р-mitrale) |
|
|
Рентгенологічні дані |
Помірне збільшення лівого передсердя та легеневої артерії |
|
|
Лікування |
Хірургічне лікування не проводиться. Хворі знаходяться на диспансерному обліку |
|
|
II стадія (легеневого застою) |
Клініка |
Хворі скаржаться на задишку під час фізичного навантаження, іноді — невелике кровохаркання, нічні напади ядухи, набряк легень. Працездатність обмежена |
|
Аускультація, фонокардіограма (ФКГ) |
Типові ознаки мітрального стенозу; акцент та розщеплення II тону над легеневою артерією |
|
|
ЕхоКГ |
Площа лівого передсердно-шлуночкового отвору 1,5–2 см2, паралельний та П-подібний рух стулок мітрального клапана. Підвищення тиску у лівому передсерді до 20–30 мм рт. ст. |
|
|
ЕКГ |
Електрична вісь відхилена вправо, мітральний зубець Р, ознаки гіпертрофії правого шлуночка |
|
|
Рентгенологічні дані |
Збільшення лівого передсердя і легеневої артерії, легеневий застій, ослаблення пульсації лівого шлуночка та підсилення — правого |
|
|
Лікування |
Порушення кровообігу відповідає I стадії за класифікацією М.Д. Стражеска та В.Х. Василенка. Вада може значно прогресувати. Показана мітральна комісуротомія |
|
|
III стадія (правошлуночкової недостатності) |
Клініка |
Відмічена стійка АГ в малому колі кровообігу з утворенням другого «бар’єра». Розвивається перевантаження правого шлуночка. Знижується частота розвитку або зникають напади серцевої астми, набряків легень (внаслідок склерозування легеневих судин, зниження легеневого кровотоку). Ознаки правошлуночкової недостатності, підвищення венозного тиску. Виражена задишка, блідість шкіри, ціаноз, фізичну роботу хворі виконувати не можуть |
|
Аускультація, ФКГ |
Аускультативні дані такі, як в попередній стадії |
|
|
ЕКГ |
Виражене відхилення електричної осі вправо, мітральний зубець Р, ознаки гіпертрофії правого шлуночка |
|
|
ЕхоКГ, рентгенологічні дані |
Площина мітрального отвору 1–1,5 см2. Виражене збільшення легеневої артерії, лівого передсердя, розширення порожнин правого шлуночка та передсердя |
|
|
Лікування |
Показання до оперативного лікування — абсолютні |
|
|
IV стадія (дистрофічна) |
Клініка |
Виражені порушення кровообігу у великому та малому колах. Їх вираженість деякою мірою зменшується на короткий термін під впливом медикаментозного лікування. Склеротичні зміни в легеневих судинах прогресують. Розширення правого шлуночка супроводжується дилатацією фіброзного кільця тристулкового клапана, розвитком його відносної недостатності. Наслідком розладів периферичного кровообігу, гіпоксії є дистрофія внутрішніх органів. Порушується серцевий ритм, виникає фібриляція передсердь, що значно погіршує гемодинаміку. Під час фізичного навантаження виникає задишка. Хворі непрацездатні і в стані спокою. Відмічаються блідість, ціаноз, атрофія м’язів, збільшення печінки. Порушення функції печінки, нирок |
|
Аускультація |
Вислуховуються шуми, зумовлені відносною недостатністю трикуспідального та пульмонального клапанів |
|
|
ЕКГ |
Прогресування ЕКГ-ознак, характерних для цієї вади |
|
|
ЕхоКГ |
У багатьох хворих виявляють кальциноз клапана, тромбоз лівого передсердя |
|
|
Рентгенологічні дані |
Подальше збільшення тіні серця, підсилення легеневого малюнка, корені легень розширені |
|
|
Лікування |
Оперативне лікування можливе після проведення підготовки хворого, але порівняно з попередньою стадією ризик підвищений у 3–4 рази |
|
|
V стадія (термінальна) |
Клініка |
Порушення кровообігу незворотні, відповідають III стадії серцевої недостатності. Значне збільшення печінки, набряки, асцит, атрофія м’язів, трофічні розлади, різні тяжкі порушення серцевого ритму. Тривалість життя хворих незначна |
|
Аускультація |
Різноманітні шуми |
|
|
ЕКГ |
Дистрофічні зміни, різні порушення серцевого ритму |
|
|
Рентгенологічні дані |
Кардіомегалія, високе стояння діафрагми, виражений застій у легенях, часто — рідина в порожнинах плеври |
|
|
Лікування |
Консервативному лікуванню хвороба майже не піддається. Хірургічне лікування не показане |
|
|
Недостатність мітрального клапана (ревматична I05.1) |
||
|
I стадія (компенсації) |
Клініка |
Порушень гемодинаміки майже немає. Визначають незначний систолічний шум на верхівці серця, незначне збільшення лівого передсердя |
|
ЕхоКГ |
Незначна (до +) регургітація на мітральному клапані |
|
|
Лікування |
Хірургічне лікування не показане |
|
|
II стадія (субкомпенсації) |
Клініка |
Зворотна течія крові в ліве передсердя зростає. Виникають його дилатація, гіпертрофія. Порушення гемодинаміки ефективно компенсуються за рахунок лівого шлуночка. Набряки легень виникають дуже рідко. Фізична активність хворих обмежена незначно: задишка з’являється під час фізичного навантаження |
|
Аускультація |
На верхівці вислуховується систолічний шум середньої інтенсивності |
|
|
ЕКГ |
Відхилення електричної осі вліво, ознаки перевантаження лівого шлуночка (в деяких випадках) |
|
|
ЕхоКГ |
Регургітація на мітральному клапані в межах 2+ |
|
|
Рентгенологічні дані |
Збільшення та підсилення пульсації лівих відділів серця |
|
|
Лікування |
Хірургічне лікування не показане. Проводиться протиревматичне лікування, обмежується фізичне навантаження |
|
|
III стадія (правошлуночкової декомпенсації) |
Клініка |
Розвивається при значній регургітації крові в ліве передсердя. Періодично виникає декомпенсація серцевої діяльності, яка усувається за допомогою медикаментозної терапії. Задишка при фізичному навантаженні менш виражена, ніж при стенозі. Працездатність обмежена |
|
Аускультація, пальпація |
Грубий систолічний шум на верхівці, який проводиться у ліву пахвову ділянку, пульсація грудної стінки в ділянці серця |
|
|
Рентгенологічні дані |
Значне збільшення та пульсація лівих відділів серця |
|
|
ЕКГ |
Ознаки гіпертрофії лівого шлуночка |
|
|
ЕхоКГ |
Регургітація на мітральному клапані більше за 2+ |
|
|
Лікування |
Хірургічне лікування показане |
|
|
IV стадія (дистрофічна) |
Клініка |
Постійні прояви правошлуночкової недостатності, які під впливом інтенсивної медикаментозної терапії тимчасово зникають. Працездатність утрачена |
|
Аускультація |
Грубий систолічний шум, шуми пов’язані з появою недостатності тристулкового клапана |
|
|
Рентгенологічні дані |
Серце значно розширене; застій у малому колі кровообігу |
|
|
ЕКГ |
Ознаки гіпертрофії лівого або обох шлуночків, фібриляція передсердь та інші порушення ритму |
|
|
ЕхоКГ |
Відображує перелічені вище порушення |
|
|
Лікування |
Хірургічне лікування показане (протезування мітрального клапана) |
|
|
V стадія (термінальна) |
Відповідає III стадії серцевої недостатності за класифікацією М.Д. Стражеска і В.Х. Василенка. Хірургічне лікування не показане |
|
|
Комбінована ревматична мітральна вада (ревматичний мітральний стеноз з недостатністю — I05.2) |
||
|
||
|
||
|
||
|
Пролапс мітрального клапана (I34.1) |
||
|
Аортальний стеноз (ревматичний I06.0) |
||
|
I стадія (повної компенсації) |
Скарги відсутні, ваду виявляють під час аускультації. На ехоКГ — невеликий градієнт систолічного артеріального тиску (САТ) на аортальному клапані в межах 26–30 мм рт. ст. Хірургічне лікування не показане |
|
|
II стадія (прихованої недостатності кровообігу) |
Клініка |
Хворі скаржаться на підвищену втомлюваність, задишку при фізичному навантаженні, запаморочення |
|
ЕКГ, рентгенологічні дані |
Ознаки збільшення та гіпертрофії лівого шлуночка |
|
|
ЕхоКГ |
Помірний градієнт САТ на аортальному клапані (до 50 мм рт. ст.) |
|
|
Лікування |
Хірургічне лікування показане |
|
|
III стадія (відносної коронарної недостатності) |
Клініка |
Скарги на біль стенокардичного характеру, запаморочення, непритомність при фізичному навантаженні. Наростання задишки в динаміці |
|
Рентгенологічні дані |
Збільшення розмірів серця, головним чином, за рахунок лівого шлуночка |
|
|
ЕКГ |
Ознаки гіпертрофії лівого шлуночка, гіпоксії міокарда, пов’язані з відносною недостатністю коронарного кровообігу |
|
|
ЕхоКГ |
Градієнт САТ вищий за 50 мм рт. ст. |
|
|
Лікування |
Хірургічне лікування показане |
|
|
IV стадія (вираженої лівошлуночкової недостатності) |
Клініка |
Прогресування симптомів, характерних для попередніх стадій, виникнення запаморочення та втрати свідомості при фізичному навантаженні. Періодичні напади пароксизмальної нічної задишки, серцевої астми, набряку легень, збільшення печінки |
|
Рентгенологічні дані |
Збільшення лівого шлуночка та інших відділів серця, застійні явища в легенях |
|
|
ЕКГ |
Порушення метаболізму міокарда та коронарного кровобігу, часто — миготлива аритмія |
|
|
ЕхоКГ |
Значне погіршення показників скоротної функції лівого шлуночка — значний градієнт САТ на аортальному клапані, кальциноз клапана (часто) |
|
|
Лікування |
Консервативне лікування дає тимчасовий ефект. Деяким хворим проводиться хірургічне лікування (питання вирішується індивідуально, з урахуванням ефективності передопераційної медикаментозної підготовки) |
|
|
V стадія (термінальна) |
Стан хворих тяжкий. Недостатність лівого та правого шлуночків прогресує. Різко виражені всі ознаки вади (суб’єктивні, об’єктивні). Лікування неефективне. Хірургічне лікування не показане |
|
|
Недостатність клапана аорти (ревматична — I06.1) |
||
|
I стадія (повної компенсації вади) |
Відмічають початкові ознаки вади, скарги відсутні. При ехоКГ — регургітація в межах 1+ на аортальному клапані. Хірургічне лікування не показане |
|
|
II стадія (прихованої серцевої недостатності) |
Клініка |
Помірне зниження працездатності хворих |
|
Аускультація |
Характерні фізикальні дані вади |
|
|
Рентгенологічні дані |
Помірне збільшення і пульсація лівого шлуночка |
|
|
ЕКГ |
Ознаки помірної гіпертрофії лівого шлуночка |
|
|
ЕхоКГ |
Регургітація на аортальному клапані в межах 2+ |
|
|
Лікування |
Хірургічне лікування не показане |
|
|
III стадія (субкомпенсації) |
Клініка |
Знижена фізична активність, виникає ангінозний біль. Мінімальний АТ — менше половини максимального |
|
Аускультація |
Діастолічний шум |
|
|
Рентгенологічні дані |
Збільшення розмірів і пульсація лівого шлуночка, аорти |
|
|
ЕКГ |
Ознаки вираженої гіпертрофії лівого шлуночка, коронарної недостатності |
|
|
ЕхоКГ |
Значна (3+ та більше) регургітація на аортальному клапані |
|
|
Лікування |
Хірургічне лікування показане |
|
|
IV стадія (декомпенсації) |
Клініка |
Виражена задишка, напади ангінозного болю при незначному фізичному навантаженні, серцева астма, збільшення печінки |
|
ЕхоКГ, ЕКГ, рентгенологічні дані |
Значна дилатація серця, відносна мітральна недостатність («мітралізація» вади), подальше погіршення функції серця, коронарного кровообігу |
|
|
Лікування |
Медикаментозне лікування малоефективне (показане хірургічне лікування) |
|
|
V стадія (термінальна) |
Прогресує недостатність лівого та правого шлуночків, дистрофічні зміни в життєво важливих органах. Недостатність кровообігу відповідає III стадії серцевої недостатності за класифікацією М.Д. Стражеска і В.Х. Василенка. Медикаментозна терапія безуспішна, хірургічне лікування не показане |
|
|
Комбінована аортальна вада |
||
|
Ревматичний аортальний стеноз із недостатністю (I06,2): |
||
|
З перевагою стенозу |
Стадії та показання до хірургічного лікування відповідають таким для аортального стенозу |
|
|
З перевагою недостатності |
Стадії та показання до хірургічного лікування відповідають таким для аортальної недостатності |
|
|
Без явної переваги |
Стадії та показання до хірургічного лікування відповідають таким для аортального стенозу |
|
|
Трикуспідальний стеноз (ревматичний — I07.0) |
||
|
Трикуспідальна недостатність (ревматична — I07.1) |
||
|
Комбінована трикуспідальна вада |
||
|
ревматичний трикуспідальний стеноз з недостатністю (I07.2) |
||
|
Поєднані вади серця |
||
|
||
|
||
|
||
|
||
|
Клапанний стеноз легеневої артерії (I37.0) |
||
|
Недостатність клапана легеневої артерії (I37.1) |
||
|
Комбінована вада клапана легеневої артерії (стеноз легеневої артерії з недостатністю клапана (I37.2) |
||
Нижче (табл. 26.14) наведена класифікація ступеня тяжкості мітрального й аортального стенозу.
Таблиця 26.14
Класифікація ступенів тяжкості мітрального й аортального стенозу
|
Ступінь тяжкості |
Площа отвору клапана, см2 |
Градієнт тиску, мм рт. ст. |
|
Аортальний клапан |
||
|
Легкий |
1,5–2,0 |
– |
|
Помірний |
1,0–1,5 |
– |
|
Тяжкий |
<1,0 |
>50 |
|
Мітральний клапан |
||
|
Легкий |
>2,0 |
<5 |
|
Помірний |
1,5–2,0 |
5–10 |
|
Виражений |
1,0–1,5 |
10–20 |
|
Тяжкий (критичний) |
<1,0 |
>20 |
Ізольоване ураження аортального клапана при ГРЛ не є характерним. Як правило, ознаки його ураження поєднуються із шумом мітральної регургітації. Після першої атаки ГРЛ РВС формуються у 20–25% дітей. При цьому переважають ізольовані вади серця, найчастіше відзначається мітральна недостатність. Недостатність аортального клапана, мітральний стеноз, мітрально-аортальна вада розвиваються значно рідше.
Пролапс мітрального клапана розвивається у 7–10% дітей, що перенесли ревматичний кардит.
Частота розвитку вад серця у підлітків, що перенесли першу атаку ГРЛ, і у дорослих. Їх діагностують у ⅓ підлітків і у 39–45% дорослих.
Періоди хвороби, в які відмічають максимальну частоту формування вад серця: 70% вад формуються в перші 3 роки від початку хвороби; повторні атаки ГРЛ посилюють клапанні ураження.
Діагностика гострої ревматичної лихоманки
У число симптомів, що свідчать проти ревматичної лихоманки, входять:
- виявлення шуму в серці у віці до 1 року;
- відсутність ознак формування вади серця після 3 і більше атак артриту або кардиту;
- вранішня скутість (в тому числі в анамнезі);
- залучення у запальний процес нових суглобів через 1–2 міс при збереженні його вираженості в уражених раніше;
- збільшення щитовидної залози.
Зміни, що виявляють при лабораторному та інструментальному обстеженні хворих ГРЛ, наведені в табл. 26.15.
Таблиця 26.15
Зміни, що виявляють при лабораторному та інструментальному обстеженні хворих на ГРЛ
|
Аналіз крові |
Підвищення ШОЕ, концентрації СРБ у крові |
|
Бактеріологічні дослідження |
У мазках із зіву виявляють (при активній інфекції і носійстві) β-гемолітичний стрептокок групи А |
|
Серологічні дослідження |
Підвищені титри антистрептолізину-О, антистрептогіалуронідази і антидезоксирибонуклеази-В. Слід звертати увагу на підвищення титрів в динаміці |
|
ЕКГ |
Дозволяє виявити (виключити) порушення серцевого ритму і провідності |
|
ЕхоКГ |
Дає можливість діагностувати ураження клапанів серця і перикарду, вивчити функціональний стан серця |
Результати інструментальних досліджень при вальвуліті мітрального й аортального клапанів представлені нижче (табл. 26.16).
Таблиця 26.16
Результати інструментальних досліджень при вальвуліті мітрального й аортального клапанів
|
Методи дослідження |
Вальвуліт мітрального клапана |
Вальвуліт аортального клапана |
|
ЕхоКГ |
Виявляють: 1) крайове потовщення передньої мітральної стулки; 2) гіпокінезію (обмеження рухливості) задньої стулки; 3) невелике пролабування стулок у кінці систоли; 4) мітральну регургітацію; 5) вигин в діастолу передньої стулки |
У 50% дітей відмічають тріпотіння в діастолу мітральних стулок. Стулки аортального клапана потовщені по краю, є транзиторний пролапс, аортальна регургітація |
|
ФКГ |
На верхівці, в V точці реєструється високочастотний систолічний шум (пансистолічний або тривалий протосистолічний). Одночасно із систолічним шумом може реєструватися мезодіастолічний шум, який швидко зникає під впливом активного лікування |
У 3–5% випадків реєструється високочастотний протодіастолічний шум вздовж лівого краю груднини, що відображає ураження аортального клапана |
|
ЕКГ |
Відзначаються ознаки гострого перевантаження лівого передсердя з мітралізацією зубця Р, початкові ознаки гіпертрофії лівого шлуночка |
Реєструються ознаки перевантаження діастоли лівого шлуночка |
|
Рентгенографія |
Відзначається мітральна конфігурація серця, згладжена талія за рахунок вушка лівого передсердя, збільшення розмірів лівого передсердя і шлуночка. Можлива поява ознак венозного застою в легенях |
Тенденція до горизонтального положення, аортальна конфігурація серця. Збільшення розмірів та пульсації лівого шлуночку, аорти |
Діагностичні критерії ГРЛ (запропоновані в 1992 р. AHA) представлені в табл. 26.17.
Таблиця 26.17
Діагностичні критерії ГРЛ
|
Великі критерії |
|
Кардит, поліартрит, хорея, кільцеподібна еритема, підшкірні ревматичні вузлики |
|
Малі критерії |
|
1. Клінічні (артралгія, лихоманка) 2. Лабораторні (зростання гострофазових реактантів: ШОЕ, СРБ) 4. Інструментальні (подовження інтервалу PQ(R)) |
|
Ознаки попередньої стрептококової інфекції |
|
Зростання гемолітичного стрептокока при бактеріологічному дослідженні матеріалу із зіву Високий титр або наростання титру антистрептококових антитіл |
Встановити діагноз ГРЛ дозволяє наявність 2 великих критеріїв і ознак попередньої стрептококової інфекції або 1 великого, 2 малих критеріїв і ознак попередньої стрептококової інфекції. Складно говорити про ревматичну природу виявленого міокардиту або перикардиту за відсутності у пацієнтів з цією патологією ознак вальвуліту.
Чинники, які слід враховувати при ранньому розпізнаванні ревматичної лихоманки
Необхідно враховувати:
- попередню стрептококову інфекцію;
- зниження працездатності, фізичної активності, що виникло після перенесеної носоглоткової інфекції;
- швидку втомлюваність після звичайного навантаження;
- появу субфебрилітету, артралгії, серцебиття, підвищеного потовиділення;
- наявність запального процесу за даними лабораторних досліджень;
- наявність кардиту за даними фізикального та інструментального обстеження.
Дані, які підтверджують наявність попередньої інфекції бета-гемолітичного стрептококу групи А. Про це свідчать виділення із зіву β-гемолітичного стрептококу групи А; позитивний тест швидкого визначення групового антигену бета-гемолітичного стрептококу групи А.
Зміни, що відбулися в клінічній картині ревматизму за останні роки, наступні:
- перебіг і прогноз хвороби став сприятливішим;
- неспецифічне запалення виражене меншою мірою;
- знизилася частота ураження серозних оболонок;
- переважають помірний і мінімальний ступінь активності запального процесу, гострі і підгострі варіанти перебігу;
- зменшилася діагностична цінність таких проявів, як кільцеподібна еритема і ревматичні вузлики;
- знизилася інформативність лабораторних тестів;
- не виявлено випадків безперервно-рецидивуючого перебігу;
- перебіг кардиту менш тяжкий;
- відсоток формування вад різко знизився;
- не відмічається серцева недостатність;
- у більшості дітей настає повне одужання.
Диференційна діагностика гострої ревматичної лихоманки
Захворювання, з якими доводиться диференціювати ГРЛ, наведені в табл. 26.18.
Таблиця 26.18
Захворювання, з якими доводиться диференціювати ГРЛ
|
Захворювання |
Коротка характеристика |
|
Нейроциркуляторна дистонія |
Характерний зв’язок хвороби зі стресами, її поступовий початок, астеноневротичний характер скарг (незадоволення вдихом, «нестача повітря», відчуття «зупинки серця», «завмирання»), вегетосудинна криза. Відсутні клінічні ознаки вальвуліту, міо- і перикардиту, ефект від протиревматичної терапії (у тому числі ГК), позитивна дія блокаторів β-адренорецепторів. Лабораторні маркери запалення нормальні. Загалом багато скарг при мінімумі об’єктивних ознак патології. Вислуховується функціональний шум, є лабільність артеріального тиску і пульсу, інверсія або сплощення зубця Т на ЕКГ. При обстеженні не вдається виявити ознаки вади серця |
|
Ідіопатичний пролапс мітрального клапана |
Конституція пацієнтів астенічна, відмічають симптоми невротизації. Виявляють ознаки вродженої дисплазії сполучної тканини (ранній розвиток плоскостопості, синдром гіпермобільності суглобів, воронкоподібна деформація грудної клітки, сколіоз грудного відділу хребта та ін.). При аускультації серця виявляють пізній систолічний шум у зоні проекції мітрального клапана, клацання всередині систоли |
|
ІЕ |
Гарячковий синдром не купірується при призначенні НПЗП, позитивна гемокультура, прогресує анемія. При ехоКГ виявляють вегетацію на клапанах і хордах, перфорації стулок, розриви хорд, міокардіальні абсцеси і ознаки застійної серцевої недостатності |
|
Неревматичний міокардит |
Існує хронологічний зв’язок з гострою носоглотковою інфекцією, відсутність артриту, виразна клінічна і ехоКГ симптоматика міокардиту, відсутність вальвуліту, повільна динаміка на тлі протизапальної терапії |
|
Кальцинований аортальний клапан |
У хворих віком старше 60 років вислуховується над аортою грубий систолічний шум, який проводиться на судини шиї. Ревматичний анамнез відсутній, характерні емболічні ускладнення. Хворі скаржаться на серцебиття, перебої в роботі серця, задишку і непритомність при фізичному навантаженні, біль за грудниною |
|
Хвороба Такаясу |
Характерні непритомні стани, вислуховуються шуми над великими артеріальними стовбурами, асиметрія артеріального тиску, пульсу на кінцівках. Відзначається переміжна кульгавість у молодих жінок, парестезії кінцівок. Верифікація діагнозу проводиться за допомогою ангіографії |
|
Анкілозуючий спондилоартрит |
Може бути виявлена аортальна недостатність. Є біль і відчуття скутості в хребті, двобічний сакроілеїт, ураження великих і середніх суглобів нижніх кінцівок, груднино-ключичних, акроміально-ключичних. Правильний діагноз встановлюється після ретельного вивчення суглобового синдрому, результатів інструментальних і лабораторних досліджень |
|
Постстрептококовий РеА |
Перебіг без кардиту, персистує близько 2 міс, недостатньо добре реагує на НПЗП, регресує без залишкових явищ. Пацієнтів слід тримати на «Д»-обліку у ревматолога з періодичним ехоКГ контролем, лабораторним обстеженням |
|
РеА іншої етіології |
Слід враховувати хронологічний зв’язок суглобової патології з попередньою кишковою і сечостатевою інфекцією, результати мікробіологічних і серологічних досліджень, наявність кон’юнктивіту, ураження шкіри, слизових оболонок. Артрит має стійкий асиметричний характер з переважним ураженням суглобів нижніх кінцівок, розвиваються бурсити п’ят, ентезопатії |
|
РА, його ювенільний варіант |
Суглобовий синдром носить стійкий поліартикулярний характер із залученням дрібних суглобів, деформацією. Відмічається тривала интермітуюча лихоманка. При синдромі Стілла виявляють еритематозно-папульозний висип, лімфаденопатію, гепато- і спленомегалію |
|
Палідромний ревматизм |
Нині розглядається як продромальний варіант РА, характеризується короткочасними нападами артриту, що повторюються через різні проміжки часу, відсутністю лабораторних ознак запалення |
|
Інтермітуючий гідроартроз |
Регулярно повторюється синовіт одного й того ж суглоба (частіше колінного) за відсутності больового синдрому і змін лабораторних показників |
У практичній роботі доводиться диференціювати ревматичний пролапс мітрального клапана з ідіопатичним. Особливості ідіопатичного пролапсу мітрального клапана наведені нижче:
- переважно відзначають у дівчаток астенічної статури;
- виявляють випадково в шкільному і підлітковому віці;
- окрім кардіальних скарг відмічають систолічний шум на верхівці серця, систолічне клацання;
- систолічний шум має пізній характер, краще виявляється в положенні стоячи, посилюється після фізичного навантаження;
- при дослідженні ехоКГ виявляють пролапс задньої стулки мітрального клапана, рідше — передньої або обох;
- зміни, що виявляють з боку серця, носять стійкий характер.
Приклади формулювання діагнозу
ГРЛ: кардит, серцева недостатність IIА (ФК III).
ГРЛ: хорея.
ХРХС: комбінована мітральна вада з переважанням недостатності, III стадія, серцева недостатність IIБ (ФК IV).
Лікування
Цілі лікування хворих на ГРЛ і ХРХС включають: 1) ерадикацію β-гемолітичного стрептококу групи А; 2) пригнічення запалення; 3) попередження формування вад серця; 4) усунення явищ серцевої недостатності у пацієнтів із вадами серця. Основні ланки комплексної етапної терапії ревматизму включають стаціонарне лікування, доліковування у місцевому ревматологічному санаторії, диспансерне спостереження.
Немедикаментозне лікування
Необхідно госпіталізувати усіх хворих на ГРЛ. Протягом перших 2–3 тиж хвороби рекомендується дотримання ліжкового режиму. Харчовий раціон повинен включати не менше 1 г білка на 1 кг маси тіла. Слід обмежити вживання кухонної солі. Пацієнтам з ГРЛ протипоказане фізіотерапевтичне лікування.
Медикаментозна терапія гострої ревматичної лихоманки і хронічної ревматичної хвороби серця
Лікування, рекомендоване при ГРЛ і ХРХС (за: Насонов Е.Л., 2010 (ред.)), наведене у табл. 26.19.
Таблиця 26.19
Лікування, рекомендоване при ГРЛ і ХРХС
|
Лікарські засоби |
Контингенти |
Дози препаратів |
|
|
Антистрептококова терапія |
|||
|
Бензилпеніцилін |
Підлітки і дорослі |
500 000–1 000 000 ОД в/м 4 рази на добу 10 днів з подальшим переходом на пеніциліни пролонгованої дії |
|
|
Діти |
100 000–150 000 ОД в/м 4 рази на добу 10 днів з подальшим переходом на пеніциліни пролонгованої дії |
||
|
Макроліди або лінкозаміди |
Призначають при непереносимості пеніцилінів |
||
|
Протизапальна терапія |
|||
|
ГК |
Підлітки і дорослі |
Преднізолон 20 мг/добу в 1 прийом вранці після їжі до досягнення терапевтичного ефекту (в середньому 14 днів) |
|
|
Діти |
Преднізолон 0,7–0,8 мг/кг в 1 прийом вранці після їжі до досягнення ефекту (в середньому 14 днів). Надалі дозу знижують на 2,5 мг кожні 5–7 днів до відміни. Тривалість курсу 1,5–2 міс |
||
|
НПЗП |
Підлітки і дорослі |
Частіше диклофенак 25–50 мг (дітям 0,7–1,0 мг/кг) 3 рази на добу призначають при вальвуліті, РА без вальвуліту, мінімальній активності процесу (ШОЕ <30 мм/год), після зниження високої активності і відміни ГК, при повторній ГРЛ на тлі РВС. НПЗП призначають до нормалізації запальної активності. В середньому курс становить 1,5–2 міс, але може бути продовжений до 3–5 міс |
|
Критерії якості лікування хворих ГРЛ включають: 1) відсутність кардіальних й артралгічних синдромів; 2) нормалізацію показників активності запального процесу; 3) нормалізацію титрів стрептококових антитіл; 4) стабільність морфо-функціональних показників ехоКГ з боку клапанів і камер серця.
Засоби лікування застійної серцевої недостатності у хворих на ГРЛ і ХРХС представлені у табл. 26.20.
Таблиця 26.20
Засоби лікування застійної серцевої недостатності у хворих на ГРЛ і ХРХС
|
Діуретики |
Петлеві |
Фуросемід |
|
Тіазидні та тіазидоподібні |
Гідрохлоротіазид Індапамід |
|
|
Калійзберігаючі |
Спіронолактон Тріамтерен |
|
|
Блокатори кальцієвих каналів |
Дигідропіридини тривалої дії |
Амлодипін |
|
Блокатори β-адренорецепторів |
Карведилол Метопролол Бісопролол |
|
|
Серцеві глікозиди |
Дігоксин |
|
Хворим із серцевою недостатністю внаслідок гострого вальвуліту недоцільне призначення кардіотонічних препаратів (хороший ефект досягається при призначенні преднізолону у дозі 40–60 мг/добу). Нітрати, що застосовуються у комплексній терапії застійної серцевої недостатності у хворих із РВС, погіршують прогноз. У пацієнтів з ХРХС і хронічною серцевою недостатністю без виражених ознак кардиту не рекомендується застосування ГК, оскільки вони посилюють міокардіодистрофію. ГК (переважно преднізолон) призначають хворим з 1) вираженим ексудативним компонентом запалення (яскраво вираженим кардитом); 2) максимальною або помірною активністю ревматичного процесу; 3) гострим (рідше підгострим) перебігом ревматичної лихоманки. При мінімальному ступені активності, в’ялому і латентному перебігу вважають більш обґрунтованим застосування НПЗП. Хворим з повторною ГРЛ на фоні РВС, що отримують ГК, в лікування включають аспартат К і Mg (3–6 таблеток на добу, 1 міс), інозин (0,6–1,2 г/добу в 3 прийоми, 1 міс), нандролон (1 мл/тиж в/м, курс 10 ін’єкцій). Тривалість курсу терапії ГК при ревматичній лихоманці в середньому триває 1,5 міс, після чого пацієнтам призначають НПЗП. При ендоміокардиті з тенденцією до затяжного перебігу первинну дозу ГК підвищують, а курс лікування подовжують. Призначення ГК хворим з РВС без ознак кардиту вважають невиправданим: ГК посилюють міокардіодистрофію.
Тривалість курсу терапії НПЗП становить 1–1,5 міс, за необхідності його продовжують до 3–5 міс (до повної нормалізації показників запальної активності). При ревматичній лихоманці у дітей (на відміну від дорослих) НПЗП поступаються за терапевтичною ефективністю гормональній терапії, особливо за наявності кардиту. Вивчається доцільність використання інгібіторів АПФ у хворих із кардитом на фоні ХРХС. Встановлено, що поєднане застосування НПЗП та інгібіторів АПФ у хворих з ревмокардитами на тлі РВС призводить до зниження судинорозширювального ефекту інгібіторів АПФ.
Хлорохін і гідроксихлорохін (йому віддають перевагу, особливо у дітей) призначають пацієнтам з первинним ревмокардитом з ураженням клапанного апарату, а також при затяжних і безперервно-рецидивуючих (сьогодні практично не виявляють) формах хвороби. Їх призначають 1 раз на добу після вечері, поєднують з ГК або підключають пізніше після виявлення тенденції до затяжного перебігу патологічного процесу.
Пацієнтам з хореєю призначають діазепам (0,006–0,01 г/добу), карбамазепін (0,6 г/добу), хлорпромазин (0,01 г/добу). Для усунення хореєподібних рухів необхідний тривалий час (до 1 року).
Санація вогнищ стрептококової інфекції. Санацію вогнищ стрептококової інфекції (хронічного тонзиліту) рекомендують проводити в умовах стаціонару.
Показання до хірургічного лікування пацієнтів із ревматичними вадами серця
До них відносяться декомпенсація вад (миготлива аритмія, легенева гіпертензія, порушення систолічної функції лівого шлуночка, хронічна серцева недостатність III–IV ФК, стенокардія). Характер оперативного втручання визначається станом (морфологічним) клапанів серця і загальним станом хворого.
Заходи первинної профілактики гострої ревматичної лихоманки
Цілі первинної профілактики ГРЛ включають: 1) підвищення природного імунітету (раціональна фізкультура і спорт, раннє загартовування, вітамінізоване харчування, максимально тривале перебування на свіжому повітрі, виключення надмірної скупченості в дошкільних установах, школах, вищих навчальних закладах, житлових приміщеннях); 2) дотримання санітарно-гігієнічних заходів зі зниження вірогідності стрептококового інфікування в дитячих колективах; 3) своєчасне й ефективне лікування гострого і хронічного рецидивуючого тонзиліту і фарингіту. Основою первинної профілактики ГРЛ є своєчасне лікування цих інфекцій верхніх дихальних шляхів, викликаних β-гемолітичним стрептококом групи А. До факторів ризику розвитку ГРЛ відносяться: перенесена гостра стрептококова інфекція та носоглоткові інфекції; несприятливі умови праці або проживання в приміщенні з підвищеною вологістю, низькою температурою повітря; вік 7–10 років; жіноча стать (хворіють в 2,6 раза частіше, ніж чоловіки); спадковість; недоношеність; вроджені аномалії сполучної тканини. Рекомендації з антимікробного лікування тонзиліту/фарингіту, викликаних β-гемолітичним стрептококом групи А (Насонов Е.Л., 2010 (ред.)), наведені в табл. 26.21.
Таблиця 26.21
Антимікробне лікування тонзиліту/фарингіту, викликаних β-гемолітичним стрептококом групи А
|
Препарати |
Групи хворих |
Дози |
Примітки |
||||
|
1. Гострий тонзиліт/фарингіт, викликаний β-гемолітичним стрептококом групи А |
|||||||
|
Препарати I ряду : |
|||||||
|
Бензатину бензилпеніцилін |
Дорослі |
2,4 млн ОД в/м |
Вводять одноразово при ГРЛ в анамнезі у хворого і близьких родичів, спалахах інфекції, викликаної β-гемолітичним стрептококом групи А, в колективах, при сумнівній сумлінності хворих |
||||
|
Діти з масою тіла <25 кг |
600 000 ОД в/м |
||||||
|
Діти з масою тіла >25 кг |
1,2 млн ОД в/м |
||||||
|
Амоксицилін |
Дорослі |
0,5 г 3 рази на добу |
Призначають усередину протягом 10 днів |
||||
|
Діти |
0,25 г 3 рази на добу |
||||||
|
Феноксиметилпеніцилін |
Дорослі |
0,5 г 3 рази на добу |
Призначають усередину протягом 10 днів за 1 год до їди. Препарат рекомендується переважно дітям раннього віку (у формі суспензії) |
||||
|
Діти з масою тіла <25 кг |
0,125 г 3 рази на добу |
||||||
|
Діти з масою тіла > 25 кг |
0,25 г 3 рази на добу |
||||||
|
Цефадроксил |
Дорослі |
0,5 г 2 рази на добу |
Тривалість прийому усередину — 10 днів |
||||
|
Діти |
30 мг/кг/добу в 1 прийом |
||||||
|
Альтернативні препарати при непереносимості β-лактамних антибіотиків |
|||||||
|
Азитроміцин |
Дорослі |
0,5 г одноразово в 1-шу добу, потім по 0,25 г/добу протягом 4 днів |
Тривалість прийому — 5 днів |
||||
|
Діти |
12 мг/кг в 1 прийом |
||||||
|
Кларитроміцин |
Дорослі |
0,25 г 2 рази на добу |
Усередину протягом 10 днів |
||||
|
Діти |
15 мг/кг/добу в 2 прийоми |
||||||
|
Мідекаміцин |
Дорослі |
0,4 г 3 рази на добу |
Вживати за 1 год до їди протягом 10 днів |
||||
|
Діти |
50 мг/кг/добу в 3 прийоми |
||||||
|
Рокситроміцин |
Дорослі |
0,15 г 2 рази на добу |
Приймати за 1 год до їди протягом 10 днів |
||||
|
Діти |
5 мг/кг/добу в 2 прийоми |
||||||
|
Спіраміцин |
Дорослі |
3 млн МО 2 рази на добу |
Усередину протягом 10 днів |
||||
|
Діти |
1,5 млн МО 2 рази на добу |
||||||
|
Еритроміцин |
Дорослі |
0,5 г 3 рази на добу |
Приймати за 1 год до їди протягом 10 днів. Можливі алергічні реакції |
||||
|
Діти |
40 мг/кг/добу в 3 прийоми |
||||||
|
Препарати резерву (при непереносимості β-лактамів і макролідів) |
|||||||
|
Лінкоміцин |
Дорослі |
0,5 г 3 рази на добу |
Усередину за 1–2 год до їди протягом 10 днів |
||||
|
Діти |
30 мг/кг/добу в 3 прийоми |
||||||
|
Кліндаміцин |
Дорослі |
0,15 г 4 рази на добу |
Тривалість прийому усередину 10 днів, запивати склянкою води |
||||
|
Діти |
20 мг/кг/добу в 3 прийоми |
||||||
|
2. Хронічний рецидивуючий тонзиліт/фарингіт, викликаний β-гемолітичним стрептококом групи А |
|||||||
|
Лікарські препарати I ряду |
|||||||
|
Амоксицилін/клавуланова кислота |
Дорослі |
0,625 г 3 рази на добу |
Усередину протягом 10 днів |
||||
|
Діти |
40 мг/кг/добу в 3 прийоми |
||||||
|
Цефуроксим |
Дорослі |
0,25 г 2 рази на добу |
Усередину відразу після їди протягом 10 днів |
||||
|
Діти |
20 мг/кг/добу в 2 прийоми |
||||||
|
Препарати резерву (при непереносимості β-лактамів) |
|||||||
|
Лінкоміцин |
Дорослі |
0,5 г 3 рази на добу |
Усередину за 1–2 год до їди протягом 10 діб |
||||
|
Діти |
30 мг/кг/добу в 3 прийоми |
||||||
|
Кліндаміцин |
Дорослі |
0,15 г 4 рази на добу |
Усередину, запиваючи склянкою води, протягом 10 днів |
||||
|
Діти |
20 мг/кг/добу в 3 прийоми |
||||||
Засобами вибору при лікуванні гострого тонзиліту, викликаного β-гемолітичним стрептококом групи А, залишаються препарати пеніциліну. Оптимальним з цієї групи є амоксицилін, він має переваги перед ампіциліном і феноксиметилпеніциліном. Феноксиметилпеніцилін (у вигляді суспензії) застосовують у дітей молодшого віку. Цефадроксил високоефективний при терапії цієї патології і добре переноситься. При непереносимості β-лактамних антибіотиків показані макроліди, які створюють високу концентрацію у вогнищі інфекції, мають високу антистрептококову активність. Лінкозаміди (лінкоміцин, кліндаміцин) призначають для лікування тонзиліту, викликаного β-гемолітичним стрептококом групи А, тільки при непереносимості β-лактамів і макролідів. Вони розглядаються як препарати 1-го ряду в профілактиці ІЕ при стоматологічних маніпуляціях. У разі хронічного рецидивуючого тонзиліту, викликаного β-гемолітичним стрептококом групи А, лікування проводять ингібіторзахищеними пеніцилінами (амоксицилін/клавуланова кислота), цефалоспоринами 2-го покоління (цефуроксим) і лінкозамідами (у разі непереносимості β-лактамних антибіотиків). Перелічені антибіотики використовують як препарати 2-го ряду при неефективній терапії гострого тонзиліту, викликаного β-гемолітичним стрептококом групи А. Сьогодні відсутні лікарські схеми, які б забезпечили 100% елімінацію β-гемолітичного стрептококу групи А з глотки. Тетрациклін, сульфаніламіди, ко-тримоксазол, хлорамфенікол не застосовують при лікуванні цієї інфекції глотки через низьку ефективність і високу резистентність. Низькою антистрептококовою активністю відрізняються і фторхінолони.
Вторинна профілактика
ЇЇ проводять з метою попередження виникнення повторних атак, а також прогресування хвороби в осіб, які перенесли ГРЛ. Проведення вторинної профілактики необхідно починати в стаціонарі після закінчення терапії. Основним лікарським засобом для вторинної профілактики ГРЛ є бензатину бензилпеніцилін. Частота введення препарату наведена в табл. 26.22.
Таблиця 26.22
Дози і частота введення бензатину бензилпеніциліну
|
Групи хворих |
Дози, ОД |
Частота введення |
|
Дорослі і підлітки |
2,4 млн |
Препарат вводять в/м 1 раз на 3 тиж |
|
Діти з масою тіла <25 кг |
600 тис. |
|
|
Діти з масою тіла >25 кг |
1,2 млн |
Тривалість проведення вторинної профілактики ГРЛ встановлюється індивідуально (табл. 26.23).
Таблиця 26.23
Тривалість проведення вторинної профілактики ГРЛ
|
Групи хворих |
Тривалість проведення профілактики |
|
Хворі, що перенесли ГРЛ, без кардиту (артрит, хорея) |
Не менше 5 років після атаки або до 18-річного віку |
|
Хворі з вилікуваним кардитом без вади серця |
Не менше 10 років після атаки або до 25-річного віку |
|
Хворі, що мають сформовану ваду серця, в тому числі оперовані |
Довічно |
Вагітним із ХРХС профілактику проводять з 8–10-го тижня вагітності до пологів, надалі — за показаннями. За результатами досліджень, проведених в Інституті ревматології РАМН і Державному науковому центрі з антибіотиків (Насонов Е.Л., 2006 (ред.)), бензатину бензилпеніцилін є ефективним препаратом. Він тривало підтримує адекватну протистрептококову концентрацію бензилпеніциліну в сироватці крові. Сьогодні застосування суміші бензатину бензилпеніциліну з новокаїновою сіллю бензилпеніциліну не рекомендується.
Профілактика інфекційного ендокардиту у пацієнтів із ревматичними вадами серця
Профілактичне призначення антибіотиків рекомендується хворим із РВС у разі проведення різних маніпуляцій, які можуть призвести до бактеріємії (екстракція зуба, аденоїдектомія, тонзилектомія, бронхоскопія, ФЕГДС, видалення поліпів у верхніх дихальних шляхах, в шлунку, маніпуляції на урогенітальному тракті та ін.). Антибіотики, які використовують для профілактики ІЕ, наведені в табл. 26.24 (за: Е.Л. Насонов, 2010 (ред.)).
Таблиця 26.24
Профілактика ІЕ у пацієнтів з РВС
|
Умови призначення |
Групи хворих |
Дози |
Примітки |
|
|
При маніпуляціях на верхніх дихальних шляхах і стравоході |
||||
|
Стандартна схема |
Дорослі |
Амоксицилін 2 г |
Усередину за 1 год до процедури |
|
|
Діти до 12 років |
Ампіцилін 50 мг/кг маси тіла |
|||
|
При неможливості застосування per os |
Дорослі |
Ампіцилін 2 г в/в або в/м |
За 30 хв до процедури |
|
|
Діти до 12 років |
Ампіцилін 50 мг/кг в/в або в/м |
|||
|
При алергії на пеніцилін |
Дорослі |
Кліндаміцин 600 мг, або Цефалексин 2 г, або Цефадроксил 2 г, або Азитроміцин 500 мг, або Кларитроміцин 500 мг |
Усередину за 1 год до процедури |
|
|
Діти до 12 років |
Кліндаміцин 20 мг/кг маси тіла, або Цефалексин 50 мг/кг, або Цефадроксил 50 мг/кг, або Азитроміцин 15 мг/кг, або Кларитроміцин 15 мг/кг |
|||
|
При алергії та неможливості застосування per os |
Дорослі |
Кліндаміцин 600 мг, або |
В/в за 30 хв до процедури |
|
|
Цефазолін 1,0 г |
В/в або в/м за 30 хв до процедури |
|||
|
Діти до 12 років |
Кліндаміцин 20 мг/кг, або |
В/в за 30 хв до процедури |
||
|
Цефазолін 25 мг/кг |
В/м або в/в за 30 хв до процедури |
|||
|
При маніпуляціях на шлунково-кишковому або урогенітальному тракті |
||||
|
Стандартна схема |
Дорослі |
Амоксицилін 2 г, або |
Усередину за 1 год до процедури |
|
|
Ампіцилін 2 г |
В/м або в/в за 30 хв до процедури |
|||
|
Діти до 12 років |
Амоксицилін 50 мг/кг, або |
Усередину за 1 год до процедури |
||
|
Ампіцилін 50 мг/кг |
В/м або в/в за 30 хв до процедури |
|||
|
При алергії на пеніцилін |
Дорослі |
Ванкоміцин 1 г |
В/в протягом 1–2 год, введення закінчити за 30 хв до процедури |
|
|
Діти до 12 років |
Ванкоміцин 20 мг/кг |
В/в протягом 1–2 год, введення закінчити за 30 хв до процедури |
||
Прогноз
ГРЛ характеризується високою частотою формування РВС, які є причиною застійної серцевої недостатності, стійкої втрати працездатності, передчасної смерті. Вірогідність розвитку РВС підвищується при пізно розпочатому лікуванні.
Глава 27. Кристалічні артропатії
Подагра
Подагра — хронічне системне захворювання, яке виникає в результаті порушення пуринового метаболізму, що призводить до підвищення рівня сечової кислоти в крові (гіперурикемія) і відкладання натрієвої солі сечової кислоти (урати) у тканинах опорно-рухового апарату та внутрішніх органах із розвитком вторинних реактивних запальних змін.
Код за МКХ-10
М-10 Подагра.
Історія вивчення подагри характеризується зміною періодів значного інтересу до даної проблеми та періодами досить тривалого забуття. Вперше клініка нападу подагри описана більше ніж 2500 років тому давньогрецьким лікарем Гіппократом. Як окрема нозологічна одиниця подагра виділена наприкінці XII ст. T. Sydengam, він же дав класичний клінічний опис подагри в книзі «Трактат про подагру». У XIII–XVIII ст. більшу частину захворювань суглобів лікарі розцінювали як подагру. Лише в другій половині XIX ст. англійський дослідник А.В. Garrod чітко відокремив РА від подагри й пов’язав її з гіперурикемією. На подагру страждали багато знаменитих людей: Олександр Македонський, Петро І, Ісаак Ньютон, Мікеланджело, Бенжамін Франклін, Вільям Пітт, Чарльз Дарвін. І це лише мала частина довгого списку імен! Захворюваність на подагру пояснювали подібністю хімічної структури сечової кислоти й такого стимулятора, як кофеїн.
Епідеміологія
У розвинених країнах подагру відмічають у 1–3% дорослого населення, а гіперурикемію виявляють у 4–20% населення. Число нових випадків подагри в рік становить 1–3 на 1000 чоловіків і 0,2 на 1000 жінок. В Україні поширеність захворювання становить 5–28 випадків на 1000 чоловіків і 1–6 — на 1000 жінок, а поширеність гіперурикемії — 15–20%. Питома вага подагри серед інших ревматичних захворювань коливається від 3 до 5%.
Найбільш поширена подагра в розвинених країнах з високим рівнем життя. Підвищення захворюваності на подагру в останні десятиліття пов’язують із поширеністю ожиріння, алкоголізму, малорухливим способом життя. Про це свідчить тісний взаємозв’язок між подагрою й метаболічним синдромом, що виявляють у 62,8% пацієнтів із подагрою, у той час як у популяції — у 25,4% осіб. Слід зазначити, що поширенню подагри сприяє те, що середній рівень сечової кислоти в організмі людини (в середньому у популяції) за останнє століття значно підвищився: з <0,21 ммоль/л (3,5 мг/дл) в 1920-х роках до 0,36–0,3 ммоль/л (6,0–6,5 мг/дл) — у 1970-х, і ця тенденція відмічається й у подальшому. Це також зумовлено поліпшенням в останні роки діагностики хвороби. Однак і сьогодні ряд авторів вважають відомості про поширеність подагри неповними. Так, за даними Інституту ревматології РАМН, діагноз подагри в перший рік захворювання встановлюють лише у 25% хворих, в інших 75% — у середньому на 7-й рік хвороби, а, за даними Peschel, діагноз подагри встановлюється в середньому лише через 4,8 року після першого нападу.
Частіше на подагру хворіють чоловіки. Співвідношення чоловіків і жінок, яких уражує ця патологія, становить 7:1. У чоловіків найбільш високий рівень захворюваності відмічають у віці 40–50 років, жінок — в 60 і більш років. У третини хворих подагру виявляють у найближчих родичів.
Етіологія, патогенез
Сечова кислота (2,6,8-триоксипурин) являє собою кінцевий продукт перетворення пуринів в організмі людини, що не метаболізується. Джерелом сечової кислоти є пуринові основи, що утворюються в результаті ендогенного біосинтезу при катаболізмі тканинних нуклеїнових кислот, а також ті, що надходять з їжею в складі клітинних нуклеопротеїнів, глутамату, глікоколу та інших сполук.
З організму людини сечова кислота виділяється в основному нирками. В капілярах клубочків вона повністю фільтрується, у звивистих канальцях першого порядку активно реабсорбується і у здорових людей виводиться у кількості близько 10% від профільтрованої. Але кількість сечової кислоти, що виводиться, може збільшуватися, коли концентрація її у плазмі крові підвищується. По-перше, її активна реабсорбція обмежується величиною її максимального транспорту, і в цих умовах частина речовини не може бути реабсорбована. По-друге, при підвищенні її концентрації у крові сечова кислота може потрапляти у просвіт канальця шляхом секреції, це відбувається у дистальних канальцях. Тобто, система активного транспорту працює, але напрямок руху сечової кислоти — у кров, а не з крові. Незначна кількість сечової кислоти виводиться з організму через шлунково-кишковий тракт і потові залози.
Загальна кількість сечової кислоти в організмі людини становить близько 1000 мг, з яких 650 мг щодоби поновляються. За добу із сечею виділяється 400–600 мг сечової кислоти. Нижня межа вмісту сечової кислоти в плазмі крові становить 0,18 ммоль/л. Рівень сечової кислоти в сироватці крові залежить від статі і віку людини: підвищується у чоловіків в пубертатний період, а у жінок — під час менопаузи, що пов’язано з впливом естрогенів у жінок репродуктивного віку на канальцеву екскрецію уратів. У чоловіків порівняно з жінками у плазмі крові міститься на 17–19% сечової кислоти більше, і період наявності гиперурикемії довший, ніж у жінок. Тому чоловіки хворіють на подагру частіше, а перший напад виникає у віці 40–50 років. Подагру рідко виявляють у чоловіків віком до 30 років, а у жінок — до настання менопаузи. Відповідно до рекомендацій Європейської ліги по боротьбі з ревматизмом зараз гіперурикемією слід вважати рівень сироваткової сечової кислоти, що перевищує 0,36 ммоль/л (6,0 мг/дл). Зміна цільового рівня пов’язана з тим, що перевищення його асоціюється з підвищенням ризику розвитку подагри у чоловіків в 4 рази, у жінок — в 17 разів.
У розвитку захворювання виділяють три основні послідовні фази:
1. Гіперурикемія і накопичення уратів.
2. Відкладання уратів у тканинах.
3. Гостре подагричне запалення.
Гіперурикемія може бути зумовлена одним з наступних механізмів: а) підвищеним синтезом сечової кислоти в організмі при нормальному виділенні нирками (метаболічний тип гіперурикемії); б) порушенням екскреції сечової кислоти нирками при нормальному синтезі (нирковий тип гіперурикемії); в) комбінацією обох механізмів (змішаний тип гіперурикемії).
Для того щоб встановити тип гіперурикемії, визначають концентрацію сечової кислоти й креатиніну в добовій порції сечі. Про підвищення утворення сечової кислоти (при звичайній дієті, що містить пуринові основи) свідчить виділення із сечею більше ніж 800 мг уратів на добу. Вміст у добовій сечі уратів менше ніж 800 мг відмічають при гіперурикемії ниркового походження.
Порушення обміну сечової кислоти може бути первинним і вторинним. Численні дослідження свідчать, що саме зменшення екскреції сечової кислоти виявляють у 90% пацієнтів із первинною подагрою. Причиною первинної гіперурикемії є генетичний дефект, у результаті якого в організмі збільшується вміст вільної сечової кислоти, зменшується — зв’язаної з α2— і γ-глобулінами. До гіперурикемії призводять вроджені дефекти ферментів, що беруть участь у метаболізмі уратів: підвищення активності фосфорибозилпірофосфатсинтетази (ФРПФ); частковий дефіцит гіпоксантингуанінфосфорибозилтрансферази (ГГФРТ) (синдром Келлі — Сигмеллера). Зазначені дефекти ферментних систем успадковуються як зчеплені з Х-хромосомою рецесивні ознаки. У таких хворих подагра виникає в більш ранньому віці (до 30 років), а також відзначається висока частота розвитку уратного нефролітіазу. Повна відсутність ГГФРТ призводить до виникнення синдрому Леша — Найхана, який характеризується затримкою психічного розвитку, прагненням до покалічення, хореоатетозом і зниженим порогом судомної активності. До цієї ж групи хвороб можна віднести недостатність глюкозо-6-фосфатази (хвороба Гірке), при якій збільшується утворення уратів у результаті підвищеного розщеплення аденозинтрифосфату (АТФ) у процесі розпаду глікогену, зумовленого гіпоглікемією. Для даного захворювання характерний молочно-кислий ацидоз, що внаслідок підвищення концентрації конкуруючих аніонів призводить до підвищення порогу секреції уратів у канальцях нирок.
Гіперурикемія може бути також наслідком надлишкового надходження екзогенних пуринів з їжею, споживання великої кількості насичених жирів у харчовому раціоні, ожиріння, надмірного фізичного навантаження, вродженої непереносимості фруктози й вживання продуктів, які містять її. У осіб, які зловживають алкоголем, збільшується інтенсивність розпаду АТФ, що сприяє утворенню уратів, підвищує вміст в організмі сечової кислоти, а при цьому виділення уратів нирками зменшується. Крім того, в деяких алкогольних напоях, зокрема в пиві, міститься велика кількість пуринів.
Недостатнє виведення уратів відмічають при захворюваннях нирок, що супроводжуються зниженням швидкості клубочкової фільтрації, діабетичному кето- і лактоацидозі, нецукровому діабеті, синдромі Бартера, зневодненні, при отруєнні свинцем (свинцева подагра); гіперпаратиреозі й гіпотиреозі, респіраторному ацидозі та інших станах.
Гіперурикемія і подагра можуть розвиватися при захворюваннях і станах, для яких характерний посилений катаболізм нуклеопротеїнів: саркоїдоз, псоріаз, мієло- і лімфопроліферативні захворювання, гемолітична анемія, поліцитемія, після рентгенотерапії, при швидкій втраті маси тіла (схудненні).
Гіперурикемія виникає при прийомі багатьох лікарських препаратів. До ліків, що блокують екскрецію сечової кислоти нирками, відносяться цитостатики, нікотинова кислота, етамбутол, ацетилсаліцилова кислота (низькі дози), піразинамід та інші. Тіазидні діуретики збільшують урикемію, потенціюють екскрецію уратів і підвищують імовірність розвитку тубулоінтерстиціального нефриту та тіазид-індукованої подагри; у петлевих діуретиків гіперурикемічна дія виражена меншою мірою, ніж у тіазидних. Застосування тіазидних діуретиків у терапевтичних дозах протягом року призводить до підвищення рівня сечової кислоти у 50% хворих. Тому у Консенсусі експертів ACCF/AHA 2011 з лікування артеріальної гіпертензії в осіб літнього віку передбачається необхідність моніторування рівня сечової кислоти та його зниження. Подагра відносно часто розвивається у хворих, які перенесли трансплантацію органів, що є результатом зменшення екскреції уратів із сечею при прийомі циклоспорину.
При подагрі обмінний фонд сечової кислоти збільшується до 2000–4000 мг, а оскільки сечова кислота і її солі поганорозчинні в рідинах організму, то при їх надлишку її солі — урати — вибірково відкладаються в тканинах суглобів, нирках, сечових шляхах, інших тканинах та індукують проліферативне гранулематозне запалення. Межа розчинності уратів, тобто рівень, вище якого утворюються кристали моноурату натрію, становить 0,41 ммоль/л (7,0 мг/дл), цей рівень раніше вважали верхньою межею нормального вмісту сечової кислоти в сироватці крові. Слід пам’ятати, що розчинність сечової кислоти в синовіальній рідині нижча, ніж у сироватці крові. Розчинність уратів знижується при рН менше ніж 7, гіпоальбумінемії, зниженні й підвищенні температури навколишнього середовища. Кристали уратів переважно відкладаються в безсудинних (хрящах), відносно слабо васкуляризованих (сухожилках, зв’язках) тканинах, дистальних периферичних суглобах та місцях, що зазнають найбільшого охолодження (вушна раковина) та тертя (ліктьові суглоби). При подагрі атаки артриту й тофуси виникають на периферії, що пояснюється тим, що в периферичних відділах тіла температура нижча, тому знижена й розчинність кристалів урату натрію.
Ризик розвитку подагри підвищується відповідно до збільшення сироваткового рівня сечової кислоти: при нормальному вмісті сечової кислоти ризик становить 5 на 1000, при рівні урикемії — 0,42–0,47 ммоль/л — 20 на 1000; 0,48–0,53 ммоль/л — 41 на 1000; 0,54–0,59 ммоль/л — 198 на 1000; більше 0,6 ммоль/л — 305 на 1000 осіб. Однак, не у всіх осіб з гіперурикемією, а лише, за даними різних авторів, у 50% розвивається клініка подагри. Цей факт свідчить про те, що в розвитку подагри патогенетичну роль відіграє не лише гіперурикемія, але й інші, ще невідомі фактори.
На сьогодні досить докладно вивчені патогенетичні механізми розвитку гострого та загострення хронічного подагричного артриту. Основне значення в цьому процесі відводиться кристалам урату натрію. У суглобі та періартикулярних тканинах утворюються преципітати кристалів урату натрію, які «покриваються» білковою оболонкою, що робить їх здатними ініціювати запальні процеси. Прозапальний потенціал кристалів уратів зумовлений їх взаємодією з моноцитами, макрофагами, синовіоцитами, остеобластами, нейтрофілами, що стимулює продукцію медіаторів запалення: цитокінів (ІЛ-1, -6, ФНП-α), хемокінів (ІЛ-8 та інших), метаболітів арахідонової кислоти, супероксидних кисневих радикалів, протеїназ та ін. Ці медіатори, кініни, компоненти комплементу індукують розвиток гострого подагричного артриту, а надходячи в кров, зумовлюють системні прояви, властиві загостренню подагри. Особливе місце в розвитку запалення при подагрі належить нейтрофілам, інфільтрація ними синовіальної оболонки суглоба розглядається як характерна ознака подагричного артриту. Нейтрофіли, що фагоцитують гострі голчаті кристали, гинуть, і це сприяє вивільненню лейкотрієнів, ІЛ-1, -8, активних кисневих радикалів, а також особливого кристалзалежного хемотаксичного фактора та великої кількості лізосомальних гідролаз. Макрофаги, які фагоцитують урати й клітинні уламки, утворюють цитокіни, ІЛ-1, -6, -8, кахексин та простагландини. Це посилює запалення та сприяє утворенню синовіоцитами колагеназ, які підтримують альтерацію.
Перші напади гострого подагричного артриту, як правило, купіруються швидко, спонтанно, без медикаментозного лікування. Важливе значення має вивчення механізмів мимовільного зворотного розвитку гострого подагричного артриту. Встановлено, що ця особливість подагричного запалення пов’язана з виробленням протизапальних механізмів, головним чином макрофагального та синовіоцитарного походження. Встановлено, що клітинна відповідь регулюється різними білками, що входять у білкову оболонку кристалів; фагоцитоз і руйнування кристалів нейтрофілами зменшує їх кількість; місцеве підвищення температури, що супроводжує процеси запалення, збільшує розчинність уратів; підвищення продукції адренокортикотропного гормону пригнічує процеси запалення; сприяє зворотному розвитку запалення й синтезу меланокортинів, що володіють потужною протизапальною активністю, викликаною у результаті активації системи гіпоталамус — гіпофіз — надниркова залоза при запаленні; на дію цитокінів (ІЛ-1 і ФНП-α), що потенціюють запальну реакцію, впливає як продукція речовин — інгібіторів цитокінів, так і цитокінів, що регулюють імунні процеси, кристали уратів індукують синтез ряду протизапальних медіаторів, що володіють властивістю пригнічувати мікрокристалічне запалення: рецепторні антагоністи ІЛ-1, -10, трансформуючий фактор росту-β, які швидко й селективно індукують експресію активованих рецепторів проліфератора пероксисом. У результаті цих процесів наступає повне спонтанне одужання.
Клініка
У розвитку клініки подагри виділяють три періоди: 1) безсимптомний; 2) поява нападів подагричного артриту та чергування нападів із періодами міжнападу; 3) наявні хронічний поліартрит, тофуси, ушкодження нирок та інших органів (табл. 27.1).
Таблиця 27.1
Клінічна класифікація подагри (АРУ, 2004)
|
I |
Клінічні стадії |
Гострий подагричний артрит |
|
Міжнападна (інтервальна) подагра |
||
|
Хронічний подагричний артрит: загострення, ремісія |
||
|
Хронічний тофусний артрит |
||
|
II |
Рентгенологічні стадії ураження суглобів |
I — великі кісти (тофуси) у субхондральній кістці та в більш глибоких шарах, іноді ущільнення м’яких тканин |
|
II — великі кісти поблизу суглоба та дрібні ерозії суглобових поверхонь, постійне ущільнення навколосуглобових м’яких тканин, іноді з кальцифікатами |
||
|
III — великі ерозії не менше ніж на ⅓ суглобової поверхні, остеоліз епіфіза, значне ущільнення м’яких тканин з кальцієвими депозитами |
||
|
III |
Ступінь функціональної недостатності |
0 (збережена) |
|
I (збережена професійна здатність) |
||
|
II (професійна здатність втрачена) |
||
|
III (втрачена здатність до самообслуговування) |
||
|
IV |
Нефролітіаз |
Подагрична нефропатія |
У преморбідний період у пацієнтів протягом декількох років, іноді десятків, відмічають безсимптомну персистуючу гіперурикемію. У 40% пацієнтів розвитку подагри передують симптоми нефролітіазу. Г.А. Захар’їн звертає увагу на характеристику осіб, у яких виникає подагра: «Це особи гарної статури, зайвого харчування й з гарним травленням, що добре їдять і тілесно недіяльні».
Гострий подагричний артрит
Першим клінічним проявом подагри у ⅔ хворих є напад гострого артриту, який має ряд типових особливостей, характерних лише для цього захворювання. Ці відмінності виявляють при опитуванні та фізикальному обстеженні хворого.
Частіше напад подагричного артриту виникає раптово, серед повного здоров’я, у нічний час або вранці. Зазвичай хворий лягає спати здоровим, а прокидається від сильного, пекучого, іноді пульсуючого болю у суглобі. Біль швидко наростає до нестерпного, підсилюється при найменшому русі, струсі ліжка, пальпації, характерний симптом «простирадла», це обумовлює повну нерухомість хворого й може нагадувати картину гострого гнійного запалення. У переважній більшості випадків це асиметричний моноартрит, для якого характерно швидка, буквально «на очах» — протягом декількох годин або доби, поява всіх ознак запалення: припухлості, яскравої гіперемії шкіри, сильного болю, підвищення місцевої температури тіла, порушення функції ураженого суглоба. Класичним проявом подагри є гострий артрит першого плеснофалангового суглоба стопи, який при першій подагричній атаці уражується у 50% випадків (надалі його ураження відмічають більше ніж у 90% хворих).
Важливою особливістю гострого подагричного артриту, що відрізняє його від артритів іншої етіології, є його швидкий повний зворотній розвиток і повне відновлення функції суглоба. Зазвичай перші атаки подагри тривають від 2–3 до 10–14 діб й закінчуються повним, часто спонтанним одужанням.
Провокувати напад подагри можуть вживання алкоголю, великої кількості продуктів, багатих пуриновими основами (м’ясо, риба, бульйон), травма, хірургічне втручання (на 5-й день післяопераційного періоду), фізичне навантаження, кровотечі, гострі патологічні стани в організмі, у тому числі інфекційні захворювання, переохолодження, прийом деяких ліків (сечогінні), променева терапія й інші. У багатьох хворих за кілька годин або навіть за 1–2 дні перед нападом відмічають продромальний період. Прояви його індивідуальні для кожного хворого й можуть характеризуватися підвищеною збудливістю або, навпаки, депресією, нервовістю, відчуттям страху, гіперсалівацією, болем в епігастрії, нудотою, печією, відрижкою, метеоризмом. Можливі підвищення артеріального тиску, поява головного болю, шуму у вухах, ознобу, парестезій, м’язових судом, симптомів ниркової коліки. Перед нападом можуть виникнути свербіж шкіри, кон’юнктивіт, озноб, лихоманка, безсоння.
У хворих з гострим подагричним артритом можуть виявляти системні прояви — озноб, лихоманка, іноді температура тіла підвищується до 38–39 °С. В аналізі крові відзначаються лейкоцитоз, прискорена ШОЕ.
Слід зазначити, що лише у 60% хворих на подагру перша гостра атака має типові клінічні прояви, у 40% артрит має особливості, пов’язані як з атиповою локалізацією, так і з атиповим характером перебігу. Виділяють наступні варіанти атипового дебюту подагри: ревматоїдоподібний, підгострий, що нагадує ревматичний або РеА, псевдофлегманозний, астенічний, позасуглобовий, періартритичний.
У цілому для подагри характерне ураження суглобів нижніх кінцівок, значно рідше до патологічного процесу залучаються суглоби верхніх кінцівок. У порядку зменшення за частототою ураження за першим плеснофаланговим йдуть такі суглоби: плесни, гомілковостопні, п’яткової кістки, колінні, променезап’ясткові, пальців кисті і ліктьові. Дуже рідко виникає артрит аксіальних суглобів, хребта.
Можливе виникнення гострого нападу подагри позасуглобової локалізації, так звана періартикулярна форма з локалізацією процесу в сухожиллях і бурсах (гостре запалення препателярної сумки, сумки ліктьового відростка, ахілова сухожилля) при інтактних суглобах. Біль та інші ознаки запалення в цих випадках частіше невиражені. Тривалість атипових нападів варіює від 3 днів до 1,5 міс.
Характер гострого подагричного артриту залежить від статі хворого. У більшості випадків у дебюті захворювання у чоловіків ушкоджуються суглоби стоп, частіше це моноартрит плеснофалангового суглоба першого пальця, а в жінок відмічають оліго- або поліартрит з більш частим ураженням суглобів кистей. Для осіб літнього віку типовий оліго- або поліартикулярний варіант початку подагри, ураження суглобів верхніх кінцівок (у тому числі суглобів кистей), швидкий розвиток тофусів.
Міжнападна (інтервальна) подагра. Для інтервальної подагри характерне чергування нападів гострого артриту з безсимптомними періодами. Якщо гіперурикемія триває, атаки артриту повторюються через 1–2 роки, з часом періоди між нападами артриту стають коротшими, а напади подагри — частішими і довшими. При повторних атаках швидкість зворотного розвитку артриту залежить від тривалості хвороби, гостроти початку, швидкості призначення й адекватності терапії.
Протягом першого року за відсутності лікування повторний напад артриту виникає у 62% пацієнтів, другого — 78%. У 4% хворих друга атака гострого подагричного артриту виникає через 5–10 років, а в 7% — може виникати більше ніж через 10 років. З кожним новим нападом у патологічний процес залучається все більше суглобів, тобто відбувається поступова генералізація суглобного синдрому, моноартрит трансформується в олігоартрит. Частіше в перші 5–6 років хвороби ураження суглобів має перебіг за типом гострого інтермітуючого олігоартриту з повністю зворотним розвитком усіх суглобових проявів і відновленням функції суглобів. Частіше до запалення залучаються суглоби нижніх кінцівок — плеснофалангові, гомілковостопні, колінні (не більше 4 суглобів), але при тяжкому перебігу можуть уражуватися всі суглоби нижніх і верхніх кінцівок, а також хребет та кульшові суглоби. У запальний процес залучаються сухожилля, зв’язки та синовіальні сумки, у тканинах з’являються поодинокі безболісні тофуси. До розвитку хронічного подагричного артриту клінічних симптомів між нападами немає. Біль, що виявляють у деяких хворих між нападами, частіше пов’язують із ОА. Поступово безсимптомні періоди зникають, і настає наступна стадія хвороби.
Хронічний подагричний артрит
Зрідка хронічний подагричний артрит виникає відразу після першого нападу подагри, частіше — після стадії інтермітуючого подагричного артриту.
Суглобовий синдром при хронічній подагрі характеризується поліартритом, відсутністю безсимптомних періодів, порушенням функції суглобів. Проміжок часу між першою атакою та розвитком хронічного артриту становить від декількох місяців до 10 і більше років. Відмінним маркером переходу артриту в хронічну форму є кількість уражених суглобів — 4 суглоби в перші 5 років, до 12 суглобів після 10 років хвороби, тобто процес із олігоартритичного трансформується в поліартритичний. Хворі скаржаться на постійний біль, обмеження обсягу рухів у суглобах. Відзначаються стійка припухлість, деформація суглобів, підвивихи, тугорухомість, укорочення пальців, анкілози і контрактури, може відмічатися внутрішньосуглобовий випіт. Деформація суглобів є наслідком деструкції хряща, руйнування епіфізарних кінців кісток, суглобових поверхонь та пов’язана з інфільтрацією уратами навколосуглобових тканин і розвитком вторинного ОА. Раніше та найчастіше виникає деструкція плеснофалангового суглоба I пальця та інших суглобів ступні, потім суглобів кистей, ліктьових, колінних суглобів. На тлі хронічного артриту можуть виникати періодичні малоінтенсивні тривалі (до 2–3 міс) загострення, з кожним загостренням у патологічний процес залучаються нові суглоби. Іноді може розвиватися «подагричний статус», для якого характерні майже безперервні, що виникають один за одним, протягом декількох місяців, інтенсивні напади артриту в одному або декількох суглобах на тлі постійного, помірно вираженого запалення. Інфільтрація суглобових тканин уратами супроводжується постійною запальною реакцією навколосуглобових тканин із розвитком хронічного тофусного артриту або уратної артропатії.
Хронічний тофусний артрит
При тривалому перебігу клінічна картина хвороби складається з 3 синдромів: хронічний подагричний артрит, синдром, зумовлений утворенням тофусів, та ураження внутрішніх органів. У стадії хронічного тофусного артриту у 70–80% пацієнтів можна виявити тофуси, що являють собою гранульоми зі скупченням у центрі кристалів урату натрію, оточених фіброзною оболонкою із включенням гігантських клітин та макрофагів. За відсутності лікування тофуси в середньому утворюються через 6–10 років після дебюту подагри, в той час як при високому ступені гіперурикемії (понад 0,09 г/л) вони можуть з’являтися через 2–3 роки. Ранню появу тофусів відмічають при деяких формах ювенільної подагри, хронічній нирковій недостатності, мієлопроліферативних захворюваннях, посттрансплантаційній (циклоспориновій) подагрі. У жінок літнього віку з нирковою недостатністю, що постійно приймають діуретики, тофуси можуть утворюватися ще до появи симптомів артриту.
Отже, тофуси є показником рівня урикемії і тривалості порушення пуринового обміну. Тофуси утворюються в будь-яких тканинах, внутрішніх органах, але найчастіше вони локалізуються в ділянці суглобів пальців кистей і ступнів, ліктьових, колінних суглобів, на вушних раковинах (рис. 27.1–27.4). Підшкірні тофуси щільні, чітко відмежовані від навколишніх тканин, підіймаються над поверхнею шкіри, розмір їх коливається від шпилькової голівки до невеликого яблука, вони безболісні. На вушній раковині вони мають вигляд горошин білого або жовтуватого кольору. Великі вузли знаходяться поблизу ліктьових, колінних суглобів, більш дрібні — на кистях, ступнях, а також у ділянці п’яткового сухожилля, сухожиль тилу кистей. Рідко тофуси виявляють на повіках, склерах, крилах носа. У жінок в постменопаузальний період вони можуть розташовуватися у вузликах Гебердена. Можлива виразка шкіри над тофусами з утворенням нориці, через яку виділяються солі сечової кислоти у вигляді білої пастоподібної або крихкої маси. Тофуси зазвичай не інфікуються, але загоюються дуже повільно. Приєднання вторинної інфекції ускладнює перебіг хвороби.
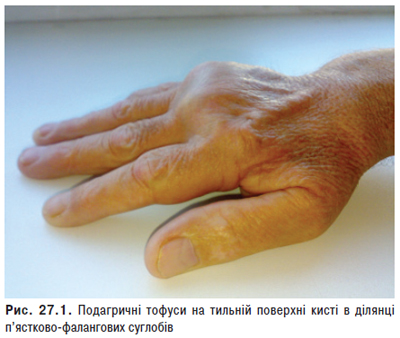
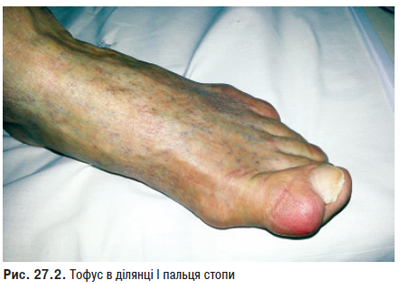
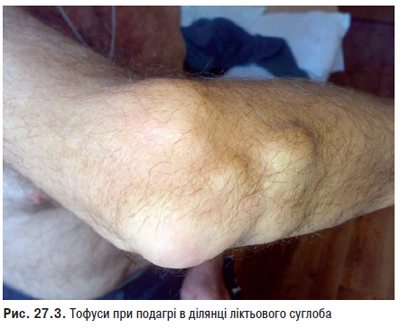
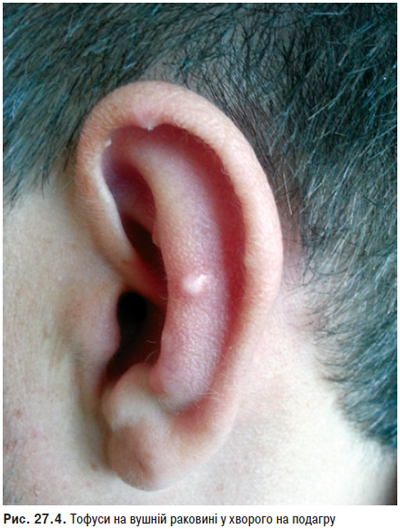
При подагрі до патологічного процесу можуть залучатися внутрішні органи. Частіше, ніж інші органи, уражуються нирки (у 25–93% хворих). Причинами подагричної нефропатії є відкладання кристалів сечової кислоти в тканині органа та судинах, а також надлишкове функціональне навантаження (необхідне посилення канальцевої екскреції), що призводить до ниркової декомпенсації. Поняття «подагрична нирка» включає хронічну та гостру уратну нефропатію, нефролітіаз.
Клінічні прояви хронічної подагричної (уратної) нефропатії зумовлені відкладенням кристалів урату натрію в мозковому шарі і пірамідках нирок та включають помірну й непостійну альбумінурію, у 20–40% пацієнтів — м’яку артеріальну гіпертензію, нефросклероз із тяжкою артеріальною гіпертензією, порушення функції нирок.
При різкому значному підвищенні концентрації сечової кислоти в крові відбувається масивна преципітація її кристалів у збірних трубочках нирок і сечоводах, що призводить до розвитку гострої ниркової недостатності. Це відмічають, наприклад, при синдромі розпаду пухлини.
Нефролітіаз виявляють у 10–25% хворих на подагру. Частота утворення каменів корелює із рівнем гиперурикемії, гиперурикозурії та кислотністю сечі. Камені, що утворюються у хворих на подагру, рентгенонегативні, складаються з уратів, оксалату і фосфату кальцію. Кристали сечової кислоти є основою для формування кальцієвих каменів.
Вісцеральні прояви подагри іноді можуть випереджати розвиток подагричного артриту. Це стосується й сечокам’яної хвороби. Подагрична нефропатія визначає прогноз перебігу хвороби: причиною смерті 20% хворих на подагру є хронічна ниркова недостатність.
Подагра супроводжується прискореним розвитком гломерулосклерозу, артеріальної гіпертензії й атеросклерозу. При тривалій гіперурикемії розвиток артеріальної гіпертензії зумовлений прегломерулярною артеріопатією та тубулоінтерстиціальним ураженням. Сечова кислота, маючи прооксидантні властивості, пригнічує синтез NO, що веде до розвитку та збільшення ендотеліальної дисфункції. На сьогодні гіперурикемія розглядається як незалежний вагомий фактор ризику серцево-судинної захворюваності й смертності, що модифікується. Подагра часто поєднується з атеросклерозом коронарних судин та ішемічної хвороби серця, які у хворих на подагру дебютують у молодшому віці, їх перебіг більш тяжкий. Доведена роль гіперурикемії як незалежного предиктора кардіоваскулярної захворюваності й смертності у пацієнтів з ішемічною хворобою серця, хронічною і гострою серцевою недостатністю, артеріальною гіпертензією, метаболічним синдромом. Характерна супутня патологія включає також дисліпідемію (частіше гіпертригліцеридемію), ожиріння, цукровий діабет.
Солі сечової кислоти виявляють в інтимі вінцевих артерій, у клапанному апараті серця (частіше в стулках мітрального клапана), міокарді й перикарді. Клінічно це проявляється мітральною недостатністю, АВ-блокадою різного ступеня, екстрасистолією, тахікардією, симптомами перикардиту, який добре купірується препаратами, що нормалізують рівень сечової кислоти в крові.
До подагричних вісцеропатій відносяться: ураження печінки внаслідок утворення подагричних гранульом (гепатопатія); алергічні прояви (кропив’янка, екзема, бронхоспастичний синдром), мігрень; ураження очей (ірити, іридоцикліти, кон’юнктивіти); ураження вен (флебіти); відкладання уратів у спинному мозку, міжхребцевих суглобах, що призводить до розвитку запалення та радикулярних уражень, виникнення люмбаго.
У жінок подагра виникає зазвичай після настання менопаузи, як правило, поєднується з ОА, артеріальною гіпертензією, хронічною нирковою недостатністю помірного ступеня, часто виникає на тлі прийому сечогінних препаратів. Частіше вже в дебюті хвороби уражуються кілька суглобів, відмічають оліго- та поліартрит. Характерне утворення тофусів в ушкоджених ОА суглобах, у тому числі у вузликах Гебердена, а також подушечках пальців.
Подагра може поєднуватися з іншими захворюваннями суглобів — ОА, РА, АС. Сама подагра супроводжується розвитком вторинного ОА.
В.М. Чепой (1990) виділяє легкий, середньої тяжкості і тяжкий варіант перебігу рецидивуючого подагричного артриту.
Легкий перебіг характеризується тим, що напади виникають рідко (1–2 рази на рік), уражуються 1–2 суглоби, активність запалення низька, навколосуглобові тофуси часто відсутні. Є незначний субхондральний остеосклероз. Концентрація сечової кислоти в крові в межах 0,47–0,53 ммоль/л.
При середньому ступені тяжкості подагричні напади виникають частіше (кожні 3–4 міс), наявні навколосуглобові тофуси, сечокам’яна хвороба. Рентгенологічно виявляють субхондральний остеосклероз, кісткові крайові узури, звуження суглобових щілин. Вміст сечової кислоти становить 0,53–0,59 ммоль/л.
При тяжкому перебігу виявляють тяжкі напади поліартриту, що супроводжуються високою температурою тіла, ознобом. Вони повторюються кожні 2–3 міс. Є безліч тофусів, на рентгенограмах — виражений субхондральний остеосклероз, великі кісти, крайові узури, остеоліз епіфізів, різке звуження суглобової щілини. Відзначають ураження нирок — сечокам’яну хворобу, нефрит, артеріальну гіпертензію, а також ішемічну хворобу серця. Рівень сечової кислоти перевищує 0,59 ммоль/л.
Лабораторне дослідження
Концентрацію сечової кислоти в сироватці крові необхідно визначати всім хворим на подагру, що важливо для уточнення терапевтичної тактики і контролю адекватності терапії. Однак слід пам’ятати, що під час гострого подагричного нападу підвищується екскреція сечової кислоти нирками, і урикемія майже у половини хворих є в межах норми, а іноді й нижче неї. Урикемію на початку лікування контролюють раз на 2–4 тиж, надалі кожні 6 міс.
У період гострого нападу в крові визначаються прискорена ШОЕ, помірний нейтрофільний лейкоцитоз, не виключений реактивний тромбоцитоз, підвищується вміст у крові СРБ, серомукоїду, α2— і γ-глобулінів. У період між нападами ці показники нормальні. Приблизно у 30% хворих виявляють у крові РФ у низьких титрах.
З біохімічних досліджень у сироватці крові слід визначати креатинін, швидкість клубочкової фільтрації, сечовину, печінкові ферменти, ліпідний спектр, у плазмі крові — глюкозу. Частіше при подагрі відмічають гіпертригліцеридемію, зниження рівня ліпопротеїнів високої щільності, збільшення рівня загального холестерину, холестерину ліпопротеїнів низької щільності. Для раннього виявлення ниркової патології систематично роблять загальний аналіз сечі, виявляють альбумінурію, визначають концентрацію креатиніну й сечової кислоти в добовій сечі. Урикозурія в нормі становить 600–800 мг/добу (1,8–3,6 ммоль/ добу), кліренс сечової кислоти — 9 мл/хв.
Дослідження синовіальної рідини
Під час гострого нападу подагри відзначають низьку в’язкість синовіальної рідини, поганий муциновий згусток, вміст лейкоцитів досягає 10–20•109/л з перевагою нейтрофілів.
Абсолютним діагностичним критерієм подагри є визначення в синовіальній рідині кристалів урату натрію за допомогою поляризаційної мікроскопії. Особливістю цих кристалів є їх голкоподібна форма, розміри 3–30 мкм, вони можуть розташовуватися поза- і внутрішньоклітинно й мають негативну подвійну променезаломлювальну властивість (зафарбовуються в жовтий колір при розташуванні паралельно осі червоного променя). Позаклітинна локалізація кристалів, як правило, відмічається в синовіальній рідині, яка взята з уражених суглобів між нападами.
У зв’язку з тим, що при бактеріальному артриті клінічні симптоми часто подібні до таких при подагрі, а в синовіальній рідині також можуть виявлятися кристали уратів, при найменшій підозрі на септичний артрит необхідно провести посів синовіальної рідини на флору.
Дослідження синовіальної оболонки
У біоптатах синовіальної оболонки, що взята під час нападу, виявляють гіперемію, набряк, клітинну інфільтрацію полінуклеарними нейтрофілами, часто з кристалами урату натрію, що фагоцитовані. При хронічному подагричному артриті виявляють ознаки проліферативного хронічного синовіту: проліферацію синовіальних ворсин, гіперваскуляризацію, периваскулярну лімфоцитарну та плазмоклітинну інфільтрацію, гігантські клітини, інкрустацію синовіальної оболонки уратами.
Дослідження вмісту тофусу
При морфологічному дослідженні вмісту тофусів у центрі виявляють кристали урату натрію, що має діагностичне значення, навколо них — зону запалення із проліферацією гістіоцитів, гігантських клітин й фібробластів. Зазвичай навколо тофуса є щільна фіброзна оболонка.
Інструментальне дослідження
До рентгенологічних ознак, типових для подагри, належать: набряк м’яких тканин, тофуси, ерозії суглобових поверхонь кісток. Причинами, що призводять до появи типових рентгенологічних ознак подагри, є накопичення в кістковій тканині кристалів сечової кислоти з розвитком запалення та некрозу. На ранніх стадіях хвороби в період гострого нападу на рентгенограмах визначається лише набряк м’яких тканин в ділянці ушкодженого суглоба. Тофуси, ерозії відносяться до типових, але пізніх ознак подагри (рис. 27.5).
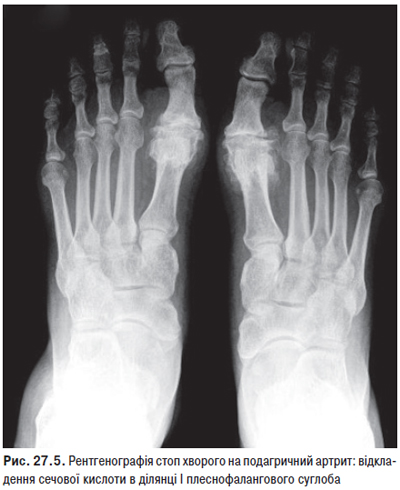
Тофуси на рентгенограмі виглядають як вогнища нерівномірного ущільнення м’яких тканин, в яких іноді відкладається кальцій. Великі кісткові тофуси розташовані субхондрально в епіфізах, вони оточені щільним валиком і мають округлу «штамповану» форму (симптом пробійника). Тофуси мають діагностичне значення і частіше локалізуються в ділянці першого плеснофалангового суглоба та дрібних суглобів кистей. Ерозії та кісти утворюються в результаті прориву тофусів назовні, частіше в порожнину суглоба. Кісти можуть утворюватися й у глибоких шарах кісткової тканини, в такому разі при руйнуванні коркової речовини кістки виявляють ерозії зі склеротичною облямівкою й нависаючими краями (симптом «здуття кісткового краю»); іноді ці ерозії називають «щурячі укуси». Остеоліз фаланг виникає за наявності множинних кіст в епіфізі фаланги й порушенні кровообігу. Результатом хронічного запалення є ущільнення м’яких навколо суглобових тканин. Як правило, крайові узури, звуження суглобової щілини не визначаються аж до пізніх стадій захворювання; також не відмічається розвиток навколосуглобового ОП, характерного для більшості артритів. Краойві остеофіти виявляють за наявності вторинного ОА.
Рентгенологічна стадія ураження суглобів при подагрі визначається відповідно до клінічної класифікації подагри (АРУ, 2004).
Спостереження свідчать, що І рентгенологічну стадію частіше виявляють через 5–9 років після першого нападу подагри, ІІ та ІІІ — через 10–15 років.
Діагноз
Класифікаційні критерії гострого подагричного артриту (Wallace S. et al., 1977)
А. Виявлення в суглобовій рідині характерних кристалів урату натрію.
Б. Наявність тофусів, вміст кристалів урату натрію в яких підтверджено за допомогою хімічного аналізу або поляризаційною мікроскопією, або наявність 6 з 12 перерахованих нижче ознак: 1) наявність в анамнезі більше ніж однієї атаки гострого артриту; 2) максимальний розвиток запалення протягом 1-ї доби; 3) моноартикулярний тип ураження; 4) почервоніння під час атаки шкіри над суглобом; 5) наявність болю і припухлості в I плеснофаланговому суглобі; 6) однобічні напади з ураженням I плеснофалангового суглоба; 7) однобічні напади з ураженням таранного суглоба; 8) виявлення вузликових утворень, схожих на тофуси; 9) гіперурикемія; 10) асиметрична припухлість суглоба, що виявляється при рентгенологічному дослідженні; 11) субкортикальні кісти без ерозій, що виявляються при рентгенологічному дослідженні; 12) негативні результати бактеріологічного дослідження суглобової рідини під час нападу артриту.
У ранній стадії подагри у 95,5% пацієнтів відмічають не менше 5 з перерахованих ознак захворювання.
Відповідно до рекомендацій експертного комітету Європейської антиревматичної ліги (2005) у кожному випадку діагноз подагри повинен бути підтверджений обов’язковою верифікацією кристалів урату натрію в синовіальній рідині або підшкірних тофусах за даними поляризаційної мікроскопії.
Якщо неможливо дослідити синовіальну рідину, дебют подагри можливо запідозрити за типовими клінічними проявами, які відрізняються високою чутливістю і специфічністю. Слід ретельно вивчити анамнез, визначити наявність провокуючих факторів (стрес, охолодження, травма, вживання алкоголю та ін.), здійснити пошук можливих тофусів (вушна раковина, суглоби), визначити рівень сечової кислоти в сироватці крові. Початок хвороби складно розпізнати у випадках, коли відсутні характерні напади з ураженням І пальця стопи й тофуси.
Диференційна діагностика
У групу захворювань, які можуть мати подібну з подагрою клінічну картину, входять: септичний артрит, пірофосфатна артропатія, інші кристалічні артрити, РеА, недиференційовані спондилоартропатії, ПсА, ОА, РА, гострий равматичний поліартрит, палідромний ревматизм, вузликова еритема.
Приклади формулювання діагнозу подагри: подагра, інтермітуюча стадія, гострий подагричний артрит, II атака з ураженням великого пальця лівої стопи, Rо стадія 0, функціональна недостатність суглобів II cт.
Подагра хронічна: поліартрит у фазі загострення з переважним ураженням суглобів правої стопи, колінних суглобів, Rо стадія II; множинні периферичні тофуси; функціональна недостатність суглобів II ст.
Лікування
Подагра належить до захворювань, які можна контролювати, запобігти рецидивам артриту та розвитку ускладнень. Але це можливо лише за умови співпраці лікаря та хворого, потребує комплексного та довготривалого лікування та є складною проблемою. Лікування подагри передбачає лікування гострого подагричного артриту, нормалізацію урикемії, запобігання рецидивам артриту, утворенню й росту тофусів, уратних конкрементів у нирках і ускладнень, лікування хронічного артриту, відновлення функції суглобів, а також лікування станів, асоційованих з подагрою (ожиріння, дисліпідемії, метаболічний синдром, артеріальна гіпертензія, серцева недостатність).
Етапи лікування подагри:
1) лікування гострого подагричного артриту;
2) патогенетична терапія — корекція гіперурикемії;
3) лікування хронічного подагричного артриту.
При лікуванні подагри використовують наступні методи: немедикаментозні та медикаментозні, сорбційні, хірургічні, санаторно-курортні.
Важливе значення має освіченість пацієнта та навчальні програми, які містять інформацію про подагру. Хворий повинен знати про наслідки гіперурикемії, фактори, які провокують напад артриту, основні клінічні ознаки хвороби, препарати, які призначаються, та інші лікувальні заходи. Бажання пацієнта дотримуватися здорового способу життя: відмова від куріння, зловживання алкоголем, зменшення маси тіла при ожирінні, раціональне та дієтичне харчування — мають вирішальне значення.
Дослідження останнього десятиліття продемонстрували, що навіть сувора малопуринова дієта зменшує вираженість урикемії лише на 0,06 ммоль/л, а добової урикозурії — не більше ніж на 200–400 мг, що є незначним для більшості хворих. Однак у пацієнтів з незначною гіперурикемією дотримання дієти може дати лікувальний ефект, а дієтичні похибки в деяких випадках можуть провокувати напад подагри, тобто, дотримання дієти необхідне всім хворим на подагру. У стаціонарі хворим призначається стіл № 6 за Певзнером.
Дієтичні рекомендації при подагрі традиційно полягають в обмеженні споживання м’ясних і рибних продуктів, напоїв, які містять цукор, фруктозу, фруктових соків, вживання алкогольних напоїв, пива. М’ясо та рибу слід вживати один раз на добу в кількості 100–150 г, при значній гиперурикемії — 2–3 рази на тиждень. Зменшують вживання насичених жирів, оскільки вони знижують екскрецію сечової кислоти нирками, збільшують частку полі- та мононенасичених жирних кислот. Хворим рекомендують, якщо немає протипоказань, випивати до 2–3 л рідини на добу. Є дані, що кава має гіпоурикемічну дію і знижує ризик розвитку подагри на 60%. Останні дослідження свідчать, що вино (сухе натуральне), чай дозволяється вживати при подагрі, реабілітовані також овочі, багаті на пурини, включаючи бобові. Встановлена протекторна дія вітаміну С, молочних продуктів, які містять мало жиру, а низькокалорійна дієта з малою кількістю вуглеводів знижує гіперурикемію на 15–20%. Умовою успішного лікування подагри при ожирінні є обов’язкове зниження маси тіла.
Лікування гострого нападу подагри
Мета цього етапу терапії — швидке та безпечне усунення болю і відновлення функції суглоба. Необхідно забезпечити повний спокій ураженої кінцівки та у першу добу місцево на ділянку ураженого суглоба проводити холодові процедури у вигляді аплікацій льоду, кріоаерозолю. Для лікування гострого нападу застосовують НПЗП, ГК та колхіцин.
НПЗП є препаратами першої лінії. Вважають, що відмінностей за ефективністю між НПЗП у разі призначення їх у перші 24–48 год після дебюту артриту немає. При гострому подагричному артриті зазвичай призначають повні терапевтичні дози неселективних НПЗП з коротким періодом напіввиведення. Частіше застосовують диклофенак 25–50 мг 4 рази на добу, німесулід 100 мг 2 рази на добу. Індометацин (25–50 мг 4 рази на добу) зараз практично не використовують через його токсичну дію. Після купірування нападу дозу знижують та припиняють прийом препарату через 2–3 дні. Специфічні інгібітори ЦОГ-2 ефективніші при тривалому перебігу гострого артриту або при хронічному поліартриті (наприклад еторикоксиб 120 мг 1 раз на добу курсом до 8 днів). Їх не рекомендується призначати пацієнтам з кардіоваскулярними факторами ризику та серцево-судинною патологією.
ГК призначають за наявності протипоказань до НПЗП або при їх недостатній ефективності. Застосовують ГК локально та системно. Внутрішньосуглобове введення ГК у перші 24 год від початку нападу ефективне у 90% хворих. При ураженні 1–2 великих суглобів або навколосуглобових сумок вводять всередину суглобів або сумок 40–80 мг метилпреднізолону або 40 мг тріамцинолону, або 3–6 мг бетаметазону; при ураженні дрібних суглобів — 20–40 мг метилпреднізолону або 5–20 мг тріамцинолону, або 1,5–3 мг бетаметазону. Якщо уражено багато суглобів, преднізолон призначають всередину в дозі 30–60 мг/добу одноразово з наступним зниженням дози протягом 7–10 днів. Слід пам’ятати, що прийом ГК per os може спровокувати розвиток РеА. У тяжких випадках вводять метилпреднізолон в/в у дозі 250–500 мг, у разі необхідності введення повторюють через 24 год. Можна застосовувати бетаметазон 7 мг в/м з повторним введенням у разі необхідності через 24 год.
Колхіцин (алкалоїд дочасника осіннього), що широко застосовувався в минулі роки, зараз використовують рідко. Це пов’язано з великою кількістю побічних ефектів при призначенні препарату у високих дозах. Так, нудоту, блювання й діарею на тлі прийому препарату відмічають у 80% хворих, тому його застосовують при протипоказаннях до призначення НПЗП та ГК. Колхіцин найбільш ефективний у перші 24 год від початку атаки. Існує два варіанти його застосування: 1-й варіант — по 0,5–0,6 мг усередину щогодини до купірування нападу або появи побічних ефектів, або до досягнення максимальної дози (6 мг, тобто 10 таблеток); 2-й варіант — в 1-й день по 1 мг 3 рази на добу після прийому їжі; 2-й день — по 1 мг вранці та ввечері, потім по 1 мг/добу. Низькі дози колхіцину (0,5–1,5 мг/добу) рекомендують призначати на початку антигіперурикемічної терапії для профілактики загострень артриту. Пацієнтам літнього віку за наявності тяжкого ураження органів травлення, а також серцевої, ниркової або печінкової недостатності препарат не призначають. Навіть у низькій дозі (0,6 мг 2 рази на добу) при нормальній функції нирок колхіцин може викликати мієлопатію та мієлосупресію. Особам віком ≥70 років дози знижують удвічі. Комбіноване застосування колхіцину і НПЗП не має переваг перед монотерапією.
У деяких країнах при гострому подагричному артриті рекомендують введення кортикотропіну.
Ведуться клінічні дослідження застосування для лікування гострого подагричного артриту імунобіологічних препаратів, що інактивують ІЛ-1 (канакінумаб — анти-ІЛ-1 моноклональні антитіла).
Антигіперурикемічна (патогенетична) терапія спрямована на зменшення вмісту сечової кислоти та її солей в організмі та попередження відкладання їх в тканинах і включає немедикаментозні та медикаментозні методи.
Зменшує урикемію: споживання їжі з низьким вмістом пуринів та дотримання інших дієтичних рекомендацій, нормалізація маси тіла, систематичні фізичні навантаження, сорбційні методи, тофектомія.
Медикаментозна терапія спрямована на нормалізацію сироваткового рівня сечової кислоти та потребує тривалого застосування ліків. Відповідно до рекомендацій EULAR, вона показана пацієнтам: 1) з повторними до 4 та більше разів на рік нападами гострого артриту; 2) з хронічним артритом; 3) за наявності тофусів.
Лікування безсимптомної гіперурикемії показане або при різкому підвищенні концентрації сечової кислоти в крові, наприклад при синдромі розпаду пухлини, або при гіперурикемії високого ступеня (концентрація сечової кислоти в сироватці крові >12 мг/дл або вміст сечової кислоти в зібраній за добу сечі >1100 мг). У 50% хворих з таким високим рівнем сечової кислоти в крові відмічають уратний нефролітіаз.
До препаратів, що знижують вміст сечової кислоти в крові, відносяться урикодепресанти та урикозуричні лікарські засоби. Ці препарати слід призначати тільки після повного завершення гострої подагричної атаки (через 2–3 тиж). Якщо до розвитку нападу хворий приймав ці ліки, режим прийому не змінюють та додатково призначають протизапальну терапію.
Антигіперурикемічну терапію вважають ефективною при досягненні цільового рівня урикемії, який не повинен перевищувати 0,36 ммоль/л (6 мг/дл), запобіганні рецидивам артриту, зменшенні кількості та зникненні тофусів, стабілізації уролітіазу.
Урикодепресанти. До препаратів цієї групи відноситься алопуринол, який інгібує фермент ксантиноксидазу, пригнічує перетворення гіпоксантину в ксантин і, таким чином, знижує синтез сечової кислоти. Він ефективний як при гіперпродукції сечової кислоти, так і при недостатній екскреції уратів нирками.
Абсолютними показаннями до застосування алопуринолу є: 1) часті напади гострого подагричного артриту (4 та більше нападів протягом року, що передує спостереженню); 2) наявність клінічних та рентгенологічних ознак хронічного подагричного артриту; 3) виявлення тофусів у м’яких тканинах та субхондральних кістках; 4) нефролітіаз; 5) розвиток у хворого на подагру ниркової недостатності; 6) рівень сечової кислоти в крові у чоловіків >0,78ммоль/л (13 мг/дл), у жінок — >0,6 ммоль/л (10 мг/дл); 7) екскреція із сечею більше ніж 1100 мг/добу сечової кислоти; 8) проведення цитотоксичної терапії або рентгенотерапії хворим з лімфопроліферативними пухлинами для профілактики уратного кризу.
У перші дні на тлі лікування алопуринолом в результаті мобілізації уратів із тканин є ймовірність розвитку гострої подагричної атаки. Щоб знизити цей ризик, а також ризик розвитку побічних реакцій, терапію алопуринолом починають із мінімальної дози препарату — 50–100 мг/добу. Потім кожні 2–4 тиж поступово підвищують дозу на 50–100 мг/добу до досягнення цільового рівня урикемії. Зазвичай під впливом алопуринолу рівень сечової кислоти знижується через 24–48 год, а нормалізується при виборі адекватної дози через 4–14 днів. Максимальна доза алопуринолу індивідуальна і зазвичай коливається від 100 до 900 мг/добу. Алопуринол у дозі 300 мг призначають в один прийом після їди, якщо доза вища, то його приймають у кілька прийомів. Необхідно пам’ятати, що через 3–4 дні після відміни алопуринолу вміст сечової кислоти в крові повертається до вихідного рівня, тому підтримувальну дозу препарату хворий повинен приймати постійно, протягом усього життя. Одночасно з алопуринолом для профілактики загострення артриту призначають низькі дози НПЗП або колхіцин усередину по 0,6 мг 1–2 рази на добу, відміняють їх через 2–3 тиж після досягнення цільового рівня урикемії. Ці ж засоби слід призначати й при відміні алопуринолу.
Протипоказаннями до призначення алопуринолу є вагітність, виразкова хвороба, ураження печінки. За наявності у хворого ниркової недостатності дозу препарату знижують відповідно до ступеня порушення функції нирок. Алопуринол в цих випадках рекомендують приймати в дозах 50–300 мг/добу залежно від кліренсу креатиніну, тому що синдром гіперчутливості має дозозалежний характер і відзначається у пацієнтів з нирковою недостатністю при дозах 200–400 мг/добу. При кліренсі креатиніну ≥90 мг/хв призначають алопуринол у дозі 300 мг/добу, >60 мг/хв — 200 мг/добу, ≥30 мг/хв — 100 мг/добу, ≤30 мг/хв — 50–100 мг/добу.
Алопуринол може викликати побічні реакції (в 2% випадках): алергічні у вигляді кропив’янки зі свербежем, макулопапульозних шкірних висипів, набряку Квінке, лихоманки, лейкопенії, ураження печінки. Тяжкий синдром гіперчутливості (відмічають рідко) залежить від дози препарату і проявляється тяжким дерматитом, васкулітом, поліорганним ураженням з летальним (у 20%) результатом. Необхідно дотримуватися обережності при одночасному прийомі антикоагулянтів (варфарин), а також азатіоприну, циклоспорину, такролімусу й інших препаратів. Одночасне застосування алопуринолу й перерахованих препаратів може призводити до негативних наслідків. При одночасному призначенні алопуринолу та азатіоприну відбувається потенціювання імуносупресивного й цитолітичного ефекту, виникає лейкопенія. Тому слід уникати їх одночасного призначення, а у разі необхідності одночасного прийому знизити дозу азатіоприну на ½. При комбінації алопуринолу з ампіциліном підвищується частота шкірних висипань, із циклофосфамідом — виникає супресія кісткового мозку.
У ситуаціях, коли алопуринол абсолютно показаний, а його прийом супроводжується розвитком шкірних реакцій, застосовують десенсибілізацію. Призначають мінімальну дозу препарату (10–25 мкг) у вигляді суспензії й поступово — кожні 3 дні — її підвищують до досягнення протягом 1 міс дози 100 мг вже у формі таблеток.
Фебуксостат (Febuxostat) (в Україні не зареєстрований) селективний інгібітор ксантиноксидази. Він більш енергійно, ніж алопуринол, знижує урикемію у пацієнтів похилого віку. Фебуксостат виводиться приблизно в рівній кількості кишечником та нирками, тому його можно призначати хворим із тяжкою нирковою недостатністю. Може спричиняти таку побічну дію: артралгії, міалгії, діарею.
Ферменти. Уриказа окиснює сечову кислоту до алантоїну, що є розчинним, та інших складових. Уриказа значно знижує урикемію та сприяє зменшенню тофусів. У США рекомбінантна немодифікована уриказа схвалена Управлінням з контролю за харчовими продуктами та лікарськими засобами США (Food and Drug Administration — FDA) для попередження гіперурикемії, що зумовлена синдромом лізису пухлини. Але ця форма урикази надзвичайно імуногенна й може викликати анафілаксію та сприяє утворенню уриказонейтралізуючих антитіл.
Урикозуричні препарати можна застосовувати в якості препаратів першого ряду у пацієнтів з первинною (ідіопатичною) формою подагри зі значним зменшенням виділення уратів нирками. Ці препарати збільшують екскрецію сечової кислоти нирками за рахунок пригнічення органічного аніонообмінника URAT1 та зниження реабсорбції уратів у проксимальних канальцях. Вони неефективні, якщо клубочкова фільтрація >60 мл/хв, протипоказані, якщо добова екскреція сечової кислоти в сечі перевищує 800 мг і відмічено нефролітіаз.
Пробенецид призначають у дозі 500 мг 2 рази на добу. Надалі дозу підвищують до 2000 мг/добу у 2 прийоми. Розвиток реакцій підвищеної чутливості нехарактерний. Є ризик розвитку кровотечі у пацієнтів, яким проводиться антикоагулянтна терапія.
Сульфінпіразон призначають у добовій дозі 400–600 мг у 4–6 прийомів. Курс лікування 7–10 днів. Може викликати погіршення перебігу гострої подагри, уролітіазу, порушення функції нирок. Препарат пригнічує агрегацію тромбоцитів, може підвищити ризик кровотечі при прийомі антикоагулянтів (особливо варфарину). Розвиток реакцій підвищеної чутливості нехарактерний. Можливі пригнічення кістковомозкового кровотворення, розвиток медикаментозної виразки.
Бензбромарон призначають в дозі 50–100 мг 1 раз на добу.
При прийомі урикозуричних препаратів для зниження ризику розвитку уратної нефропатії й утворення каменів рекомендується пити не менше ніж 2 л рідини на добу, контролювати рН сечі та приймати цитрати, які змінюють рН сечі на лужну.
Лозартан (антагоніст рецепторів ангіотензину II) виявляє урикозуричний ефект, але він менш значний, ніж у вищеперерахованих лікарських засобів. Вважають за доцільне його призначення пацієнтам з незначною гіперурикемією, а також хворим з коморбідними станами: артеріальна гіпертензія, метаболічний синдром. Особливо лозартан показаний при порушенні пуринового обміну, індукованого прийомом тіазидних діуретиків. У якості препаратів, що коригують дисліпідемію, призначають фенофібрат й аторвастатин, які виявляють також урикозуричний ефект.
Основна мета лікування тофусної подагри — значне зниження концентрації сечової кислоти в сироватці крові, що створює умови для резорбції уратів з тофусів. Позитивний результат у більшості хворих досягається при тривалому лікуванні алопуринолом через 6–12 міс постійної терапії: рідшають напади артриту, знижується їх інтенсивність, розм’якшуються та розсмоктуються тофуси. Концентрацію сечової кислоти в сироватці крові необхідно підтримувати як мінімум на 1,0 мг/дл нижче верхньої межі норми (6,0 мг/дл). Профілактичний прийом колхіцину в таблетках або НПЗП знижує частоту розвитку подагричних атак у таких хворих. Усунення тофусів зменшує загальну кількість уратів в організмі, тому хірургічне видалення їх також має значення.
У хворих, що перенесли трансплантацію органів, часто виникає подагра. Причиною її розвитку є прийом пацієнтами циклоспорину, який зменшує екскрецію уратів із сечею. Існують труднощі у лікуванні гострого нападу подагри й нормалізації концентрації сечової кислоти у сироватці крові хворих, що перенесли трансплантацію органів. Це зумовлено прийомом циклоспорину й наявністю ниркової недостатності, які є відносним протипоказанням до призначення НПЗП. Найбільш безпечним методом купірування гострих подагричних атак є призначення кортикостероїдів, у тому числі внутрішньосуглобове введення. Комбінація колхіцину з азатіоприном може викликати розвиток тяжкої невропатії. Хворому можна призначити алопуринол у дозі, що відповідає ступеню порушення функції нирок, якщо він не приймає азатіоприн, оскільки ця комбінація препаратів залишається небезпечною в плані розвитку тяжкої лейкопенії й апластичної анемії, навіть якщо дозу азатіоприну на період прийому алопуринолу знизити на 50–75%. Урикозуричні препарати часто неефективні у таких пацієнтів внаслідок низької клубочкової фільтрації (<50 мл/хв).
З інших методів лікування при подагрі використовують системну ензимотерапію відповідно до загальноприйнятих схем лікування; сорбційні методи лікування (проводят 4–5 сеансів плазмаферезу з інтервалом у 2– 3 дні); тофектомію.
Фізіотерапевтичні процедури протипоказані під час гострого нападу. При зменшенні вираженості клінічних проявів обережно призначають ультрафіолетове опромінення суглоба, УВЧ, калій-літієвий электрофорез, аплікації диметилсульфоксиду. В період ремісії показані: ультразвук на суглоби, фонофорез з гідрокортизоном; теплолікування — грязі, парафін, озокерит; водолікування — радонові, сірководневі, йодобромні хлоридно-натрієві ванни.
Санаторно-курортне лікування проводиться у Трускавці, Сочі, П’ятигорську, Цхалтубо, Карлових Варах, Маріанске Лазне та ін.
Прогноз. Ураження суглобів при подагрі не впливає на тривалість життя, але суттєво впливає на її якість. Поганий прогноз зумовлений ураженням нирок і розвитком хронічної ниркової недостатності, яка є причиною смерті 25–41% хворих на подагру. Серед інших причин смерті можна відзначити кардіальні й мозкові судинні катастрофи, що зумовлені атеросклеротичним ураженням.
До критеріїв ефективності лікування хворих подагрою відносяться: зменшення концентрації в крові сечової кислоти й частоти подагричних атак; зниження частоти прийому й доз НПЗП, колхіцину, ГК; розсмоктування тофусів; відсутність прогресування нефролітіазу.
Хвороба відкладення кристалів пірофосфату кальцію дигідрату
Хвороба відкладення кристалів пірофосфату кальцію дигідрату (синонім — пірофосфатна артропатія (ПА)) — захворювання, яке зумовлене утворенням та відкладенням в сполучній тканині кристалів пірофосфату кальцію дигідрату.
Код за МКХ-10
М11 Інші кристалічні артропатії.
М11.2 Інший хондрокальциноз.
М11.8 Інші уточнені кристалічні артропатії.
Епідеміологія
В основному уражуються особи похилого віку, з однаковою частотою чоловіки та жінки. Відкладення кристалів пірофосфату кальцію за рентгенологічними даними у людей віком 65–74 роки відмічають у 15% випадків, серед 75–84-річних — у 36%, старше 84 років — у 50% (Барскова В.Г., Кудаева Ф.М., 2010).
Профілактика
Специфічні заходи профілактики не застосовуються.
Класифікація пірофосфатної артропатії
Загальноприйнятої класифікації не існує. Виділяють три основні форми ПА: генетичну (сімейну), вторинну і первинну (ідіопатичну, спорадичну) (Бунчук Н.В., 1997) (табл. 27.2). Описані також форми хвороби, зумовлені травмою або хірургічним втручанням.
Таблиця 27.2
Основні форми пірофосфатної артропатії
|
Форми |
Характеристика |
|
Сімейна (генетична) |
Для неї характерна рання (у віці 30–40 років) поява симптомів хвороби, їх значна вираженість, генералізоване ураження суглобів. Захворювання успадковується за аутосомно-домінантним типом і уражує в основному чоловіків |
|
Вторинна |
Виникає на тлі спадкових і придбаних метаболічних захворювань: гіперпаратиреоз, сімейна гіпокальціурична гіперкальціємія, гемохроматоз, гемосидероз, гіпофосфатазія, хвороба Коновалова — Вільсона, гіпомагнезіємія. Вважають, що для утворення кристалів пірофосфату кальцію має значення підвищення рівня заліза, міді, кальцію, зниження — магнію. Кристали пірофосфату кальцію виявляють у синовіальній рідині у пацієнтів із хворобою Педжета, акромегалією, охронозом, ОА, подагрою, гіпотиреозом, амілоїдозом, гіпермобільністю суглобів, при травмах і ушкодженнях суглобів |
|
Ідіопатична |
У більшості випадків причина розвитку ПА залишається невідомою |
Етіологія і патогенез захворювання
Передбачається, що для формування зон кристалізації необхідні метаболічні порушення в хрящовій тканині і структурні зміни компонентів матриксу. Зокрема, для появи кристалів пірофосфату в хрящах (гіалінових, фіброзних) і у волокнистій сполучній тканині зв’язок та сухожиль необхідне локальне підвищення концентрації неорганічного пірофосфату кальцію, а також первинні зміни матриксу. До накопичення неорганічного пірофосфату в суглобовому хрящі може призвести порушення активності ряду ферментів (зокрема підвищення активності пірофосфогідролаз), зниження активності лужної фосфатази (ЛФ) і неорганічної пірофосфатази.
З іншого боку, зменшується вміст таких структурних компонентів, як протеоглікани і колаген, фрагментація колагенових фібрил, аномалії в розщепленні гексоз. Відносно збільшується вміст кератансульфату, хондроїтин-6-сульфату, зменшується — хондроїтин-4-сульфату. У міжклітинній речовині середньої зони хряща біля колагенових волокон з’являються перші відкладення кристалів. У сухожиллях, зв’язках, синовіальній мембрані кристали утворюються біля зон фіброзно-хрящової метаплазії. Переважний розвиток пірофосфатної артропатії в другій половині життя, можливо, пов’язаний із розвитком подібних дистрофічних змін сполучної тканини.
Запалення в суглобі починається, коли кристали проникають у порожнину суглоба, синовіальну рідину (проявів немає доти, поки кристали знаходяться в хрящовій тканині). Наслідком запалення, що виникає, є ушкодження хряща суглоба і подальший вихід кристалів у суглобну порожнину (так звана гіпотеза петлі, яка сама затягується).
Патоморфологічна картина пірофосфатної артропатії
Особливості патоморфологічної картини ПА наведені в табл. 27.3.
Таблиця 27.3
Особливості патоморфологічної картини при ПА
|
Кальцифікація у фіброзно-хрящових структурах — менісках колінних суглобів, капсулі, гіаліновому хрящі, сухожиллях і зв’язках (у тому числі хрестоподібній зв’язці колінного суглоба) |
|
Агрегати кристалів пірофосфату кальцію в гіаліновому хрящі розташовуються в середній і поверхневій зонах, у фіброзному — безсистемно |
|
Депозити кристалів покриті капсулою із колагенових волокон |
|
Структура матриксу, що оточує кристали, може бути нормальною |
|
Найбільш значні відкладення кристалів відмічають в ділянках дегенерації суглобового хряща |
|
Матрикс, що оточує агрегати кристалів, збіднений протеогліканами |
|
У ділянці депозитів кристалів відзначають ознаки хондроїдної метаплазії |
|
У синовіальній оболонці кристали розташовуються у поверхневому шарі |
|
При гострому запаленні відмічають набряк, інфільтрацію нейтрофілами, при хронічному — мононуклеари, проліферацію фібробластів |
Особливості клінічної картини пірофосфатної артропатії
Перебіг захворювання тривалий час може бути прихованим. Нерідко безсимптомний перебіг можливий у осіб літнього віку. Клінічні прояви, що розвиваються, зумовлені запаленням (мікрокристалічним) синовіальної оболонки та дегенеративно-дистрофічними змінами тканин суглобів. Найчастіше клініка захворювання може нагадувати ОА або подагру. У ряді випадків виявляють хронічне запалення суглобів.
Варіанти перебігу пірофосфатної артропатії
Виділяють псевдоартроз, псевдоподагру, псевдоревматоїдний артрит, псевдоневропатичну артропатію, псевдоанкілозуючий спондиліт та інші варіанти (табл. 27.4). Слід зазначити, що чіткий поділ на окремі варіанти можливий не завжди.
Таблиця 27.4
Деякі клінічні варіанти перебігу ПА
|
Клінічні варіанти ПА |
Характеристика |
|
Псевдоостеоартроз |
Відзначають біль у суглобах при рухах, дефігурацію суглобів, обмеження рухів. Ознаки запалення (з випотом у порожнину суглобів) приєднуються часто або присутні постійно. Зазвичай вони мало виражені. Під час нападів можливе незначне запалення багатьох суглобів. Уражуються 2–4 суглоби, насамперед — колінні, променезап’ясткові, кульшові. Рідше уражуються гомілковостопні, ліктьові, п’ястково-фалангові (II і III пальців) суглоби. При рентгенологічному дослідженні виявляють ознаки, характерні для ОА, а також хондрокальциноз менісків, гіалінового хряща, кальциноз капсули, синовіальної мембрани. Варто враховувати, що запалення суглобових тканин більш виражене при хондрокальцинозі в порівнянні з ОА. На пізніх стадіях хвороби уражуються сухожилля і періартикулярні тканини |
|
Псевдоподагра |
Для цього варіанта характерний нападоподібний розвиток запалення суглобів (як при подагричному артриті). Зазвичай уражується один або кілька суглобів (на 1-му місці — колінні суглоби). Можуть відзначатися підвищення температури тіла, лейкоцитоз, збільшення ШОЕ. Напад гострого артриту триває 1–2 тиж і повністю купірується. Частота рецидивів нападів становить від 1–2 на рік до декількох протягом місяця. При повторних нападах завжди, крім інших, уражується суглоб, з якого починалося захворювання. Найбільш характерним є інтермітуючий перебіг з періодами загострення і зменшення вираженості симптомів. Прогресування з тяжким ураженням суглобів відзначають відносно рідко. Поступово збільшується вираженість рентгенологічних змін, хондрокальциноз стає більш вираженим і розповсюдженим. Може відзначатися кальциноз м’яких тканин, синовіальних сумок і сухожиль. На противагу подагрі переважно уражуються колінні суглоби |
|
Псевдоревматоїдний артрит |
Можливе стійке запалення декількох (іноді багатьох) суглобів кистей і стоп. При цьому ерозії кісток, РФ у крові і деформація суглобів відсутні |
|
Псевдоанкілозуючий спондиліт |
Іноді розвиваються кальциноз міжхребцевих дисків, дегенеративно-дистрофічні зміни хребта, відзначають біль й обмеження рухів у різних відділах. Клінічна картина нагадує АС, але сакроілеїт і синдесмофіти відсутні |
|
Псевдоневропатична артропатія |
У деяких випадках розвивається деструкція одного або декількох (рідше) великих суглобів. Зміни подібні до так званих невропатичних артропатій (сирингомієлія, спинна сухотка) |
Діагностика пірофосфатної артропатії
При підозрі на ПА рекомендується досліджувати рентгенологічно певні анатомічні ділянки. Так, варто враховувати, що в типових випадках кальцифікації піддаються меніски колінних, променезап’ясткових суглобів (трикутний хрящ між дистальним відділом ліктьової кісти і першим рядом кісток зап’ястка), лобковий симфіз. На рентгенограмах виявляються вузькі лінійні тіні, які слідують за контурами суглобних відділів кісток на невеликій відстані від них (прояв кальцинозу гіалінового суглобового хряща). Кальциноз гіалінового хряща може бути розповсюдженим. Найчастіше зміни виявляють у кульшових і колінних суглобах. Визначається локальне звапнення капсули, синовіальної мембрани, хрящових «губ» суглобів, фіброзних ділянок міжхребцевих дисків. Кальциноз виявляють також у суглобах, з боку яких клінічні прояви не відмічено.
Характер дегенеративних змін, що виявляються при рентгенологічному дослідженні. Ці зміни типові для ОА і включають звуження суглобної щілини, остеофіти, склероз і кісти в субхондральних відділах кісток. Їх виявляють у суглобах, ураження яких нехарактерне для ОА — дрібних суглобах кистей і стоп, променезап’ясткових, ліктьових, гомілковостопних. Можливе виявлення кальцифікації сухожиль м’язів (литкового, триголового, замикаючого), кортикальних ерозій стегнової кісти, а також деструкції суглобів, що нагадує ішемічний некроз або невропатичну артропатію. Часто виявляють субхондральні кісти в дуговідросткових та крижово-клубових суглобах, кальцифікацію міжхребцевих дисків.
Відмінності рентгенологічних ознак хондрокальцинозу від ознак, характерних для ОА. Додатково до ознак, характерних для ОА (субхондральний остеосклероз, крайові остеофіти, звуження суглобної щілини), відмічають відкладення кальцію в поверхневих відділах хрящової тканини. Ці відкладення мають різний характер — у вигляді зерен, смуг, гомогенних скупчень. Вапно відкладається і по краю міжхребцевих дисків. У тих випадках, коли хвороба починається в дитячому віці, розвиваються анкілоз хребта і множинні ураження суглобів. У тяжких випадках виявляють остеоліз.
Методи, які використовують для ідентифікації кристалів пірофосфату кальцію. Найчастіше застосовують поляризаційну мікроскопію. Необхідно враховувати, що кристали пірофосфату кальцію можуть бути виявлені у деяких пацієнтів з подагрою, ОА, іншими захворюваннями суглобів. Тому в процесі діагностики варто враховувати етіологію захворювання, можливий зв’язок з ендокринними і метаболічними порушеннями (гіперпаратиреоз, гемохроматоз, хвороба Вільсона та ін.), провести ряд біохімічних досліджень (визначити вміст у крові кальцію, заліза, магнію, міді, ЛФ).
Лабораторні зміни, що виявляють при ПА. У період нападів виявляють нейтрофільний лейкоцитоз, збільшення ШОЕ, вмісту СРБ, α2— і γ-глобулінів, серомукоїду.
Характерні зміни в синовіальній рідині. У ній виявляють велику кількість мікрокристалів пірофосфату кальцію, що знаходиться як у вільному стані, так і всередині фагоцитів. Характерним є виражений цитоз (5000–40 000 у мкл) за рахунок полінуклеарів.
Гістологічні зміни, що відмічають у синовіальній оболонці. У біоптаті виявляють відкладення кристалів пірофосфату кальцію, заліза й ознаки реактивного запалення.
Діагностичні критерії ПА. Діагностичні критерії хвороби відкладення кристалів пірофосфату кальцію дигідрату (McCarty D., 1972) включають:
1. Виявлення в синовіальній рідині, біоптатах тканин, на секції кристалів пірофосфату кальцію дигідрату з верифікацією їх структури.
2. Виявлення за допомогою методу поляризаційної мікроскопії оптичних властивостей, характерних для кристалів пірофосфату кальцію дигідрату.
3. Наявність на рентгенограмах типових ознак хондрокальцинозу.
4. Гострий артрит з ураженням великих суглобів (особливо колінних).
5. Хронічний артрит, що супроводжується гострими атаками, з ураженням колінних, кульшових, променезап’ясткових, ліктьових, плечових або п’ястково-фалангових суглобів.
Діагноз вважається достовірним за наявності першого критерію або поєднання другого і третього. Він визнається ймовірним у випадках, коли виявляють тільки кристали пірофосфату кальцію дигідрату або хондрокальциноз на рентгенограмах. Діагноз є можливим за наявності четвертого або п’ятого критерію (тобто характерних клінічних проявів).
Діагностичні ознаки хондрокальцинозу. При проведенні диференційної діагностики необхідно враховувати наступні ознаки:
- наявність хондрокальцинозу у родичів;
- гострі напади артриту з ураженням колінного або іншого великого суглоба;
- нормальна концентрація сечової кислоти в крові;
- виявлення кристалів пірофосфату кальцію в синовіальній рідині та біоптатах синовіальної оболонки;
- наявність відкладення кальцію в ділянці суглобової капсули при рентгенологічному дослідженні.
Диференціація ПА і подагри. Основні відмінності цих захворювань наведені в табл. 27.5.
Таблиця 27.5
Основні відмінності подагри та ПА
|
Ознаки |
Подагра |
ПА |
|
Стать |
Переважно (95%) хворіють чоловіка |
Однаково часто хворіють чоловіки і жінки |
|
Фактори, що провокують напади |
Надлишкове вживання їжі, алкоголю, фізична травма |
Травма |
|
Характерне ураження суглобів |
Плесно-фаланговий суглоб I пальця |
Великі суглоби (у першу чергу колінні) |
|
Періартикулярні зміни |
Тофуси |
Фіброзні зміни |
|
Рентгенологічні ознаки |
Симптом «пробійника» (великі кісти в епіфізах) |
Субхондральні кісти невеликого розміру |
|
Зміни в біоптатах синовіальної оболонки |
Виявляють кристали сечової кислоти |
Виявляють кристали пірофосфату кальцію |
|
Дослідження синовіальної рідини |
Наявні кристали сечової кислоти |
Наявні кристали пірофосфату кальцію |
|
Реакція на лікування |
Купірує напад колхіцин |
Напади купірують НПЗП |
Через асоціацію ПА з гемохроматозом, гіперпаратиреозом, гіпотиреозом, подагрою, гіпофосфатазією, гіпомагнеземією, сімейною гіпокальціуричною гіперкальціємією, акромегалією, охронозом хворим із вперше виявленими кристалами пірофосфату кальцію необхідно визначити рівень в крові кальцію, фосфору, магнію, заліза, феритину, церулоплазміну, ЛФ, гормонів щитоподібної залози.
Лікування пірофосфатної артропатії
Патогенетична терапія відсутня. У період загострення призначають НПЗП, колхіцин, внутрішньосуглобове або в/в введення ГК (при стійких ексудативних явищах). Постійний прийом колхіцину в дозі 0,5–0,6 мг 1–3 рази на добу призначають пацієнтам з частими повторними нападами псевдоподагри. За наявності ознак псевдоартрозу великих суглобів застосовують ті ж методи лікування, що і при ОА. Функціональний стан суглобів поліпшують фізіотерапевтичні методи лікування — фонофорез гідрокортизону, ультразвук, діатермія, індуктотермія та ін. Рекомендують масаж регіональних м’язів, парафінові та грязьові аплікації, сірководневі, радонові ванни.
Специфічних методів лікування не існує. Безсимптомний варіант хвороби терапії не потребує. Лікування таких супутніх захворювань, як гіпотиреоз, гіперпаратиреоз, гемохроматоз не супроводжується розсмоктуванням кристалів пірофосфату кальцію (іноді можливе зменшення кількості нападів). При значних дегенеративних змінах в суглобах можливе ендопротезування.
Прогноз
Він є відносно сприятливим. При 5-річному спостереженні пацієнтів із ПА негативна динаміка відмічена тільки у 27% хворих (Барскова В.Г., Кудаева Ф.М., 2010), у 11% з них проведене хірургічне втручання.
Глава 28. Остеоартроз
ОА* — гетерогенна група захворювань різної етіології зі схожими біологічними, морфологічними, клінічними проявами і наслідком, в основі яких лежить ураження всіх компонентів суглоба, в першу чергу хряща, а також субхондральної кістки, синовіальної оболонки, зв’язок, капсули, оточуючих суглоб м’язів.
*У більшості посилань зарубіжних авторів використовується термін «остеоартрит» (osteoartritis), що підкреслює роль запального компонента в патогенезі хвороби.
Код за МКХ-10
М.15–М.19 Артроз.
Епідеміологія
ОА — найчастіше захворювання суглобів в багатьох популяціях, яке зазвичай маніфестує у осіб віком старше 40 років. Велике епідеміологічне дослідження в США виявило, що рентгенологічні ознаки ОА принаймні однієї суглобової групи, наявні у ⅓ дорослих віком 25–74 років, причому поширеність ОА має тенденцію до збільшення з віком. Так, серед осіб віком старше 65 років хворобу діагностували у 50%, а віком старше 75 років — у 80% випадків відповідно. Клінічний ОА, при якому наявна головна ознака захворювання — біль в суглобах протягом більшості днів попереднього місяця, діагностовано у 12% пацієнтів, включених у дане дослідження. За оцінками епідеміологів, діагноз «ОА» встановлений лікарями у більше 20 млн дорослих в США. Показники поширеності маніфестного ОА в Шотландії зростали від 5% у віковій групі 40–50 років до 25% серед осіб віком до 70 років. У Швеції поширеність клінічного ОА колінних, кульшових суглобів і суглобів кистей у віковій групі 50–79 років становила 5,8%.
За даними епідеміологічного дослідження, проведеного в 1980-ті роки в СРСР, поширеність ОА (встановленого і вірогідного) серед міських жителів віком 15 років і старше становила 6,43%. Найбільш частою локалізацією ураження периферичних суглобів були кисті, хребет, колінні і кульшові суглоби. Частота ОА колінних суглобів, як і ураження іншої локалізації, є різною і залежить як від популяції, що вивчається, так і від епідеміологічного методу, який використовувався. Поширеність рентгенологічно підтвердженого ОА колінних суглобів коливається в межах від 2,9% серед жінок віком 45–65 років до 7,7–14,3% серед осіб віком 45–49 років.
Діагноз «ОА» раніше часто обґрунтовували тільки на основі рентгенологічних критеріїв. Частота болю в колінних суглобах за наявності рентгенологічних ознак ОА, за даними різних досліджень, становила 40–80% із підвищенням частоти клінічних симптомів у осіб віком старше 50 років. Результати дослідження, проведеного у Швеції, продемонстрували, що серед осіб віком 70–79 років зі скаргами на біль у суглобах, ураження колінних суглобів діагностували у 30–43% жінок і 15–25% чоловіків, і що саме колінні суглоби найчастіше уражені у представників обох статей. Поширеність ОА суглобів кистей в європейських популяціях становить близько 10% у осіб віком 40–49 років і 92% — у віці понад 70 років (>90% у жінок і 80% — чоловіків). Цікавим є факт, що больовий синдром виявляли лише у 9% чоловіків і у 26% жінок зі стадіями III–IV за Кеllgren — Lawrence. Поширеність ураження кульшових суглобів серед осіб віком старше 55 років значно нижча, ніж колінних суглобів і суглобів кистей. ОА кульшових суглобів III і IV стадії визначали у 8,4% жінок і 3,1% чоловіків у Великобританії, Швеції і Нідерландах, поширеність первинного коксартрозу в цій же віковій групі становила 3,4 і 3,0% відповідно. Статистичні дані щодо ОА в Україні наведені у розділі «Ревматичні хвороби в Україні: стан проблеми та шляхи її вирішення».
Фактори ризику
Думка про те, що ОА є групою хвороб, що відрізняються за локалізацією ураження суглобів, але мають ознаки загального патологічного процесу, який призводить до суглобової недостатності, з’явилася внаслідок аналізу факторів ризику патології за умови різної локалізації процесу. Наприклад, фактори ризику ОА кульшових і колінних суглобів мають чіткі відмінності:
- при ОА кульшових суглобів відмінність за статтю, частотою вроджених дефектів розвитку відсутня;
- ОА колінних суглобів відрізняється більш частим ураженням осіб жіночої статі, характеризується попереднім травматичним ушкодженням суглобів.
Крім того, отримано дані, що група факторів ризику ураженння пателофеморального відділу колінних суглобів відрізняється від факторів ураження медіального тібіофеморального відділу (перші більше пов’язані зі спадковою схильністю і наявністю вузликів на кистях, другі частково зумовлені наявністю надмірної маси тіла та попередніми хірургічними втручаннями).
Згідно із сучасними уявленнями, ОА виникає в результаті взаємодії численних генетичних факторів і чинників навколишнього середовища. Відповідно до цього хвороба має мультифакторіальний патогенез з багатьма визнаними факторами ризику (табл. 28.1).
Таблиця 28.1
Фактори ризику розвитку ОА
|
Ендогенні фактори |
Екзогенні фактори |
|
Вік |
Травми |
|
Стать |
Професійна діяльність |
|
Дефекти розвитку |
Спортивна активність |
|
Спадкова схильність |
Надмірна маса тіла |
Поширеність ураження артрозом суглобів кистей, кульшових і колінних суглобів драматично збільшується як серед чоловіків, так і серед жінок віком 50–80 років, проте причини, через які вік — один з найсильніших факторів ризику ОА, остаточно не визначені. З одного боку, можливо, що хондроцити у процесі старіння втрачають здатність до поповнення або відновлення матриксу суглобового хряща, втраченого в результаті пошкодження або нормального обміну, і в результаті призводять до розвитку дефіциту (як у випадку ОП). З іншого — суглобовий матрикс, що старіє, може ставати чутливішим до нормальних кумулятивних мікропошкоджень. І в такому разі відновні механізми клітин не здатні компенсувати цю підвищену чутливість.
На сьогодні існують дані епідеміологічних досліджень на користь участі статевих гормонів, в першу чергу естрогену, у розвитку ОА. Вони включають високу частоту цієї патології у жінок, яка підвищується під час менопаузи, асоціацію генералізованої форми ОА з можливими маркерами впливу ендогенних статевих гормонів, у тому числі гінекологічні операції, маса кістки й ожиріння.
У жінок в період постменопаузи з ОА колінних, кульшових суглобів, суглобів кистей і поліостеоартрозом відмічають збільшену МЩКТ, яку не пояснюють ані ожирінням, ані більш повільною втратою кістки у жінок з ОА в період менопаузи. Вища кісткова маса — результат надлишку естрогену, може викликати збільшення механічного впливу на хрящ під час навантаження суглоба. Ожиріння, у свою чергу, також пов’язують з вищими рівнями ендогенного естрогену у жінок в період постменопаузи. Доведено, що надмірна маса тіла підвищує ризик ОА колінних, кульшових суглобів, суглобів кистей у жінок, але за рахунок яких механізмів це відбувається: за рахунок механічної дії власної маси тіла на хрящ, більш високого рівня естрогену або інших системних впливів, — остаточно невідомо. Можливо, що естроген має різний вплив, який залежить від часу виникнення менопаузи і патогенетичної стадії ОА. Високий рівень естрогену може викликати підвищення ризику захворювання у жінок в пременопаузальний період або безпосередньо, або через збільшену кісткову масу, але в той же час сповільнювати розвиток і прогресування захворювання в період постменопаузи і у жінок старшого віку.
Нерозпізнані у новонароджених дисплазії сполучної тканини і підвивихи кульшових суглобів часто викликають розвиток тяжкого ОА в зрілому віці. На думку деяких вчених, близько 80% усіх випадків так званої ідіопатичної форми цієї хвороби кульшових суглобів пов’язано із нерозпізнаними вадами розвитку, такими як дисплазії і підвивихи. У той же час високу поширеність ОА кульшових суглобів в Європі і США не можна пояснити відомою частотою аномалій розвитку.
Спадкову схильність виявляють частіше при генералізованій формі ОА. Описано декілька великих родин з генералізованим ОА і ПА з аутосомно-домінантним типом успадкування і раннім (10 і 50 років) віком дебюту захворювання. Відома родинна форма хвороби з ураженням очей — синдром Стіклера, також характеризується аутосомно-домінантним типом успадкування. Нещодавно на основі рентгенологічного обстеження кистей і колінних суглобів 500 пар близнюків віком 45–70 років встановлено, що конкордантність у 130 пар монозиготних близнюків удвічі вища, ніж у дизиготних, частка генетичних чинників становила до 65%.
Природа генетичного впливу не встановлена. З одного боку, можливо, патологія зумовлена структурними дефектами (наприклад колагену) або змінами в метаболізмі хряща чи кістки, з іншого — можливий генетичний вплив на відомі фактори ризику, наприклад ожиріння. Для виділення гена або генів, що беруть участь в патогенезі ОА, необхідні подальші дослідження.
Важливу роль травми в етіології ОА визнають багато дослідників, але в той же час це не просто посттравматична хвороба. Фактори ризику, ймовірно, чинять додатковий вплив: наприклад, хворі із системним фактором ризику, що виявляється у розвитку ОА дистальних міжфалангових суглобів кистей, мають підвищений ризик ураження колінних суглобів після меніскектомії.
Науково доведено зв’язок професійних факторів та спортивного навантаження з розвитком ОА. Так, у фермерів і футболістів відзначають підвищений відносний ризик ураження кульшових суглобів, а надмірне навантаження на окремі суглоби асоціюється з підвищеним відносним ризиком ОА цих суглобів (наприклад плечі та лікті в осіб, що подають м’яч у бейсболі, гомілковостопні суглоби у танцюристів балету, п’ястково-фалангові суглоби у боксерів, колінні — у баскетболістів).
Дослідження популяцій ОА постійно демонструють, що особи з надмірною масою тіла мають вищий ризик ураження цією патологією колінних суглобів, аніж при нормальній масі тіла. Згідно з результатами епідеміологічного дослідження, проведеного в США, у жінок з надмірною масою тіла і індексом маси тіла >30, але <35 ризик розвитку ОА у 4 рази вищий порівняно із жінками з індексом маси тіла до 25. Для чоловіків з такою ж надмірною масою тіла ризик підвищувався в 4,8 раза порівняно з чоловіками з нормальною масою тіла.
Більше того, якщо надмірну масу тіла відзначають у віці 37 років, коли ОА є вкрай рідкісним явищем, підвищується ризик ураження колінних суглобів у віці до 70 років. В осіб з надмірною масою тіла відмічають не тільки високий ризик розвитку ОА колінних суглобів, але, як показали тривалі спостереження, і високий ризик прогресування хвороби. При надмірній масі тіла існує більший ризик розвитку ОА кульшових суглобів, хоча ця асоціація не така сильна, як у разі ураження колінних суглобів. У близнюкових дослідженнях, проведених у Лондоні, теж виявили асоціацію надмірної маси тіла з ОА суглобів кистей, зокрема з ураженням основи I пальця кисті. Надмірна маса тіла — фактор ризику клінічного ураження колінних суглобів у жінок, а зменшення маси тіла на 5 кг у жінок з індексом маси тіла вище середнього, в свою чергу, знижувало ризик розвитку ОА на 50%. Крім того, особи з надмірною масою тіла мають вищий ризик прогресуючого перебігу захворювання.
Класифікація
Виділяють дві основні форми ОА: первинний (ідіопатичний) і вторинний, що виникає на фоні різних захворювань.
I. Первинний (ідіопатичний)
А. Локалізований.
- Суглоби кистей.
- Суглоби стоп.
- Колінні суглоби.
- Кульшові суглоби.
- Хребет.
- Інші суглоби.
Б. Генералізований (≥3 груп суглобів).
- З ураженням дистальних і проксимальних міжфалангових суглобів.
- З ураженням великих суглобів.
- Ерозивний.
ІІ. Вторинний
A. Посттравматичний.
Б. Вроджені, набуті або ендемічні хвороби (хвороба Пертеса, синдром гіпермобільності та ін.).
B. Метаболічні хвороби.
- Охроноз.
- Гемохроматоз.
- Хвороба Вільсона.
- Хвороба Гоші.
Г. Ендокринопатії.
- Акромегалія.
- Гіперпаратиреоз.
- Цукровий діабет.
- Гіпотиреоз.
Д. Хвороба відкладення кальцію (фосфату кальцію, гідроксиапатит).
Е. Невропатії (хвороба Шарко).
Ж. Інші захворювання (аваскулярний некроз, РА, хвороба Педжета та ін.).
Рентгенологічна класифікація
Класифікацію Кеllgren тa Lawrence (1957) використовують для визначення рентгенологічної стадії ОА:
0 стадія — відсутні зміни, характерні для ОА.
1-ша стадія — сумнівні зміни (сумнівне звуження суглобової щілини (СЩ) та можливі крайові остеофіти).
2-га стадія — мінімальні прояви (визначені остеофіти та можливе звуження СЩ).
3-тя стадія — помірні прояви (множинні остеофіти, визначене звуження СЩ, ознаки склерозу, можлива деформація країв кістки).
4-та стадія — виражені зміни (великі остеофіти, виражене звуження СЩ, виражений склероз, визначена деформація країв кістки).
Патогенез
Тривалий час ОА відносили до дегенеративних захворювань суглобів і вважали, що оскільки хрящ — це тканина, не здатна до оновлення і відновлення, то остеоартрозний процес — наслідок старіння і дегенерації хряща. В даний час відбулися зміни в розумінні патогенетичних процесів, що відбуваються при ОА. Цю хворобу більше не розглядають як простий наслідок старіння і дегенерації хряща. Патологічні зміни при ОА, мабуть, — результат активних процесів, більшість з яких за своєю природою можуть бути швидше репаративними, аніж деструктивними.
Патологічний процес уражує не тільки суглобовий хрящ, в якому виникають дегенеративні процеси, фібриляція, утворення тріщин, ерозій, що спричинює повну втрату хряща, але й інші складові суглоба, включаючи субхондральную кістку, зв’язки, капсулу, синовіальну мембрану і періартикулярні м’язи.
ОА — це, в першу чергу, хвороба суглобового хряща. На ранніх стадіях хвороби хрящ стає товщим, ніж в нормальному суглобі, але при прогресуванні процесу відбувається його поступове стоншення. Хрящ стає більш рихлим, можуть виникати глибокі тріщини, що тягнуться до кістки. Часто такі зміни відмічають тільки в одній частині суглобової поверхні — у ділянці, що несе навантаження. Проте, навіть якщо хрящ в інших ділянках виглядає інтактним, то біохімічно він не є нормальний. І хоча деструкція і втрата суглобового хряща, — центральні ознаки ОА, зміни субхондральної кістки теж важливі. Дійсно, при ідіопатичному ОА досі залишається незрозумілим, первинні зміни виникають в суглобовому хрящі чи в субхондральній кістці, проте найбільш виражені макроскопічні зміни зазвичай виявляють саме в суглобовому хрящі. Метаболічні процеси, що відбуваються в матриксі, а саме синтез і деградація всіх його компонентів, у здорових осіб перебувають у стані рівноваги. Забезпечують нормальний перебіг обмінних процесів хрящової тканини основні клітини хряща — хондроцити. Вони беруть участь у регуляції синтезу і деградації всіх компонентів хрящового матриксу. Вважають, що в основі патогенезу ОА лежить переважання катаболічних процесів над анаболізмом, що зумовлено саме патологією хондроцитів. Функціональна активність цих клітин регулюється надзвичайно великою кількістю біологічно активних медіаторів, і саме вони синтезують численні медіатори, що беруть участь в регуляції обміну хрящової тканини як в нормі, так і при патології.
Можливо, що під впливом, наприклад, надмірної механічної сили або іншого імпульсу, хондроцити починають продукувати запальні медіатори, такі, як ІЛ-1, -6, -8, -17, -18, які викликають підвищення катаболічної активності хондроцитів. Так, доведено, що під дією прозапального цитокіну ІЛ-1 хондроцити синтезують протеолітичні ферменти (агреканази і матриксні металопротеїнази), що викликають деградацію колагену і протеоглікана хряща. Крім того, характерною особливістю хондроцитів при ОА є гіперекспресія ЦОГ-2 та індукованої форми синтетази монооксиду азоту (фермент, що регулює утворення оксиду азоту, який здійснює токсичний вплив на хрящ).
Протеолітичні процеси, у свою чергу, викликають синтез хондроцитами факторів росту, таких, як кістковий морфогенний протеїн-2, кістковий морфогенний протеїн-7, інсуліноподібний фактор росту 1-го типу, трансформуючий фактор росту-β та інші, які стимулюють синтез макромолекул матриксу і пригнічують продукцію катаболічних ферментів. Імовірно, що баланс між цитокинами і факторами росту визначає співвідношення синтезу і деградації екстрацелюлярного матриксу в хрящі людини in vivo.
Активація синтезу в субхондральній кістці інсуліноподібного фактору росту 1-го типу, трансформуючого фактору росту-β, а також ІЛ-1 і ІЛ-6 і простагландину Е2 призводить до підвищення активності металопротеїнази й активатора плазміногену, сприяє росту остеофітів і підвищенню жорсткості субхондральної кістки, яке, в свою чергу, стимулює деградацію суглобового хряща, викликає підвищення локальної секреції факторів росту хондроцитами, замикаючи при цьому патологічне коло.
Клінічна картина
Біль і обмеження функції суглоба — головні клінічні симптоми ОА. Перші прояви хвороби пацієнти часто відчувають як дискомфорт, на що вони в здебільшого не звертають уваги, особливо якщо це перший епізод, за яким виникає тривалий безсимптомний період. Проте при ретельному розпитуванні хворого нерідко можливо встановити момент виникнення болю, який і слід вважати маніфестацією патології. На початкових стадіях біль при ОА носить механічний характер, тобто виникає при рухах і проходить у спокої, що відрізняє його від болю при запальних артропатіях, при яких збільшення вираженості болю виникає саме у спокої. Ритм та інтенсивність болю можуть змінюватись у міру прогресування ОА. Так, при вираженому ураженні нижніх кінцівок біль виникає при зміні положення тіла вночі на відміну від запального болю, що виникає в другій половині ночі.
На пізніх стадіях хвороби характериним є постійний біль, особливо у разі ураження кульшових суглобів. Іноді інтенсивність болю змінюється залежно від температури, вологості повітря й атмосферного тиску, який впливає на тиск у суглобовій порожнині. Механізм виникнення болю при ОА остаточно не встановлено. Оскільки суглобовий хрящ не інервується і, отже, не є чутливим до болю, його виникнення пов’язане із розвитком патологічних змін в нехрящових структурах суглоба. Основні причини болю, імовірно, полягають у появі трабекулярних мікропереломів, кісткового венозного стазу, внутрішньомедулярної гіпертензії, наявності хронічного синовіту, посиленні тиску на субхондральну кістку, виникненні спазму навколосуглобових м’язів, дегенеративних змін у інтраартикулярних зв’язках, а також подразненні остеофітами оточуючих тканин. Збільшення вираженості болю відбувається при приєднанні запального процесу і появі випоту в суглобі, який легко діагностувати при клінічному огляді. Про наявність «запального» компоненту свідчить раптове, часто безпричинне, посилення болю а також його поява у нічний час, посилення ранкової скутості в ураженому суглобі.
Ранкова скутість при ОА в цілому розвивається на пізніх стадіях. Спочатку вона дискретна і виникає переважно після періодів нерухомості суглобів — «феномен гелю». Скутість нетривала, зазвичай менше 30 хв, що є відмінною рисою від запальних артритів.
Окрім болю, характерною рисою ОА є крепітація в ураженому суглобі, що виникає при виконанні активних рухів у ньому. При прогресуванні захворювання через біль, а також появу рефлекторного спазму м’язів може виникнути обмеження функціональної здатності суглоба аж до утворення сухожилково-м’язових контрактур. Функціональна недостатність досить варіабельна і залежить від ступеня вираженості болю і скутості, наявності або відсутності випоту у ньому. Виникають порушення рухової активності: ходіння, стояння, ходіння вгору і вниз по сходах, сексуального життя, тощо. Ступінь цих порушень використовують як параметри вимірювання вираженості функціональної недостатності.
При ОА відмічають ураження періартикулярних тканин: періартрити, тендиніти, атрофія періартикулярних м’язів. Причини і механізми больового синдрому при ОА багатогранні, а зв’язок між вираженістю болю і рентгенологічними змінами нерідко відсутній.
Діагностика
Анамнез
Запропоновано враховувати наступні особливості ураження суглобів при ОА:
- поступовий початок болю;
- збільшення вираженості болю в положенні стоячи або при навантаженні;
- виникнення болю у спокої свідчить про приєднання запального компоненту;
- припухлість суглоба за рахунок невеликого випоту або потовщення синовіальної оболонки;
- ранкова скутість триває менше 30 хв, приєднання запального компоненту призводить до подовження тривалості ранкової скутості;
- крепітація при активному русі в суглобі;
- обмеження активних і пасивних рухів у суглобі;
- атрофія навколишніх м’язів;
- поступовий розвиток деформації кінцівок (варусна деформація колінних суглобів, «квадратна» кисть, вузлики Гебердена і Бушара відповідно у дистальних і проксимальних міжфалангових суглобах кистей);
- ОА, як правило, не супроводжують такі загальні симптоми, як зменшення маси тіла, стомлюваність або втрата апетиту.
Фізикальне обстеження
Клінічна оцінка суглобів у пацієнтів з ОА включає огляд суглобів, пальпацію, визначення амплітуди рухів, вимірювання окружності суглобів. Огляд хворого проводять в положеннях пацієнта стоячи і лежачи.
При огляді суглоба можна виявити:
- припухлість і згладженість контурів у зоні ураженого суглоба;
- внутрішньосуглобовий випіт;
- дефігурацію або деформацію суглоба;
- м’язову атрофію;
- нестабільність зв’язкового апарату;
- патологічне встановлення і вкорочення кінцівки (на стороні ураження).
Методом пальпації виявляють:
- болючість суглоба й періартикулярних тканин;
- наявність крепітації і хрускоту в суглобах;
- наявність вільної рідини в суглобі;
- гіпертрофовану синовіальну оболонку;
- стан зв’язкового апарату і тонус м’язів.
Для визначення функції суглобів встановлюють обсяг і якість активних і пасивних рухів в суглобах, за допомогою яких можна виявити всі резерви рухової функції суглоба.
Лабораторні дослідження
Лабораторних ознак, патогномонічних для даної хвороби, не існує. Проте лабораторні дослідження слід проводити з наступною метою:
- диференційна діагностика (при ОА відсутні запальні зміни в клінічному аналізі крові, не виявляють РФ, концентрація сечової кислоти в сироватці крові в межах норми);
- перед початком лікування (загальний аналіз крові і сечі, креатинін у сироватці крові, рівень сироваткових трансаміназ) для виявлення можливих протипоказань до призначення препаратів.
Дослідження синовіальної рідини слід проводити тільки за наявності синовіту з метою диференційної діагностики. Для ОА характерний незапальний характер синовіальної рідини: прозора, в’язка, з концентрацією лейкоцитів <2000 кл/мм3.
Інструментальні дослідження
Характерні рентгенологічні ознаки ОА (рис. 28.1):
- звуження СЩ;
- субхондральний остеосклероз;
- остеофітоз.
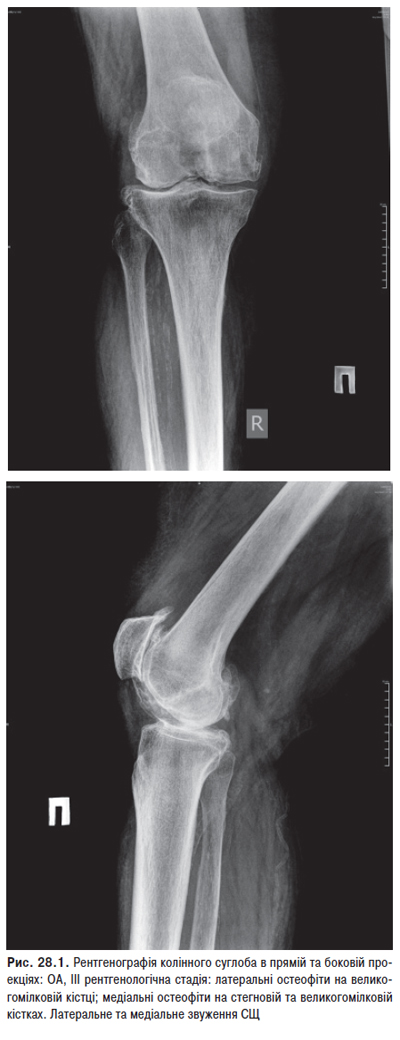
Стадію захворювання визначають переважно за класифікацією Kellgren — Lawrence. Нові інструментальні методи (спектроскопія ядерно-магнітного резонансу, КТ, остеосцинтиграфія, УЗД суглобів) застосовують для вивчення характеру ураження всіх складових суглоба, але не для оцінки ефективності лікування, оскільки для цього вони нестандартизовані.
Критерії діагностики
Діагноз «ОА» встановлюють на основі клінічних і рентгенографічних критеріїв ACR (табл. 28.2).
Таблиця 28.2
Діагностичні критерії ОА (за: Altman R. et al., 1991)
|
Клінічні критерії |
Клінічні, лабораторні і рентгенологічні критерії |
|
Колінні суглоби |
|
|
1. Біль і |
1. Біль і |
|
2а. Крепітація |
2. Остеофіти або |
|
2б. Ранкова скутість <30 хв |
3а. СР, характерна для ОА (або вік >40 років) |
|
2в. Вік >38 років або |
|
|
3а. Крепітація |
3б. Ранкова скутість <30 хв |
|
3б. Ранкова скутість <30 хв |
3в. Крепітація |
|
3в. Кісткові розростання або |
|
|
4а. Відсутність крепітації |
|
|
4б. Кісткові розростання |
|
|
Чутливість — 89% |
Чутливість — 94% |
|
Специфічність — 88% |
Специфічність — 88% |
|
Кульшові суглоби |
|
|
1. Біль і |
1. Біль і не менше двох критеріїв з трьох |
|
2а. Внутрішня ротація <15° |
2а. ШОЕ <20 мм/ч |
|
2б. ШОЕ <15 мм/ч |
2б. Остеофіти |
|
(або згинання в кульшовому суглобі >115°) або |
2в. Звуження СЩ |
|
3а. Внутрішня ротація <15° |
|
|
3б. Ранкова скутість <60 хв |
|
|
3в. Вік >50 років |
|
|
3г. Біль при внутрішній ротації |
|
|
Чутливість — 86% |
Чутливість — 89% |
|
Специфічність — 75% |
Специфічність — 91% |
|
Суглоби кистей |
|
|
1. Тривалий біль або скутість |
|
|
2. Кісткові розростання в двох або більше суглобів з 10, що оцінюються |
|
|
3. Менше двох п’ястково-фалангових суглобів є припухлими |
|
|
4а. Кісткові розростання, що включають 2 і більше дистальних міжфалангових суглобів (2-й і 3-й дистальні міжфалангові суглоби можуть братися до уваги в двох критеріях: 2 та 4а) або |
|
|
4б. Деформація одного або більше суглобів з 10, що оцінюються* |
|
|
Чутливість — 93% |
|
|
Специфічність — 91% |
|
*ІІ і ІІІ дистальні міжфалангові суглоби; ІІ і ІІІ проксимальні міжфалангові суглоби; І зап’ястно-п’ястковий суглоб обох кистей.
Враховуючи діагностичні критерії встановлення правильного діагнозу зазвичай не викликає труднощів. Проте кожну клінічну ситуацію необхідно проаналізувати з позиції можливості вторинного походження ОА (див. «Класифікація»).
Лікування
Мета лікування. Основні завдання лікування ОА — зменшення вираженості болю, корекція функціональної недостатності суглобів, обмеження прогресування захворювання і, насамкінець, поліпшення якості життя хворих. Необхідно також забезпечити розуміння пацієнтом свого захворювання і вміння керувати ним, а також навчити використовувати засоби захисту суглобів. Метод лікування у конкретного хворого необхідно розробляти з урахуванням тяжкості і поширеності ураження суглобів, загального статусу, а також наявність супутніх захворювань. Цього можна досягнути немедикаментозними, фармакологічними і хірургічними методами.
Показання до госпіталізації
Лікування хворих на ОА слід проводити в амбулаторних умовах, за винятком необхідності проведення хірургічного втручання або наявності вираженого болю в суглобах.
Немедикаментозне лікування
Нефармакологічні методи включають освіту хворих і фізичні методи. Застосування освітніх програм для хворих дозволяє підсилити дію лікарських засобів. Помірному зменшенню вираженості болю і поліпшенню функції суглобів сприяє навчання пацієнтів та їх родичів навичкам проведення щоденних тренувань. Особливе значення має соціальна підтримка хворих. Такі заходи повинні бути складовою лікувального плану для хворих на ОА. Доведеною є ефективність таких освітніх програм у плані зменшення вираженості болю, частоти візитів до лікаря, поліпшення якості життя і підтримки функцій суглобів.
Фізичні вправи при ОА сприяють зниженню інтенсивності болю і збереженню функціональної активності суглобів. ЛФК проводять без статичних навантажень (сидячи, лежачи, в басейні) — вона не повинна викликати біль у суглобах. Необхідними є вправи, спрямовані на зміцнення м’язів, наприклад, при ОА з ураженням колінних суглобів слід використовувати вправи для зміцнення сили чотириголового м’яза стегна, що зумовлює достовірне зменшення вираженості болю, збереження і збільшення обсягу рухів в суглобах (наприклад «велосипед») і поліпшення загального аеробного стану (наприклад ходіння по рівній місцевості в помірному темпі). Такі аеробні навантаження сприяють також зменшенню маси тіла ваги.
Корисними є вправи в басейні, їзда на велосипеді, лижний спорт, використання тренажерів (наприклад тредміл). Інтенсивність і частота повторень рухів на заняттях повинні визначатися станом хворого і вираженістю больового синдрому. Обсяг рухів збільшують поступово. У більшості хворих проведення занять самостійно в домашніх умовах викликає такий же ефект, як і під наглядом фахівця з ЛФК.
Ортопедичну корекцію застосовують для зменшення навантаження на суглоби. Так, при плоскостопості хворим на ОА слід носити взуття на низькому широкому підборі з супінаторами (устілки, що підтримують склепіння стопи і знижують навантаження на суглоби).
При ураженні колінних суглобів рекомендують носити наколінники, ортези, що фіксують коліно у вальгусному положенні, використання ортопедичних устілок з підведеним на 5–10° латеральним краєм. Вказані пристрої зменшують навантаження на медіальні відділи колінного суглоба, проявляють знеболювальний ефект і покращують функцію суглобів.
Для зменшення навантаження при ОА суглобів нижніх кінцівок рекомендують ходіння з паличкою, яка майже на 50% зменшує навантаження на кульшовий суглоб. Тростину слід носити в руці, протилежній найбільш ураженому суглобу. При двобічній локалізації хвороби рекомендують ходіння з милицями канадського типу. Застосування ортезів і шинування 1-го зап’ястко-п’ясткового суглоба сприяє усуненню підвивиху і поліпшенню функції кисті. Цей метод включений в європейські рекомендації з ведення хворих на ОА суглобів кистей.
Локальне використання поверхневого холоду або тепла може спричиняти невеликий знеболювальний ефект у окремих хворих. Ультразвукова терапія призводить до помірного зниження інтенсивності болю при ОА. Черезшкірна електростимуляція нервів має доведений, але нетривалий анальгезивний ефект. Достовірних свідчень на користь ефективності рефлексотерапії на сьогодні немає, тому її слід рекомендувати особам, що надають перевагу нетрадиційним методам лікування.
Медикаментозне лікування
На сьогодні прийнята класифікація протиартрозних препаратів залежно від їх фармакологічного впливу, яка включає декілька класів лікарських засобів (табл. 28.3).
Таблиця 28.3
Фармакотерапія ОА
|
Препарати, що модифікують симптоми захворювання |
|
|
Швидкодіючі |
Повільнодіючі |
|
Анальгетики |
Хондроїтин сульфат |
|
НПЗП |
Глюкозамін |
|
Трамадол |
Неомилені сполуки сої/авокадо |
|
Діацереїн |
|
|
Гіалуронова кислота |
|
До першого належать симптоматичні препарати швидкої дії, а саме анальгетики і НПЗП, здатні зменшувати вираженість болю, припухлість, скутість і покращувати функцію суглобів. Виділяють групу препаратів, що модифікують структуру захворювання, хоча для жодного препарату даного класу хондропротекторна дія у людини не підтверджена. Проміжне положення займають симптоматичні препарати сповільненої дії.
Симптоматичні препарати швидкої дії
«Прості» анальгетики (наприклад парацетамол) рекомендують як препарати першого ряду для зменшення вираженості болю при ОА, особливо у хворих з непостійним болем помірної інтенсивності без ознак запалення. Основна перевага парацетамолу — його відносно низька токсичність для верхніх і нижніх відділів ШКТ, особливо порівняно з НПЗП, які часто призначають особам літнього віку. Дозу парацетамолу підбирають індивідуально, але не більше 4 г/добу 9-разовий прийом до 650 мг), оскільки вищі дозові режими супроводжуються розвитком ускладнень з боку ШКТ. У дозі, що рекомендується, доведеною є безпека застосування парацетамолу при ОА протягом 2 років. Парацетамол не рекомендують призначати пацієнтам, що зловживають алкоголем.
НПЗП — найбільш широко застосовувані в клінічній практиці лікарські засоби. Разом з тим фармакоепідеміологічні дані демонструють не тільки їх широке використання, але часто і неправильне застосування НПЗП у хворих, особливо старшого віку із супутньою патологією, що супроводжується більш частим розвитком різних побічних реакцій. НПЗП при ОА призначають у разі неефективності парацетамолу, а також за наявності ознак запалення. Доведених фактів про перевагу знеболювальних та протизапальних властивостей одного НПЗП над іншим не отримано.
Таким чином, вибір конкретного НПЗП визначає перш за все його безпеку в конкретних клінічних умовах. Наприклад, серед клініко-фармакологічних властивостей індометацину слід брати до уваги його негативну дію на метаболізм суглобового хряща при ОА.
Ускладнення з боку ШКТ — один з найсерйозніших побічних ефектів НПЗП, що виникає найчастіше в перші 3 міс його прийому. Відносний ризик їх виникнення (має дозозалежний характер) варіює серед різних НПЗП. Мінімальний ризик розвитку шлунково-кишкової кровотечі викликають селективні по відношенню до ЦОГ-2 НПЗП. Їх слід призначати за наявності факторів ризику розвитку побічних явищ з боку ШКТ:
- вік старше 65 років;
- наявність в анамнезі виразкової хвороби або шлунково-кишкової кровотечі;
- одночасний прийом ГК або антикоагулянтів; наявність тяжких супутніх захворювань.
Прийом будь-яких НПЗП може призводити до підвищення ризику розвитку серцево-судинних катастроф. Тому при призначенні НПЗП необхідно ретельно оцінювати кардіоваскулярні фактори ризику. У пацієнтів, що приймають низькі дози ацетилсаліцилової кислоти, слід уникати прийому НПЗП (ібупрофен, індометацин і напроксен), що знижують його кардіопротекторний ефект. Будь-які НПЗП можуть викликати дестабілізацію артеріального тиску (АТ) (в першу чергу систолічного) у пацієнтів, що приймають антигіпертензивні препарати, і навіть у пацієнтів з нормальним АТ. У осіб віком старше 55 років, що отримують НПЗП, необхідно проводити ретельний моніторинг АТ і за необхідності модифікувати антигіпертензивну терапію.
Неселективні і селективні НПЗП можуть викликати порушення функції нирок.
НПЗП у хворих на ОА застосовують тільки в період посилення больового синдрому на відміну від їх систематичного прийому у разі запальних артритів. Доза НПЗП при ОА нижча, аніж при артритах. Постійний прийом в низьких дозах НПЗП, про ефективність якого є наукові публікації, на наш погляд, є неприйнятним.
Знеболювальні препарати центральної дії опіоїдного ряду застосовують протягом короткого періоду з метою купірування сильного болю та за умови неефективності парацетамолу або НПЗП, а також у разі неможливості призначення оптимальних доз цих лікарських засобів.
При ОА трамадол призначають в дозі 50–200 мг/добу. Відносно часті побічні реакції при застосуванні препарату (нудота, запаморочення, закреп і сонливість) можна суттєво зменшити шляхом титрування дози (поступово підвищуючи дозу на 25 мг протягом 3 днів).
Велика кількість побічних реакцій, особливо серед осіб старшого віку, більшість з яких приймають ліки з приводу супутніх захворювань, що потребує врахування взаємодії препаратів, більш прийнятними для використання є локальні засоби лікування ОА. З цією метою використовують мазі, гелі, креми на основі НПЗП. Локальні форми НПЗП, що використовуються для лікування ураження колінних суглобів і суглобів кистей, є досить ефективними та безпечними.
Введення ГК в порожнину суглоба показане у випадку ОА із симптомами запального процесу синовіту. Найчастіше внутрішньосуглобові введення виконують у колінні суглоби. Ефект даного лікування проявляється зменшенням вираженості болю і симптомів запалення, а тривалість становить від 1 тиж до 1 міс. Серед препаратів використовують тріамцинолон (20–40 мг), метилпреднізолон (20–40 мг), бетаметазон (2–4 мг). Частота введення не повинна перевищувати 2–3 рази на рік в один суглоб. Частіше введення не рекомендуються, беручи до уваги високу імовірність прогресування деструкції суглобового хряща.
Симптоматичні препарати повільної дії
Повільнодіючі симптоматичні препарати для лікування при ОА займають проміжне місце: з одного боку, вони володіють вираженим впливом на біль і функціональний стан суглобів, як і НПЗП, з іншого — виявляють деякі «хондропротекторні» властивості. Ефективність застосування при ОА цілого ряду препаратів даної групи вважається доведеною. Відмінні особливості цих препаратів — час настання ефекту, зазвичай через 2–8 тиж від початку лікування, і тривалість ефекту — протягом 2–3 міс після припинення лікування.
На сьогодні хондроїтин сульфат, глюкозамін (глюкозаміну сульфат 750*, глюкозаміну сульфат натрію хлорид*), неомилені сполуки авокадо/соя і препарати гіалуронової кислоти внесені до рекомендацій EULAR для лікування ОА колінних, кульшових суглобів і суглобів кистей. Зазначені лікарські засоби виявляють симптоматичний ефект і, можливо, здатні впливати на перебіг хвороби.
Хондроїтін сульфат і глюкозамін є сульфатованими глікозаміногліканами, які розміщені в екстрацелюлярному матриксі суглобового хряща. При пероральному прийомі вони добре абсорбуються і досягають високих концентрацій в порожнині суглоба. Механізм їх дії остаточно не встановлений. Доведено, що хондроїтин сульфат і глюкозамін мають власну протизапальну активність.
Проведено більше 20 контрольованих досліджень ефективності хондроїтин сульфату і глюкозаміну. Продемонстровано, що лікування цими препаратами викликає зниження больового синдрому і поліпшення функцій суглобів, що є аналогічним порівняно з іншими симптоматичними препаратами («простими» анальгетиками, НПЗП), при цьому їх безпека виявилася порівнянною з плацебо.
Докази на користь структурно-модифікуючої дії хондроїтин сульфату і глюкозаміну наведені в декількох тривалих подвійних сліпих плацебо-контрольованих дослідженнях у хворих на ОА. Встановлено, що терапія хондроїтин сульфатом в дозі 800 мг/добу протягом 2 років мала статистично достовірний стабілізувальний вплив на ширину СЩ у хворих на гонартроз. На підставі аналізу результатів лікування хондроїтин сульфатом протягом 2 років у 622 хворих на гонартроз описано сповільнення прогресування захворювання у пацієнтів, що приймали хондроїтин сульфат, порівняно з групою, що отримувала плацебо (дослідження STOPP). Беручи до уваги результати 11 рандомізованих контрольованих досліджень, трьох опублікованих метааналізів та останніх рекомендацій EULAR, встановлено, що тривале застосування хондроїтин сульфату є безпечним, добре переноситься, достатньою мірою контролює больовий синдром і збільшує обсяг рухів у суглобах у хворих на ОА з ураженням колінних суглобів.
Застосування хондроїтин сульфату протягом 3 років при ОА суглобів кистей чинило захисний вплив на ризик появи «нових» ерозій.
Структурно-модифікуюча дія глюкозамін сульфату в добовій дозі 1500 мг підтверджена в двох подвійних сліпих рандомізованих плацебо контрольованих 3-річних дослідженнях порівняння у 212 і 202 пацієнтів з гонартрозом. Аналіз результатів свідчить, що через 3 роки у хворих, що приймають глюкозамін сульфат, не виявлено статистично значущого звуження СЩ на відміну від пацієнтів, що отримували плацебо.
Зважаючи на дані про те, що хондроїтин сульфату і глюкозамін сульфат характеризуються різною фармакологічною дією на метаболізм хряща, відносно нещодавно з’явилися результати досліджень з вивчення ефективності поєднаного застосування цих препаратів при ОА. На підставі багатоцентрового подвійного сліпого плацебо-контрольованого клінічного дослідження, що включало 1583 пацієнти з ОА колінних суглобів, встановлено, що комбінація хондроїтин сульфату і глюкозаміну гідрохлориду* виявилася більш ефективною відносно зниження інтенсивності больового синдрому в підгрупі хворих на ОА із вираженим болем в колінних суглобах порівняно з плацебо.
Біологічне походження препаратів на основі хондроїтин сульфату та глюкозаміну потребує особливої уваги стосовно біодоступності та біоеквівалентності цих лікарських засобів, а також наявності білкових речовин, які можуть викликати алергічні та аутоімунні реакції.
Неомилені сполуки авокадо/соя екстрагують з авокадо і сої у співвідношенні 1:2. Дослідження, проведені in vitro, показали їх здатність пригінчувати активність ІЛ-1 та індуковану ним продукцію строміелізину, ІЛ-6, -8, простагландину Е2 і колагенази, а також стимулювати синтез колагену хондроцитами хряща.
Неомилені сполуки авокадо/соя зменшують вираженість болю у спокої і при ходінні, покращують функціональний стан колінних і кульшових суглобів. Препарат добре переноситься пацієнтами, має високий профіль безпеки. Існують припущення про його можливий структурно-модифікуючий вплив, однак даний факт потребує подальшого дослідження.
Похідні гіалуронової кислоти застосовують для внутрішньосуглобового введення. На сьогодні використовують 2 похідні цієї речовини: низькомолекулярні (мол. маса 500–730 кДа) і високомолекулярні (мол. маса 6 МДа). Вони здатні зменшувати вираженість болю в колінних суглобах і покращувати їх функціональну здатність, при цьому ефект триває 3–12 міс.
Внутрішньосуглобове введення гіалуронової кислоти може бути особливо корисним хворим, що мають протипоказання для прийому НПЗП або при недостатній їх ефективності.
Особливо обнадійливим виглядає застосування препаратів гіалуронової кислоти у хворих з високим ризиком розвитку побічних реакцій на НПЗП:
- пацієнти літнього віку;
- наявність виразки або кровотечі в анамнезі;
- супутня терапія ГК.
Необхідно враховувати і протипоказання до проведення терапії препаратами гіалуронової кислоти, які включають інфекційні ураження шкіри, порушення її цілісності в зоні введення, наявність алергічних реакцій на тваринний білок. Описані поодинокі випадки розвитку псевдоподагричного нападу протягом декількох годин або днів після ін’єкції, але такі реакції виникають вкрай рідко, а їх наявність у анамнезі, так само як і наявність хондрокальцинозу, не є протипоказаннями для такого лікування.
*Необхідно також враховувати при призначенні лікарських засобів на основі хондроїтин сульфату та глюкозаміну відсутність цукрового діабету чи порушення толерантності до глюкози, а також продуктивних і проліферативних процесів молочної залози та органів малого тазу.
Діацереїн — препарат з доведеним (в дослідженнях in vitro) пригнічувальним впливом на активність ІЛ-1, описаним повільнодіючим, персистуючим, симптоматичним ефектом у хворих на ОА з ураженням колінних, кульшових суглобів. Результатами серії рандомізованих контрольованих досліджень і двох метааналізів описано достовірний анальгезивний ефект препарату у хворих на ОА, проте доведеним є факт більш частого виникнення діареї (основного побічного явища препарату) серед пацієнтів, що приймають діацереїн.
Засоби, що чинять антирезорбтивний вплив на кістку
Думка про те, що антиостеопоротичні препарати наділені потенційним структурно-модифікуючим впливом при ОА, виникла після описання хондропротекторних властивостей алендронату, кальцитоніну та естрогенів у серії експериментальних досліджень на тваринах. Прийом естрогенів та алендронату асоціювався з меншим ступенем типових для ОА змін субхондральної кістки, а також патологічних змін, зумовлених набряком кісткового мозку серед жінок старшого віку порівняно з тими, хто не приймав антирезорбтивних лікарських засобів. У той же час результати рандомізованих контрольованих досліджень є суперечливими і недостатніми для підтвердження цієї гіпотези.
Так, у рандомізованому контрольованому дослідженні з оцінки ефективності різних дозових режимів ризедронової кислоти (5; 15; 35 або 50 мг/добу), незважаючи на доведений дозозалежний вплив препарату на зниження рівня біомаркерів деградації хряща (С-кінцевого перехресного телопептиду колагену ІІ типу), достовірного впливу препарату на швидкість прогресування ОА (за оцінкою ступеня звуження СЩ) не виявлено. Дані щодо здатності знижувати рівні С-кінцевого перехресного телопептиду колагену ІІ типу продемонстровані при терапії стронцію ранелатом поряд зі зниженням рентгенологічного прогресування ОА та суттєвим впливом на розвиток ОП за загальноприйнятими клініко-лабораторними показниками, при замісній гормональній терапії та прийомі селективних модуляторів естрогенових рецепторів.
Кальцитонін лосося в дозі 1 мг/добу протягом 84 днів у хворих на ОА колінних суглобів асоціювався з достовірним зниженням рівнів С-кінцевого перехресного телопептиду колагену ІІ типу, неоепітопу колагену ІІ типу, матриксної металопротеїнази-13, а також з статистично достовірним покращенням показника альгофункціонального індексу Лекена.
Структурно-модифікуюча терапія ОА
В Україні стронцію ранелат* зареєстрований для лікування ОА: структурно-модифікуюче лікування ОА колінних, кульшових суглобів та хребта для зменшення прогресування руйнування хряща і зменшення вираженості клінічних симптомів.
*В Україні стронцію ранелат зареєстрований під торговою назвою Стромос.
Стронцію ранелат відрізняється своїм достовірним впливом як на симптоми, та і на структуру захворювання. Такий ефект стронцію ранелату можливий завдяки унікальному механізму його дії.
In vitro стронцію ранелат:
- стимулює формування хрящового матриксу в здоровому та пошкодженому ОА суглобі людини без стимуляції резорбції хряща;
- знижує активність резорбції кісткової тканини в субхондральній кістці людини.
Фармакологічні дослідження in vivo продемонстрували, що стронцію ранелат зменшує розвиток макроскопічних пошкоджень виростків стегна та верхньої суглобової поверхні великогомілкової кістки, а також враженість синовіту та склерозу субхондральної кістки.
Це призводить до позитивного впливу стронцію ранелату як на суглобовий хрящ, так і на субхондральну кістку.
У ході дослідження III фази рівень біохімічних маркерів руйнування хряща (СТХ II у сечі) достовірно знизився при застосуванні стронцію ранелату порівняно з таким при застосуванні плацебо.
Ефективність стронцію ранелату доведена в широкомасштабному багатоцентровому міжнародному рандомізованому подвійному сліпому плацебо-контрольованому дослідженні SEKOIA, спрямованому на визначення переваг застосування стронцію ранелату (у дозі 1 та 2 г/добу) порівняно з плацебо для попередження прогресування руйнування суглобового хряща упродовж 3 років у чоловіків та жінок з ОА колінного суглоба. Загалом у дослідженні взяли участь 1683 пацієнти, дорослі чоловіки та жінки із симптоматичним первинним ОА колінного суглоба відповідно до клінічних та рентгенологічних критеріїв ACR. Середня тривалість спостереження становила 29,8 міс.
Дослідження продемонструвало достовірне зниження середнього показника звуження ширини суглобового простору (ЗШСП) після 3 років лікування в групі пацієнтів, які застосовували стронцію ранелат у дозі 2 г/добу, порівняно з показником у групі плацебо. У відносному вираженні показник ЗШСП був на 26% меншим при застосуванні стронцію ранелату в дозі 2 г/добу порівняно з плацебо.
У пацієнтів, які приймали стронцію ранелат упродовж 3 років, за даними рентгенографії виявляли зменшення втрати суглобового хряща приблизно через 1 рік терапії. Ефект був достовірним з першого року лікування.
Частка пацієнтів із відповідними клінічними проявами прогресування руйнування хряща (ЗШСП >0,5 мм) в групі пацієнтів, які приймали стронцію ранелат у дозі 2 г/добу, достовірно зменшилася на 23% (р=0,012) порівняно з такою у групі плацебо.
Відносний ризик рентгенологічної та клінічної прогресії, яка характеризувалася проявами прогресування руйнування хряща (ЗШСП >0,5 мм) та недостатнім зниженням рівня болю в колінному суглобі (<20% за субшкалою болю анкети WOMAC), достовірно зменшився на 44% (р=0,008) у групі пацієнтів, які отримували стронцію ранелат у дозі 2 г/добу.
Покращення показника ЗШСП при застосуванні стронцію ранелату супроводжувалося зменшенням вираженості клінічних симптомів перебігу ОА. Відзначалася тенденція до покращення загальної оцінки за шкалою WOMAC в групі пацієнтів, які застосовували стронцію ранелат у дозі 2 г/добу, порівняно із групою плацебо (р=0,071).
При проведенні пост-хок аналізу встановлено достовірне поліпшення показників субшкали болю за анкетою WOMAC у групі пацієнтів, які застосовували стронцію ранелат у дозі 2 г/добу (р=0,028).
У пацієнтів із ОА колінного суглоба та діагностованим супутнім ОА кульшового суглоба, які брали участь у дослідженні SEKOIA, при застосуванні стронцію ранелату відмічали як покращення структури колінного суглоба та зменшення вираженості симптомів перебігу ОА колінного суглоба, так і зменшення вираженості болю в колінному суглобі, особливо в групі застосування препарату в дозі 2 г.
Пост-хок аналіз включав об’єднані дані досліджень SОТІ та ТROPOS за участю 1105 жінок із ОП та супутнім рентгенологічно підтвердженим ОА хребта на момент включення у дослідження. Оцінку прогресування ОА хребта проводили за загальним показником Лейна. Оцінка показала зменшення прогресування ОА хребта на 42% у пацієнтів, які застосовували стронцію ранелат, порівняно із групою плацебо, після 3 років застосування препарату.
Через 3 роки лікування кількість пацієнтів із погіршеним показником ЗШСП достовірно зменшилася на 33% у групі пацієнтів, які застосовували стронцію ранелат, порівняно із групою плацебо (Інструкція для медичного застосування препарату Стромос. Реєстраційне посвідчення № UA/4943/01/01)*.
*Для отримання повної інформації необхідно ознайомитись з інструкцією для медичного застосування препарату.
Хірургічне лікування
Ендопротезування суглобів показане:
- при ОА з вираженим больовим синдромом, що не підлягає консервативному лікуванню;
- при серйозному порушенні функцій суглоба (до розвитку значних деформацій, нестабільності суглоба, контрактур і м’язової атрофії).
Ендопротезування кульшових суглобів призводить до зменшення вираженості болю, поліпшення рухової функції і підвищення якості життя хворих на ОА. Тривалість ефекту становить близько 10 років, частота інфекційних ускладнень і повторних операцій — 0,2–2,0% на рік. Найкращі результати ендопротезування відмічені у хворих віком 45–75 років, з масою тіла <70 кг, з високим соціальним рівнем життя.
Втім, відносно осіб літнього віку теж отримано позитивні результати ендопротезування. Проте таким пацієнтам необхідний більш тривалий період госпіталізації, нагляду і реабілітації, що в результаті спричинює суттєве зростання вартості лікування. Тривале спостереження за особами молодого віку (молодше 45 років) показало, що у них (особливо серед тих, хто займається важкою фізичною працею) частіше виникає потреба у проведенні повторної операції. Крім того, необхідність в повторному оперативному втручанні підвищується в осіб з надмірною масою тіла. Місце остеотомії й артроскопії в лікуванні ОА продовжують уточнювати.
Наслідок захворювання й ефективність терапії оцінюють в динаміці за показниками болю в суглобах, тривалістю ранкової скутості, функціональною активністю (індекс Lequesne, WOMAC) і якістю життя хворого (анкета SF-36).
Lequesne Index — включає оцінку болю у спокої і при ходінні (5 питань), максимально прохідної відстані (1 питання) і повсякденної активності (4 питання). Бальну оцінку кожного питання підраховують і отримують показник тяжкості захворювання:
1–4 — легкий ОА;
5–7 — помірний ОА;
8–10 — тяжкий ОА;
11–13 — дуже тяжкий ОА;
14 — вкрай тяжкий ОА.
Western Ontario and McMaster OA (WOMAC) scale — опитувальник введений в практику в 1986 р., складається з питань для самостійної оцінки пацієнтом вираженості болю (у спокої і при ходьбі — 5 питань), скутості (тривалість і вираженість — 2 питання) і функціональної недостатності у повсякденній діяльності (17 питань). Оцінку проводять за шкалою ВАШ у сантиметрах — від 0 (немає симптомів/обмежень) до 10 (максимальна вираженість симптомів/обмежень), потім всі показники підсумовують.
Dreiser Functional Index for hand arthropathies (F1HOA) — оцінює функціональний стан кисті за 10 параметрами за допомогою бальної оцінки:
0 — можливо без утруднення;
1 — можливо з невеликим утрудненням;
2 — можливо із значним утрудненням;
3 — неможливо.
Australian/Canadin Osteoarthritis Hand Index (AUSCAN) — опитувальник, який заповнюється хворим з метою оцінки болю (5 питань), скутості (1 питання) і функції суглобів кисті (9 питань). Оцінку проводять аналогічно індексу WOMAC.
Анкета SF-36 — оцінює вплив емоційного і фізичного стану пацієнта при виконанні роботи або звичайній повсякденній діяльності.
Прогноз
Прогноз для життя у хворих на ОА в цілому сприятливий. Проте ця хвороба в багатьох країнах займає одне з провідних місць серед причин, що приводять до інвалідності, що визначає його соціальну значущість, особливо враховуючи те, що ОА вважається першою причиною ендопротезування суглобів. Смертність після операцій з приводу ОА становить 1%, в основному при ендопротезуванні кульшового суглоба, в зв’язку з тромбожировою емболією.
Глава 29. Остеопороз
Визначення та епідеміологія остеопорозу
ОП — прогресуюча системна хвороба скелета, що характеризується зменшенням маси кістки і порушенням структури (мікроархітектоніки) кісткової тканини, які призводять до підвищення крихкості кістки та ризику виникнення переломів.
Значне поширення ОП, у тому числі серед осіб середнього працездатного віку, зумовлює велике соціально-економічне значення даної проблеми. Тому до числа пріоритетних напрямів, що рекомендує ВООЗ для детального вивчення у рамках «Декади кісток і суглобів» (The Bone and Joint Decade 2000–2010), віднесений ОП поряд з РА, ОА, болем у нижній ділянці спини і травмами (WHO, 1999).
Великий інтерес до ОП викликаний, насамперед, високою поширеністю серед населення як самого захворювання, так і його наслідків (переломи периферичних кісток і хребта), які стають причиною тимчасової непрацездатності, інвалідності, а також підвищеної смертності. Згідно з прогнозами, серед осіб європеоїдної раси зумовлені ламкістю переломи кісток виникнуть за роки життя приблизно у 50% жінок і 20% чоловіків віком старше 50 років. Установлено, що у жінок європеоїдної раси ризик перелому шийки стегна протягом життя становить 1:6, що перевищує ризик розвитку раку молочної залози (1:9).
Переломи стегнової кістки — найтяжче ускладнення ОП. Вони потребують госпіталізації хворого та зумовлюють тяжкий ступінь непрацездатності та підвищення смертності. Більшість переломів стегнової кістки виникає після падінь: 80% відзначається у жінок, а 90% — серед осіб віком старше 50 років. Частота переломів стегнової кістки має значну варіабельність серед населення різних країн. Скоригована за віком частота цих переломів найвища у країнах Скандинавії та Північної Америки, а на півдні Європи вона майже у 7 разів нижча; в усіх країнах вона нижча у сільських, ніж у міських районах. Переломи стегнової кістки частіше відмічають у худих і субтильних осіб, ніж в огрядних з гіперстенічною статурою, крім того, низький індекс маси тіла також є істотним чинником ризику. Довжина стегнової кістки є кількісною ознакою, що пов’язана з ризиком виникнення перелому. Створюється враження, що частота переломів стегнової кістки підвищується у північних широтах, в осіб європеоїдної раси та у популяціях з більшою кількістю людей похилого віку. Переломи проксимального відділу стегнової кістки підвищують смертність на 6–37% (залежно від попереднього стану здоров’я хворого) причому 20% летальних наслідків відзначають протягом першого місяця після перелому.
Одним з найпоширеніших типів переломів при ОП є переломи хребців. У 1 з 4 жінок у США й 1 з 8 жінок у Європі віком 50 років і старше відмічають рентгенологічні ознаки перелому, принаймні, одного хребця. Частота переломів хребців у жінок підвищується з віком, сягаючи максимальних значень у групі осіб віком 75 років і старше, тоді як у чоловіків така тенденція відсутня. Переломи найчастіше локалізуються у середньогрудному відділі хребта на рівні Т6—Т8, нижньогрудному — Т11—Т12 та у поперековому відділі — L1.
Більш висока частота розвитку ОП в жінок пояснюється тим, що у них початково (конституційно) менша міцність хребців через їх менший розмір (нижча маса в грамах), більша середня тривалість життя та більша втрата кісткової маси протягом життя (внаслідок розвитку менопаузи). Діагноз переломів кінцівки встановлюють за принципом — кістка або переламана, або ні. Ці типи переломів зазвичай є наслідком легкої травми (звичайне падіння) і можуть бути анатомічно відновлені. І навпаки, переломи хребців є наслідком прогресування основного захворювання і можуть виникати без травматичного впливу, при цьому анатомічно відновити хребець вже неможливо. Більше того, ці переломи часто належать до категорії клінічно «німих». Встановлено, що в ⅔ випадків переломи хребців не діагностуються ані клінічно, ані рентгенологічно. Ось чому особливо актуальна рання ефективна терапія ОП.
Частота випадків перелому зап’ястка підвищується в жінок європеоїдної раси віком 45–60 років, потім стабілізується або збільшується незначно. Більшість переломів кісток зап’ястка відзначають у жінок, 50% з яких старше 65 років. Частота випадків таких переломів у чоловіків низька й з віком підвищується незначно.
У пацієнтів з будь-яким типом переломів через підвищену ламкість кісток зростає ризик переломів іншого типу (рис. 29.1). Переломи тіл хребців підвищують ризик їх повторних переломів у 10 разів. Переломи стегна й передпліччя призводять до подібного ризику переломів у тих самих місцях.
Несприятливі наслідки остеопоротичних переломів можна підрозділити на три великі категорії: смертність, захворюваність і фінансові витрати. Вплив переломів на виживаність залежить від їхнього типу. Переломи стегна найчастіше призводять до підвищення смертності (10–20% протягом першого року після перелому, найвищий ризик смерті у перші 6 міс, потім він поступово знижується). Зниження виживаності складно пояснити прямим впливом перелому, а підвищення смертності пов’язано, можливо, з наявністю та розвитком супутніх захворювань.
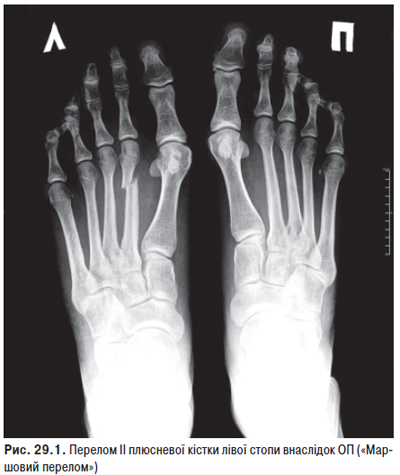
Крім того, чинниками, що зумовлюють розвиток ОП, можуть бути:
1. Генетичні/антропометричні фактори:
- літній вік;
- етнічна приналежність (європеоїдна й монголоїдна раси);
- низька маса тіла;
- сімейний анамнез переломів;
- поліморфізм гена рецептора D (BB-генотип);
- поліморфізм гена естрогенових рецепторів.
2. Гормональні чинники:
- жіноча стать;
- рання менопауза;
- пізній початок менструацій;
- аменорея;
- безпліддя.
3. Спосіб життя/особливості харчування:
- паління;
- недостатнє або надлишкове фізичне навантаження;
- зловживання кофеїном;
- алкоголізм.
4. Супутні хвороби:
- ендокринні;
- ревматичні;
- гематологічні/онкологічні;
- інші.
5. Лікарські засоби/хірургічні операції:
- оваріоектомія;
- гормони щитоподібної залози;
- ГК;
- хіміотерапія та ін. (табл. 29.1).
Таблиця 29.1
Чинники ризику ОП і переломів кісток у жінок європеоїдної раси у постменопаузальний період
|
Основні чинники ризику |
|
|
Додаткові чинники ризику |
|
Незважаючи на те, що ОП і остеопоротичні переломи розглядаються як патологія, що характерна для жінок у постменопаузальний період і у літньому віці, ОП нерідко виявляють і в чоловіків (табл. 29.2).
Таблиця 29.2
Чинники ризику ОП у чоловіків
|
Ступінь ризику |
Причина |
|
Високий ризик |
|
|
Помірний ризик |
|
|
Більш рідкісні причини |
|
При оцінці ризику переломів і прийнятті рішення про доцільність медикаментозної терапії поряд з показниками МЩКТ слід враховувати такі запропоновані ВООЗ чинники ризику:
- наявність раніше перенесеного перелому;
- застосування ГК;
- сімейний анамнез переломів;
- паління;
- зловживання алкоголем;
- низька маса тіла.
Переважна більшість випадків захворювання (до 85%) належить до первинного ОП. Однак серйозною медичною проблемою є і вторинний ОП, що виникає при різних захворюваннях (ревматичних, хронічній обструктивній хворобілегень і бронхіальній астмі, захворюваннях нирок, органів травлення), після тривалої іммобілізації і тривалого прийому ряду препаратів (ГК, імуносупресорів, протисудомних препаратів, гормонів щитоподібної залози та ін.).
Епідеміологія, патогенез та клінічні особливості ГК-індукованого остеопорозу
З 1948 р. з моменту відкриття ГК Хенчем і Кендаллом їх широко застосовують у клінічній практиці. І на сьогодні ми не уявляємо собі лікування хворих без застосування ГК, особливо при ревматичних хворобах.
ГК широко застосовують при ревматичних, бронхолегеневих, ендокринних та шкірних хворобах, при трансплантації тощо.
Але побічні ефекти, які виникають внаслідок прийому ГК, змусили переглянути показання до їх призначення та більш ретельно вивчати побічні ефекти. Переломи — найчастіший побічний ефект, викликаний прийомом цих препаратів (табл. 29.3).
Таблиця 29.3
Побічні ефекти внаслідок прийому ГК
|
Побічні ефекти |
Лікування ГК (n=112) |
Без лікування ГК (n=112) |
|
Переломи кісток |
21 (19%) |
8 (7%) |
|
Катаракта |
17 |
5 |
|
Інфекції |
14 |
4 |
|
ШКТ кровотечі, виразки |
11 |
4 |
|
Цукровий діабет |
8 |
3 |
|
Оперізувальний лишай |
8 |
1 |
|
Інфаркт міокарда |
4 |
4 |
|
Інсульт |
6 |
1 |
|
Глаукома |
1 |
1 |
|
Летальний кінець |
2 |
2 |
Частота прийому ГК (перорально та парентерально) не відрізняється у чоловіків і жінок, але збільшується з віком.
Метааналіз проспективних популяційних досліджень, проведених у різних країнах світу, показав, що у віці до 30 років приймали ГК 3%, а у віці 80 років — 5,2% пацієнтів. У Великобританії 0,5% населення системно приймають ГК більше 3 міс, а у віковій групі ≥55 років — 1,4%. Крім того, за даними B. Ettinger та співавторів (2001), близько 1% дорослого населення Великобританії приймають ГК (>70 років — 2,4%). У США ГК приймають 1–3% чоловіків та жінок віком близько 50 років.
Про ризик розвитку ГК-індукованого ОП (ГКіОП) відомо, що його відмічають майже у 50% пацієнтів, яким проводилася довготривала терапія. У великому когортному дослідженні у Великобританії виявлено, що відносна частота клінічного вертебрального перелому у пацієнтів, які отримували високі дози ГК, становила 5,18. Відносна частота клінічного вертебрального перелому у пацієнтів, що отримували середні дози, становила 2,6 з відносною частотою перелому кісток кульшового суглоба та невертебрального перелому 1,6 та 1,3 відповідно. Виявлено, що частота перелому залежить від дози, причому у разі припинення терапії ГК частота переломів поверталася до рівня початку лікування.
ГКіОП — це ОП, що пов’язаний з прийомом ГК.
Код за МКХ-10
М81.4 Медикаментозний остеопороз.
М80.4 Медикаментозний остеопороз з патологічним переломом.
ГКіОП — друга (після постменопаузального ОП) найбільш часта форма ОП і сама розповсюджена форма вторинного ОП (останнє пов’язано зі зниженням МЩКТ та підвищеним ризиком перелому у всьому скелеті. Відомо, що особливого впливу зазнають місця зі значною кількістю губчастих кісток, а саме хребет та ребра.
Інгаляційні ГК, менш ймовірно, викликають системні ефекти, порівняно з пероральними ГК, однак вони можуть спричиняти адреналову супресію і зменшувати МЩКТ при застосуванні у високих дозах.
Ризик перелому, характерний для ГКіОП:
- від дози ГК, як кумулятивної, так і щоденної, залежить втрата кісткової маси;
- відомо, що при короткотривалій терапії втрата кісткової маси є зворотною у разі припинення лікування, тоді як довготривала терапія спричинює стійке зниження МЩКТ, що підвищує ймовірність переломів;
- необхідне визначення МЩКТ — як початково, до терапії ГК, так і швидкість її подальшої втрати.
Втрата кісткової маси та руйнування кісток залежать від типу та дози ГК і проявляється найбільш виражено протягом перших 6 міс лікування.
Клінічні фактори, що підвищують категорію ризику розвитку ГкіОП (Grossman J.M. et al., 2010):
- низький індекс маси тіла;
- переломи стегна у батьків;
- паління;
- вживання ≥3 доз алкогольних напоїв на день;
- висока добова доза ГК;
- висока кумулятивна доза ГК;
- використання в/в пульс-терапії ГК;
- зниження показника центральної МЩКТ, що перевищує найнижчі достовірні значення.
Загальна характеристика ГКіОП:
- швидке зниження МЩКТ, особливо в перші 6–12 міс терапії ГК;
- після припинення прийому ГК можливе відновлення МЩКТ;
- прийом ГК в дозі >5–7,5 мг/добу протягом року підвищує ризик переломів кісток приблизно вдвічі;
- найбільш типові переломи хребців і стегнової кістки;
- при ГКіОП ризик переломів меншою мірою залежить від зниження МЩКТ, ніж при постменопаузальному ОП.
Механізми, залучені до розвитку ГкіОП (рис. 29.2), наступні:
- ГК викликають зниження функції остеоцитів та підвищення їх апоптозу, зниження диференціації та функції остеобластів та збільшують їх апоптоз, а також підвищують активацію остеокластів та зменшують їх апоптоз, що призводить до підвищення резорбції кісток, зниження формування та якості кістки, зменшення кісткової маси, що обумовлює ризик переломів;
- впливають на нейроендокринну систему: знижують синтез гормону росту, інсуліноподібного фактора росту-1 та трансформуючого фактора росту-β, що призводить до погіршення формування кістки, підвищення ризику переломів;
- знижують рівень статевих гормонів, що зумовлює підвищення кісткової резорбції, зменшення кісткової маси та підвищення ризику переломів;
- безпосередньо впливають на метаболізм кальцію, а саме: зменшують кишкову абсорбцію, підвищують ниркову екскрецію, внаслідок чого виникає негативний баланс кальцію, що також підвищує кісткову резорбцію;
- знижують експресію до вітаміну D і синтез паратиреоїдного гормону;
- знижують синтез PgE2 і тим самим негативно впливають на репарацію кісткової тканини;
- безпосередньо впливають на м’язи, викликають лізис міофібрил, внаслідок чого розвиваються міопатія, м’язова слабкість, що призводить до ризику падінь та підвищення ризику переломів.
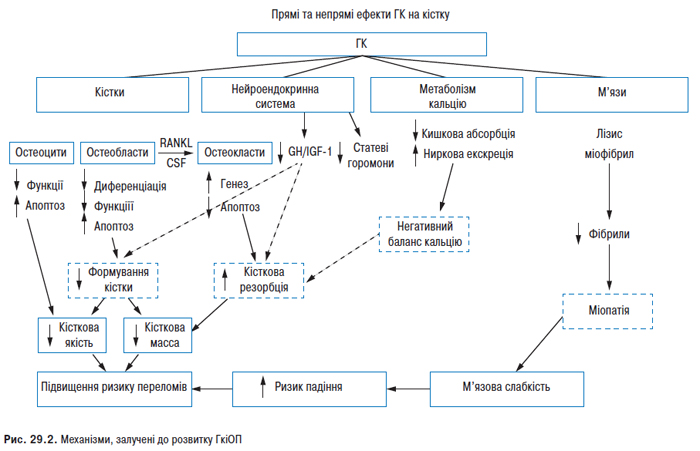
Експериментально встановлено зниження синтезу остеобластами RANKL/OPG (зниження остеопротегерину) під дією дексаметазону.
Крім того, експериментально також встановлено пригнічення апоптозу остеокластів ГК.
Дані щодо впливу на кістку та інші системи та тканини є підґрунтям для вибору антиостеопоротичної терапії, що спрямована як на пригнічення резорбції, так і на формування нової повноцінної кістки.
Вивчення ОП вимагає, з одного боку, більш детальних досліджень причин і механізмів розвитку хвороби, з іншого — цілеспрямованого і раціонального застосування лікувальних і профілактичних заходів, що спрямовані на попередження його розвитку і прогресування.
Морфологія кісткової тканини
Кісткова система являє собою живу тканину, що метаболічно активна протягом усього життя. Кісткова тканина — основа для м’язів і руху, у ній зберігаються мінерали, особливо кальцій, вона є захисним і живильним середовищем для кісткового мозку. Скелетна система морфологічно адаптована до механічних навантажень, вона одночасно є і легкою, і міцною.
Для розуміння патогенезу ОП фундаментальне значення мають нормальна структура кісткової тканини (гістологія) і процес її постійної перебудови (ремоделювання).
З морфологічної точки зору існує два основних типи кісткової тканини: кортикальна, чи компактна, що становить 80% скелета, і губчаста, чи трабекулярна, що становить 20% скелета.
Трубчасті кортикальні кістки здатні витримувати велике навантаження, що тисне, не ламаючись, але не можуть занадто сильно згинатися. Губчасті трабекулярні кістки менш витривалі стосовно граничного навантаження, але мають більшу гнучкість, ніж кортикальні.
Кістки складаються з органічного матриксу, неорганічного матриксу і клітин. Органічний матрикс, що становить 1/3 маси кістки, утворений колагеновими білками і протеогліканами (табл. 29.4).
Таблиця 29.4
Компоненти кісткової тканини
|
Органічний матрикс |
Колагенові волокна Протеоглікани |
|
Неорганічний матрикс |
Кристали гідроксиапатиту |
|
Клітини |
Остеокласти Остеобласти Остеоцити |
Неорганічний матрикс, що становить ⅔ маси кістки, складається із солей кальцію у формі кристалів гідроксиапатиту.
Обмін у кістковій тканині
Клітини кісткової тканини — це остеокласти, остеобласти, остеоцити та клітини, що вистилають.
Остеокласти — це багатоядерні гігантські клітини, що походять з клітин системи фагоцитуючих мононуклеарів кісткового мозку або периферичної крові (моноцитів, макрофагів), які після злиття 3–4 клітин у середовищі кісткової тканини диференціюються у зрілі остеокласти. Остеокласти здатні розчиняти мінералізований кістковий матрикс.
Остеобласти — це маленькі одноядерні клітини, що синтезують і відкладають остеоїд (який переважно складається з колагену) у кісткових заглибленнях, сформованих раніше остеокластами. Через кілька діб органічний матрикс (колаген, протеоглікани), утворений остеобластами на поверхні кістки, мінералізується шляхом відкладення кальцію і фосфору, що формують кристали гідроксиапатиту. Остеобласти походять із мезенхіальних стовбурних клітин сполучної тканини кісткового мозку. Остеобласти здатні змінювати свої функціональні і морфологічні характеристики і після того, як вони оточуються створеним ними мінералізованим кістковим матриксом, можуть перетворюватися у клітини, що вистилають, які розташовуються на поверхні кістки, або в остеоцити.
Остеоцити — це клітини полігональної форми з довгими відростками, що утворюють лакуну (порожнину) для цих клітин. Кожен остеоцит за допомогою канальців зв’язаний із сусідніми остеоцитами: так утворюється клітинний синцитій.
Вважається, що остеоцити розташовуються у ділянках найбільшої механічної напруги кістки (наприклад у зонах мікроушкоджень) і відіграють певну роль у гомеостазі кальцію, здійснюючи періостеоцитарну резорбцію.
Моделюванням кістки називають процес утворення кістки заново на кісткових поверхнях, там, де кісткової тканини раніше не було; цей процес відбувається у період росту.
Ремоделюванням кістки називають гармонізований сполучений процес, що складається з резорбції старої кісти остеокластами та наступної заміни новою кісткою, що утворена остеобластами. Обидва процеси відбуваються в одному і тому самому місці, підтримуючи, таким чином, структурну цілісність кістки.
Цикл ремоделювання кістки складається із послідовних фаз, що зазначені нижче (рис. 29.3):
1) активація остеокластів;
2) фаза резорбції кістки;
3) фаза реверсії — активації остеобластів;
4) фаза формування кістки;
5) фаза мінералізації кісткового матрикса;
6) фаза спокою.
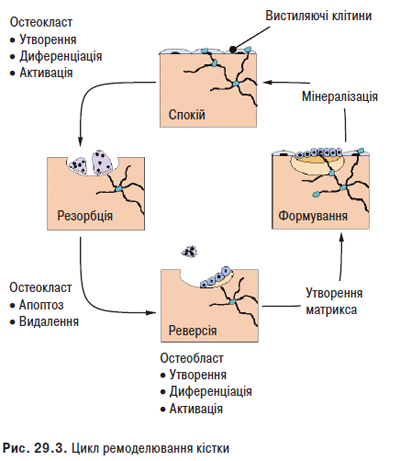
Маса кістки людини на більш пізніх етапах життя залежить від піку кісткової маси, що досягається у період внутрішньоутробного розвитку, у дитинстві та статевому дозріванні, а також від наступної швидкості її втрати.
Величина піку маси кісткової тканини — це максимальна маса кістки, що досягається протягом життя людини.
Важливість величини піку маси кісткової тканини для подальшої міцності кістки вперше виявлена в одномоментних дослідженнях, результати яких свідчать, що якщо у 30-річної людини кісткова маса відповідає верхній границі вікової норми, то існує велика імовірність її збереження також у межах вікової норми й у 70 років.
Вік досягнення піку маси кісткової тканини розрізняють залежно від ділянки скелета і методу оцінки. При використанні методу двохенергетичної рентгенівської абсорбціометрії (dual-energy X-ray absorbtiometry — DEXA) показано, що максимальна «поверхнева» кісткова щільність у хребті і шийці стегнової кісти досягається у віці до 30 років, тоді як кісткова маса всього скелета — на 6–10 років пізніше.
На пік маси кісткової тканини впливає безліч факторів, що взаємодіють (генетичні, гормональні, аліментарні, фізичні, навколишнього середовища) (рис. 29.4).
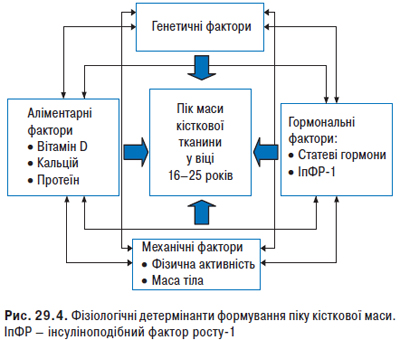
Діагностичні ознаки остеопорозу
Відповідно до рекомендацій робочої групи ВООЗ), стан, при якому Т-критерій становить 2,5 SD і більше (тобто МЩКТ на 2,5 SD відхиляється від нормального середнього значення для дорослих молодих людей), класифікується як ОП, а коли Т-критерій відхиляється від середнього значення більше ніж на -1,0 SD, але менше ніж на 2,5 SD — як остеопенія (табл. 29.5).
Таблиця 29.5
Класифікація ОП за даними кісткової денситометрії (ВООЗ, 1994)
|
Норма |
Т-критерій > –1,0 SD |
|
Остеопенія |
–2,5 SD <Т-критерій < —1,0 SD |
|
ОП |
Т-критерій <–2,5 SD |
Оскільки остеопенія значно поширеніша, ніж ОП, більшість переломів відмічають у популяції пацієнтів з остеопенією.
Сучасний підхід до діагнозу ОП складається з ретельного збору анамнезу, фізикального обстеження, а також проведення загальних і спеціальних лабораторних досліджень.
Діагностика та диференційна діагностика ГК-індукованого остеопорозу
Клінічне обстеження: зменшення росту на 2 см — грудний кіфоз; скарги на біль у спині; поява відчуття стомленості в спині у положенні сидячи або стоячи.
Інструментальне обстеження: рентгенологічне дослідження грудного та поперекового відділів хребта у прямій та боковій проекції.
DEXA. Для ранньої діагностики ГКіОП перевага надається визначенню МЩКТ хребта; для оцінки ризику переломів — МЩКТ проксимального відділу стегна.
Для регулярного контролю кісткової структури в повсякденну практику увійшли методи оцінки МЩКТ, зокрема ультразвукова денситометрія.
На сьогодні для вимірювання МЩКТ активно використовуються DEXA і кількісна комп’ютерна томографія (ККТ).
Найперші зміни, пов’язані з ГКіОП, відмічають в поперековому відділі хребта через значну кількість губчастої кістки. Рекомендується проведення вимірювання DEXA поперекового відділу хребта (передньозаднє сканування) та шийки стегна, коли пацієнти вперше починають терапію ГК або невдовзі після цього, оскільки випромінювання при ККТ сильніше за DEXA (рис. 29.5, 29.6).
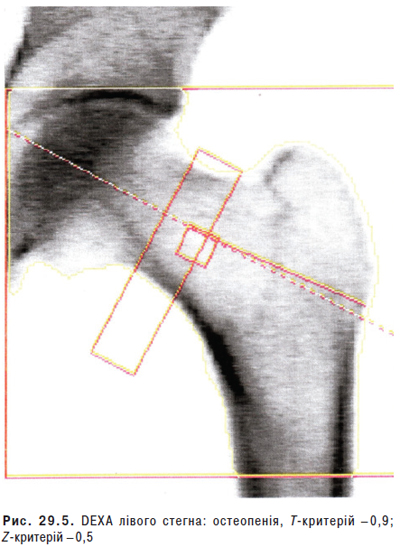

Частота втрати кісткової маси хребців варіює від 5 до 10% на рік у пацієнтів, які приймали високі дози ГК. Відповідно рекомендується проводити повторні DEXA-сканування кожні 12 міс.
При порівнянні частоти виникнення переломів у групах плацебо в декількох клінічних дослідженнях виявлена значно вища частота вертебральних переломів у пацієнтів з ГКіОП, порівняно з постменопаузальним ОП, незважаючи на вищу вихідну МЩКТ при ГКіОП.
При визначенні МЩКТ методом DEXA мають значення такі показники:
- кістковий мінеральний компонент (bone mineral content, BMC) — кількість мінералізованої тканини (г) при скануванні кісток зазвичай визначають за довжиною сканувального шляху (г/cм);
- МЩКТ (bone mineral density, BMD) — зазвичай оцінюють кількість мінералізованої кісткової тканини на сканованій площі — (г/cм²).
При ККТ виконується трипросторове вимірювання МЩКТ, яке визначає кількість мінералізованої кісткової тканини на об’єм кістки (г/см³), проте для даного методу діагностики характерне велике променеве навантаження порівняно з технологією DEXA, що робить двомірне визначення МЩКТ (г/cм²) при DEXA більш поширеним. Після отримання результату індивідуальна МЩКТ порівнюється з референсною базою даних, оцінка МЩКТ відбувається за Т- і Z-критеріями. DEXA — найбільш точний метод моніторингу терапії, інтервали між динамічними дослідженнями становлять 12–24 міс.
Т-критерій — кількість стандартних відхилень вище або нижче середнього показника піку кісткової маси молодих осіб з найбільшою кістковою масою. Він зменшується паралельно з поступовим зниженням кісткової маси при збільшенні віку обстежуваних осіб.
Z-критерій — кількість стандартних відхилень вище або нижче середнього показника для осіб аналогічного віку.
Визначення ОП розроблено ВООЗ в 1994 р. для жінок європеоїдної раси і базується на визначенні МЩКТ за Т-критерієм. У 2007 р. Міжнародним товариством з клінічної денситометрії запропонована нова інтерпретація результатів денситометрії за Т- і Z-критеріями. У разі застосування пацієнтом препаратів для лікування ОП, що здатні поглинати рентгенівські промені сильніше, ніж атоми кальцію, при вимірюванні МЩКТ методом DEXA виникає помилка, що вимагає застосування додаткових коефіцієнтів перерахунку. На основі експериментальних досліджень на приматах виведена формула:
Кінцева МЩКТ = виміряна МЩКТ поперекових хребців/1+0,061ЧBSC клубової кістки (%),
де BSC — вміст стронцію в клубовій кістці, визначений при кістковій біопсії і виражений в молярних відсотках вмісту стронцію.
ККТ, особливо периферична, є зручним методом, що дає можливість виявляти порушення кісткової структури навіть на початкових стадіях. Променеве навантаження при цьому набагато нижче, ніж при звичайній рентгенографії органів грудної клітки.
МРТ — перспективний метод, особливо для оцінки стану кісток периферичного скелета, який продовжує розвиватися як у технічному відношенні, так і в плані отримання зображення, що надалі може дозволити визначати не лише кількісну інформацію, але й відомості про якість кісткової структури.
Для верифікації діагнозу використовується алгоритм діагностики ОП (табл. 29.6).
Таблиця 29.6
Алгоритм діагностики ОП
|
Клінічні прояви |
Обстеження |
||
|
Чи був недавно перелом? |
Був |
Перелом хребта |
Диференційний діагноз |
|
|||
|
|||
|
|||
|
|||
|
Перелом проксимальної ділянки стегнової кістки |
Визначення кісткової маси та біохімічних маркерів кісткового метаболізму |
||
|
Інші переломи (наприклад зап’ястка) |
|||
|
Не було |
Підозра на ОП |
||
|
Чинники ризику ОП |
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
Відсутність усього, що перелічено вище |
Обстеження не проводиться |
||
Класифікацію диференційної діагностики наведено в табл. 29.7.
Таблиця 29.7
Диференційна діагностика ГКіОП
|
Первинний ОП
|
Остеомаляція
|
|
Ендокринні розлади
|
Захворювания гемопоетичної системи
|
|
Захворювання сполучної тканини
|
Захворювания ШКТ
|
|
Порушення функції нирок
|
Медикаментозна терапія
|
|
Різні інші причини
|
Міжнародна асоціація остеопорозу (IOF) для скринігу пацієнтів пропонує використовувати 1-хвилинний тест для оцінки чинників ризику ОП.
Тест Міжнародної асоціації остеопорозу для оцінки факторів ризику остеопорозу
1. Сімейний анамнез
- У когось із ваших батьків виявляли ОП або перелом внаслідок незначного удару?
- У когось із ваших батьків відмічали порушення постави у зрілому віці («овдовілий горб»)?
2. Анамнез
- Вам 40 років або більше.
- У Вас були переломи кісток після незначних ударів або падінь у зрілому віці?
- Ви часто падаєте (більше 1 разу за минулий рік) або боїтеся падінь через слабкість?
- Ваш ріст зменшився більше ніж на 3 см (після 40 років)?
- У Вас низька маса тіла (індекс маси тіла менше 19 кг/м2).
- Ви коли-небудь приймали препарати кортикостероїдів (кортизон, преднізолон) більше 3 міс?
- Ви хворієте на РА?
- У Вас виявлено підвищення функції щитоподібної або паращитоподібної залози?
Для жінок:
- У Вас наступила менопауза у віці до 45 років?
- У Вас відсутня менструація 12 міс і більше, що не пов’язано з вагітністю, менопаузою або видаленням матки?
- Ви перенесли операцію з видалення яєчників у віці до 50 років без наступного прийому гормонозамісної терапії?
Для чоловіків:
- Ви страждали від імпотенції, зниження лібідо чи інших симптомів, пов’язаних зі зниженням рівня тестостерону?
Ваш спосіб життя:
- Ви вживаєте алкоголь більше безпечної кількості (більше, ніж еквівалент 20 мл спирту на добу)?
- Ви палите або палили коли-небудь?
- Ви приділяєте менше 30 хв на день фізичним вправам (роботі у саду, у будинку та ін.)?
- Ви споживаєте мало молочних продуктів (внаслідок алергії або з інших причин) без замісного прийому препаратів кальцію?
- Ви мало часу проводите на свіжому повітрі й сонці (менш 10 хв на день) без замісного прийому вітаміну D?
Позитивна відповідь на будь-яке запитання не свідчить про наявність у пацієнта ОП, це означає наявність клінічно підтверджених факторів ризику, що можуть призвести до остеопоротичних переломів. Подальше ведення хворого залежить від лікаря (подальше дослідження та/або призначення терапії).
У спеціалізованих лабораторіях використовуються методи визначення біохімічних маркерів кісткового метаболізму.
Лабораторне обстеження:
- загальний аналіз крові (при значній ШОЕ — виключити мієломну хворобу);
- рівень кальцію, фосфору, ЛФ, альбуміну (виключити гіперпаратиреоз та остеомаляцію);
- йодовмісні гормони щитоподібної залози (виключити гіпертиреоз);
- загальний тестостерон у чоловіків (виключити гіпогонадизм);
- вітамін D;
- рівень фолікулостимулюючого гормону, лютеїнізуючого гормону й естрадіолу при аменореї (виявлення супутньої нозології).
Специфічні маркери кісткового метаболізму представлені у табл. 29.8.
Таблиця 29.8
Специфічні маркери кісткового метаболізму
|
Показники кісткоутворення |
Показники резорбції кістки |
|
Кров + ЛФ (загальна активність і кісткова фракція) + Остеокальцин (BGP) Проколагеновий пептид I типу (PICP) |
Кров Тартратрезистентна кисла фосфатаза (TRAP) Вільна 7-карбоксиглутамінова кислота |
|
Сеча Гідроксипролін, що не діалізується |
Сеча + Відношення загального гідроксипроліну і гідроксипроліну, що діалізується, та креатиніну + Відношеня кальцію та креатиніну Метаболіти піридину Гідроксилізилпіридинолін Лізилпіридинолін Глікозиди гідроксилізину — лактозил-гідроксилізин — глюкозил-лактозил-гідроксилізин |
Позначка «+» стосується показників, які найчастіше використовуються у клінічній практиці.
*Перевагу надають дослідженню проби сечі, що зібрана протягом 2 год вранці натще.
Механізми розвитку остеопорозу
Для визначення вибору адекватної терапії ОП важливе значення має розуміння механізмів його розвитку. Важливим патогенетичним механізмом втрати кісткової маси в жінок у постменопаузальний період є дефіцит естрогенів. У нормі естрогени відіграють центральну роль у фізіологічному ремоделюванні, а дефіцит естрогенів після менопаузи призводить до порушення балансу ремоделювання. Такий дисбаланс зумовлює прогресуючу втрату трабекулярної кістки, у тому числі через збільшення остеокластогенезу, що, очевидно, є результатом продукції остеокластогенних прозапальних цитокінів (ІЛ-1 і ФНП-α), активність яких під впливом естрогенів знижується.
Безпосередній вплив естрогенів на прискорення апоптозу остеокластів також зумовлений збільшенням продукції трансформуючого фактору росту-b.
В останні роки відзначається швидкий прогрес у розумінні клітинних основ ремоделювання, зокрема, встановлено, що активатор рецептора нуклеарного фактора каппа-В (RANK), його ліганд (RANKL) і псевдорецептор остеопротегерин є ключовими регуляторами кісткової резорбції остеобластами in vitro та in vivo (рис. 29.7). Остеобласти конституційно експресують на своїй поверхні RANKL, який взаємодіє зі своїм рецептором, що розпізнає RANK, який експресується на попередниках остеокластів і стимулює диференціювання останніх. Взаємодія RANKL з RANK на зрілих остеокластах приводить до активації й підвищення їх виживаності. Остеопротегерин, що знаходиться у мікросфері кісткової тканини, в основному, секретується остеобластами і стромальними клітинами та блокує взаємодію RANKL з RANK і тим самим діє як фізіологічний регулятор процесів відновлення кісткової тканини. (рис. 29.8) Естрогени також проявляють частину своїх антирезорбтивних ефектів у кістці, стимулюючи експресію остеопротегерину в остеобластах.
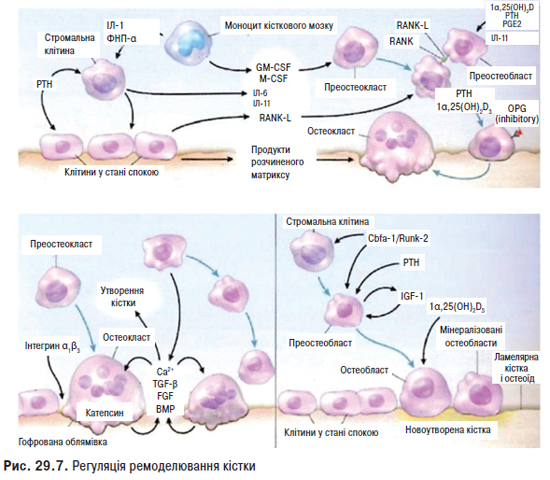
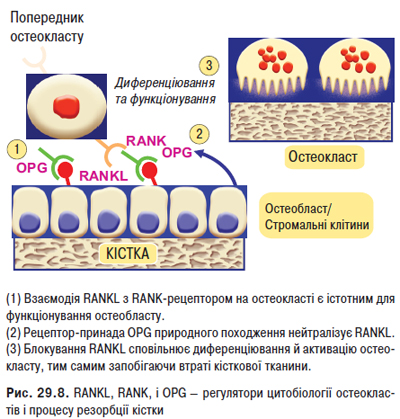
Крім того, при зниженні рівня естрогенів (або їх відсутності) збільшується пул Т-клітин, що секретують ФНП-α, а також зменшується вміст трансформуючого фактора росту-b. При цьому моноцити секретують у підвищеній кількості ІЛ-1. ФНП-α стимулює синтез макрофагального колонієстимулюючого фактора (М-КСФ) і RANKL, що сприяє збільшенню кількості остеокластів. RANKL і ІЛ-1 також підвищують виживаність остеокластів, запобігаючи їх апоптозу.
(1) Взаємодія RANKL з RANK-рецептором на остеокласті є істотним для функціонування остеобласту.
(2) Рецептор-принада OPG природного походження нейтралізує RANKL.
(3) Блокування RANKL сповільнює диференціювання й активацію остеокласту, тим самим запобігаючи втраті кісткової тканини.
На сьогодні відкриті й інші нові важливі гени і шляхи диференціації та функції остеобластів — білок LRP5, рецептори Frizzled і Kremen, білок Dickkopf, склеростин (продукт гена SOST) тощо, що розкривають механізми ремоделювання кісткової тканини, але мають поки що в основному теоретичне значення (табл. 29.9).
Таблиця 29.9
Майбутні лікарські засоби, що базуються на новітніх механізмах фармакологічної дії
|
Лікарський засіб |
Ефект |
|
Види терапії на основі антитіл, що спрямовані проти:
Інгібітори катепсину K Кальцієміметики (цинакальцет) Лептин |
Антирезорбтивний Анаболічний Анаболічний Антирезорбтивний Антирезорбтивний? Анаболічний? |
Сучасні підходи до лікування остеопорозу
Сприйнятливість кістки, а також системних і місцевих гормонів до фармакологічних препаратів надає безліч можливостей для створення лікарських препаратів, що сприятливо впливають на кісткову тканину. Розробка і синтез нових лікарських засобів може базуватися на:
- блокаді RANKL за допомогою антитіл;
- розробці інгібіторів катепсину К, протеази цистеїну, що вибірково експресуються в остеокластах і беруть участь у розщепленні колагену, відіграючи ключову роль у резорбції кістки;
- розробці кальцієміметиків, призначених для зв’язування з кальційчутливими рецепторами у клітинах паращитоподібних залоз.
Крім того, лептин (гормон, отриманий з адипоцитів), який регулює процес відтворення і метаболізму ліпідів, також є потенційною мішенню для створення нового лікарського засобу.
Препарати, які застосовують у теперішній час, мають один із трьох механізмів дії:
- гальмування резорбції кістки (антирезорбтивні чи антикатаболічні препарати);
- стимуляція кісткоутворення (анаболічні препарати), наприклад, терипаратид і інші форми паратиреоїдного гормону;
- препарати нового покоління, що комбінують обидва механізми (є лише один препарат даного покоління — стронцію ранелат).
Профілактика та лікування ГК-індукованого остеопорозу на основі клінічних рекомендацій EULAR та ACR
Первинна профілактика
Важливе місце в профілактиці ГКіОП займають освітні програми з метою своєчасного виявлення осіб з підвищеним ризиком розвитку ГКіОП та пропаганда здорового способу життя. З метою профілактики ГКіОП рекомендуються регулярні фізичні вправи, корисні для осіб будь-якого віку. Особливо важливі вправи, спрямовані на тренування рівноваги, які знижують ризик падінь.
Вторинна профілактика
Попередження переломів за наявності ГКіОП, а саме: прийом лікарських засобів, профілактика падінь, носіння протекторів стегна (за показаннями).
Здоровий спосіб життя, а саме повноцінне харчування, підвищена рухова активність, виконання фізичних вправ, корекція маси тіла. Відмова від куріння та зловживання алкоголем.
Кальцій — один з основних мінералів, який відіграє значну роль у підтримці міцності скелету.
Важливе місце займає корекція харчування хворих на ГКіОП, а саме:
- достатній прийом кальцію (мінімум 1200 мг/добу),
- вітаміну D (мінімум 800 МО/добу),
- білка (1–1,2 г/мг/добу),
- обмеження вживання солі та кави.
На сьогодні встановлено, що вагомим чинником у патогенезі падінь є дефіцит D-гормону (кальцитріолу) в організмі пацієнтів з ГКіОП. D-гормон відіграє важливу роль як у диференціації, проліферації клітин скелетних м’язів, так і в реалізації Са2+-залежних механізмів, що є одними з центральних у процесі м’язового скорочення. Слід відмітити, що відновлення рівня D-гормону є центральною ланкою в профілактиці падіння та спонтанних малотравматичних переломів. Відновлення можливе шляхом призначення нативного вітаміну D або його активних метаболітів.
Попередження спонтанних падінь за допомогою активних метаболітів набагато ефективніше, ніж при використанні нативного вітаміну D, що підтверджує ряд великих досліджень (STOP/IT) і метааналізи.
Сучасні підходи до лікування ГКіОП передбачають урахування рекомендацій EULAR та ACR.
Рекомендації EULAR щодо застосування ГК при ревматичних хворобах підтверджені доказовою базою (2007) (табл. 29.10, рис. 29.9).
Таблиця 29.10
Рекомендації EULAR щодо застосування ГК при ревматичних хворобах
|
Рекомендації |
Ступінь впевненості рекомендацій |
|||
| ВАШ 100 (95% ДІ) | А + В% | Рівень доказовості | ||
|
6А |
Якщо пацієнт почав приймати преднізолон з дози >7,5 мг/добу та продовжує приймати цей препарат більше 3 міс, йому слід призначити додатково Са та вітамін D |
95 (91–99) |
100 |
ІА |
|
6В |
Антирезорбтивна терапія бісфосфонатами для зниження ГКіОП повинна ґрунтуватися на факторах ризику, в тому числі на вимірюванні МЩКТ |
96 (92–99) |
93 |
ІВ–ІІІ |
|
6 |
Повна пропозиція (6А+6В) |
95 (89–100) |
||
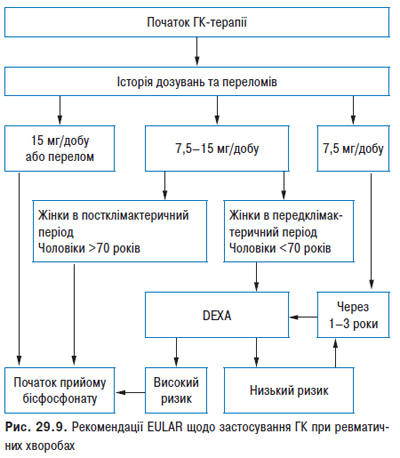
Рекомендації АСR (2010) з профілактики та лікування ГКіОП (вік ≥50 років, тривалість терапії ГК ≥3 міс) (Grossman J.R. et al., 2010):
- вживання кальцію в дозі 1200–1500 мг/добу (у вигляді добавки + пероральний прийом);
- вживання вітаміну D у вигляді добавки.
Пацієнти низького ризику:
- алендронат для прийому ≥7,5 мг/добу за преднізолоном
або
- ризедронова кислота для прийому ≥7,5 мг/добу за преднізолоном
або
- золедронова кислота для прийому ≥7,5 мг/добу за преднізолоном.
Пацієнти помірного ризику:
- алендронат незалежно від дози ГК
або
- ризедронова кислота незалежно від дози ГК
або
- золедронова кислота для прийому ≥7,5 мг/добу за преднізолоном.
Пацієнти високого ризику:
- алендронат або ризедронова кислота
або
- золедронова кислота
або
- терипаратид*.
*Не зареєстрований в Україні.
Рекомендації АСR (2010) з профілактики та лікування ГКіОП (вік до 50 років зі спонтанними переломами в анамнезі) (Grossman J. et al., 2010):
- 1–3 міс терапії ГК/нерепродуктивного віку:
- алендронат при прийомі ≥5 мг/добу за преднізолоном
або
- ризедронова кислота при прийомі ≥5 мг/добу за преднізолоном
або
- золедронова кислота при прийомі ≥7,5 мг/добу за преднізолоном
-//- /репродуктивного віку — недостатньо даних для рекомендацій
- ≥3 міс терапія ГК/нерепродуктивного віку:
- алендронат при прийомі будь-якої дози за преднізолоном
або
- ризедронат при прийомі будь-якої дози за преднізолоном
або
- золедронова кислота при прийомі будь-якої дози за преднізолоном
або
- терипаратид* при прийомі будь-якої дози за преднізолоном
- -//- /репродуктивного віку:
- алендронат при прийомі ≥7,5 мг/добу за преднізолоном
або
- ризедронова кислота при прийомі ≥7,5 мг/добу за преднізолоном
або
- терипаратид* при прийомі ≥7,5 мг/добу за преднізолоном.
Усім пацієнтам, що отримують ГК разом з бісфосфонатами, обов’язково необхідно проводити корекцію дефіциту D-гормону і відновлення балансу Са з використанням вітаміну D (800 МО/добу). Проте, оскільки додавання вітаміну D з Са не запобігає втраті кісткової маси у пацієнтів, які одержували терапію ГК в середньотерапевтичних і високих дозах, для них слід надавати перевагу активним метаболітам вітаміну D з/без Са. Активні метаболіти вітаміну D можуть бути рекомендовані як профілактика ГКіОП. Комбінація бісфосфонат + активний метаболіт вітаміну D (1 мкг/д) ефективніша в попередженні переломів, ніж комбінація вітамін D + бісфосфонат або монотерапія (табл. 29.11).
Таблиця 29.11
Ефективність відновлення рівня D-гормону при різних типах D-дефіциту
|
I тип |
II тип |
III тип |
|
|
Тип D-дефіциту |
Первинний дефіцит вітаміну D |
Вторинний дефіцит D-гормону |
D-гормоно- резистентність |
|
Ефективність терапії вітаміном D |
+ |
– |
– |
|
Ефективність терапії альфакальцидолом |
+ |
+ |
+ |
Бісфосфонати на сьогодні є препаратами першої лінії в лікуванні і профілактиці ГКіОП. Механізм дії: фізико-хімічне зв’язування з гідроксиапатитом на резорбтивній поверхні; пряма дія на остеобласти з порушенням їх утворення, метаболізму і функціональної активності, індукції апоптозу і, як наслідок, пригнічення кісткової резорбції. Здатність бісфосфонатів пригнічувати патологічну резорбцію кісткової тканини визначає їх лікувальну дію при ОП.
Для лікування і профілактики ГКіОП в Україні із зареєстрованих препаратів застосовується золедронова кислота.
Золедронова кислота. Включення в лікування ГКіОП золедронової кислоти 5 мг на рік ґрунтується на дослідженні HORIZON GIO, в якому показано переважаючу ефективність золедронової кислоти над ризедроновою кислотою (статистично достовірна), як у групі лікування так і в групі профілактики (рис. 29.10, 29.11).
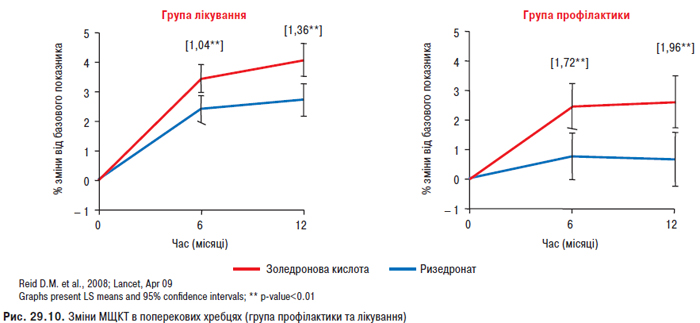
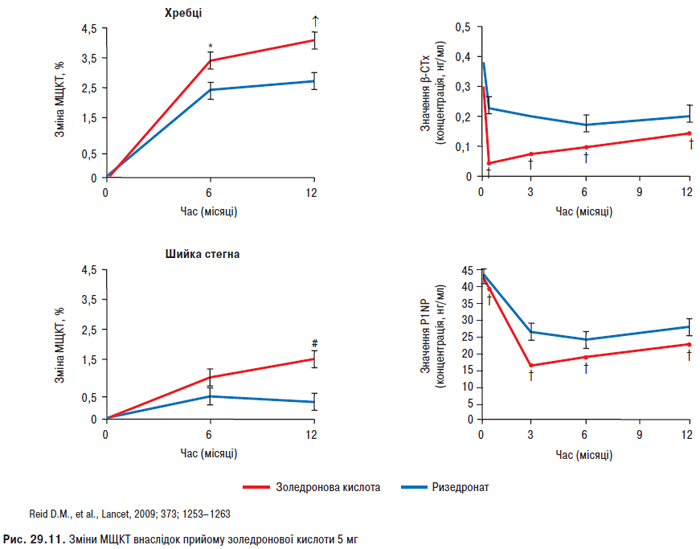
Дослідження HORIZON GIO дозволило зробити висновки щодо золедронової кислоти 5 мг:
- має перевагу в порівнянні з ризедроновою кислотою щодо покращення МЩКТ протягом 12 міс;
- для обох субпопуляцій продемонстрували високий профіль безпеки та лікування і в цілому добре переносилася;
- загальна частота серйозних побічних ефектів зіставна в обох групах порівняння;
- не зафіксовано збільшення порушення функції нирок після лікування золедроновою кислотою протягом 1 року.
Застосування золедронової кислоти 1 раз на рік та відсутність необхідності стояти або ходити протягом 30 хв (що необхідно при прийомі пероральних бісфосфонатів) призводить до високої комплаєнтності (прихильності) прийому даного препарату.
Профілактика ГКіОП показана всім пацієнтам, яким планується довготривала (>3 міс) терапія ГК в дозі >5 мг/добу.
Первинна — попередження розвитку ОП у пацієнтів із системною ГК терапією >7,5 мг/добу більше 3 міс.
Вторинна — попередження втрати кісткової маси і переломів у пацієнтів зі зниженою МЩКТ (>1–1,5 од.) та/або з переломами в анамнезі.
Принцип дії стронцію ранелату істотно відрізняється від інших сучасних антиостеопоротичних препаратів. Більшість доступних засобів є антирезорбтивними агентами (бісфосфонати, селективні модулятори естрогенових рецепторів, кальцитонін, деносумаб). Значення, ефективність і побічна дія цієї групи препаратів детально представлені у сучасній літературі.
З 90-х років ХХ ст. численними експериментальними, а потім клінічними дослідженнями показано, що солі стронцію, які розглядалися як кісткостимулювальні агенти, мають антирезорбтивну дію. Створений на основі стронцію і ранелової кислоти унікальний препарат стронцію ранелат має подвійний механізм дії: він підвищує формування кістки та у той же час знижує її резорбцію, регенеруючи кісткову тканину.
При терапії стронцію ранелатом відзначається мінімальне накопичення стронцію у кістковій тканині, що зумовлено його взаємодією з мінеральним компонентом кістки. Умовно можна виділити дві ділянки мінерального матриксу, де накопичуються іони стронцію.
Перша ділянка — це поверхневий шар кристала гідроксиапатиту і тісно пов’язана з ним оболонка, що складається з гідратованих іонів кальцію, карбонату і фосфату. Абсорбуючись на поверхні кристала, іони стронцію випадковим чином заміщають іони кальцію, що включені у кристалічну решітку верхніх шарів мінералу. Такі заміни у нормі постійно відбуваються з іонами кальцію та іншими іонами, що входять у структуру кристала (ізоіонні заміни), як у процесі його росту, так і на поверхні вже сформованого мінералу.
Другою ділянкою розподілу іонів стронцію у кристалі гідроксиапатиту є глибокий шар мінералу. Проте, кристал має більшу тропність до кальцію, ніж до стронцію, тому в біологічному гідроксиапатиті навіть при високих концентраціях Sr кількість замін Са на Sr завжди невелика. Виявлено, що 13-тижневий прийом стронцію у високій дозі — 750 мг/кг/добу — приводив до заміщення менше одного з 10 атомів кальцію в кристалічній решітці. В експериментах із дослідження кісткової тканини тварин і людей, що одержували препарати стронцію, показано, що кристали, до складу яких входить стронцій, мають більш правильну форму і більш стабільні. Одночасно з цим встановлено, що включення стронцію не позначається на розмірі кристала, параметрах кристалічної решітки і ступені упорядкованості структури.
Стронцію ранелат має унікальну здатність регенерувати кістку завдяки роз’єднанню процесів кісткоутворення і резорбції. Результати досліджень свідчать, що стронцію ранелат діє і на остеобласти, збільшуючи реплікацію їх попередників і приводячи до утворення кістки in vitro, і на преостеокласти, сприяючи зменшенню їх диференціації і реплікації в остеобласти, і на остеобласти — призводячи до збільшення їх апоптозу.
У проведених дослідженнях (рис. 29.12) з урахуванням нових даних установлено, що:
- стронцію ранелат зменшує диференціацію і знижує резорбтивну активність остеокластів за рахунок підвищення експресії остеопротегерину (OPG);
- стронцію ранелат пригнічує експресію RANKL, завдяки чому також знижується активність і диференціація остеокластів;
- стронцію ранелат збільшує апоптоз остеокластів через вплив на кальційчутливі рецептори;
- вплив стронцію ранелату на диференціацію остеобластів відбувається завдяки його агоністичному впливу на кальційчутливі рецептори, що приводить до стимуляції кісткоутворення;
- стронцію ранелат збільшує відповідь остеобластів також за допомогою інших катіон-чутливих рецепторів.
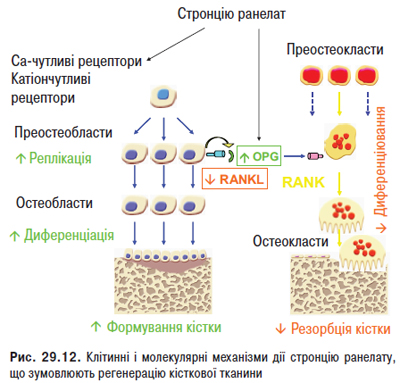
Таким чином, стронцію ранелат взаємодіє з кісткою як із живою тканиною, завдяки чому досягається його висока ефективність у запобіганні переломам.
Збереження нормальної фізіологічної мінералізації кістки поряд з відновленням порушеного балансу процесу ремоделювання кістки, що тонко регулюється, викликає формування нової, більш міцної кісткової тканини.
Для обґрунтування застосування будь-якого інноваційного препарату в клінічній практиці необхідне патогенетичне обґрунтування дії даного препарату, а також факти доказової медицини.
Підтвердженням унікального механізму дії стронцію ранелату стали експериментально-клінічні дослідження, в яких встановлено, що стронцію ранелат сприяє регенерації кістки й одночасно поліпшує якість кісткової тканини.
Результати досліджень біоптатів кісткової тканини, отриманих у пацієнток, які приймали стронцію ранелат у ході клінічних досліджень, демонструють, що кісткова тканина, синтезована на фоні терапії стронцію ранелатом, характеризується нормальною компактною структурою і має гарні якісні показники. Стронцію ранелат не порушує кристалічної структури кістки і процесу її мінералізації. Тривимірні мікроКТграми біоптатів кісток підтверджують істотний вплив стронцію ранелату на мікроархітектоніку губчастої і компактної речовини кістки.
Під впливом стронцію ранелату відбувається збільшення як кортикального, так і трабекулярного шарів кістки: збільшення кількості трабекул на 14% (р=0,05), зменшення міжтрабекулярних проміжків на 16% (р=0,04), збільшення товщини компактної речовини кістки на 18% (р=0,08) (рис. 29.13).
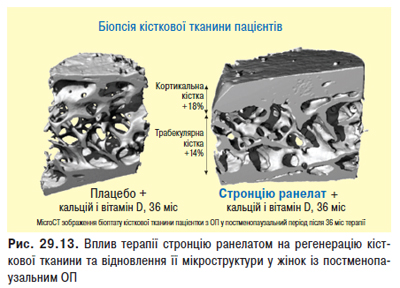
В експерименті доведено, що на фоні застосування стронцію ранелату кістка на 54% стає більш міцною і стійкою до навантаження, а за своїми якісними характеристиками відповідає більш молодому віку. Отже, під впливом стронцію ранелату відбувається високоякісна регенерація кісткової тканини. Саме цей факт пояснює його ефективність у профілактиці остеопоротичних переломів будь-якої локалізації незалежно від поєднання чинників ризику і тривалості попереднього лікування.
В експериментальному дослідженні (Li Y.F. et al., 2010) на щурах було доведено, що стронцію ранелат прискорює консолідацію переломів. На тлі прийому стронцію ранелату відзначено виражене прискорення формування кістки, підвищення МЩКТ і біомеханічної міцності, а також покращання макроструктурних властивостей кісткової тканини відносно контрольної групи оваріоектомованих щурів. Граничне навантаження збільшилося на 211 і 61,4% (р<0,01), а загальна кількість кісткової речовини мозолі — на 74,8 і 79,3% (р<0,01) через 4 і 8 тиж після перелому відповідно. Прийом стронцію ранелату також сприяв скороченню відновлювального періоду, оскільки був пов’язаний з посиленням остеогенезу через 4 тиж після перелому та формуванням більш зрілої, компактної кістки, відзначеної на гістологічних зрізах у місці перелому через 8 тиж (рис. 29.14).
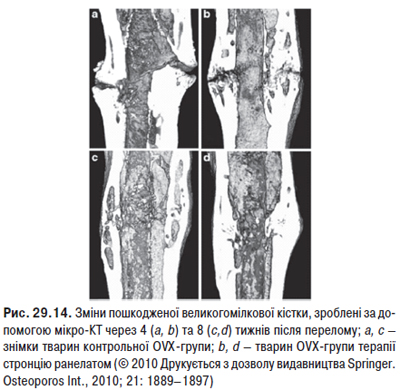
Нові дані щодо впливу стронцію ранелату на структуру кісткової тканини представлені професором Буссе під час 29-го Конгресу Американського товариства з вивчення кісткової тканини і мінерального обміну. У пацієнтів, які тривалий час отримували терапію бісфосфонатами і були переведені на стронцію ранелат, за результатами парних біопсій, проведених до початку і через 1 рік терапії стронцію ранелатом, відзначено достовірне збільшення об’єму кістки на 30% і товщини трабекул на 10%. Також зафіксовано збільшення активних ділянок формування кістки і позитивний вплив стронцію ранелату на її якість. Ці результати підтверджують подвійний механізм дії стронцію ранелату і його унікальну властивість регенерувати кісткову тканину.
На тлі терапії стронцію ранелатом відзначали значне покращення мікроархітектури кістки навколо імпланту (збільшення відношення об’єму кісткової тканини до загального об’єму кістки на 36%, потовщення трабекул — на 13% і підвищення їх щільності — на 23%), більш виражену пластинчасту структуру кістки і збільшення площини контакту імпланту з кісткою (+19%). Також зафіксовано істотний позитивний вплив стронцію ранелату на біомеханічні властивості кісткової тканини як кортикального (еластичність +11,6%; твердість +13%), так і трабекулярного шарів (еластичність +7%; твердість +16,5%) (Maimoun L. et al., 2010).
Застосування стронцію ранелату спирияє посиленню механічної фіксації імпланта і покращенню мікроархітектури кістки навколо нього.
Ефективність стронцію ранелату також доведена при лікуванні чоловіків з ОП за результатами оцінки основного критерію ефективності після 1 року застосування стронцію ранелату у дворічному подвійному сліпому плацебо-контрольованому дослідженні за участю 243 пацієнтів з високим ризиком виникнення переломів.
Усі пацієнти отримували щодня добавки кальцію (1000 мг) та вітаміну D (800 ОД).Статистично значуще підвищення МЩКТ у групі пацієнтів, які застосовували стронцію ранелат, порівняно з групою пацієнтів, які застосовували плацебо, відмічали з 6-го місяця лікування.
Після 12 міс лікування виявляли статистично значиме підвищення середнього значення МЩКТ поперекового відділу хребта (основного показника ефективності) (5,32%; р<0,001). Схожі результати отримано у основному дослідженні III фази щодо запобігання переломів у жінок у період постменопаузи.
Після 12 міс лікування відзначали статистично значиме підвищення МЩКТ шийки стегна та МЩКТ стегнової кістки (р<0,001).
У результаті проведення III етапу дослідження STRATOS (STRontium Administration for the Treatment of Osteoporosis) визначена ефективна доза препарату стронцію ранелату — 2 г/добу щодня на ніч, при застосуванні якої досягається найкраща ефективність профілактики переломів поряд з відмінною переносимістю лікування хворими.
У багатоцентровому рандомізованому плацебо-контрольованому дослідженні SOTI (Spinal Osteoporosis Therapeutic Intervention) за участю 1649 жінок вірогідно доведено, що стронцію ранелат значно знижує ризик розвитку переломів хребців уже протягом першого року лікування. Крім того, доведена довгострокова (більше 10 років) ефективність стронцію ранелату.
Надійний захист від переломів хребців продемонстрований незалежно від ступеня тяжкості ОП, віку, наявності або відсутності переломів в анамнезі, індексу маси тіла, сімейного анамнезу та інших чинників ризику: у пацієнток без переломів хребців в анамнезі стронцію ранелат забезпечує зниження відносного ризику розвитку вертебральних переломів на 48% (р<0,001) у хворих, які мали хоча б один вертебральний перелом в анамнезі, стронцію ранелат забезпечував зниження ризику розвитку нових переломів хребців на 45% (р<0,001).
Дані, отримані в результаті багатоцентрового рандомізованого плацебо-контрольованого дослідження TROPOS (Treatment Of Peripheral Osteoporosis) за участю близько 5000 жінок, підтвердили високу ефективність стронцію ранелату в попередженні периферичних переломів, про що свідчать результати дослідження. Через 3 роки лікування досягнуто зниження ризику переломів стегнової кістки на 36% у пацієнток з ОП віком старше 74 років. Ці дані підтверджені і через 5 років терапії — зниження ризику розвитку переломів стегнової кістки на 43% у пацієнток, які належали до групи високого ризику.
Стронцію ранелат дозволяє досягти зниження ризику переломів у пацієнток з постменопаузальним ОП незалежно від віку, маси тіла, тяжкості захворювання та наявності інших чинників ризику, що є клінічним підтвердженням регенерації кісткової тканини при застосуванні препарату (табл. 29.12).
Таблиця 29.12
Ефективність стронцію ранелату у запобіганні переломам хребців незалежно від чинників ризику
|
Вік |
МЩКТ |
Наявність переломів у анамнезі |
|||||||
|
<70 років |
↓ ризику = –37% |
Т-балів (стегно, хребці) > –2,5 |
↓ ризику = –72% |
0 |
↓ ризику = –48% |
||||
|
70–80 років |
↓ ризику = –42% |
Т-балів (стегно, хребці) ≤ –2,5 |
↓ ризику = –39% |
1 |
↓ ризику = –45% |
||||
|
>80 років |
↓ ризику = –32% |
≥2 |
↓ ризику = –33% |
||||||
|
Сімейний анамнез ОП |
МЩКТ |
Паління |
|||||||
|
Так |
↓ ризику = –31% |
МЩКТ ≤ 25,46 кг∕м² |
↓ ризику = –37% |
Так |
↓ ризику = –40% |
||||
|
Ні |
↓ ризику = –44% |
МЩКТ ≤ 25,46 кг∕м² |
↓ ризику = –44% |
Ні |
↓ ризику = –40% |
||||
Переломи хребців при ОП проявляються прогресуючою деформацією скелета, зменшенням росту та болем у спині. Саме ці чинники можуть істотно знизити якість життя. За допомогою спеціальних анкет для пацієнток з ОП (SF-36 і QUALIOST) встановлено, що стронцію ранелат істотно поліпшує якість життя таких хворих. Порівняно з контрольною групою у пацієнток, що одержували стронцію ранелат, на 20% скоротилося зменшення росту, а третина з них позбулися болю у спині. Пацієнтки відзначали поліпшення фізичного й емоційного самопочуття.
У дослідженнях ІІІ етапу продемонстровані відмінна переносимість лікування і сприятливий профіль безпеки стронцію ранелату. Препарат добре переносився хворими, тому що на відміну від бісфосфонатів не викликав подразнення верхніх відділів ШКТ. У цілому частота розвитку побічних ефектів не відрізнялася від плацебо, при цьому вони зазвичай були короткочасними і мали легкий або середній ступінь тяжкості. Побічними ефектами, які виявляли найчастіше, були нудота (6,6% у групі лікування та 4,3% — у групі плацебо), діарея (6,5% проти 4,6% для плацебо)*. Зазначені побічні ефекти відмічали на початковій стадії лікування, надалі вони зникали без відміни прийому препарату.
*Для отримання повної інформації необхідно ознайомитись з інструкцією для медичного застосування препарату.
Рекомендована добова доза становить 2 г стронцію ранелату (1 однодозовий пакет, вміст якого перед застосуванням розчиняють у склянці води).
Стронцію ранелат рекомендується приймати перед сном, бажано не раніше ніж через 2 год після прийому їжі. Вміст одного пакетика слід висипати у склянку, додати води та розмішати гранули до утворення суспензії, яку слід випити відразу ж після її приготування.
З огляду на подвійний механізм дії стронцію ранелату, що спрямований на регенерацію кісткової тканини, його ефективність у запобіганні як хребцевим, так і периферичним переломам і безпеку, даний препарат може бути важливою альтернативою антирезорбтивній терапії й зайняти місце препарату першої лінії у лікуванні постменопаузального ОП.
Стронцію ранелат зайняв гідне місце серед препаратів для лікування постменопаузального ОП й рекомендований як препарат першої лінії в клінічних посібниках Бельгії, Болгарії, Італії, Латвії, Німеччини, Угорщини, Франції. Стронцію ранелат також включено до Американських клінічних рекомендацій з лікування остеопорозу (ICSI Health Care Guide-lines for Diagnosis and Treatment of Osteoporosis, www.icsi.org). Нові Європейські рекомендації з діагностики та ведення остеопорозу у жінок в постменопаузальний період (European Guidance for the diagnosis and management of osteoporosis in postmentopausal women, 2008) рекомендують стронцію ранелат як препарат першого вибору для лікування ОП у жінок в постменопаузальний період з найбільш доведеною ефективністю у попередженні як переломів хребців, так і невертебральних переломів незалежно від переломів в анамнезі та інших факторів ризику.
Результатом розробки нової концепції — синтезу таргетного препарату для лікування постменопаузального ОП — стало відкриття специфічного людського моноклонального антитіла з високим ступенем афінності до лігандів рецептора активатора ядерного фактора kB (RANKL). Деносумаб — ефективний інгібітор кісткової резорбції. Зв’язуючи RANKL, подібно до остеопротегерину, деносумаб запобігає взаємодії RANK з RANKL, знижуючи таким чином диференціацію, активність і життєздатність остеокластів, пригнічуючи загалом кісткову резорбцію (Головач И.Ю., 2013).
У міжнародному рандомізованому плацебо-контрольованому дослідженні III фази FREEDOM було оцінено ефективність та безпеку деносумабу для запобігання переломам у 7868 жінок із постменопаузальним ОП (Cummings S. et al., 2009).
Введення деносумабу підшкірно 2 рази на рік упродовж 3 років дозволило знизити відносний ризик переломів хребців на 68%, невертебральних — на 20%, переломів стегна — на 40% порівняно з групою плацебо. Терапія деносумабом упродовж 36 міс супроводжувалася підвищенням МЩКТ хребців поперекового відділу на 9,2%, стегнової кістки — на 6,0%.
Деносумаб знижував рівнень маркера кісткової резорбції СТХ (С-телопептиду) на 86% через 1 міс після ін’єкції та на 72% — перед повторною ін’єкцією (через 6 міс лікування).
Кількість небажаних побічних реакцій, а також випадків призупинення терапії через небажані реакції була приблизно однаковою. Також не відзначали істотної різниці у кількості пацієнтів з онкологічними і серцево-судинними захворюваннями. Не зареєстровано жодного випадку остеонекрозу нижньої щелепи, проте підвищувався ризик підшкірних інфекцій (целюліту).
Отримані дані 6-річного спостереження за пацієнками з постменопаузальним ОП, які отримували деносумаб у дослідженні FREEDOM. МЩКТ хребців і стегна прогресивно підвищувалася, досягши +15,2 і +7,5% відповідно в кінці 6-го року лікування (Henry G., 2013).
У прямих порівняльних відкритих багатоцентрових дослідженнях продемонстровано перевагу деносумабу перед бісфосфонатами (ібандронова кислота, ризедронова кислота, алендронова кислота) за критерієм підвищення МЩКТ поперекових хребців і стегнової кістки за 1 рік терапії (табл. 29.13) (Kendler D.L. et al., 2010; Recknor C. et al., 2012; Roux C. et al., 2012).
Таблиця 29.13
Порівняння впливу деносумабу і деяких бісфосфонатів на МЩКТ за 1 рік терапії
|
Хребет |
Стегнова кістка |
|
|
Деносумаб vs ібандронова кислота |
+4,1 vs + 2,1 |
+2,2 vs +0,9 |
|
Деносумаб vs ризедронова кислота |
+3,4 vs + 1,1 |
+2,0 vs +0,5 |
|
Деносумаб vs алендронова кислота |
+3,0 vs +1,85 |
+1,9 vs 1,05 |
p<0,0001 для всіх порівнянь.
На сьогодні деносумаб є препаратом 1-ї лінії терапії постменопаузального ОП у жінок, включений до клінічних рекомендацій Міжнародної асоціації остеопорозу (International Osteoporosis Foundation — IOF), Національних асоціацій США, Великобританії, Канади, країн Євросоюзу, Азії та Африки.
Глава 30. Захворювання кісток
Гіпертрофічна остеоартропатія
Визначення. Гіпертрофічна остеоартропатія (ГОАП) — синдром, який включає зміни дистальних фаланг у вигляді барабанних паличок та нігтів у вигляді скла годиника, потовщення довгих кісток кінцівок, синовіт великих та середніх суглобів.
Код за МКХ-10
М89.4 Інша гіпертрофічна остеоартропатія.
Хвороба Марі — Бамбергера, пахідермоперіостоз.
Етіологія та патогенез. Причини розвитку ГОАП невідомі. Вважають, що виникнення «барабанних паличок» зумовлене розвитком сполучнотканинних елементів, набряком м’яких тканин і окістя. В результаті морфологічних змін товщають нігтьові фаланги, нігті стають опуклими. Вважають також, що, можливо, в кровообіг потрапляють неідентифіковані субстанції, які викликають проліферацію кісткової тканини в трубчастих кістках і розростання сполучної тканини і дрібних судин в дистальних відділах фаланг. Зворотний розвиток процесу відмічають після радикального видалення пухлин, корекції вроджених вад серця, лікування ІЕ. Припускають, що ця субстанція руйнується, інактивується в легенях (створення штучного шунтування крові з правих відділів в легені у собак викликає розвиток ГОАП). Шунтування крові виявляють при пухлинах легені, поліпах тонкого кишечнику, цирозі печінки. У інших випадках субстанція накопичується в значній кількості. При синіх вадах є аномальні тромбоцити, які взаємодіють з ендотелієм дрібних судин, при цьому виділяються фактори росту фібробластів, що індукують прояву ГОАП. Вважають, що розвиток «барабанних паличок» є початковою стадією ГОАП, проте, при раку легені вони можуть бути єдиним паранеопластичним симптомом (тобто без артритичних проявів, синовіту). Це відмічають у 22–30% хворих на рак легені, «барабанні палички» виникають за місяці і роки до появи клінічної симптоматики пухлини.
Класифікація ГОАП. Виділяють наступні форми патології: ідіопатичну (пахідермоперіостоз), вторинну (синдром Марі — Бамбергера), локалізовану (табл. 30.1).
Клініка ГОАП. Дистальні фаланги набувають вигляду барабанних паличок, а нігті — скла годинника (рис. 30.1).
Таблиця 30.1
Форми ГОАП
|
Ідіопатична (пахідермоперіостоз) |
Причина розвитку невідома, формується в дитинстві і відмічається рідко. Виділяють 2 варіанти хвороби: 1) з розвитком в перші роки життя; 2) з розвитком у віці 15 років |
|
Вторинна (синдром Марі — Бамбергера) |
Виявляють при захворюваннях серця («сині» вади, ІЕ), середостіння (ахалазія та пухлини стравоходу, тимома), легень (туберкульоз, бронхоектази, полікістоз, легеневий фіброз, злоякісні пухлини (первинні та метастатичні), артеріовенозні фістули, мезотеліома плеври), печінки (хронічний активний гепатит, цироз печінки, рак, стан після трансплантації печінки), кишечнику (хвороба Крона, неспецифічний виразковий коліт, поліпоз, злоякісні пухлини, хронічні інфекції), інших хворобах — злоякісних пухлинах різних локалізацій, гіпертиреозі, таласемії |
|
Локалізована форма |
При цій формі зміни відзначають не в усіх 4 кінцівках, а в 1–2 (при геміплегії, аневризмах, ІЕ, відкритій артеріальній протоці) |
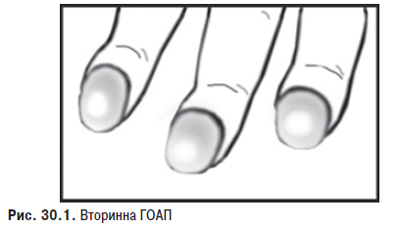
Больових відчуттів у суглобах і кістках немає. Ці явища можуть розвинутися за декілька місяців або років. При пухлинах вони випереджають їх клінічні прояви. Осалгії, артралгії можуть розвинутися вже на тлі змін, що сталися (зрідка біль передує ГОАП). Через багато років розвивається циліндрична форма передпліч або гомілок («гомілка слона», «гомілка у вигляді пляшки шампанського»). Причиною болю можуть бути синовіти колінних, промене-зап’ясткових, гомілковостопних суглобів (кількість випоту, як правило, незначна). Синовіт розвивається в результаті змін в епіфізах кісток. Відзначається також гіпергідроз шкіри в ділянці кистей і стоп, сальна, жирна шкіра (себорея).
Особливості первинної форми ГОАП (пахідермоперіостоз, сімейна або ідіопатична ГОАП). Вона формується в дитинстві і відмічається рідко. Відзначають 2 варіанти хвороби: при першому вона розвивається в перші роки життя, при другому — у віці 15 років. Переважають чоловіки (8,9:1), в 38% випадків виявляють спадковий характер хвороби. Характерні для ГОАП зміни іноді доповнюються змінами шкіри: відзначають її складчастість у вигляді «мозкових» складок на лобі, щоках (cutis verticis gyrata), іноді в ділянці скальпа («скальп бульдога»). Шкіра потовщена, потові та сальні залози гіпертрофовані (себорея), відзначають виражений гіпергідроз шкіри. Потовщення періосту і кортикального шару кісток у цих хворих більш виражене. Суглоби можуть товщати за рахунок періосту епіфізів.
Захворювання, при яких відзначається вторинна ГОАП. Окрім пухлин різної локалізації, вона може супроводжувати інші захворювання:
- нагноювальні хвороби легень і плеври (абсцес, бронхоектатична хвороба, емпієма плеври);
- вроджені «сині» вади серця;
- полікістоз легень;
- ІЕ;
- легеневий фіброз;
- первинний біліарний цироз печінки;
- артеріовенозні фістули;
- захворювання органів травлення, що супроводжуються діареєю (виразковий коліт, хвороба Крона, туберкульоз кишечнику, хронічна дизентерія, множинний поліпоз кишечнику, рак товстої кишки, рак печінки);
- мезотеліому плеври,
- захворювання середостіння (пухлину стравоходу, тимому, ахалазію стравоходу);
- туберкульоз;
- гіпертиреоз;
- лімфогранулематоз;
- таласемію.
При хронічних запальних захворюваннях легень ГОАП розвивається повільно, а артритичні явища відмічають дуже рідко. Для раку характерне поєднання «барабанних паличок» з остеоартропатичними змінами. Не виявлено розвитку ГОАП при неускладненому туберкульозі легень.
Діагноз. Дані, які дозволяють встановлювати діагноз ГОАП, є наступними:
- характерні зміни фаланг пальців кистей та стоп (периметр фаланги біля основи нігтя більший за периметр дистального міжфалангового суглоба), ніготь протискується при пальпації;
- при рентгенографії в проксимальних і дистальних ділянках діафізів велико-, малогомілкової, променевої кісток відмічають розростання кісткової тканини в ділянці окістя (періостит);
- при пахідермоперіостозі зміни виникають і в епіфізах;
- зміни кісток двобічні і симетричні;
- вони прогресують, число уражених кісток збільшується, товщає кортикальний шар;
- можливі зміни в лопатках, ключицях, ребрах, хребті, черепі;
- при рентгенографії кистей і стоп виявляють поступове розсмоктування кісткової тканини дистальних фаланг;
- зміни в суглобах при ГОАП не виявляють, також відсутні зміни в аналізах крові.
Зміни, які відзначають у хворих на ГОАП при лабораторному й інструментальному обстеженні: збільшення ШОЕ, РФ не виявлено; синовіальна рідина прозора, цитоз до 2000 в мкл, вміст нейтрофілів <50%. При рентгенографії виявляють субперіостальне утворення кісткової тканини в дистальних відділах діафізів кісток передпліччя і гомілок, рідше — у фалангах. Ураження симетричне.
Діагноз первинної ГОАП встановлюють тільки після виключення захворювань, які викликають вторинну ГОАП.
Захворювання, з якими диференціюють ГОАП. Первинну форму ГОАП диференціюють з акромегалією (зміни шкіри нагадують це захворювання). Локальні періостальні зміни кісток виявляють при:
- пухлинах кісток,
- остеомієліті;
- синдромі Рейтера,
- васкулітах,
- ПсА;
- хронічному венозному стазі;
- гіпертиреозі (періостит розвивається в ділянці кистей і стоп).
Захворювання (стани), які супроводжуються дифузним періоститом. До них належать:
- гіпервітаміноз А;
- парапротеїнемії;
- флюороз;
- нейрофіброматоз;
- ниркова остеодистрофія;
- гіперфосфатемія;
- інфантильний кортикальний гіперостоз.
Лікування при ГОАП. Видалення пухлини, корекція вади серця, лікування ІЕ супроводжується регресією змін, характерних для ГОАП, якщо вони наступили нещодавно (через декілька місяців вони вже не зникають). При артралгіях, осалгіях призначають НПЗП, гіпергідрозі — атропін і його похідні.
Остеонекроз
Визначення. Термін «остеонекроз» означає омертвіння кістки в результаті порушення її кровопостачання. З цієї причини застосовується термін «аваскулярний некроз кістки» (кістковий інфаркт). Інший термін — «асептичний некроз» — означає, що патологічний процес має неінфекційний характер.
Код за МКХ-10
М87 Остеонекроз.
М87.0 Ідіопатичний асептичний некроз кістки.
М87.3 Інший вторинний остеонекроз.
М87.9 Остеонекроз, неуточнений.
Етіологія та патогенез. Причина ішемії кісткової тканини не встановлена. У зв’язку з ідіопатичним остеонекрозом в США щорічно проводиться до 50 тис. операцій з протезування суглобів. У хворих розвивається некроз губчатої речовини і кісткового мозку (фаза некрозу може тривати тижні й місяці), субхондральна фрагментація епіфіза, його деформація, вторинне ураження хряща, деформуючий артроз. Найчастіше відмічають первинні асептичні некрози у хворих з ювенільним остеохондрозом: голівки стегнової кістки (хвороба Легга — Калве — Пертеса, яка виникає у період 3–12 років), відтинаючий остеохондрит дистального епіфіза стегна (хвороба Кеніга), асептичний некроз човноподібної кістки стопи (хвороба Келера I), голівок плеснових кісток, частіше II, III і IV (Келера II), напівмісячної кістки зап’ястка (хвороба Кінбека), горба великогомілкової кістки (хвороба Осгуда — Шлаттера). Вважають, що головну роль в патологічному процесі відіграє ішемічний некроз первинних або вторинних центрів ендохондральної осифікації. Деякі випадки можуть бути пов’язані зі стресом або пораненням. Більшість цих хвороб виникає спорадично, хоча відмічають також сімейні форми.
До причин розвитку некрозів кісток відносять хронічну мікротравму, надмірне механічне навантаження (при уроджених дисплазіях, плоскій стопі тощо), аномалії окостеніння, порушення обміну, нервової трофіки та ін. Ділянки остеонекрозу можуть бути єдиними і множинними. Вони можуть розташовуватись у будь-якій частині кістки. Звідси поділ остеонекрозу на медулярний та кортикальний:
- медулярний — некроз захоплює губчасту речовину і кістковий мозок;
- кортикальний — некроз знаходиться в субперіостальних і субхондральних ділянках компактної речовини трубчастих кісток.
До захворювань, при яких розвивається остеонекроз, належать:
- лікування ГК;
- травма;
- опіки;
- променева терапія;
- СЧВ;
- РА;
- подагра;
- хронічне запалення суглобів;
- поліцитемія;
- хронічні хвороби печінки;
- панкреатит, ілеїт, коліт;
- саркоїдоз;
- хвороба Гоше;
- гіперліпідемії;
- атеросклероз;
- гемоглобінопатії;
- серповидно-клітинна анемія;
- коагулопатії;
- тромбоемболія;
- повітряна емболія;
- пухлини.
Виявлено достовірний зв’язок розвитку остеонекрозів з прийомом ГК, особливо при тривалому застосуванні у високих дозах. Частота розвитку остеонекрозів знижується після зниження дози ГК. До факторів ризику його розвитку відноситься надмірне вживання алкоголю, паління. У 15% пацієнтів причин виникнення остеонекрозу не виявляють (ідіопатична форма). У деяких сім’ях відмічають аутосомно-домінантний тип успадкування остеонекрозу голівки стегнової кістки, мутацію гену колагену II типу.
Клініка. Клінічна картина остеонекрозу залежить від місця розташування некротизованої ділянки. Кортикальні (субхондральний і субперіостальний) остеонекрози супроводжуються болем і можуть призводити до переломів:
- субперіостальний (на місці некрозу формується нова кісткова тканина (шляхом «повзучого заміщення»);
- субхондральний (хрящ може відриватися у порожнину суглоба, що призводить до дистрофічних процесів, загоєння повільне);
- медулярний (клінічних проявів немає, виявляють випадково при рентгенологічному обстеженні).
Розвиток некрозів кісток, що утворюють великі суглоби, супроводжується сильним болем. Біль може передувати рентгенологічним змінам. Вони зменшуються у спокої і збільшуються після фізичного навантаження. Рухливість ураженого суглоба обмежується, може відзначатися локальна болючість при пальпації. При ураженні голівки стегнової кістки, що відзначається при ревматичних захворюваннях, основним клінічним проявом є резистентний до лікування сильний біль. Функція кульшового суглоба значно порушується, розвивається інвалідизація хворих. З часом уражена кінцівка коротшає. Виражений больовий синдром і деформація суглоба розвиваються при некрозі виростків стегнової і великогомілкової кісток. Симптоматика некрозів кісток іншої локалізації (розвиваються рідко) менш яскрава.
Діагностика. При підозрі на розвиток остеонекрозу застосовують рентгенографію, МРТ, сцинтиграфію (табл. 30.2).
Таблиця 30.2
Інформативність рентгенографії, МРТ, сцинтиграфії при остеонекрозі
|
Рентгенографія |
На ранній стадії остеонекрозу зміни відсутні. У фазу репаративного процесу кістка набуває плямистого вигляду через наявність суміжних ділянок підвищеної і зниженої рентгенівської щільності. Знижену щільність відмічають у ділянках, що піддані резорбції і заміщенню фіброзною тканиною, підвищена — в зонах з переважанням остеосклерозу. Внаслідок компресії губчастої кістки під субхондральною кістковою пластинкою (при остеонекрозі голівки стегнової кістки) з’являється тонка рентгенонегативная лінія, розташована паралельно суглобовій поверхні (симптом серпа). Пізніше настає деформація ураженої кістки, розвивається вторинний ОА (рис. 30.2, 30.3) Одночасно із змінами у вогнищі некрозу часто формується протрузія вертлюжної западини |
|
МРТ |
Це найбільш чутливий метод, що дозволяє виявити ознаки ураження кісткового мозку до появи інших змін. Метод використовують при підозрі на розвиток остеонекрозу і відсутності рентгенологічних змін |
|
Сцинтиграфія з технецієм (99mТс) |
Дозволяє виявити патологічні зміни раніше, ніж рентгенографія. Ізотоп фіксується в зоні ушкодження кістки через підвищену васкуляризацію й активізацію остеогенезу. У центрі некрозу виявляється «холодна» пляма — зона відсутності або зниження накопичення ізотопу |
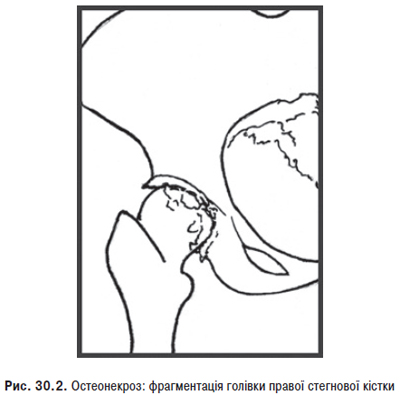
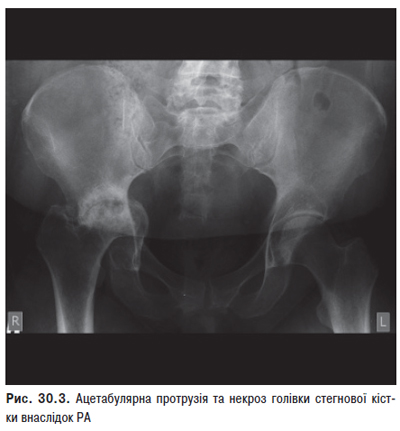
Лікування. Основним та радикальним методом лікування остеонекрозу є хірургічне втручання, проте його виконання можливе не у всіх хворих. Метою консервативного лікування є купірування больового синдрому, уповільнення прогресу кісткових змін. Призначають НПЗП, проводять розвантаження суглобів за допомогою милиць. Біль зменшується при проведенні лазеротерапії. Рекомендується лікування системного ОП, атеросклерозу, поліпшення мікроциркуляції. Доказів ефективності вазодилататорів, засобів, які знижують рівень ліпідів, аналогів простацикліну, антикоагулянтів, бісфосфонатів, гіпербаричної оксигенації, екстракорпоральної ударно-хвильової терапії на сьогодні немає. З метою збереження голівки стегнової кістки, відстрочення ендопротезування застосовують неваскуляризовану кісткову пластику, васкуляризовану кісткову пластику, ендопротезування, артропластику суглобових поверхонь.
Профілактика остеонекрозів. Заходи з їх профілактики не розроблені.
Глава 31. Фіброміалгії
Фіброміалгія — це синдром, який проявляється дифузним болем, підвищеною стомлюваністю скелетних м’язів, наявністю характерних болючих точок.
Код за МКХ-10
М79.7. Фіброміалгія.
Епідеміологія
У популяції розповсюдженість фіброміалгії становить 2–3,3%, при цьому переважно хворіють жінки (в 5–10 разів частіше порівняно з чоловіками). Серед жінок віком 20–40 років розповсюдженість фіброміалгії становить близько 4%, віком 55–64 роки — 8%. У дітей віком 9–15 років вона досягає 1,2–6,2%. Проблема має велике соціально-економічне значення: головний біль невстановленого генезу, хронічний біль в ділянці плечового пояса, нижній ділянці спини є головною причиною втрати працездатності на виробництві. Лікування цього захворювання в США щорічно обходиться в 1 млрд дол. США.
Етіологія і патогенез захворювання
Етіологія і патогенез захворювання не встановлені. Передбачається роль вірусної інфекції (вірусів Епштейна — Барр, Коксакі, герпесу), захворювання може виникнути після фізичної травми, психологічного стресу. Ймовірні (можливі) патогенетичні механізми фіброміалгії наведені в табл. 31.1.
Таблиця 31.1
Можливі патогенетичні механізми фіброміалгії
|
Порушення мікроциркуляції, мікронабряк в м’язах під впливом серотоніну, брадикініну, лейкотрієнів, простагландинів, ІЛ-1, що супроводжуються підвищеною чутливістю больових рецепторів м’язів |
|
Вивільнення з гладких клітин гістаміну, серотоніну під впливом субстанції Р і нейрокіну (що виділяються з периферичних закінчень чутливих нейронів) з розвитком набряку нервових закінчень і болю |
|
Порушення сприйняття болю внаслідок недостатньої продукції ендогенних опіатоподібних субстанцій |
|
Зниження напруження кисню в м’язовій тканині, гіпоксія, які призводять до болю в м’язах |
|
Поява болю внаслідок гіпертонусу м’язів |
Тригерні точки, причини їх розвитку. Встановлено, що гостре напруження м’язів, їх тривале скорочення або надмірне стомлення при багаторазових скороченнях призводять до перенапруження з наступним розвитком тяжів в м’язі і болісних вузликів (точок). Вони отримали назву «м’язового мозолю», «м’язового ревматизму», «несуглобового ревматизму», «фіброзиту», «міогелезу», «м’язового ішіасу», «ідіопатичної міалгії». Термін «міофасціальні тригерні точки», «тригерні точки» використовується з 50-х років минулого століття, проте в літературі продовжують з’являтися нові назви: стиснута точка, вузликовий фіброміозит, м’язові ущільнення або індурація, інтерстиціальний міофіброзит, міофасцит та ін.
Патофізіологічна природа ущільнених тяжів, що пальпуються, і тригерних точок. Встановлено, що м’язові волокна тугого тяжа, що проходить через зону тригерної точки, не проявляють електроміографічної активності у стані спокою. Тугі тяжі зберігаються після настання смерті аж до появи трупного задубіння. Хоча при пальпації м’яз відчувається як напружений і не піддається розтягуванню, відсутність електроміографічної активності вказує на те, що він не знаходиться в спастичному стані. Тугий тяж зникає після певного лікування тригерних точок. Без відповідного лікування тригерні точки можуть зберігатися десятки років, обмежуючи рухи, періодично викликаючи активність, напади відбитого болю.
Гостре напруження м’яза, хронічне навантаження, пов’язане з тривалим скороченням, або надмірне стомлення при багаторазових скороченнях можуть призвести до перевантаження скорочувальних елементів з ушкодженням тканин, виходом іонів Са++ із саркоплазматичного ретикулума без їх зворотного поглинання. Нормальне надходження енергії від АТФ і надлишок Са++ ініціюють і підтримують стійку контрактуру залучених в патологічний процес м’язових волокон. Це супроводжується неконтрольованим метаболізмом у зоні ураження, сильним локальним звуженням судин, зниженням кровотоку, скороченням м’язових волокон. Група тугих м’язових волокон пальпується у вигляді щільного тяжа. М’язова контрактура без прояву електричної активності може бути також результатом майже повного виснаження АТФ: без АТФ кінці міозинових філаментів не можуть звільнитися від зв’язку з актиновими філаментами і саркомер стає ригідним. Для забезпечення стійкості контрактури необхідно, щоб центральна ділянка АТФ-дефіцитної контрактури була оточена зоною інтенсивного метаболізму, що повністю утилізував АТФ.
Причина виникнення болю в ділянці тригерних точок. За допомогою декількох механізмів в зону тригерних точок виділяються речовини, які сенсибілізують нервові закінчення: гістамін, серотонін, киніни, простагландини. Джерелом серотоніну є тромбоцити, які виділяються при крововиливі в ділянці тригерних точок. Окрім сенсибілізації, серотонін викликає локальну ішемію. У пошкодженій тканині з’являються дегранульовані гладкі клітини, що виділяють гістамін. Зниження кровотоку, ішемія супроводжуються підвищенням концентрації молочної і піровиноградної кислоти, що сприяє виділенню простагландинів. Таким чином, сенсорне і моторне гіперподразднення ділянки тригерних точок веде до локальної болючості.
«Спонтанний відбитий біль». Біль від скелетного м’яза, де є тригерні точки, поширюється певною зоною, що інервується одним із сегментів спинного мозку. Кожен м’яз має специфічну для нього зону розподілу відбитого болю або патерн. Знаючи зону віддзеркалення болю або патерн, можна визначити, де знаходиться (у якому м’язі) сама тригерна точка, і потім проводити лікування. Атлас патернів при ураженні різних м’язів людського тіла наведений в 2-му томі монографії Дж.Г. Тревелла і Д.Г. Сімонса «Міофасціальні болі».
Різновиди тригерних точок, відмінність активних і латентних тригерних точок. Виділяють активні і латентні тригерні точки (табл. 31.2).
Таблиця 31.2
Характеристика тригерних точок
|
Різновиди |
Характеристика |
|
Активні |
Активна тригерна точка — це фокус підвищеного подразнення у м’язі або його фасції, що проявляється болем. Біль відбивається в характерні для цього м’яза ділянки у спокої або при рухах. Активна тригерна точка перешкоджає повному розтягуванню м’яза, послаблює м’язову силу, дає відбитий біль у відповідь на стиснення |
|
Латентні |
Латентні тригерні точки не викликають біль (болючість відмічають тільки при пальпації), але можуть бути причиною обмеження рухів і слабкості ураженого м’яза. Латентна тригерна точка може зберігатися протягом багатьох років після травми, періодично викликаючи гострий біль при незначному перерозтягуванні, перевантаженні або переохолодженні м’яза. Таким чином, як активна, так і латентна тригерна точка викликають дисфункцію |
|
Сателітні |
Сателітна міофасціальна тригерна точка — це фокус гіперподразнення у м’язі або його фасції, який стає активним внаслідок розташування його в зоні феномену, відбитого від іншої тригерної точки |
|
Вторинні |
Вторинна міофасціальна тригерна точка — гіперподразнена ділянка, яка виникає у м’язі або його фасції при перевантаженні, коли він виконує функцію м’яза, що має первинну тригерну точку |
Контингенти, у яких формуються тригерні точки. У нормі в м’язах немає ущільнених тяжів, тригерних точок, вони неболісні при пальпації, не викликають судомних реакцій і не відбивають біль при стисканні. Тригерні точки можуть сформуватися у пацієнта будь-якого віку і статі. Найчастіше їх виявляють у жінок середнього віку, що ведуть сидячий спосіб життя. Вони часто звертаються за медичною допомогою з приводу міофасціального болю. Міофасціальні тригерні точки є також основним джерелом болю в скелетних м’язах у дітей. Максимум тригерних точок відзначають у середньому віці. В осіб старшого віку у міру зниження рухової активності переважно відмічають м’язову ригідність і обмеження рухів, викликані латентними точками. Активні тригерні точки рідко формуються у робітників, що мають постійне фізичне навантаження, і часто розвиваються у людей, які лише зрідка переносять фізичні перевантаження.
М’язи, в яких найчастіше локалізуються тригерні точки. Активні тригерні точки найчастіше виявляють у м’язах шиї, плечового пояса, тазової ділянки, жувальних м’язах. Частими місцями локалізації тригерних точок є трапецієвидний, драбинчатий, груднино-ключично-соскоподібний м’яз, що піднімає лопатку і квадратний м’яз попереку.
Характер відбитого болю. Біль, відбитий від міофасціальних тригерних точок, зазвичай має тупий і тривалий характер. Часто він відчувається в глибині м’яза, його інтенсивність варіює від відчуття дискомфорту до жорстокого, сильного болю. Біль можуть відмічати у спокої або тільки при рухах. Відбитий біль може бути викликаний посиленим натисненням пальця на тригерну точку або пенетрацією тригерної точки ін’єкційною голкою. Чим чутливіша тригерна точка, тим сильніший і стійкіший відбитий нею біль, більше його поширення.
Поширення болю від тригерних точок. Не завжди міофасціальний біль розподіляється в межах того ж дерматома, міотома і склеротома. Є багато винятків, коли біль поширюється в ділянці, яка інервується іншими сегментами.
Чинники, що активують тригерні точки, наведені в табл. 31.3.
Таблиця 31.3
Чинники, що активують тригерні точки
|
Тригерні точки активуються при різкому м’язовому перевантаженні, фізичній перевтомі, прямому ушкодженні або охолодженні м’яза |
|
Зазвичай пацієнти пов’язують виникнення болю з різким перевантаженням або травмою, яка мала місце декілька місяців або років тому |
|
Первинні тригерні точки розвиваються у м’язах, які піддаються сильним повторним або тривалим скороченням |
|
Тригерні точки також активуються іншими тригерними точками, вісцеральними захворюваннями, артритами, емоційними розладами |
|
Сателітні тригерні точки розвиваються в м’язах, які лежать в зонах больової іррадіації від уражених внутрішніх органів (інфаркт міокарда, пептична виразка, жовчнокам’яна хвороба, ниркова колька) |
|
Латентні тригерні точки можуть активуватися під час і після вірусного захворювання, при тривалому знаходженні м’яза в скороченому стані (наприклад під час сну), охолодженні м’яза, особливо якщо він знаходиться в стані втоми або ригідності після фізичного навантаження |
|
Тригерну точку активують різке скорочення м’яза, його стиснення |
|
Сила стиснення, необхідна для активації латентної тригерної точки і провокації клінічного больового синдрому, залежить від ступеня тренованості ураженого м’яза: чим він витриваліший до фізичних вправ, тим нижче сприйнятливість його тригерних точок до активуючих впливів |
Особливості тригерних точок у травмованому м’язі. Після травми травмовані тканини загоюються, а м’язи «навчаються» уникати болю, оскільки активні тригерні точки розвивають здатність обмежувати рухи свого м’яза. У результаті розвиваються хронічний м’язовий біль, ригідність і дисфункція. У спокої та за відсутності провокуючих чинників активні тригерні точки можуть спонтанно перейти в латентний стан. При цьому больовий синдром зникає, проте випадкова реактивація тригерних точок навіть через декілька років викликає біль. Якщо зберігається постійна активність тригерних точок, що не піддається лікуванню, то це свідчить про те, що захворювання з фази нервово-м’язової дисфункції перейшло в дистрофічну фазу.
Активні міофасціальні тригерні точки можуть супроводжуватися вегетативними проявами (локальний спазм судин, підвищене потовиділення, сльозотеча, нежить, слинотеча); пропріорецептивними розладами (порушення рівноваги, запаморочення, дзвін у вухах, порушення сприйняття ваги предметів, що знаходяться в руках); порушенням моторної координації.
Чинники, що сприяють виникненню тригерних точок, представлені у табл. 31.4.
Таблиця 31.4
Чинники, що сприяють виникненню тригерних точок
|
1. |
Структурні невідповідності (зокрема асиметрія при різній довжині ніг) |
|
2. |
Напруження м’язів при незручних меблях, неправильній позі, неправильному навантаженні м’язів, їх стисканні ременями, тісним одягом, білизною |
|
3. |
Порушення обміну речовин: недостатність вітаміну В1, В6, В12, фолієвої й аскорбінової кислоти, Са, К, Fe, Mg |
|
4. |
Гіпотиреоз, подагра, гіпоглікемія різного походження |
|
5. |
Психологічні чинники: депресія, тривожність і м’язове напруження |
|
6. |
Інфекція: вірусні захворювання, особливо простий герпес, синусити, парадентальний абсцес, інвазія паразитами (лямбліоз, амебіаз та ін.) |
|
7. |
Алергічний риніт |
|
8. |
Ушкодження нервів |
Особливості тригерних точок при дискогенному радикуліті. При дискогенному радикуліті біль може бути пов’язаний не лише зі стисненням нервів, але і з існуванням тригерних точок. Навіть після добре виконаної ламінектомії хворі продовжують відчувати сильний біль. Тому для повного одужання хворих необхідне виявлення й інактивація міофасціальних тригерних точок.
Морфологічні зміни в м’язах. У біоптатах м’язів при світловій мікроскопії змін не виявляють, при електронній мікроскопії фіксують ультраструктурні порушення міофібрил.
Клініка фіброміалгії
Більшість хворих (близько 88%) скаржаться на дифузний кістково-м’язовий біль (табл. 31.5).
Таблиця 31.5
Клініка фіброміалгії
|
Анамнез |
Анамнез тривалий, прояви різноманітні, лабораторні та інструментальні маркери відсутні, а тому період до верифікації діагнозу значний |
|
|
Фактори ризику |
До них відносять спадковість, жіночу стать, емоційні травми, стреси, наявність хронічного регіонарного больового синдрому |
|
|
Тригерні фактори |
Психологічний стрес, інфекції, гормональні порушення, гіповітаміноз |
|
|
Клінічні прояви |
Біль |
Характерним і основним проявом фіброміалгії є розповсюджений скелетно-м’язовий біль без вираженого неврологічного розподілу. Біль має персистуючий характер, його локалізація та інтенсивність постійно варіюють. Інтенсивність болю зростає під впливом стресу, тривоги, холодної погоди, фізичного перевантаження. Другим характерним проявом фіброміалгії є болючі точки, які знаходять пальпаторно або за допомогою долориметра. Больовий поріг на механічні стимули знижений і становить до 1,8 кг/см2 при 6–8 кг/см2 у контролі. З перебігом хвороби кількість болючих точок збільшується |
|
Алодинія |
Біль виникає на фізіологічні стимули |
|
|
Гіпералгезія |
Має місце підвищена чутливість до больових стимулів |
|
|
Втома, швидка стомлюваність |
Вираженість втоми змінюється протягом доби, максимально вона виражена вранці та ввечері. Хворі відчувають себе комфортно тільки в період від 10:00 до 14:00. Стан погіршують психологічний стрес, порушення сну. Фізичні перевантаження призводять до появи втоми на наступний день після них. Відмічається швидка стомлюваність: у хворих знижуються сила м’язів (на 50–60% порівняно зі здоровими людьми) та м’язова витривалість |
|
|
Порушення сну |
Сон порушений у 80% хворих, не відновлює їх сили навіть при достатній тривалості. Він носить поверхневий характер |
|
|
Скутість |
Має генералізований характер та відмічається у 76–92% хворих. Максимуму цей симптом сягає вранці та після періоду фізичного спокою. Зазвичай він корелює із вираженістю болю. Типовим для фіброміалгії є суб’єктивне відчуття припухання в ділянці суглобів, яке не підтверджується при обстеженні |
|
|
Когнітивна дисфункція |
У хворих порушуються концентрація уваги (у 95%), пам’ять (у 93%). Ці прояви корелюють із больовим синдромом, на них частково впливають порушення сну |
|
|
Коморбідні стани |
Синдром подразненого кишечнику |
Виявляють у 32–66% хворих |
|
Мігрень |
Реєструється у 50% хворих |
|
|
Первинна дисменорея |
Відмічають у 40% хворих жінок |
|
|
Інтерстиціальний цистит |
Проявляється дизурією, болем у ділянці сечового міхура, таза |
|
|
Синдром неспокійних ніг |
Відмічають у 30–50% хворих, є причиною переривання сну, посилює прояви фіброміалгії |
|
|
Інші прояви |
Нерідко виявляють гіпервентиляційний синдром, лабільність артеріального тиску, кардіалгію, синдром Рейно, порушення серцевого ритму, сухість в очах, роті |
|
|
Асоціація з іншими захворюваннями |
Ревматичні захворювання |
Симптоми фіброміалгії відмічають у 17–40% хворих із СЧВ, у 17–57% з РА, 24% з ПсА, у 31% з АС та аутоімунним тиреоїдитом. Не встановлено підвищення ризику розвитку у хворих з фіброміалгією системних захворювань сполучної тканини |
|
Психічні розлади |
Ризик розвитку тривожних розладів у хворих на фіброміалгію в 5 разів вищий, ніж у популяції. В 14–36% випадків відзначається депресія |
|
|
Інфекційні захворювання |
Розвиток фіброміалгії виявляють у 5–19% хворих, інфікованих вірусом гепатиту С |
|
|
Фізикальні дані |
Пальпаторно та за допомогою долориметра виявляють характерні діагностичні точки, які розташовані у місцях з’єднання фіброзної та м’язової тканини. При пальпаторному обстеженні застосовують зусилля до 4 кг. Відповідь розцінюють як позитивну у разі виникнення болю |
|
|
Лабораторні дослідження |
Самостійного значення рутинні лабораторні параметри не мають. Біохімічні показники нормальні |
|
|
Інструментальні обстеження |
Їх проводять за наявності супутньої патології. Для виявлення порушень сну проводять полісомнографічне дослідження. Обговорюються можливості використання МРТ |
|
Больові точки переважно розташовуються у шийному і поперековому відділах хребта, рідше (у 20–23%) в ділянці нижньощелепно-скроневого і гомілковостопного суглобів. У більшості пацієнтів виявляють 5–8 больових точок. Найбільш типова їх локалізація: трапецієподібні м’язи; м’язи спини; медіальна поверхня ліктьових і колінних суглобів; ділянка кульшових суглобів.
Фіброміалгія супроводжується розвитком сутулості, причому при випрямленні спини зникає як спонтанний біль, так і біль при натисканні. Окрім вищенаведених симптомів у хворих на фіброміалгію відмічають ряд інших (табл. 31.6).
Таблиця 31.6
Симптоми, що відмічають у хворих на фіброміалгію
|
Відчуття втоми у м’язах, млявість, підвищена втомлюваність |
|
Біль у м’язах ділянки плечей, шиї, кінцівок |
|
Виявлення болісних точок з обох боків тіла, чутливість яких посилюється в холодну погоду і після емоційних стресів |
|
Щільні болючі вузлики в крижовій ділянці і верхній частині сідниць |
|
Скутість в м’язах, яка відзначається в ранкові і вечірні години і залежить від кількості болючих точок |
|
Відчуття набряку кистей і стоп |
|
Відчуття втоми, що зберігається після сну |
|
Часті прояви у пацієнтів нейроциркуляторної дистонії, функціональної диспепсії, синдрому подразненої товстої кишки і подразненого сечового міхура, дисменореї |
|
Частий головний біль, депресія, відчуття тривоги, порушення сну |
|
Підвищена метеочутливість |
|
Відсутність істотних змін при лабораторних дослідженнях (можливе збільшення ШОЕ до 20 мм/год) |
Діагностика фіброміалгії
Для дослідження больових точок застосовують долориметр, проте пальпаторне дослідження дає аналогічні результати, і тому набуло переважного поширення. Діагностичні критерії фіброміалгії, запропоновані ACR (1990), наведені в табл. 31.7.
Таблиця 31.7
Діагностичні критерії фіброміалгії, запропоновані ACR (1990)
|
Поширений біль в анамнезі |
|
Визначення: біль розглядається як «поширений» за наступних умов: біль у лівій половині тіла, біль у правій половині тіла, біль вище і нижче талії. Додатково мають відмічати біль в осьовому скелеті (потилиці або передній частині грудної клітки або в грудному відділі хребта або нижній частині спини). У цьому визначенні біль у плечах і сідницях вважають як біль на тому та іншому боці. Біль в нижній частині спини вважається болем в нижньому сегменті. Тривалість болю перевищує 3 останні місяці. Наявність іншої клінічної патології не виключає діагноз фіброміалгії |
|
Вказівка на біль при пальпації в 11 з 18 точок наступної локалізації |
|
Визначення: при пальпації мають бути болючими щонайменше 11 з 18 наступних точок: потилиця: двобічна, місця прикріплення субокципітальних м’язів; передньо-нижня ділянка шиї: двобічна, в передніх частинах міжпоперечних зв’язок (поперечних відростків) від С5 до С7; трапецієподібна: двобічна, середина верхнього краю трапецієподібного м’яза; надостна: двобічна, на початку м’яза вище ості лопатки ближче до медіального краю; друге ребро: двобічна, латеральніше реберно-груднинного зчленування, верхня поверхня; зовнішній виросток: двобічна, на 2 см дистальніше виростка плечової кістки; ділянка сідниць: двобічна, у верхньо-зовнішньому квадранті сідниці, в першій м’язовій складці; великий вертлюг: двобічна, задня поверхня вертлюга; коліно: двобічна, у медіального жирового валика медіальної щілини колінного суглоба |
Пальпаторне дослідження слід проводити з силою 4 кг. Пальпована точка розглядається як позитивна, якщо пацієнт оцінює пальпацію як болючу. «Чутлива» пальпація не вважається болючою. У клінічній практиці допустима діагностика лікарем фіброміалгії і без повного набору болючих точок. При встановленні діагнозу рекомендується враховувати наявність таких «малих» симптомів, як втомлюваність, порушення сну, скутість, синдром подразненого кишечнику. Методика обстеження хворих з міофасціальним болем наведена в табл. 31.8.
Таблиця 31.8
Методика обстеження хворих з міофасціальним болем
|
Спостереження за станом хворих |
Важливу інформацію може дати спостереження за хворим: як він заходить в кабінет, як сідає, знімає одяг. Пацієнти з болючими активними тригерними точками здійснюють повільні щадні рухи, уникають рухів, які можуть розтягнути уражений м’яз. Слід звернути увагу, чи рухає хворий руками повною мірою, як повертає голову (одну або увесь торс), як сидить (прямо або зігнувшись), чи не спотворено його обличчя |
|
Опитування хворого |
При опитуванні необхідно, щоб хворий сидів у зручній позі, ноги спиралися на підлогу (підставку), руки знаходилися на підлокітниках. При такій розслабленій позі він зможе оцінити роль м’язового напруження в провокації болю. На прохання лікаря хворий обкреслює пальцем больову зону на своєму тілі, і лікар відмічає її контур на спеціальному бланку (з намальованими контурами тіла людини). Окрім точного встановлення місця болю, лікар уточнює дату її появи, в якому напрямі розвивався біль (вгору, вниз, упоперек). Якщо хворий говорить, що у нього усе болить, треба поступово запитувати про кожну групу м’язів. Потім точний малюнок больових зон лікар зіставляє з відомими больовими патернами окремих м’язів. Для виявлення м’язових тяжів і тригерних точок застосовують зигзагоподібну і глибоку пальпацію |
|
Зигзагоподібна пальпація |
Лікар, поперемінно зміщуючи кінчик пальця то в один, то в інший бік упоперек м’язових волокон, рухає його уздовж м’яза. При такій пальпації деякі тригерні точки виявляються у вигляді вузликів. Але все таки зазвичай їх виявляють уздовж пальпованого тяжа як максимально болючі |
|
Глибока пальпація |
Шкіру над м’язом змащують мазю. Великі (чи вказівні) пальці обох рук глибоко занурюють з двох боків тяжа на одному кінці м’яза і проводять уздовж нього ковзаючий рух у напрямку до тригерних точок, тобто здійснюють рух, що «доїть». Це рух, що «доїть», слід проводити у напрямі венозного відтоку. При такій глибокій пальпації тугого тяжа в ділянці тригерної точки відчувається грудка або вузлик |
Таким чином, для виявлення м’язових тяжів і тригерних точок застосовують зигзагоподібну і глибоку пальпацію. Зигзагоподібна пальпація виявляє тугий тяж, що включає тригерну точку, а глибока пальпація виявляє саму тригерну точку у вигляді вузлика.
Прийоми, що полегшують пошук тригерних точок. Полегшити пошук тригерних точок допоможе лікареві знання ряду фактів (табл. 31.9).
Таблиця 31.9
Інформація, знання якої полегшує пошук тригерних точок
|
1. |
Зазвичай рух, пов’язаний із розтягуванням ураженого м’яза, обмежений. При спробі збільшити амплітуду цього руху виникає сильний біль |
|
2. |
Збільшення вираженості болю в м’язі, що має активні тригерні точки, відмічають при активному або пасивному її розтягуванні |
|
3. |
Біль посилюється при подоланні м’язом, що скорочується, сили зовнішньої дії |
|
4. |
Максимальна скорочувальна сила ураженого м’яза ослаблена |
|
5. |
У зоні відбитого від міофасціальних точок болю виявляють глибоку болючість і порушення чутливості |
|
6. |
Тригерна точка при пальпації відчувається як чітко обмежена ділянка з гострою болючістю. Ця болючість значно менше виражена в декількох міліметрах від точки |
|
7. |
У безпосередній близькості від тригерної точки пальпуються напружені м’язові волокна |
|
8. |
Натиснення пальцем на активну тригерну точку викликає «симптом стрибка» (хворий сильно здригається) |
|
9. |
Щипкова пальпація викликає локальну судомну відповідь. Особливо таку відповідь відмічають з боку груднино-ключично-соскоподібного, великого грудного, дельтовидного, широкого м’яза спини, великого сідничного м’яза. До судомної відповіді залучаються м’язові волокна, що знаходяться в зоні ущільненого (тугого) тяжа |
|
10. |
Помірний безперервний тиск на тригерну точку викликає або посилює біль у відбитій зоні |
|
11. |
У деяких хворих шкіра проявляє виражений дермографізм або ознаки панікуліту (позитивною є проба катання шкіри). Найчастіше ці прояви виявляють у верхній ділянці грудей, попереково-крижовій ділянці |
При обстеженні хворих з міофасціальним болем виявляють підвищення активності ЛДГ у крові. При термографічному дослідженні шкіри над активними тригерними точками виявляють зону діаметром 5–10 см з підвищеною температурою тіла. Може бути збільшена шкірна провідність над тригерною точкою.
Фіброміалгію необхідно диференціювати з РПМ; дерматоміозитом; гіпо- і гіпертиреозом; періоститом; слабо вираженим суглобовим синдромом. Фіброміалгію диференціюють також із гіпотиреозом, паранеопластичним синдромом, синдромом хронічної втоми.
Перебіг фіброміалгії
Хвороба має хронічний перебіг, ремісії виникають зрідка, симптоматика прогресує на початку хвороби. З часом прояви хвороби можуть зменшуватися, особливо в осіб старшого віку. Погіршення стану хворих наступає після перевантаження (фізичного, емоціонального), переохолодження, пошкодження мяких тканин, інфекцій, зміни гормонального статусу, при недостатній тривалості сну. Стан хворих погіршується зимою, покращується влітку.
Лікування фіброміалгії
Для інактивації тригерних точок використовують ряд методів лікування (табл. 31.10).
Таблиця 31.10
Методи лікування, що використовуються для інактивації тригерних точок
|
1. |
Розтягування й анестезія охолодженням (хлоретилом) з наступним розтягуванням м’яза до його повної довжини. Про ефективність процедури свідчить збільшення обсягу руху. Слід врахувати, що надлишкове охолодження м’яза може призвести до посилення тригерної точки |
|
2. |
Компресія тригерних точок з метою викликати в ній ішемію і гіпоксію. Використовуються пальцьовий, змішаний і вібраторний масаж |
|
3. |
Проколювання тригерних точок як з одночасною ін’єкцією ізотонічного розчину натрію хлориду, новокаїну, так і без них |
|
4. |
Застосування ультразвуку слабкої інтенсивності, що чинить тепловий вплив на глибокі шари м’язової тканини і сприяє інактивації тригерних точок |
Інактивація міофасціальної тригерної точки ин’єкційним методом. В якості передін’єкційної підготовки хворий протягом тижня щодня приймає 500 мг аскорбінової й ацетилсаліцилової кислоти. Пацієнт знаходиться в горизонтальному положенні, перед ін’єкцією його слід попередити про можливий спалах відбитого болю і судорожне скорочення м’яза. М’яз розтягують, щоб волокна, що містять тригерні точки, туго натягувалися. Це дозволить утримувати тригерні точки в стійкому положенні. Для ін’єкції тригерну точку фіксують, притискуючи її двома розведеними пальцями. У тригерні точки вводиться стерильний 0,5% новокаїн з ізотонічним розчином до тих пір, поки ділянка не стане безболісною. Після ін’єкції проводять анестезію охолодженням і пасивне розтягування м’яза. Для зменшення постін’єкційної болючості на м’яз на декілька хвилин накладають гарячий компрес. Через декілька хвилин після накладення компресу хворий здійснює рухи, що усувають залишкову ригідність. Потім хворому необхідно регулярно робити вправи з пасивного розтягування м’язів. При сильному постін’єкційному болю призначають парацетамол.
Метод ішемічної компресії. Розслаблений м’яз розтягують до появи відчуття дискомфорту, потім стискують тригерні точки великим (найбільш сильним пальцем) до появи переносимого болю (хворий не повинен напружувати м’яз, оскільки це захищає тригерні точки від стиснення). Стиснення тригерних точок із силою 9–13 кг продовжувати протягом 1 хв. Після зігрівання м’яза гарячим компресом й активного її розтягування процедуру можна повторити.
Як альтернативний метод болючу точку в м’язі стиская сам хворий на 7–10 с кілька разів на добу протягом декількох днів. При глибокому заляганні ураженого м’яза можливе застосування стиснення ліктем.
Масаж тригерних точок. Хворий розташовується в зручній позі, щоб уражені м’язи були розслаблені й помірно розтягнуті. Шкіру над м’язами змащують олією. Лікар зануреними в м’яз двома великими пальцями повільно (8 мм/с) ковзає уздовж неї від дистального кінця у напрямку до тригерної точки, здійснюючи рух, що неначе доїть. Тиск поступово збільшують. Можна застосовувати також кругову розминку м’язової тканини в ділянці тригерної точки.
Метод інактивації тригерної точки ультразвуком. Використовують 2 методи лікування (табл. 31.11).
Таблиця 31.11
Методика інактивації тригерних точок ультразвуком
|
1. |
Навколо тригерної точки здійснюють кругові рухи спрямованого ультразвуку інтенсивністю 0,5 Вт/см2. Круговий рух здійснюють за 1–2 с |
|
2. |
Впливають на тригерні точки ультразвуком інтенсивності, що змінюється. Спочатку інтенсивність ультразвуку підвищують до больового порогу (1,5 Вт/см2), потім знижують наполовину. Через 2–3 хв інтенсивність поступово підвищують до первинного больового порогу |
Інактивація тригерних точок вологою парою. Волога пара здатна розслаблювати м’язи і зменшувати напруженість волокон в ділянці тригерних точок. Вона знижує інтенсивність відбитого болю і локальну болісність при стисканні. Накладення вологого гарячого компресу може привести до одужання м’яза протягом 72 год без додаткових процедур.
Застосування ГК при інактивації тригерних точок. При запальних процесах в сполучній тканині, що перешкоджають лікуванню тригерних точок, найбільш ефективним є введення кортикостероїдів за схемою: лікування преднізолоном починають з добової дози 60 мг, потім протягом тижня дозу щодня знижують на 10 мг. Підтримувальну дозу можна залишити на декілька тижнів. Після усунення запального процесу можна застосувати наведені вище методи інактивації тригерних точок.
Усунення надмірної післяпроцедурної м’язової болючості в ділянці тригерних точок. В ділянку тригерної точки вводять 1 мл (4 мг) дексаметазону, змішаного з 1 мл 2% розчину новокаїну.
Інші засоби лікування, які застосовують у хворих на фіброміалгію. Характер фармакотерапії, яку застосовують у хворих на фіброміалгію (Насонов Е.Л., 2011 (ред.)), представлено у табл. 31.12.
Таблиця 31.12
Фармакологічні засоби, які застосовують у хворих на фіброміалгію
|
Групи препаратів |
Препарати, дози |
Примітки |
|
Анальгетики |
Трамадол, початкова доза 50 мг/добу з подальшим підвищенням до 200–250 мг/добу |
Впливає на норадренергічні і серотонінергічні шляхи, вираженість болю |
|
Трамадол 37,5 мг + парацетамол 325 мг |
||
|
Трициклічні антидепресанти |
Амітриптилін, початкова доза 10 мг за 1–2 год до сну з подальшим підвищенням до 50–75 мг/добу |
Зменшує психологічний дискомфорт, інтенсивність болю |
|
Антагоністи серотонінових 5-НТ3-рецепторів |
Тропісетрон, 5 мг/добу |
Знижує інтенсивність болю, вираженість вегетативних симптомів, психологічного дискомфорту, коригує сон |
|
Селективні інгібітори зворотного захоплення серотоніну |
Флуоксетин, 20 мг/добу |
Призначають при виражених порушеннях емоційної сфери. Можна комбінувати з низькими дозами трициклічних антидепресантів |
|
Циталопрам, 20–40 мг/добу |
||
|
Сертралін, 50–200 мг/добу |
||
|
Неселективні інгібітори зворотного захоплення серотоніну |
Дулоксетин, 60–120 мг/добу |
Знижує інтенсивність больового синдрому, число больових точок |
|
Мілнаципран, 25–150 мг/добу |
Знижує інтенсивність больового синдрому, ступінь стомлюваності, порушень емоційної сфери |
|
|
Венлафаксин, 75 мг/добу |
Знижує вираженість основних проявів фіброміалгії |
|
|
Інгібітори моноамінооксидази |
Моклобемід 450–600 мг/добу |
Помірний знеболювальний ефект |
|
Антагоністи допамінових рецепторів |
Праміпексол: початкова доза 0,25 мг/добу в подальшому підвищується до 4,5 мг/добу (приймають увечері) |
Позитивно впливає на інтенсивність больового синдрому, втому, функціональну активність |
|
Міорелаксанти |
Циклобензаприн, початкова доза 10 мг на ніч з подальшим підвищенням до 30 мг/добу в 2 прийоми |
Зменшує вираженість втоми, больового синдрому, покращує якість сну |
|
Антиконвульсанти |
Прегабалін, 450 мг/добу |
Зменшують ступінь вираженості болю, порушень сну, втоми, синдрому «неспокійних» ніг |
|
Габапентин, 300–400 мг/добу |
Характер немедикаментозного лікування фіброміалгії наведений в табл. 31.13.
Таблиця 31.13
Немедикаментозне лікування фіброміалгії
|
Навчання пацієнтів |
Індивідуальні заняття та в групах, встановлення контактів, зв’язків між хворими. Обговорення патофізіологічних механізмів фіброміалгії |
|
Когнітивно-поведінкова терапія |
Її мета — позбавити хворого відчуття некурабельності хвороби, сформувати стратегію поведінки, яка б сприяла контролю пацієнтом свого захворювання, зменшенню негативного впливу на його життя, модифікації та формуванню здорового, життєстверджувального способу життя. У реалізації програми повинні брати активну участь усі члени родини |
|
Фізичні тренування. Гідротерапія |
Включають тренування сили, гнучкості, аеробні вправи. Головна мета фізичних тренувань — збереження функціональної активності м’язів, попередження мікротравматизації, підвищення продукції β-ендорфінів, зниження симпатичної активності, поліпшення психологічного статусу, сну. Слід притримуватися таких рекомендацій, як індивідуальний підбір вправ, помірна інтенсивність, регулярність виконання, поступове збільшення навантаження. Гідротерапія сприяє релаксації, редукції перевантаження суглобів, вазодилатації, аналгезії. Зменшує вираженість болю, покращує функціональний стан хворих. При збільшенні вираженості міалгій слід тимчасово знизити інтенсивність тренувань |
|
Масаж, фізіотерапія |
Призначають за індивідуальними показаннями |
У комплексну терапію фіброміалгії включають курси ЛФК, психологічної релаксації (психотерапії), аутогенні і фітнес-тренування, плавання, прогулянки, навчальні програми, фізіотерапевтичні методи (в тому числі лазеротерапію, ультразвук, фонофорез, теплові процедури), масаж (легкий), гомеопатичне лікування (Rhus toxicodendron). Окрім лікарів-ревматологів до ведення хворих слід залучати психологів (психіатрів), терапевтів.
В табл. 31.14 наведено інструменти, які використовують для оцінки динаміки проявів фіброміалгії на тлі лікування.
Таблиця 31.14
Інструменти, які використовують для оцінки динаміки проявів фіброміалгії на тлі лікування
|
Оцінка інтенсивності болю за ВАШ |
|
|
Опитувальник Fibromyalgia Impact Questionnaire (FIQ) |
Опитувальник (заповнює пацієнт) складається із 10 пунктів, 4–10 з яких являють собою шкали, які оцінюють вплив проявів фіброміалгії на працездатність, інтенсивність болю, стомлюваності, скутості, тривоги, депресії, якість сну за останній тиждень. Градація від 0 до 10, максимально можливе число балів — 100 |
|
Опитувальник Mc Gill Pain Questionnaire (заповнює пацієнт) |
Дозволяє характеризувати в динаміці інтенсивність болю, його якісні особливості: емоціональний та сенсорний компоненти |
|
Brief Pain Inventory |
Оцінюється інтенсивність болю за останні 24 год. Градація від 0 до 10 балів (10 — максимально виражений біль, 0 — він відсутній). Оцінюється вплив болю на загальну активність пацієнта, його настрій, здатність рухатися, роботу, ставлення до людей, сон, здатність радіти життю |
|
Tender point Count |
Відзначають кількість діагностичних болючих точок. Мінімальне значення стимулу, яке обстежуваний сприймає як больове відчуття, є порогом больової чутливості. За допомогою долориметра вимірюють больову чутливість; больовий поріг виражають в одиницях відношення сили тиску до площі наконечника. Він дозволяє контролювати ефективність дії анальгетиків. Середній поріг больової чутливості розраховують як середнє значення показника за 18 діагностичними точками. Градація при пальпаторній оцінці: 0 — біль відсутній, 1 — слабкий біль (ствердна відповідь при запитанні), 2 — фізична, вербальна реакція на тиск, поява гримаси, 3 — виражений біль (рух назад, відсмикування) |
|
Оцінка розладів ментального статусу |
|
|
Опитувальник Mini International |
Скринінг психопатології |
|
Опитувальник Hamilton |
Оцінка вираженості депресії |
|
Оцінка вираженості тривоги |
|
|
Оцінка сну |
|
|
Опитувальник Leeds |
Оцінка (кількісна та якісна) на тлі медикаментозного лікування |
|
Пітсбургський індекс якості сну |
Якість сну, його порушення за останній місяць |
|
Опитувальник шпиталю Saint Mary |
Об’єктивна систематична оцінка концентрації і сну за останні 24 год |
|
Sleep Quality assessment (SQA) |
Оцінка відновлювального ефекту сну |
|
Полісомнографічне дослідження |
|
Профілактика
Первинна профілактика не розроблена, вторинна — включає виявлення та корекцію факторів ризику, які викликають розвиток та хронізацію болю в скелеті, м’язах. До третинної профілактики належать: лікування проявів фіброміалгії, реабілітаційні заходи (з метою підвищення якості життя хворих, функціональної здатності), попередження загострення хвороби.
Прогноз
Суттєво знижується працездатність, мають місце значні матеріальні затрати. Для життя прогноз сприятливий.
Глава 32. Хвороба Педжета
Хвороба Педжета — захворювання, при якому відбувається посилена патологічна перебудова кісткової тканини з чергуванням процесів резорбції остеокластами та її новоутворення. Формується дезорганізована мозаїчна структура кісткової тканини з ділянками дрібнопористої і трабекулярної будови. Також відмічається підвищена васкуляризація уражених ділянок кісток, фіброз кісткового мозку. Посилена резорбція кісткової тканини відбувається на ранніх стадіях хвороби, потім процеси резорбції змінюються новоутворенням кісткової тканини, хаотичним заміщенням дефектів.
Код за МКХ-10
М88 Хвороба кісток Педжета (деформуючий остит).
М88.0 Ураження черепа при хворобі Педжета.
М88.8 Ураження інших кісток при хворобі Педжета.
М88.9 Хвороба Педжета (кісток), неуточнена.
М88. Викривлення хребта при хворобі кістки (деформуючий остит).
Епідеміологія хвороби
У чоловіків хворобу Педжета виявляють частіше, ніж у жінок (3:2). Починаючи з 50-річного віку, захворюваність подвоюється кожні 10 років, досягаючи майже 90% до 90-річного віку. Переважно уражується населення північноєвропейського походження. Захворювання не виявляють в Африці, Індії, на Далекому та Ближньому Сході. У США воно переважно поширене на півночі країни. Поширеність хвороби становить 5% у Великобританії і 1–3% в США.
Етіологія
Існує припущення, що хвороба Педжета розвивається в результаті вірусної інфекції: в остеокластах уражених ділянок кістки виявлені внутрішньоклітинні включення, схожі на нуклеокапсиди параміксовірусів. Відзначається генетична детермінованість захворювання: хворобу Педжета у родичів хворих відмічають в 7 разів частіше, ніж у популяції. Особливо високий ризик розвитку захворювання виявляють у тих випадках, коли у родича воно проявилося в ранньому віці та/або його перебіг був тяжким. Не виявлено чіткого зв’язку з антигенами групи HLA. У своєму розвитку хвороба Педжета проходить 3 стадії:
I Початкова (остеомієлітична, стадія руйнування).
II Активна (змішана, що поєднує остеоліз та остеогенез).
III Неактивна (остеосклеротична, остеопластична).
Патогномонічною картина стає на останній стадії: кісткова тканина за своїм видом нагадує мозаїку з товстих кісткових балок, які складаються з грубих волокон.
Клініка
Клінічні прояви хвороби Педжета приведені в табл. 32.1.
Таблиця 32.1
Характеристика клінічних проявів хвороби Педжета
|
Клінічні прояви |
Характеристика |
|
Больовий синдром в результаті ураження кісток |
Больовий синдром відмічають у 80% випадків, турбує біль у суглобах — колінних, кульшових, в хребті. Відзначається гіпертермія м’яких тканин над ураженими ділянками кістки, зумовлена підвищеною васкуляризацією. Зміни розвиваються в одній або декількох кістках скелета. Моноосальна форма відмічається в 20%, поліосальна — у 80% випадків. При моноосальній формі найчастіше уражуються великогомілкові та клубові кістки. У цілому найчастіше уражуються (в порядку зменшення) кістки тазу → поперековий відділ хребта → стегнова кістка → грудний відділ хребта → великогомілкова і плечова кістки |
|
Деформації кісток і суглобів |
Розвиваються у задавнених випадках. Характерне потовщення кісток черепа і дугоподібне викривлення кісток гомілки (див. рис. 31.1) |
|
Спонтанні переломи |
Відзначається схильність до патологічних (спонтанних) переломів. Частіше відзначають переломи стегнової, великогомілкової, плечової та кісток передпліччя |
|
Клініка остеоартриту |
Розвивається при локалізації ураження кісткової тканини поблизу суглобів |
|
Неврологічні прояви |
Інсульти (як результат стиснення кровоносних судин), стиснення нервів (черепних, корінців спинномозкових) і спинного мозку, порушення анатомо-топографічних співвідношень між кістками черепа і головним мозком, глухота (результат осифікації сухожилля стремена або стиснення слухового нерва), головний біль. Хворі відмічають біль у спині, бувають випадки синдрому кінського хвоста. Стиснення спинного мозку виникає при компресійних переломах хребців зі зміщенням, звуженні каналу хребта. Ураження спинного мозку може бути наслідком стиснення артерій, які живлять спинний мозок. При звуженні каналу хребтової артерії виникає ішемія головного мозку. |
До ускладнень хвороби Педжета відносяться: збільшення розмірів серця, застійна серцева недостатність, артеріальна гіпертензія, стенокардія, розвиток остеогенної саркоми (0,2–1,0%), фібросаркоми, доброякісних гігантоклітинних пухлин, гіперкальціємія, гіперкальціурія, нефрокальциноз. Для серцевої недостатності є характерним високий серцевий викид.
Діагноз
Інформативність лабораторних показників при хворобі Педжета. Найбільш показовим є підвищення активності ЛФ і вмісту остеокальцину у сироватці крові. Ці показники є маркерами процесів новоутворення кісткової тканини, особливо ЛФ. За їх концентрацією можна судити про тяжкість захворювання і ступінь активності патологічного процесу. Рівень показників залежить від локалізації процесу: високі показники маркерів визначаються при ураженні кісток черепа, нижчі — при ураженні інших кісток (рис. 32.1). Підвищена активність ЛФ в крові виявляється і у пацієнтів з безсимптомним перебігом хвороби. При цій патології відмічають підвищене виведення гідроксипроліну і піридиноліну з сечею, зумовлене посиленою резорбцією кісткової тканини. Часто виявляється гіперкальціємія, особливо за наявності переломів, або в період тривалої іммобілізації.
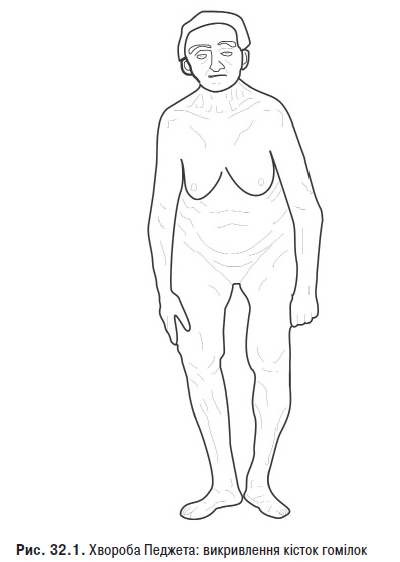
Зміни на рентгенограмах та сцинтиграмах пацієнтів з хворобою Педжета. Для діагностики, оцінки тяжкості ураження і поширеності патологічного процесу використовують рентгенографію, сцинтиграфію кісток скелета з технецієм (табл. 32.2).
Таблиця 32.2
Інформативність рентгенографії та сцинтиграфії у пацієнтів з хворобою Педжета
|
Метод дослідження |
Інформативність |
|
Рентгенографія |
На рентгенограмах кісток скелета виявляють ознаки остеолізу, новоутворення кісткової тканини або поєднання ушкоджень. Характерним є потовщення кортикального шару та прилеглих трабекул. Краї діафізів довгих трубчастих кісток остеолітично змінені, шорсткі (симптом «листя трави»). Відмічають рентгенологічний симптом «полів капелюха» або «тазового кільця» — виражене потовщення трабекул в ділянці з’єднань клубової і лонної, клубової та сідничої кісток з боку внутрішньої поверхні таза. Типовою є також наявність на рентгенограмах черепа «вогнищевого» або «пластівчастого» ОП (osteoporosis circumscripta, симптом «ватних кульок») — ділянок надлишкового руйнування кісткової речовини в кістках черепа, а також потовщення кісток склепіння черепа. На рентгенограмі кісток тазу відмічають груботрабекулярну перебудову кісткової структури, ділянки склерозу і потовщення кортикального шару. Остеолітичні зміни прогресують повільно |
|
Сцинтиграфія |
Сцинтиграфію скелета використовують для оцінки поширеності ураження й ефективності лікування, що проводиться. Для неї характерне формування зон підвищеного захоплення радіоактивної речовини. На оглядовій сцинтиграмі тіла визначається підвищене накопичення радіоізотопу в кістках черепа, таза, хребцях поперекового відділу хребта, стегнових, великогомілкових кістках, лопатках і проксимальних відділах плечових кісток. При цьому захворюванні до процесу не залучаються прилеглі кістки, немає «метастазування» у віддалені зони скелета |
Результати представлених методів дослідження відрізняються один від одного: близько 12% ушкоджень, видимих на сцинтиграмах, не визначаються при рентгенографічному дослідженні, а 6% ушкоджень, що виявляють на рентгенограмах, відсутні на сцинтиграмах.
Диференціація хвороби Педжета. Хворобу Педжета диференціюють із лімфомами, метастазами злоякісних пухлин, зокрема аденокарциномою передміхурової залози (відмічають схожі зміни в хребцях). Проте, на відміну від цих захворювань, при хворобі Педжета уражені хребці збільшуються в розмірах. Слід враховувати характерну для хвороби Педжета груботрабекулярну перебудову кісткової структури, а також перехід від остеолізу до склерозування кісткової тканини, що не характерно для метастатичних уражень. Слід пам’ятати, що вогнища підвищеного накопичення радіоізотопного препарату на сцинтиграмі відмічають при цілому ряді захворювань, зокрема при остеомієліті, метастазах, артриті, переломах.
Методи лікування хвороби Педжета
Лікування хвороби Педжета починають з використання кальцитоніну і бісфосфонатів, які знижують активність остеокластів (табл. 32.3).
Таблиця 32.3
Препарати, які застосовують при хворобі Педжета
|
Кальцитонін |
Застосовують у разі вираженого остеолізу, при локалізації патологічного процесу в кістках, що несуть вагове навантаження, сильному болю, наявності неврологічної симптоматики. Його призначають підшкірно в дозі 100 MО щодня до отримання клінічного поліпшення і зниження біохімічних показників. Надалі дозу знижують до 50–100 МО через день або 3 рази на тиждень. Симптоми хвороби купіруються через декілька тижнів від початку лікування, але після припинення введення препарату рецидивують. Більше ніж у 25% хворих відзначається «феномен плато», оскільки в організмі виробляються нейтралізуючі антитіла до кальцитоніну лосося. У таких випадках ефективною є заміна препарату на кальцитонін людини |
|
Бісфосфонати |
Ефективною є етидронова кислота в дозі 5–10 мг/кг/добу (таблетки по 200 і 400 мг). Її прийом супроводжується поліпшенням стану, зниженням у 2 рази концентрації в крові біохімічних маркерів резорбції кістки. Етидронову кислоту приймають протягом 6 міс з наступною 6-місячною перервою, оскільки вона пригнічує процеси кісткоутворення. У деяких хворих можливе парадоксальне збільшення вираженості больового синдрому на тлі лікування з розвитком вогнищевої або генералізованої остеомаляції. Хороший ефект дає памідронова кислота — бісфосфонат другого покоління, який вводять парентерально. Вона не впливає на мінералізацію новоутвореної кісткової тканини |
|
Інші засоби |
НПЗП застосовують для купірування больового синдрому, у тому числі, пов’язаного із вторинним ОА. При стисненні нервів, необхідності збільшити рухливість в суглобі проводять хірургічні втручання |
Прийом двонатрієвої етидронової кислоти може супроводжуватись абдомінальними спазмами, діареєю, гіперфосфатемією, посиленням болю в кістках і навіть підвищенням частоти переломів. Памідронову кислоту вводять в/в у дозі 60–90 мг в ізотонічному розчині хлориду натрію або водному розчині глюкози. При цьому використовуються різні схеми лікування: щоденно протягом 3–5 днів; 1 раз на тиждень протягом 5–6 тиж; 1 раз з щомісячними контрольними обстеженнями. Повторне лікування супроводжується прогресуючою резистентністю. Алендронова кислота призначається перорально в дозі 40 мг/добу протягом 6 міс. Ризедронову кислоту призначають перорально 30 мг/добу протягом 2 міс, тілудронат — 400 мг протягом 3 міс. Золедронова кислота в дозі 4 мг в/в (вводять протягом 15 хв) дає більш тривалий ефект, ніж ризедронова. Хворим призначають також анальгетики, НПЗП, препарати кальцію та вітаміну D3.
Побічні ефекти при застосуванні кальцитоніну і бісфосфонатів. У 20% хворих введення кальцитоніну супроводжується припливами крові до обличчя, нудотою і транзиторною гіпокальціємією. Цього можна уникнути, якщо почати лікування з низьких доз (25–50 MО) і поступово їх підвищувати. До побічних дій бісфосфонатів відносяться грипоподібні симптоми, подразнення ШКТ, субфебрилітет, транзиторна лейкопенія. Якщо дозу бісфосфонатів поступово підвищувати кожні 1–2 тиж, то ці побічні ефекти не з’являються.
Прогноз
Тривала антирезорбтивна терапія призводить до пригнічення функції багатоядерних остеокластів, запобігає появі нових патологічно змінених клітин. Відбувається поступова загибель заражених параміксовірусами багатоядерних остеокластів, настає тривала ремісія захворювання або повне одужання.
Глава 33. Інфекційний ендокардит
ІЕ — запальне ураження клапанів серця, ендокарда, зрідка — ендотелія аорти та крупних артерій, обумовлене прямою інвазією інфекційного збудника. Його перебіг супроводжується розвитком імунопатологічних проявів, можливою генералізацією септичного процесу.
Код за МКХ-10
I33.0. Гострий та підгострий інфекційний ендокардит.
М03.6 Артропатія при інфекційному ендокардиті (I33.0).
Епідеміологія
Частота захворювання коливається від 1 до 11,6 випадків на 100 тис. населення. Відмічають значне підвищення захворюваності (за 1982–1996 рр. — у 8 разів). Причиною зростання є стан здоров’я населення України (зниження імунної реактивності, підвищення вірулентності збудників внаслідок нераціональної антибіотикотерапії), поліпшення діагностики хвороби, підвищення кваліфікації лікарів. 60–90% хворих становлять чоловіки, вік більшості хворих — до 40 років. У віковій популяції старше 60 років співвідношення чоловіків та жінок становить 8:1. Відзначають значне підвищення частоти первинного клапанного ІЕ. Вторинний ендокардит виявляють у 6–25% випадків. Кількість хворих на ІЕ у похилому віці в останні роки збільшилась з 5 до 29%. В 10% випадків ІЕ виникає повторно. В останні роки при клапанній патології атеросклеротичного дегенеративного генезу частота виявлення ІЕ підвищилася на 50% (раніше становила 10–32%). Виділено внутрішньолікарняний (госпітальний) ІЕ, який виникає внаслідок лікувальних та діагностичних маніпуляцій, використання центральних венозних катетерів, катетеризації артерій. Причинами ятрогенного ІЕ можуть бути екстракція зуба, протезування клапанів серця, гемодіаліз, терапія ГК, хіміотерапія, інфікування в/в катетерів, хірургічні втручання, постін’єкційні абсцеси тощо.
Профілактика інфекційного ендокардиту
Відповідно до рекомендацій комітету експертів Європейського кардіологічного товариства профілактику слід проводити хворим, у яких ІЕ розвивається частіше порівняно з популяцією (група помірного ризику), а також асоціюється з високою летальністю (група високого ризику) (табл. 33.1).
Таблиця 33.1
Групи ризику розвитку ІЕ
|
Групи високого ризику |
ІЕ в анамнезі |
|
Штучні клапани серця (в тому числі біопротези, алотрансплантати) |
|
|
«Сині» вроджені вади серця (тетрада Фало, транспозиція крупних артерій та ін.) |
|
|
Прооперовані системні легеневі шунти |
|
|
Групи помірного ризику |
Набуті вади серця |
|
Неоперовані вроджені вади серця (відкрита артеріальна протока, первинний дефект міжпередсердної та міжшлуночкової перегородки, коарктація аорти, двостулковий аортальний клапан) |
|
|
Пролапс мітрального клапана з регургітацією та/або потовщенням стулок |
|
|
Гіпертрофічна кардіоміопатія (ГКМП) |
Антибіотикопрофілактику проводять при втручаннях, які супроводжуються бактеріємією потенціальними збудниками ІЕ. Показання до антибіотикопрофілактики ІЕ та її схеми наведені (за: Насонов Е.Л., 2011 (ред.)) відповідно в табл. 33.2 та 33.3.
Таблиця 33.2
Показання до антибіотикопрофілактики ІЕ
|
Ротова порожнина |
Стоматологічні маніпуляції з ризиком пошкодження ясен, слизової оболонки |
|
Дихальні шляхи |
Аденоїдектомія та/або тонзилектомія |
|
Операції з порушенням цілісності слизової оболонки |
|
|
Бронхоскопія (яка виконується жорстким бронхоскопом) |
|
|
ШКТ |
Дилатація стриктури стравохода |
|
Склеротерапія варикозно розширених вен стравохода |
|
|
Оперативні втручання на жовчних шляхах з причини обтурації |
|
|
Урогенітальний тракт |
Біопсія передміхурової залози або сечовивідних шляхів |
|
Трансуретральна резекція передміхурової залози |
|
|
Цистоскопія на фоні інфекції сечових шляхів |
|
|
Літотрипсія |
|
|
Дилатація уретри |
|
|
Гінекологічні маніпуляції за наявності інфекції |
Таблиця 33.3
Схеми антибіотикопрофілактики ІЕ
|
Маніпуляції в ротовій порожнині, на стравоході, дихальних шляхах |
Стандартна схема |
Рer os амоксицилін 2,0 г за 1 год до процедури |
|
При неможливості прийому per os |
Ампіцилін 2,0 г в/в або в/м за 30 хв до процедури |
|
|
При алергії на пеніциліни |
Per os кліндаміцин 600 мг або азитроміцин 500 мг, або кларитроміцин 500 мг за 1 год до процедури |
|
|
При алергії на пеніциліни та неможливості прийому per os |
Кліндаміцин 600 мг в/в за 30 хв до процедури |
|
|
Маніпуляції на ШКТ та урогенітальному тракті |
Група високого ризику |
В/м або в/в ампіцилін 2,0 г + в/м або в/в гентаміцин 1,5 мг/кг за 30 хв до процедури; через 6 год — в/м або в/в ампіцилін 1,0 г або амоксицилін 1,0 г per os |
|
Група високого ризику + алергія на пеніциліни |
Ванкоміцин 1,0 г в/в протягом 1–2 год + гентаміцин 1,5 мг/кг в/в або в/м (введення закінчують за 30 хв до процедури) |
|
|
Група помірного ризику |
Per os амоксицилін 2,0 г за 1 год до процедури або ампіцилін 2,0 г в/м або в/в за 30 хв до процедури |
|
|
Група помірного ризику + алергія на пеніциліни |
В/в ванкоміцин 1,0 г протягом 1–2 год (введення закінчують за 30 хв до процедури) |
В Інституті серцево-судинної хірургії застосовують наступний режим антибіотикотерапії для профілактики ІЕ (Кнышов Г.В. и соавт., 2012): за відсутності алергії на пеніциліни — ампіцилін + оксацилін 2 г перорально за 30–60 хв до процедури; у разі наявності алергії на пеніциліни — кліндаміцин 600 мг перорально за 30–60 хв до процедури; альтернативний режим включає цефалексин 2 г в/в, цефазолін або цефтріаксон 1 г в/в.
Етіопатогенез інфекційного ендокардиту
В минулому столітті у 83–96% випадків ІЕ з крові висівався зеленкуватий стрептокок. За даними різних авторів, сьогодні його частка становить 40–70%, стафілококів — 15–40%, грамнегативних бактерій — до 10%, грибкової мікрофлори — до 4,6%. Патологія, яка супроводжується розвитком ІЕ, наведена в табл. 33.4.
Таблиця 33.4
Патологія, яка супроводжується розвитком ІЕ
|
Етіологія |
Фонова патологія |
|
Стрептококові ІЕ (групи А, В, С, D, G) |
Виникають при хірургічних маніпуляціях в ротовій порожнині, тонзилітах, фарингітах, синуситах, стрептококових пневмоніях (особливо в післяопераційний період) |
|
Стафілококові ІЕ |
Виявляють при операціях на серці, імплантації штучного водія ритму, застосуванні інвазивних інструментальних методів дослідження, тривалому використанні інтравенозних катетерів, програмному гемодіалізі, інфікуванні та нагноєнні ран, утворенні абсцесів, наркоманії |
|
Ентерококові ІЕ |
Виникають при сечостатевій інфекції, патології ШКТ, акушерсько-гінекологічних операціях, катетеризаціях сечового міхура, використанні внутрішньоматкових контрацептивів |
|
ІЕ, викликані 2 та більше збудниками |
Відмічають у пацієнтів з імунодефіцитами (у хворих з пухлинами, які приймають цитостатики) |
При співіснуванні декількох збудників відмічають галопуючий перебіг, полівальвулярне ураження із залученням міокарда. Причиною виникнення ІЕ у наркоманів є золотистий стафілокок (65–71,4%), зеленкуватий стрептокок (2,5–16,4%), ентерокок (2,5%), грамнегативні бактерії (2–8%), гриби (5%), декілька мікроорганізмів (5–13%). Ранній ІЕ протезованого клапана викликають стафілококи (51–56%), грамнегативні бактерії (14–17%), гриби (9%). ІЕ у хворих з електрокардіостимуляторами викликають переважно стафілококи (66,7–93,5%). Частота бактеріємії при деяких операціях, маніпуляціях представлена у табл. 33.5.
Таблиця 33.5
Частота бактеріємії при деяких операціях, маніпуляціях
|
Хірургічні втручання на періодонті — 80% |
Фіброгастродуоденоскопія — 4% |
|
Видалення зуба — 82% |
Іригоскопія — 10% |
|
Зняття зубного каменя — 40% |
Мітральна комісуротомія — 0,4–2,3% |
|
Тонзилектомія — 38% |
Анулопластика мітрального клапана — 4% |
|
Простатектомія за наявності бактеріурії — 57% |
Протезування клапанів — 1,2–9,5% |
|
Простатектомія при стерильній сечі — 11% |
Катетеризація серця — 0,94 на 1000 випадків |
|
Видалення сечового катетера після урологічної операції — 50% |
Встановлення катетера Свана — Ганца — 2,4–9,3% |
|
Фізіологічні пологи — 7% |
Інтравенозні катетери — 0,8–0,85% |
|
Бронхоскопія — 15% |
Регулярний гемодіаліз — 1,75–5,1% |
|
Дилатація стравохода — 45% |
Імплантація штучного водія ритму серця — 1% |
|
Колоноскопія — 5% |
Наркоманія — 1,5–2/1000 на рік |
Основою патогенезу ІЕ є порушення внутрішньосерцевої гемодинаміки; травматизація ділянок клапанного та пристінкового ендокарду; бактеріємія; зниження резистентності організму; патологія внутрішньосерцевих структур вродженого та набутого характеру. Джерела інфекції та фактори, що полегшують адгезію мікроорганізмів, наведені в табл. 33.6.
Таблиця 33.6
Джерела інфекції та фактори, що полегшують адгезію мікроорганізмів
|
Бактеріємія |
Джерела бактеріємії |
Вогнища хронічної інфекції; інвазивні дослідження: бронхоскопія, гастроскопія, колоноскопія, хірургічні втручання і, перш за все, тонзилектомія, аденоїдектомія, дренування гнійників, процедури в ротовій порожнині (екстракція зуба або декількох, дентальні кровотечі та ін.), бактеріємія при захворюваннях легень |
|
Фактори, що полегшують адгезію мікроорганізмів |
Локальні фактори |
Морфологічні зміни клапанного апарату (вроджені або набуті), зміни серцевої гемодинаміки (при вадах серця), механічні та біологічні протези |
|
Загальні фактори |
Зміни системи природної резистентності (імуносупресивна терапія, наркоманія, алкоголізм, похилий вік) |
Причиною локальних гемодинамічних порушень є висока швидкість патологічного струменя крові (регургітація на мітральний або аортальний клапан); перехід крові з камери з високим тиском в камеру з низьким тиском (дефект міжшлуночкової перегородки), вузьке сполучення між двома камерами серця або ділянками магістрального судинного русла з виникненням перепаду тиску (дефект міжшлуночкової перегородки, коарктація аорти). Наявність перелічених умов призводить до локальних пошкоджень міокарда, активації тромбогенезу. За наявності бактеріємії виявляють інфікування ділянок пошкодженого міокарда, фіксацію збудника до ендотелію, розвиток бородавчато-виразкового ендокардиту. Слід відмітити, що мікроорганізми здатні інфікувати інтактні ендотеліоцити шляхом взаємодії специфічних глікопротеїдів, ендотеліального фагоцитозу. І в цьому випадку відзначають пошкодження ендотелію, оголення субендотеліального колагену, агрегацію тромбоцитів, полімеризацію фібрину, формування вегетацій. До цих подій призводить ряд імунологічних змін, які виявляють у хворих з ІЕ : пригнічення Т-системи лімфоцитів; гіперфункція В-системи; поліклонова гіпергаммаглобулінемія з високими титрами IgM та IgG; продукція аутоантитіл (кріоглобуліни, РФ, антиміокардіальні антитіла); порушення в механізмі активації комплементу; утворення ЦІК, депозиція їх в тканинах (гломерулонефрит, артрит, міокардит).
Морфологія інфекційного ендокардиту
ІЕ проявляється розвитком вегетацій, що розташовані на клапанних структурах та розповсюджуються на парієтальний ендокард. Вегетації утворені тромбами, нитками фібрину, еритроцитами, лейкоцитами, зруйнованою тканиною клапанів, бактеріальними масами. Вони бувають поодинокими, множинними, розміром від 1 мм до декількох сантиметрів, різної форми (частіше гроноподібної), сірого кольору, різної щільності. Характеристики вегетацій залежать від типу збудника, переважання структурних компонентів, стадії хвороби. Найбільші розміри вегетацій відмічають при грибковій етіології ІЕ. При ураженні мітрального та трикуспідального клапанів вегетації поширюються на стінки передсердь, хорди з їх розривом. При ураженні аортального клапана запальний процес переходить на синуси Вальсальви, формуються аневризми, які прориваються в передсердя або шлуночки. Ураження стулок клапанів супроводжується їх деструкцією, відривом, перфорацією. У 20–30% випадків навколо фіброзного кільця формується абсцес, який глибоко проникає в міокард, а в ряді випадків — проривається в порожнину перикарда.
Класифікація інфекційного ендокардиту
Класифікація ІЕ, запропонована у 2004 р. Європейським кардіологічним товариством, наведена в табл. 33.7.
Таблиця 33.7
Класифікація ІЕ (Європейське кардіологічне товариство, 2004)
|
За станом серця |
ІЕ природних клапанів: первинний (на інтактних клапанах) або вторинний (на уражених раніше клапанах) |
|
ІЕ клапанного протеза: ранній (до 1 року після операції) та пізній |
|
|
За локалізацією |
ІЕ з ураженням мітрального клапана |
|
ІЕ з ураженням аортального клапана |
|
|
ІЕ з ураженням тристулкового клапана |
|
|
ІЕ з ураженням клапана легеневої артерії |
|
|
ІЕ з ураженням двох або більше клапанів |
|
|
Пристінковий ІЕ |
|
|
За мікробіологічною характеристикою |
Залежно від збудника (стрептококовий, стафілококовий, грибковий ІЕ і т.д.) |
|
ІЕ з негативною гемокультурою |
|
|
Серологічно негативний ІЕ |
|
|
ІЕ, негативний за ПЛР |
|
|
Гістологічно негативний ІЕ |
|
|
Особливі форми ІЕ |
ІЕ наркоманів |
|
Нозокоміальний/госпітальний ІЕ (розвивається через 3 доби та більше від моменту госпіталізації або в результаті інвазивних маніпуляцій в стаціонарі, перенесених в останні 6 міс) |
|
|
ІЕ у осіб похилого віку |
|
|
ІЕ при хронічному гемодіалізі |
В табл. 33.8 представлена класифікація ІЕ, запропонована в 1999 р. Об′єднаним пленумом кардіологів та кардіохірургів України.
Таблиця 33.8
Класифікація ІЕ
|
I |
Активність процесу |
Активний, неактивний |
|
II |
Ендокардит природних клапанів |
1) Первинний; 2) вторинний (набуті та вроджені вади серця, травма, чужорідні тіла) Ендокардит протезний |
|
III |
Локалізація |
Аортальний, мітральний, трикуспідальний клапан, клапан легеневої артерії, ендокард передсердь або шлуночків |
|
IV |
Збудник |
Грам(+), грам(-), L-форми, рикетсії, гриби |
|
V |
Стадія клапанної вади; стадія серцевої недостатності |
|
|
VI |
Ускладнення |
|
У табл. 33.9 наведена класифікація ІЕ, включена в Європейські рекомендації щодо ведення пацієнтів з інфекційним ендокардитом (2009).
Таблиця 33.9
Класифікація ІЕ
|
I. За локалізацією інфекції, наявністю або відсутністю внутрішньосерцевих чужорідних матеріалів |
||||
|
1. Лівобічний ІЕ |
На фоні природного клапана |
|||
|
На фоні протезного клапана: ранній (<1 року після операції); пізній (>1 року після операції) |
||||
|
2. Правобічний ІЕ |
||||
|
3. ІЕ у осіб з імплантованим обладнанням (електрокардіостимулятор, імплантований кардіовертер-дефібрилятор) |
||||
|
II. За способом набуття |
||||
|
1. Ятрогенний ІЕ |
Нозокоміальний |
ІЕ розвивається у пацієнтів, госпіталізованих більше ніж за 48 год до появи ознак (симптомів) ІЕ |
||
|
Ненозокоміальний |
Ознаки та/або симптоми ІЕ виникли менше ніж через 48 год після госпіталізації у пацієнтів, які до цього отримували медичну допомогу у вигляді: |
|||
|
а) амбулаторного в/в лікування, гемодіалізу або в/в фармакотерапії менше 30 днів до встановлення ІЕ |
||||
|
б) госпіталізації до відділення інтенсивної терапії в період менше 90 днів до встановлення ІЕ |
||||
|
в) перебування в хоспісі або будинку для осіб похилого віку |
||||
|
2. Негоспітальний ІЕ |
Ознаки та/або симптоми ІЕ з’явилися менше ніж через 48 год після госпіталізації у пацієнтів, які не відповідають критеріям ятрогенної інфекції |
|||
|
3. ІЕ наркоманів |
ІЕ у ін’єкційних наркоманів за відсутності інших можливих шляхів інфікування |
|||
|
III. Активний ІЕ |
||||
|
ІЕ зі стійкою лихоманкою та позитивним бактеріологічним дослідженням крові |
||||
|
або при хірургічному втручанні — морфологічно активний запальний процес |
||||
|
або позитивна динаміка на фоні лікування антибіотиками |
||||
|
або гістопатологічне доведення активності ІЕ |
||||
|
IV. Рецидивуючий ІЕ |
||||
|
Рецидив |
Повторення епізодів ІЕ, спричинених тим самим мікроорганізмом менше ніж через 6 міс після виникнення першого епізоду |
|||
|
Реінфекція |
а) інфікування іншим мікроорганізмом |
|||
|
б) повторення епізодів ІЕ, спричиненого тим самим мікроорганізмом, більше ніж через 6 міс після першого випадку |
||||
Приклад формулювання діагнозу: первинний ІЕ аортального клапана стафілококової (S. еpidermidis) етіології, активна фаза, недостатність аортального клапана IV ступеня, серцева недостатність IIА, ФК III; гостре порушення мозкового кровообігу (2.01.2012).
Клініка інфекційного ендокардиту
У абсолютної більшості пацієнтів хвороба розвивається раптово. У 10–15% випадків цьому передують: інфікування рани; операції з гнійно-септичними ускладненнями; катетеризація порожнин серця; застосування катетерів, тривала в/в терапія; наркоманія. Клінічна картина формується із таких ознак:
1) інфекційний процес;
2) ураження клапанів;
3) емболії периферичних судин;
4) імунні ускладнення септичного процесу.
При гострому перебігу першим проявом хвороби є раптове підвищення температури тіла до 38–39 °С з ознобами, підвищеним потовиділенням. Температурна крива має гектичний характер. Температура менш виражена при підгострому перебігу. Інтоксикаційний синдром проявляється головним болем, болем у м’язах та суглобах. Величина температури тіла залежить від характера збудника і найбільш високою вона є при стафілококовій інфекції. Температура тіла може бути нормальною при прийомі протизапальних засобів, антибіотиків, а також у осіб похилого віку.
Серцеві шуми з’являються ще до руйнування стулок клапанів. Їх причиною є міокардит, дилатація шлуночків, розширення фіброзних кілець. Після деструкції стулок, хорд характер шуму різко змінюється, іноді — раптово. При первинному ІЕ серцевий шум з’являється, при вторинному — змінюються його локалізація і характер. Шуми у серці відмічають у 85–90% випадків та змінюються в процесі прогресування хвороби. При підгострому ІЕ їх виявляють у 95% хворих. Шум може не вислуховуватися у ранні строки хвороби, особливо при гострому перебігу. При ураженні парієтального ендокарда і в ряді випадків — тристулкового клапана шум не вислуховується навіть при тривалому динамічному спостереженні. На фоні мітрального стенозу і стенозу клапана легеневої артерії ІЕ розвивається рідко. Діагностичне значення має систолічний шум мітральної та трикуспідальної недостатності, а також протодіастолічний шум регургітації крові через аортальний клапан. Недостатність клапана легеневої артерії розвивається надзвичайно рідко. Слід враховувати, що протодіастолічний шум аортальної недостатності може бути дуже тихим та коротким внаслідок швидкого підвищення діастолічного тиску у лівому шлуночку. Аортальна регургітація розвивається гостро, тому характерні зміни артеріального тиску та дилатація лівого шлуночка можуть бути відсутніми, що ускладнює діагностику. Наявність регургітації крові через тристулковий клапан дозволяє (за відсутності шуму в ділянці мечовидного відростка) підтвердити пульсацію шийних вен та печінки. Слід враховувати появу «нових» шумів, особливо протодіастолічного по лівому краю груднини, а також підвищення інтенсивності «старих» шумів.
Розміри серця визначаються наявністю або відсутністю серцевої патології, тривалістю перебігу ендокардиту. Для розвитку серцевої недостатності необхідний тривалий перебіг інфекційного процесу. При розриві стулок аортального клапана, відриві хорд мітрального клапана серцева недостатність може виникнути в перші тижні хвороби (у цих випадках розвивається гостра серцево-судинна недостатність). Серцева недостатність, як правило, є результатом ураження, руйнування клапанів, але може бути і наслідком міокардиту, перикардиту або інфаркту міокарда. Емболічні ускладнення за клінічними даними діагностують у 30–58% хворих. Але в дійсності процент ураження значно вищий, оскільки емболії судин нирок, селезінки у більшості випадків не діагностуються за життя. Найчастіше уражуються судини головного мозку (паралічі, втрата свідомості в результаті емболії, розвитку дифузного менінгоенцефаліту, сонливість, запаморочення, диплопія, м’язові судоми, субарахноїдальні крововиливи, абсцеси мозку, гнійний менінгіт), периферичних артерій кінцівок (гангрена), сітківки ока (раптова втрата зору, геморагії на очному дні (п’ятна Рота)), легеневої артерії (при ураженні трикуспідального клапана). Для грибкового ІЕ характерні емболи великого розміру, які можуть закривати просвіт великих судин. При емболії крупних артерій можуть утворюватися мікотичні аневризми з тенденцією до розривів.
Характерні клінічні прови ІЕ наведені в табл. 33.10.
Таблиця 33.10
Характерні клінічні прови ІЕ
|
Слизові оболонки |
У 10–15% хворих відмічають петехії з локалізацією на кон’юнктиві, слизових оболонках щік та м’якого піднебіння. Вони з’являються групами та зникають через 2–3 дні. Причиною появи є мікроемболії судин, ДВЗ-синдром, підвищення проникності капіллярів. Петехії непатогномонічні для ІЕ: їх можуть виявляти при васкулітах різної етіології, хворобах системи крові, септичних станах. Ураження слизових оболонок може бути причиною носових кровотеч, гематурії |
|
Шкіра |
З’являються петехії на шкірі дистальних відділів кінцівок — гомілок та стоп, лінійні геморагії під нігтьовими пластинками рук та ніг (у 20%). Останні розташовуються у поздовжному напрямку, не сягаючи країв нігтя (до 2 мм в довжину). Причиною їх появи є мікроемболії капілярів нігтьового ложа. Вузлики Ослера розвиваються у 10–15% хворих з підгострим ІЕ та менше 10% у хворих з гострою формою. Ці болючі підшкірні вузлики діаметром 2–5 мм (іноді 10 мм) локалізуються на подушечках пальців рук та ніг. Зрідка їх відмічають у ділянці thenar та hypothenar. Шкіра над ними гіперемована, як правило, вони зникають через 2–3 доби. Вузлики являють собою запалення м’яких тканин навколо септичних емболів в дистальних артеріолах. При біопсії у вузликах виявляють інфекційні збудники. Розвиток вузликів Ослера не є специфічним: вони можуть з’являтися при гемолітичній анемії, СЧВ. На пальцях рук можуть розвинутись еритематозні макулопапульозні висипання, схильні до виразкування (плями Джейнуея) |
|
М’язи |
Міалгія |
|
Суглоби |
Розвиваються поліартралгія, артрит (моно-, оліго-, полі-), метастатичний гнійний артрит, бактеріальний дисцит, гіпертрофічна остеоартропатія (у 15–52%), остеомієліт |
|
Легені |
Уражуються у 75–89% хворих внаслідок гематогенної дисемінації, макро- та мікроемболій системи легеневої артерії септичними емболами, застійних явищ. До легеневих проявів належать: пневмонія з абсцедуванням; абсцес легені; емпієма плеври; пневмоторакс; гідроторакс; гострий респіраторний дистрес-синдром (набряк легень некардіогенного походження). Пневмонія є проявом (ознакою) ІЕ, а не ускладненням. Характерними проявами є множинні вузлові та сегментарні легеневі інфільтрати (ознаки септичної емболії судин легень) |
|
Серцево-судинна система |
Розвивається ураження судин — розповсюджений проліферативний ендоваскуліт, який призводить до ураження міокарда, нирок, ЦНС, селезінки, шкіри. У 5,1—16% хворих відмічають ІМ, а в осіб, що померли, — у 40–60% випадків. Інфаркти невеликого розміру, зрідка — множинні. Причинами розвитку ІМ є емболія гілок коронарних артерій; коронарний тромбоз; мікотична аневризма; коронарит; зниження тиску в стенозованих коронарних артеріях; прикриття вустя коронарної артерії вегетацією. У дрібні та крупні гілки коронарних артерій потрапляють тромботичні частинки із клапанних вегетацій, розвиваються дрібно- та крупновогнищеві інфаркти з розвитком аневризм. Клініка ІМ не відрізняється від такої при ішемічній хворобі серця. Але частіше відсутній ангінозний синдром та розвивається рання серцева недостатність. Симптоматика ІМ може бути повністю відсутньою. До ознак ураження міокарда при ІЕ відносяться: невідповідність між вираженістю вади та серцевою недостатністю; прогресуючий характер декомпенсації з переважанням правошлуночкової або тотальної недостатності; порушення ритму та провідності (шлуночкова, передсердна і політопна екстрасистолія, алоритмія, фибриляція та тріпотіння пересердь, стійка тахікардія, порушення АВ-провідності, внутрішньошлуночкової провідності, блокада ніжок пучка Гіса, зміни зубця Т). Зрідка вислуховується шум тертя перикарда. У результаті розповсюдження запального інфільтрату в глибину міокарда виникають міжм’язові внутрішньосерцеві абсцеси. Клінічно вони проявляються порушеннями ритму та провідності аж до повної АВ-блокади. Для діагностики абсцесів необхідна ехоКГ, особливо черезстравохідна |
|
Нервова система |
Розвиваються ішемічний (емболія судин головного мозку), геморагічний інсульти (розрив мікотичної аневризми, септичний артеріїт, піогенний некроз судин); енцефалопатія (у 27% хворих), абсцес мозку (мікроабсцес), менінгіт. Виділяють форми з ураженням нервової системи: 1) коматозну (інсульт); 2) психопатологічну (психоз); 3) енцефаломієлітичну (головний біль, нудота, блювання, психічні розлади). Моноплегія у хворих з лихоманкою та шумами в серці змушує підозрювати наявність у них ІЕ |
|
Нирки |
Відзначають наступний характер ураження: інфаркт нирки (у 2/3 пацієнтів); вогнищевий гломерулонефрит; дифузний інтра- та екстракапілярний гломерулонефрит (до 15% випадків); абсцес нирки (рідко); тубулоінтерстиціальний нефрит (як ускладнення терапії антибіотиками); амілоїдоз (у 3–5% хворих при підгострому ІЕ); гостра ниркова недостатність; хронічна ниркова недостатність. До розвитку гостої ниркової недостатності призводять: двобічна емболія ниркових артерій; білатеральний кортикальний некроз (асоційований з грамнегативною флорою; ускладнення антибактеріальної терапії — гострий канальцевий некроз, гострий інтерстиціальний нефрит; порушення ниркової гемодинаміки (серцева недостатність з низьким викидом). Виділяють також такий характер ураження, як інфекційно-токсична нейропатія. Особливостями ураження нирок є мало виражена артеріальна гіпертензія при дифузному гломерулонефриті, а також можливість усунути явища ниркової недостатності за допомогою раціональної антибіотикотерапії. Амілоїдна дистрофія нирок супроводжується набряками, гіпопротеїнемією, гіперхолестеринемією, протеїнурією, циліндрурією |
|
Печінка |
Розвиваються такі ураження: інфекційно-токсичний гепатит; венозний застій; тромбоемболія судин, інфаркт (зрідка); абсцес печінки (мікробна емболія) |
|
Підшлункова залоза |
Відмічають наступні ураження: тромбоемболія судин; крововиливи, некрози тканин залози; дистрофічні, атрофічні зміни; гострий панкреатит (зрідка) |
|
Селезінка |
Її розміри збільшені у 23–57% хворих (до ери антибіотиків — у 98%). Виявляють інфаркт селезінки, абсцеси, розриви. Можлива поява гикавка, болю в животі (лівому підребер’ї), лівобічного плевриту, шуму тертя очеревини. Збільшення селезінки пов’язано із гіпертрофією пульпи, утворенням інфарктів та абсцесів |
Виділяють псевдотифозну форму ІЕ (описав її Буйо), для якої характерні поступовий початок, блювання, діарея, біль у животі, затьмарення свідомості, підвищення температури тіла. Клінічні прояви ІЕ залежать від етіології хвороби (табл. 33.11).
Таблиця 33.11
Особливості клініки ІЕ залежно від етіології
|
Збудник |
Особливості клініки ІЕ |
|
Стафілокок |
Викликає ІЕ з гострим початком, гектичною лихоманкою, частими гнійно-септичними та тромбоемболічними ускладненнями. Розвитку стафілококового ІЕ сприяють хірургічні маніпуляції, операції на серці, крупних судинах, гемодіаліз, катетеризація вен. У більшості хворих стафілокок уражує інтактний до цього клапан. Характерною є перфорація клапанів, їх деструкція, розвиток серцевої недостатності. Типовими є ураження мозку (емболії, абсцес, гострий менінгіт), геморагічні висипи на шкірі з переходом в некроз, нагноєння. З причини розвитку інфарктів нирок, абсцесів, гнійних нефритів хворих помилково госпіталізують в урологічні відділення. Перебіг ІЕ може маскуватися під пневмонії з розвитком легеневих абсцесів, множинних інфарктів. Розвитком тяжких ускладнень (тромбоемболії, абсцедування міокарда, легень, нирок, селезінки) супроводжується ІЕ, викликаний білим стафілококом |
|
Ентерококи |
Перебіг ІЕ вкрай злоякісний, з множинними емболіями, резистентністю до терапії. До патологічного процесу залучаються мітральний та аортальний клапани |
|
Грамнегативні мікроорганізми |
Перебіг ендокардиту гострий та тяжкий, з множинними емболіями, серцевою недостатністю, резистентною до терапії. У наркоманів можливе одночасне ураження аортального і трикуспідального клапанів. ІЕ, викликаний грамнегативними мікроорганізмами, виникає після операцій на ШКТ, сечостатевих органах, при застосуванні антибіотиків широкого спектра дії, цитостатиків. При сальмонельозному ІЕ залучаються попередньо пошкоджені мітральний і аортальний клапани з їх деструкцією, утворенням передсердних тромбів. Сальмонели можуть уражувати ендотелій з утворенням аневризм. Ураження ендокарда, яке майже не піддається антибактеріальній терапії, притаманне кишковій паличці. Особливо тяжкий ендокардит з клінікою септичного шоку викликає синьогнійна паличка |
|
Гриби |
Роль грибів як збудників ІЕ зростає, що зумовлено широким використанням антибактеріальних засобів, ГК, збільшенням кількості наркоманів. Гриби мають тропність до ендо-, міо- та перикарду і можуть викликати розвиток панкардитів. ІЕ характеризується високою активністю процесу, схильністю до тромбоемболічних ускладнень, формуванням гігантських крихких вегетацій, мікотичних аневризм. Переважно уражується аортальний клапан. Типовою є емболічна оклюзія крупних артерій, особливо ніг. Гриби ростуть всередині судин, викликають гнійні процеси в артеріях, тромбози, інфаркти. Характерними є розвиток увеїту, ендофтальміту, швидкопрогресуючий перебіг, висока смертність. Для визначення грибів в крові необхідна спеціальна техніка культивування. Медикаментозне лікування ефекту не дає |
|
Стрептококи |
ІЕ можуть викликати стрептококи груп A, B, C, D, G. Вони викликають деструкцію клапанів, залучення міокарда до патологічного процесу, розвиток абсцесів, інфаркту міокарда. Залишається частим збудником ІЕ зеленкуватий стрептокок. ІЕ, викликаний ним, характеризується помірною активністю патологічного процесу, підгострим перебігом, ундулюючою лихоманкою, частими імунокомплексними ускладненнями — васкулітом, нефритом, міокардитом |
|
Анаеробна флора |
Для ІЕ характерні висока активність патологічного процесу, рефрактерність до терапії, висока смертність (до 46%). До особливостей ІЕ належать розвиток тромбофлебітів, високий ризик артеріальних емболій |
|
Коринебактерії (дифтероїди) |
Характерна стертість клінічних симптомів ІЕ, схильність до розвитку міокардиту, нефриту, васкулітів |
|
Гонококи |
Викликаний ним ІЕ відрізняється щоденною двохвильовою лихоманкою, тривалим перебігом. Можливе ураження клапанів легеневої артерії. На сьогодні гонококовий ІЕ відмічають дуже рідко |
|
Бруцели |
Часто до патологічного процесу залучається аортальний або тристулковий клапан. Розвиваються аневризми синуса Вальсальви з переходом інфекції на стінку передсердь, шлуночків, міокард, перикард. Ураження міжшлуночкової перегородки призводить до порушення провідності |
|
Рикетсії |
ІЕ частіше розвивається у чоловіків. Його перебіг може бути без лихоманки або з мінімальним підвищенням температури тіла. Уражується аортальний або мітральний клапан, у хворих розвиваються артрити, зрідка — тромбофлебіти. У 1/3 хворих відмічають мікрогематурію, піурію. ШОЕ може підвищуватися до 70 мм/год |
|
Віруси |
Доведено, що вони можуть бути причиною розвитку ІЕ. З тканини ендокарда виділяють віруси Коксакі В1, В2, В3, В4; в крові підвищується титр антитіл до них |
Полімікробний ІЕ, викликаний декількома мікроорганізмами, виявляють при імуносупресивних станах (лікування цитостатиками, зловживання наркотиками тощо), характеризується галопуючим перебігом, полівальвулярним ураженням, залученням міокарда, швидким розвитком серцевої недостатності. Частіше уражується аортальний клапан. За даними Інституту серцево-судинної хірургії імені М.М. Амосова, частота ураження при ІЕ різних клапанів становить: аортального — 62,7%, мітрального — 19,7%, трикуспідального — 5,1%, аортального + мітрального — 12,2%, клапана легеневої артерії — 0,4% (Бешляга В.М. и соавт., 2003). У початковій стадії з’являється систолічний шум по лівому краю груднини і в 5-й точці. Він пов’язаний із вегетаціями, які звужують вустя аорти. Потім в динаміці з’являється протодіастолічний шум в 5-й точці та над аортою, який посилюється у вертикальному положенні та в позиції на лівому боці. У міру руйнування стулок клапанів шум наростає, збільшується зона його розповсюдження. Слабшає 2-й тон над аортою, знижується діастолічний артеріальний тиск. При ізольованому ураженні мітрального клапана слід звертати увагу на динаміку наростання систолічного шуму, ослаблення 1-го тону. У наркоманів діагностичне значення мають поява та наростання в динаміці систолічного шуму над мечоподібним відростком, який посилюється на висоті вдиху. Але слід пам’ятати, що в 50% випадків шум з’являється на пізніх стадіях хвороби. Клапани легеневої артерії уражуються зрідка, в основному у пацієнтів з вродженими вадами серця. При первинному ІЕ ізольована вада серця формується протягом 1,5–3 тиж (ревматична вада — 3–5 міс).
ІЕ з ураженням правого серця. Наявні у наркоманів панікуліти, септичні флебіти призводять до виникнення ІЕ правого серця, ураження правого передсердно-шлуночкового клапана. До цього можуть також призвести периферичні та центральні катетери, трансвенозні провідники. Джерелом інфекції є шкіра (золотистий стафілокок, кандіда альбіканс), розчини, що інфузуються (синьогнійна паличка, серація). Найчастіше виділяють золотистий стафілокок. Відмічають виражену лихоманку (протягом декількох тижнів), біль в грудній клітці (плеврального характеру), кровохаркання, виділення мокротиння, задишку при фізичному навантаженні, слабкість, анорексію, шум недостатності правого АВ-клапана (посилюється під час вдиху), пульсація шийних вен та печінки. Для ІЕ правих відділів серця характерні повторні епізоди тромбоемболії легеневих артерій та пневмонії. Часто виявляють легку жовтяницю, тривале підвищення температури тіла та негативну гемокультуру. ІЕ правих відділів найкраще діагностується за допомогою трансторакальної ехоКГ.
ІЕ протезованих клапанів. ІЕ виявляють в 20% усіх випадків протезованих клапанів. Частота його продовжує підвищуватися. Діагностика ІЕ протезованого клапана складніша, ніж ІЕ природного клапана. Інфікування протезованих клапанів відбувається у 2–3% хворих протягом року після операції та у 0,5% пацієнтів кожного наступного року. 30% випадків ІЕ відмічають в 1-й рік протягом 2 міс після операції. Причиною його є занесення бактерій із протезом та з місця розрізу. Цю ранню інфекцію часто викликають мікроорганізми, резистентні до антибіотиків. Наслідком є розвиток септичного шоку, розрив клапана, міокардит. В ранній післяопераційний період джерелом бактеріємії можуть бути сечовивідні шляхи, рани, легенева інфекція, септичні флебіти. Під час транзиторної бактеріємії відбувається колонізація бактеріями протеза, місця його прикріплення. Хворим з імплантованими клапанами необхідно призначати антибактеріальні засоби під час проведення будь-яких процедур. Також слід проводити ретельне лікування навіть у випадках легких інфекційних захворювань. Прогноз є більш сприятливим при розвитку інфекційного процесу, викликаного стрептококом, через 2 та більше місяців. Інфікування протезованих клапанів супроводжується такими ж симптомами, як і ураження природних. Все-таки більш часто уражується кільце клапана, виникають його розриви, інфекція пенетрує в міокард. Наслідком цих процесів можуть бути:
- розвиток абсцесу міокарда;
- порушення провідності;
- поява аневризми синуса Вальсальви;
- виникнення фістули у праві відділи серця або в перикард;
- подовження інтервалу Р–Q(R);
- виникнення вперше блокади лівої або правої ніжки пучка Гіса (при інфікуванні клапана аорти воно свідчить про залучення до процесу міжшлуночкової перегородки, виникнення абсцесу);
- поява непароксизмальної функціональної тахікардії, АВ-блокади 2–3-го ступення із вузькими комплексами QRS (блокада може бути наслідком абсцесу серця);
- розвиток стенозу протезованих клапанів (внаслідок звуження кільця клапана вегетаціями або обмеження екскурсії клапана);
- частковий розрив протезованого клапана (з появою шуму регургітації при аускультації або реєстрація аномальної позиції клапана, його зміщення при рентгеноскопії, ехоКГ).
Інфікування протезованого клапана (ускладнене стенозом, розривом, серцевою недостатністю), повторні емболії, резистентність до антибактеріальної терапії, ознаки включення в патологічний процес міокарда, рецидив бактеріальної інфекції після адекватного лікування антимікробними препаратами, наявність грибкової інфекції вимагають невідкладного хірургічного втручання. При розвитку ІЕ у хворих, що отримують антикоагулянти для профілактики емболії, їх прийом слід продовжувати при високому ризику повторної емболії. У таких випадках ризик розвитку крововиливу в мозок сягає 36% з подальшою 80% летальністю.
Фази ІЕ наведені в табл. 33.12.
Таблиця 33.12
Фази ІЕ
|
Інфекційно-токсична фаза |
||
|
Критерії |
Клінічні |
Лихоманка, озноб, шкірні висипи (геморагічні, пустульозні, некротичні), міалгії, артралгії, поява та зміна характеру шумів (формування вади) |
|
Лабораторні |
Позитивний результат бактеріологічного дослідження гемокультури, підвищення титру антитіл до бактеріальних (грибкових) антигенів, збільшення ШОЕ |
|
|
Імунозапальна фаза |
||
|
Критерії |
Клінічні |
Дифузний гломерулонефрит, гепатит, міокардит, спленомегалія, васкуліти (капілярити), артралгії, артрити, потовщення кінцевих фаланг пальців, лихоманка, що реагує на ГК |
|
Лабораторні |
ЦІК, імунокомплексні депозити в нирках, міокарді, судинах, тромботичних вегетаціях; поява РФ, антиглобулінового фактора; гіпергаммаглобулінемія; збільшення вмісту в крові фібронектину, СРБ; посилення реакції бласттрансформації з фітогемаглютиніном та бактеріальними антигенами; визначення антитіл проти тканин (нирок, печінки, сердця тощо); ознаки анемії |
|
|
Дистрофічна стадія (поліорганної недостатності) |
||
|
Вона проявляється тяжким незворотним ураженням внутрішніх органів, неефективністю лікування. До проявів цього синдрому відносяться: 1) респіраторний дистрес-синдром; 2) гостра ниркова недостатність; 3) гостра печінкова недостатність; 4) ДВЗ-синдром; 5) порушення функції ЦНС |
||
Деякі критерії ступеня активності ІЕ представлені в табл. 33.13.
Таблиця 33.13
Деякі критерії ступеня активності ІЕ
|
Симптоматика |
Ступінь активності |
||
| 1-й | 2-й | 3-й | |
|
ШОЕ, мм/год |
До 20 |
20–40 |
40 та більше |
|
Гаммаглобуліни, % |
19–15 |
20–25 |
25–45 |
|
Фібриноген, г/л |
4–5 |
5–6 |
6–10 |
Особливості клініки ІЕ в похилому та старечому віці включають: 1) ранній розвиток застійної серцевої недостатності; 2) нерідко перебіг без підвищення температури тіла; 3) більш часті неврологічні ускладнення: тромбоемболія судин головного мозку, розриви мікотичних аневризм; 4) кардіотоксичну дію антибіотиків, що застосовуються; 5) резистентність до багатьох антибіотиків. Деякі автори виділяють критерії ІЕ в похилому та старечому віці (Gants N., 1991), які включають: 1) лихоманку із серцевою недостатністю; 2) лихоманку із цереброваскулярними розладами; 3) лихоманку з нирковою недостатністю; 4) лихоманку та біль у попереку; 5) анемію невстановленого походження; 6) шум у серці, який тільки розвинувся; 7) госпітальну інфекцію з лихоманкою у хворих з в/в катетерами. Виділяють ряд клінічних масок ІЕ (табл. 33.14).
Таблиця 33.14
Клінічні маски ІЕ
|
Церебральні |
Гостре порушення кровообігу мозку; менінгіт; менінгоенцефаліт (геміплегія, афазія, атаксія, головний біль, психічні розлади) |
|
Судинні |
Клініка системного васкуліту, тромбангіїту (геморагічний синдром, тромбоемболічні ускладнення) |
|
Ниркові |
Симптоми дифузного гломерулонефриту, інфаркту нирки (геморагія, протеїнурія, гіпертензивний криз) |
|
Очні |
Раптова втрата зору, неврит зорового нерва, петехії на кон’юнктиві, очному дні |
|
Легеневі |
Інфаркт-пневмонія, плеврит, кровохаркання, набряк легень |
|
Гематологічні |
Прояви анемії |
|
Суглобові |
Артрит, остеомієліт |
|
Кишкові |
Псевдотифозна форма ІЕ: блювання, діарея, біль у животі, жовтяниця |
|
Гострої інфекції |
Клініка гострого інфекційного захворювання (грип, малярія, тиф тощо): лихоманка, підвищене потовиділення |
Ускладнення ІЕ представлені нижче (табл. 33.15).
Таблиця 33.15
Ускладнення ІЕ
|
Внутрішньосерцевий абсцес |
Необхідна операція з дренуванням порожнини, часто з протезуванням клапана |
|
Гнійний перикардит |
Необхідне хірургічне втручання, дренування порожнини |
|
Дисемінована інфекція |
Частіше відмічають при стафілококовому ІЕ, необхідне дренування абсцесу |
|
Мікотичні аневризми |
Слід звертати увагу на постійний головний біль, вогнищеву неврологічну симптоматику. Необхідна МРТ головного мозку, черевного відділу аорти, синусів Вальсальви, артерій брижі |
|
Стійка лихоманка |
Необхідно шукати абсцес, повторне інфікування (особливо за наявності в/в катетерів). Виключити медикаментозну алергію (еозинофілію без лейкоцитозу) |
|
Емболічні інфаркти |
Вони супроводжуються наступним розвитком абсцесів |
|
Ішемія та інфаркти міокарда |
Емболія коронарних артерій супроводжується аритміями, дисфункцією шлуночків, розвитком інфаркту міокарда |
Діагностика інфекційного ендокардиту
У процесі діагностики слід враховувати наявність у хворих з підозрою на ІЕ факторів ризику розвитку цієї патології (табл. 33.16).
Таблиця 33.16
Фактори ризику розвитку ІЕ
|
Фактори, що сприяють бактеріємії |
В/в вживання наркотиків → стоматологічні, хірургічні, гастроентерологічні, урогінекологічні маніпуляції; венозні катетери (особливо в центральних венах) → вогнища інфекції в організмі → рани, травми |
|
Фонові фактори |
Протези клапанів → ІЕ в минулому → вроджені та набуті вади серця → ГКМП → пролапс мітрального клапана |
|
Супутні стани та хвороби |
Наркоманія, токсикоманія, у тому числі алкоголізм → ВІЛ-інфекція; лікування імуносупресантами → онкопатологія → цукровий діабет → похилий вік; дистрофія |
Клінічні симптоми з’являються в середньому через 2 тиж з моменту виникнення бактеріємії. Гострий ІЕ розвивається в основному на інтактних клапанах, перебіг (тривалість 1,5–2 міс) характеризується яскравою клінічною картиною сепсису, швидким розвитком деструкції і перфорації стулок клапанів, серцевою недостатністю, множинними тромбоемболіями. Без хірургічного втручання часто настає смерть. Картина підгострого ІЕ розвивається поступово (протягом 2–6 тиж) і триває 3–4 міс з різним ступенем вираженості симптомів. Його перебіг більш сприятливий. Слід враховувати, що гострий ІЕ може в подальшому набути підгострого перебігу.
До загальноприйнятих критеріїв діагностики ІЕ відносяться критерії, розроблені в Duke University Medical Center — Модифіковані критерії діагностики ІЕ (Li J. et al., 2000) (табл. 33.17).
Таблиця 33.17
Модифіковані критерії діагностики ІЕ
|
Морфологічні критерії |
1. Визначення мікроорганізмів в культурі або при гістологічному дослідженні вегетацій, вегетацій-емболів, внутрішньосерцевих абсцесів |
|
|
2.Патологічні зміни: вегетації або внутрішньосерцевий абсцес з гістологічним підтвердженням активного ендокардиту |
||
|
Клінічні критерії |
Основні |
1. Позитивний результат бактеріологічного дослідження крові з ростом типових збудників ІЕ (Str. viridans, Str. bovis, HACEK-група, позалікарняні штами S. aureus, Enterococcus spp. за відсутності джерела гною) в двох різних пробах крові, взятих з інтервалом 12 год, або в усіх трьох чи в більшості проб з 4 та більше посівів крові, взятих протягом 1 год. Одноразовий позитивний результат позитивного бактеріологічного дослідження крові для виявлення Coxiella burnetii або титр IgG до C. burnetii більше 1:800 |
|
2. Ознаки ІЕ, виявлені при ехоКГ: вегетації на клапанних або підклапанних структурах; абсцес міокарда, фіброзного кільця; дисфункція протезованого клапана; клапанна недостатність, що вперше виникла. Черезстравохідну ехоКГ рекомендується проводити пацієнтам з підозрою на ІЕ протезованого клапана |
||
|
Додаткові |
1. Попереднє ураження клапанів або ін’єкційна наркоманія |
|
|
2. Лихоманка вище 38 °С |
||
|
3. Судинні симптоми: артеріальні емболії, інфаркти легень, мікотичні аневризми, внутрішньочерепні крововиливи; симптом Лукіна — Лібмана, плями Джейнуея |
||
|
4. Імунологічні прояви: гломерулонефрит, вузлики Ослера, плями Рота, позитивна проба на РФ |
||
|
5. Позитивний результат бактеріологічного дослідження, який не відповідає основним критеріям, або високі титри антитіл до можливих збудників ІЕ |
||
|
ІЕ достовірний |
Якщо наявні 2 основних критерії або 1 основний та 3 додаткових або 5 додаткових |
|
|
ІЕ можливий |
Якщо наявні 1 основний і 1 додатковий або 3 додаткових критерії |
|
|
Діагноз ІЕ виключений |
а) якщо прояви, характерні для ІЕ, вкладаються в альтернативний діагноз, або |
|
|
б) симптоми ІЕ зникають протягом 4 і менше днів антибіотикотерапії, або |
||
|
в) відсутні будь-які патоморфологічні дані про ІЕ при хірургічному втручанні або аутопсії |
||
Критерії Дюка базуються на клінічних, ехокардіографічних проявах та мікробіологічних посівах, мають високу чутливість і специфічність (близько 80%) у діагностиці ІЕ. Але вони не замінюють клінічну оцінку лікаря.
Діагностика ІЕ включає лабораторні методи (аналіз крові та сечі, біохімічні, імунологічні (IgM, G, ЦІК, рівень комплементу, СРБ, РФ), у тому числі серологічні, дослідження та інструментальні методи діагностики (рентгенографія органів грудної клітки, ехоКГ, черезстравохідна ехоКГ, ЕКГ, МРТ, ангіографія, УЗД та КТ органів черевної порожнини). У хворих на ІЕ знижується в крові рівень комплементу (у 5–40% хворих, особливо за наявності дифузного гломерулонефриту), через 2–3 тиж хвороби з’являється РФ (у 60–70% хворих). Збільшення ШОЕ виявляють у 90% хворих, анемію — у 70–90%, лейкоцитоз — у 10–30%, високий титр ЦІК — у 65–100%, підвищення креатиніну — у 10–30%, протеїнурію — у 50–60%, мікрогематурію — у 30–59%. Рентгенографія органів грудної клітки дозволяє виявити розширення тіні серця, застій у малому колі кровообігу, інфаркти легень. Вегетації візуалізуються через 2 тиж після встановлення діагнозу ІЕ. В М-режимі можлива візуалізація вегетацій, які перевищують 3–5 мм. Одномірна ехоКГ виявляє вегетації у 55% хворих, двомірна — у 70–80%, черезстравохідна — у 90%. Відсутність вегетацій не виключає ІЕ. Виявлення вегетацій — ознака підвищеного (в 3 рази) розвитку ускладнень. Коли звичайна ехоКГ не виявляє ІЕ, додатково виконують черезстравохідне дослідження датчиком з високою частотою сканування. За допомогою трансторакальної та трансстравохідної ехоКГ В.М. Бешляга та співавтори (2003) виявляють вегетації у 99% хворих. При виявленні ІЕ безумовні переваги (порівняно з М-режимом) має двомірна ехоКГ. Малі (або «молоді») вегетації краще виявлють при трансстравохідній ехоКГ ехоКГ. За розмірами вегетації поділяють на малі — до 0,5 см, середніх розмірів — 0,5–1,0 см, великі — більше 1,0 см. Іноді вегетації розміром 0,2–0,3 см не можуть виявити навіть за допомогою черезстравохідної ехоКГ. Успіх візуалізації вегетацій залежить від їх рухливості: плоскі та фіксовані вегетації виявляти досить складно. При значній давності хвороби вегетації виявляють на стулках клапанів з двох боків і на хордах. Відбувається частковий або повний відрив хорд з пролапсом стулок і регургітацією. Іноді вегетації переходять на кінці папілярних м’язів. У випадках запущеної хвороби виявляють перфорацію стулок, повну їх деструкцію, параклапанні абсцеси, фістули (виникають при прориві абсцесів), а вегетації можуть бути відсутніми внаслідок їх відриву. При гострому ураженні (до 1 міс) вегетації рихлі, зниженої ехогенності, високорухливі (за своєю ехогенністю вони близькі до крові). Все це вимагає проведення ехоКГ в динаміці. Абсолютною ознакою ІЕ вважають поєднання регургітації з макроанатомічними ознаками хвороби (Бешляга В.М. и соавт., 2003). МРТ дозволяє виявити параклапанні абсцеси, інфаркти та абсцеси мозку, кровотечі із мікотичних аневризм. Виявити ці аневризми (частіше вони виникають при підгострому ІЕ (у 15–20% хворих)) дозволяє ангіографія, але її застосовують дуже рідко. Абсцеси міокарда виникають при гострому ІЕ у 20% хворих, при їх локалізації в нижній частині міжшлуночкової перегородки раптово може виникнути блокада лівої ніжки пучка Гіса, збільшитись інтервал Р–R. За допомогою УЗД, КТ черевної порожнини виявляють інфаркти та абсцеси внутрішніх органів.
ЕхоКГ у хворих з підозрою на ІЕ дозволяє виявити інтракардіальні маси на клапанах та оточуючих тканинах, внутрішньосерцеві абсцеси, параклапанну регургітацію, патологічні сполучення (свищі) між порожнинами серця; провести аналіз функції клапанів серця та стан внутрішньосерцевої гемодинаміки; оцінити загрозу системних емболічних ускладнень. Повторні трансторакальні та трансстравохідні ехоКГ проводять при появі нових шумів, емболії, персистуючій лихоманці, серцевій недостатності, абсцесі серця, АВ-блокаді; для виявлення ускладнень, контролю за розміром вегетацій. Трансторакальну ехоКГ проводять після завершення антибактеріальної терапії з метою оцінки морфологічного стану клапанних структур та гемодинаміки. В усіх випадках ІЕ, які вимагають хірургічного лікування, проводять інтраопераційну ехоКГ.
До основних методів діагностики ІЕ, окрім ехоКГ, відноситься бактеріологічне дослідження (посів крові). Слід враховувати, що для росту ряду збудників (деяких штамів стрептококів, коринебактерій, гемофільної палички, актинобацилусу тощо) необхідний тривалий період (7–21 день). Для росту інших збудників (гриби, бруцела, мікоплазма, хламідія, гістоплазма, легіонела, коксієла барнетті, деякі штами стрептококів) необхідні спеціальне середовище та методи культивування. При посіві крові необхідно брати не менше 6 порцій крові по 10 мл (від 10 до 30 мл) з інтервалами 5–10 хв із різних вен. Позитивний результат в одній порції не має діагностичного значення. При гострому ІЕ рекомендують взяти кров протягом 1 год і після цього розпочати антибіотикотерапію. При підгострому ІЕ кров беруть протягом 2 діб. При негативному результаті можна відміняти антибіотики на 2 доби, а потім повторювати посів. При негативних посівах іноді антибіотики відміняють на 7–10 і більше днів, якщо після цього знову підвищується температура тіла, то це свідчить про правильність діагнозу.
ІЕ диференціюють з іншими ендокардитами: 1) ревматичним; 2) аутоімунним (Лібмана — Сакса) при СЧВ; 3) фібропластичним еозинофільним Лефлера-2; 4) асептичним (при уремії, раку, анемії). Диференційну діагностику проводять також із ГРЛ (табл. 33.18); тромбоемболією гілок легеневої артерії у хворих з вадами серця за наявності серцевої недостатності; інфекційними (туберкульоз, сепсис), аутоімунними захворюваннями (СЧВ, ВП, поліміозит/дерматоміозит); злоякісними пухлинами (лімфомами) з паранеопластичними реакціями; інсультом; абсцесом мозку; менінгітом; міксомою передсердя, небактеріальним тромбендокардитом.
Таблиця 33.18
Диференціація ІЕ з деякими іншими хворобами
|
Патологія |
Відмітні особливості |
|
Лихоманка невизначеного генезу |
В усіх випадках слід проводити повне обстеження хворого для виключення (підтвердження) ІЕ |
|
ГРЛ |
Існування хронологічного зв’язку зі стрептококовою інфекцією глотки, підтвердженою лабораторно; повільне формування вади серця з переважним ураженням мітрального клапана; швидкий зворотний розвиток симптомів (клінічних та лабораторних); мігруючий симетричний поліартрит. Запідозрити розвиток вторинного ІЕ у хворого з ревматичною вадою серця можна при появі високої температури тіла після медичних маніпуляцій (переважно стоматологічних); швидкого формування нової вади з розвитком серцевої недостатності; появи петехій на шкірі і слизових оболонках; відсутності ефекту від протизапальної терапії |
|
СЧВ |
Алопеція, еритема на обличчі, фотосенсибілізація, виразки в порожнині рота або носа; перикардит; високі титри АНФ, антитіл до нативної ДНК; розвиток ендокардиту Лібмана — Сакса на пізніх стадіях хвороби. У 5–7% випадків можливий розвиток ІЕ на фоні ендокардиту Лібмана — Сакса |
|
АФС |
Артеріальні та/або венозні тромбози різної локалізації; тромбоцитопенія; ниркові, неврологічні, кардіологічні, шкірні розлади; патологія клапанів, яка зумовлена тромботичними вегетаціями (не відрізняються від таких при ІЕ); гемокультура відсутня |
|
Ювенільний ідіопатичний артрит (синдром Стілла) |
Плямисто-папульозний висип на шкірі грудей, живота, рук і ніг; лімфаденопатія; гепатоспленомегалія; ексудативний плеврит; перикардит; суглобовий синдром, синовіт, залучення шийного відділу хребта |
|
Геморагічний васкуліт (хвороба Шенляйна — Геноха) |
Геморагічна пурпура поєднана з ураженням нирок, нерідко — суглобів; патологія клапанів серця і гемокультура відсутні |
|
Хвороба Кавасакі |
Переважно дитячий чи підлітковий вік осіб монголоїдної раси; двобічний кон’юнктивіт; лихоманка протягом 5 днів, резистентна до антибіотиків; гіперемія, набряк, сухість губ; малиновий язик; ураження слизової оболонки порожнини рота та глотки; збільшення лімфатичних вузлів шиї; поліморфний висип на шкірі тулуба; вальвуліт з формуванням клапанної недостатності |
Лікування інфекційного ендокардиту
Усі хворі з ІЕ підлягають госпіталізації. Призначається тривала антимікробна терапія та хірургічне видалення інфікованих тканин. Основним методом лікування ІЕ є призначення комбінації антибактеріальних препаратів. Принципи антибіотикотерапії ІЕ включають її етіотропність; бактерицидну дію антибіотиків; парентеральний (переважно в/в) шлях введення; врахування існування супутніх джерел інфекції; безпеку медикаментозного режиму (врахування можливих побічних ефектів); достатню тривалість. Показання до призначення медикаментозної терапії включають: відсутність ознак серцевої недостатності, нестабільності гемодинаміки, порушення провідності; відсутність ознак неконтрольованості інфекційного процесу, метастатичних вогнищ інфекції, великих вегетацій; стабільну функцію нирок, ІЕ клапанів правих відділів серця. При проведенні антибактеріальної терапії ІЕ слід враховувати результати посіву крові, дотримуватись режиму дозування, частоти введення. При стрептококовій етіології ІЕ рекомендується проводити антибактеріальну терапію не менше 4 тиж, стафілококовій — не менше 6, викликаному грамнегативною флорою — не менше 8. Загалом вважається, що терапія ендокардиту природного клапана повинна тривати 2–6 тиж, протезованого — щонайменше 6. У деяких випадках антибіотикотерапія може тривати декілька місяців. Якщо після операції з клапанів виділяють культуру мікроорганізмів, то розпочинають новий повний курс лікування. При виборі препарата орієнтуються на визначену при останньому посіві чутливість мікроорганізмів. Причини резистентності до антибіотиків включають пізній початок лікування, високу вірулентність бактерій, наявність мікробних асоціацій, похилий вік хворих, паралельне ураження декількох клапанів, протезний ендокардит, повторні гемодіалізи. Сучасні схеми антибактеріальної терапії ІЕ наведені в табл. 33.19–33.21.
Таблиця 33.19
Початкове емпіричне лікування ІЕ (до або без ідентифікації збудника)
|
Антибіотик |
Доза, шляхи введення |
Тривалість лікування, тиж |
Клас, рівень доказовості |
Примітки |
|
ІЕ природних клапанів або пізній ІЕ протезних клапанів |
||||
|
Ампіцилін/сульбактам або амоксицилін/клавуланат + гентаміцин |
12,0 г/добу в/в в 4 прийоми; 12,0 г/добу в/в в 4 прийоми; 3 мг/кг/добу в/в або в/м в 2–3 прийоми |
4–6 4–6 4–6 |
IIb, c IIb, c |
При негативній культурі крові здійснюють консультацію інфекціоніста |
|
Ванкоміцин + гентаміцин + ципрофлоксацин |
2,0 г/добу в/в в 2 прийоми; 3 мг/кг/добу в/в або в/м в 2–3 прийоми; 1000 мг/добу per os в 2 прийоми |
4–6 4–6 4–6 |
IIb, c |
Слід враховувати, що у хворих, які не переносять бета-лактами, ципрофлоксацин не завжди діє на Bartonella spp., тому додають доксициклін |
|
Ранній ІЕ протезного клапана |
||||
|
Ванкоміцин + гентаміцин + рифампіцин |
2,0 г/добу в/в в 2 прийоми; 3 мг/кг/добу в/в або в/м в 2–3 прийоми; 1200 мг/добу per os в 2 прийоми |
6 2 |
IIb, c |
У разі відсутності клінічного поліпшення проводять хірургічне втручання або розширюють спектр антибіотиків, які діють на грамнегативні збудники |
Таблиця 33.20
Схеми емпіричної антимікробної терапії ІЕ при негативному результаті бактеріологічного дослідження
|
Перебіг ІЕ |
Клапани |
Основна схема |
Альтернативна |
|
Гострий |
Природні |
Оксацилін в/в по 2,0 г 6 разів на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж |
Ванкоміцин в/в по 1,0 г 2 рази на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж |
|
Підгострий |
Природні |
Ампіцилін/сульбактактам в/в по 3,0 г 4 рази на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж |
Ванкоміцин в/в по 1,0 г 2 рази на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж або цефтріаксон в/в по 2,0 г 1 раз на добу 4 тиж |
|
Протези |
Ванкоміцин в/в по 1,0 г 2 рази на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж |
Римфампіцин в/в по 0,3–0,45 г 2 рази на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж |
|
|
ІЕ в ін’єкційних наркоманів |
Природні |
Оксацилін в/в по 2,0 г 6 разів на добу 4 тиж + гентаміцин в/в по 1 мг/кг 3 рази на добу 4 тиж |
Ванкоміцин в/в по 1,0 г 2 рази на добу, 4 тижні + гентаміцин в/в по 1 мг/кг 3 рази на добу, 4 тиж |
Таблиця 33.21
Етіотропна терапія ІЕ
|
Зеленкуватий та інші стрептококи, високочутливі до пеніцилінів (МЩКТ ≤0,1 мкг/мл) |
Бензилпеніцилін в/в 16–20 млн ОД/добу за 4–6 введень протягом 4 тиж або цефтріаксон в/в або в/м 2,0 г 1 раз на добу 4 тиж |
||
|
Бензилпеніцилін в/в 16–20 млн ОД/добу за 4–6 введень протягом 2 тиж + гентаміцин в/в або в/м 240 мг/добу в 2–3 введення протягом 2 тиж |
|||
|
Цефтріаксон в/в або в/м 100 мг/кг 1 раз на добу протягом 2 тиж + гентаміцин в/в або в/м 240 мг/добу в 2–3 введення протягом 2 тиж |
|||
|
Зеленкуватий та інші стрептококи, помірно чутливі до пеніцилінів (МЩКТ 0,1–0,5 мкг/мл) |
Бензилпеніцилін в/в 20–30 млн ОД/добу за 4–6 введень протягом 4 тиж + гентаміцин в/в або в/м 160–240 мг/добу в 3 введення протягом 2 тиж |
||
|
Цефазолін в/в або в/м 8–10 г/добу за 3–4 введення протягом 4 тиж + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення протягом 2 тиж |
|||
|
Ентерококи |
Ампіцилін в/в 12 г/добу в 4–6 введень протягом 4–6 тиж + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення протягом 4 тиж |
||
|
Полірезистентні ентерококи |
E. faecalis |
Іміпенем+циластатин в/в 2–4 г/добу в 2–4 введення протягом 8 та більше тижнів + ампіцилін в/в 12 г/добу в 4–6 введень 8 та більше тижнів |
|
|
E. faecium |
Лінезолід в/в 1200 мг/добу в 2 введення 8 та більше тижнів |
||
|
Стафілококи, чутливі до метициліну |
ІЕ природних клапанів |
Оксацилін в/в 8–12 г/добу в 4–6 введень протягом 6 тиж + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення протягом 3–5 днів |
|
|
ІЕ клапанних протезів |
Оксацилін в/в 8–12 г/добу в 4–6 введень протягом 6 та більше тижнів + рифампіцин per os 300 мг/добу в 2 прийоми 6 та більше тижнів + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення протягом 2 тиж |
||
|
Стафілококи, резистентні до метициліну |
ІЕ природних клапанів |
Ванкоміцин в/в по 1–2,0 г/добу в 2–4 введення (вводити протягом 1–2 год) 6 тиж |
|
|
ІЕ клапанних протезів |
Ванкоміцин в/в по 1–2,0 г/добу в 2–4 введення (вводити протягом 1–2 год) 6 тиж + рифампіцин per os 300 мг/добу в 2 прийоми 6 та більше тижнів + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення 2 тиж |
||
|
Група HACEK |
Цефтріаксон в/в або в/м 2,0 г 1 раз на добу 4–6 тиж |
||
|
Цефотаксим в/в або в/м 6–8 г/добу в 3 введення 4–6 тиж |
|||
|
Pseudomonas spp. |
Цефтазидим в/в 6–8 г/добу в 3 введення 6 тиж + тобраміцин в/в або в/м 5–8 мг/кг на добу в 2 введення 6 тиж |
||
|
Іміпенем+циластатин в/в 2–4 г/добу в 2–4 введення, 6 тиж + тобраміцин в/в або в/м 5–8 мг/кг на добу в 2 введення 6 тиж |
|||
|
Enterobacteriaceae |
Цефотаксим в/в або в/м 6–8 г/добу в 3 введення, 4–6 тиж + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення протягом 4–6 тиж |
||
|
Іміпенем + циластатин в/в 2–4 г/добу в 2–4 введення протягом 6 тиж + гентаміцин в/в або в/м 160–240 мг/добу в 2–3 введення протягом 4–6 тиж |
|||
|
Гриби |
Амфотерицин В в/в 1 мг/кг 1 раз на добу 6–8 тиж + флуцитозин per os 150 мг/кг на добу в 4 прийоми 6–8 тиж |
||
|
Після протезування клапана |
Флуконазол per os 200–400 мг/добу в 1 прийом не менше 6 міс |
||
Примітка: до групи HACEK входять Haemophilus spp., Actinobacillus actinimycetem comitans, Cardiobacterium hominis, Eikinella spp., Kingella kingae.
У зв’язку з високою вартістю лікування хворих на ІЕ (близько 47 тис. дол. на курс) нині застосовують короткотривалі схеми лікування ІЕ. Показанням до їх застосування є висока чутливість збудника до пеніциліну (МПК <0,1 мг/л). Критеріями відбору хворих для призначення короткотермінових схем лікування вважаються: ІЕ природного клапана; вегетації не більше 5 мм (при ехоКГ); відсутність аортальної недостатності; відсутність ускладнень (серцева недостатність, тромбоемболії, порушення провідності); чіткий клінічний ефект антибактеріальної терапії протягом 7 днів з нормалізацією температури тіла, поліпшенням самопочуття.
При стрептококовій етіології ІЕ використовують наступні схеми: 1) бензилпеніцилін (натрієва сіль) в/в по 3 млн ОД 6 разів на добу 2 тиж + гентаміцин в/в по 3 мг/кг 1–3 рази на добу 2 тиж або 2) цефтріаксон в/в 2,0 г 1 раз на добу 2 тиж + нетилміцин в/в по 4 мг/кг 1–2 рази на добу 2 тиж або 3) оксацилін в/в по 2,0 г 6 разів на добу 2 тиж + гентаміцин в/в 3 мг/кг 1–3 рази на добу 2 тиж.
Для 2-тижневої терапії не застосовують ванкоміцин + гентаміцин, оскільки вони повільно знищують мікроорганізми. Комбінацію оксациліну з аміноглікозидами використовують при ІЕ, викликаному золотистим стафілококом, чутливим до метициліну, в разі ураження клапанів правих відділів серця та за відсутності емболічних ускладнень.
Варіанти антибіотикотерапії, які застосовуються в Інституті серцево-судинної хірургії ім. М.М. Амосова (Книшов Г.В. та співавт., 2012), наведені в табл. 33.22.
Таблиця 33.22
Режими антибіотикотерапії при різній етіології ІЕ
|
Емпірична антибіотикотерапія ІЕ нативних клапанів серця |
|
|
Рекомендований режим |
Ампіцилін 2 г кожні 4 год в/в + оксацилін 2 г кожні 4 год в/в + амікацин 7,5 мг кг кожні 12 год в/в |
|
Альтернативний режим 1 |
Ванкоміцин 15 мг/кг кожні 12 год в/в + амікацин 7,5 мг/кг кожні 12 год в/в |
|
Альтернативний режим 2 |
Тейкопланін 12 мг/кг кожні 24 год в/в + амікацин 7,5 мг/кг кожні 12 год в/в |
|
Режими антибіотикотерапії ІЕ, викликаного Strept. viridans і Strept. bovis |
|
|
Цефтріаксон 2 г/добу в/в, в/м протягом 4 тиж + амікацин 7,5 мг/кг кожні 12 год в/в, в/м протягом 2 тиж або |
|
|
ванкоміцин 15 мг/кг кожні 12 год в/в або тейкопланін 12 мг/кг кожні 24 год в/в 4 тиж |
|
|
Режими антибіотикотерапії ІЕ, викликаного Enterococcus |
|
|
Уназин 3 г кожні 6 год в/в 4–6 тиж + амікацин 7,5 мг/кг кожні 12 год в/в, в/м 2 тиж або |
|
|
ванкоміцин 15 мг/кг кожні 12 год в/в, в/м 4–6 тиж + уназин 3 г кожні 6 год в/в 4–6 тиж або |
|
|
тейкопланін 12 мг/кг кожні 24 год в/в 4–6 тиж + амікацин 7,5 мг/кг кожні 12 год в/в 4–6 тиж |
|
|
Режими антибіотикотерапії ІЕ, викликаного Staphylococcus |
|
|
Метацилінчутливі Staphylococcus |
Оксацилін 2 г кожні 4 год в/в 4–6 тиж + амікацин 7,5 мг/кг кожні 12 год в/в, в/м 2 тиж або |
|
цефазолін 2 г кожні 8 год в/в 4 тиж + амікацин 7,5 мг/кг кожні 12 год в/в, в/м 2 тиж або ванкоміцин 15 мг/кг кожні 12 год в/в 4–6 тиж або тейкопланін 12 мг/кг кожні 24 год в/в 4–6 тиж |
|
|
Метацилінрезистентні Staphylococcus |
Ванкоміцин 15 мг/кг кожні 12 год в/в 4–6 тиж або тейкопланін 12 мг/кг кожні 24 год в/в 4–6 тиж |
|
Емпірична терапія пізнього ІЕ протезованого клапана серця (Staph. epidermidis, Staph. areus, Strept. vіridans, Enterococci) |
|
|
Ванкоміцин 15 мг/кг кожні 12 год в/в 6 тиж + амікацин 7,5 мг/кг кожні 12 год в/в, в/м 2 тиж + рифампіцин 300 мг 3 рази на добу per os 6 тиж або |
|
|
тейкопланін 12 мг/кг кожні 24 год в/в 6 тиж + амікацин 7,5 мг/кг кожні 12 год в/в, в/м 2 тиж + рифампіцин 300 мг 3 рази на добу per os 6 тиж |
|
Показання до призначення ГК включають: 1) алергічну реакцію на антибіотики; 2) бактеріальний шок; 3) нефрит, що швидко прогресує. Слід враховувати негативні ефекти ГК: більш високу летальність при їх використанні; частий розвиток тромбоемболічних ускладнень (в 2 рази частіше); зниження фагоцитарної активності лейкоцитів; швидше руйнування клапанів. Альтернативою ГК є детоксикація за допомогою плазмаферезу. При цьому курс лікування становить 3–5 процедур з видаленням близько ¾ об’єму циркулюючої плазми. Показання включають: аутоімунні прояви; альтернатива ГК; тяжко хворі на ІЕ; випадки інфекції, що не купірується при підготовці до протезування клапана. Плазмаферез без поєднання з антибіотиками до вилікування ІЕ не приводить.
До загальних показань до хірургічного лікування включено: 1) серцеву недостатність, що прогресує (розвиток аортальної або мітральної недостатності, поява нападів серцевої астми, периферичних набряків); 2) неефективність антимікробної терапії; 3) формування абсцесу міокарда або фіброзного кільця; 4) ІЕ протезованого клапана (особливо ранній ІЕ механічного клапана); 5) повторні тромбоемболії; 6) грибковий ендокардит. Оптимальна передопераційна підготовка становить 4 тиж. Вона включає імунотерапію, інфузії гіперімунної плазми, нормального імуноглобуліну людини для в/в введення. Антистафілококову та антисиньогнійну плазму вводять в/в крапельно в дозі 125–250 мл щоденно або через день, 4–6 інфузій на курс. Хірургічні операції на серці можливі і після перенесеного ішемічного інсульту. При цьому операції не відміняють при розвитку серцевої недостатності, неконтрольованої інфекції, збереження високого ризику емболій. Операції відкладають на 1 міс у разі внутрішньочерепного крововиливу. При ІЕ, зумовленому наявністю кардіостимулятора, лікування включає довготривалу антибіотикотерапію та вилучення кардіостимулятора. Кардіостимулятор вилучають навіть за відсутності визначеного джерела інфекції, тобто при підозрі на ІЕ. Шляхи вилучення включають черезшкірну та хірургічну екстракцію. Першу проводять навіть при великих вегетаціях, другу — за неможливості черезшкірного вилучення або при деструктивному ІЕ трикуспідального клапана, уже великих вегетаціях (>25 мм). Реімплантацію проводять через проміжок часу від декількох днів до тижнів. Не рекомендується проведення тимчасової стимуляції. Перед імплантацією проводиться рутинна антибіотикотерапія. Правобічний ІЕ у наркоманів часто рецидивує, тому, як правило, здійснюється консервативне лікування.
Після проведення пластики клапанів серця призначають варфарин протягом 3 міс, після імплантації механічних протезів клапанів — пожиттєво. Оптимальна доза варфарину є індивідуальною, контроль здійснюють за допомогою визначення міжнародного нормалізованого відношення (МНВ). Рівень необхідного МНВ залежить від конструктивних особливостей протезів і коливається в межах 2–3,5. Для пацієнтів віком старше 70 років МНВ становить 1,6–2,5 (у середньому 2,0). Алгоритм підбору дози варфарину наведений у табл. 33.23.
Таблиця 33.23
Алгоритм підбору дози варфарину, яка забезпечує терапевтичний діапазон МНВ (за: Кнышов Г.В. и соавт., 2012)
|
Перші 2 дні |
Призначають 2 таблетки (5 мг) один раз після вечері |
|
|
3-й день |
Вранці визначають МНВ |
|
|
МНВ <1,5 |
Підвищити добову дозу на 1/2 таблетки. Визначити МНВ через 1–2 дні |
|
|
МНВ 1,5–2,0 |
Підвищити добову дозу на 1/4 таблетки. Визначити МНВ через 1–2 дні |
|
|
МНВ 2,0–3,0 |
Залишити добову дозу без змін. Визначити МНВ через 1–2 дні |
|
|
МНВ 3,0–4,0 |
Знизити добову дозу на 1/4 таблетки. Визначити МНВ через 1–2 дні |
|
|
МНВ >4,0 |
Пропустити 1 прийом, потім добову дозу знизити на 1/2 таблетки. Визначити МНВ через 1–2 дні |
|
|
4–5-й день |
Вранці визначити МНВ. Дії відповідають алгоритму 3-го дня. Якщо підбір дози займає більше 5 днів, то подальша кратність визначення МНВ — 1 раз на 2 дні з використанням алгоритму 3-го дня |
|
При двох послідовних показниках МНВ в цільовому діапазоні наступні визначення проводять через тиждень. Після цього дозу вважають підібраною і в подальшому МНВ визначають 1 раз на місяць.
Оцінка ефективності лікування. При правильному виборі антибіотиків покращення наступає протягом 3–10 діб. Відсутність позитивної динаміки протягом цього часу свідчить про неадекватність вибраних лікарських засобів. Загалом критерії ефективності лікування включають покращення загального стану, стійку нормалізацію температури тіла, негативні результати бактеріальних досліджень. Одужання вважається таким, якщо через 1 рік температура тіла, ШОЕ нормальні, не висівається збудник з крові. Рецидиви хвороби розрізняють ранні (в перші 2–3 міс після завершення лікування) та пізні (через 3–12 міс). Повторний ІЕ розвивається: 1) в термін до одного року, якщо виділений другий збудник (не той, що виділявся спочатку); 2) через 1 рік і більше після завершення лікування.
Прогноз
Внутрішньолікарняний рівень смертності становить 10–26%. Він залежить від характера збудника: при ІЕ, викликаному зеленкуватим стрептококом, летальність становить 4–9%, гемолітичним стрептококом — 13,8%, ентерококом — 16–35%, стафілококом — 25–47%, грибами — 5% (Чазов Е.И., Беленков Ю.Н., 2005 (ред)). Виживаність хворих після виписки зі стаціонару протягом 1 року становить 94%, 5 років — 75% (при хірургічному лікуванні (1 клапан) — 77–89%, 2 клапана — 61,6%), 10 років — 33%. Факторами ризику смерті у віддалений період є серцева та ниркова недостатність, похилий вік, неврологічні ускладнення. До предикторів несприятливого прогнозу у пацієнтів з ІЕ відносяться: 1) характеристика пацієнта: ІЕ протезованого клапана, похилий вік, серцево-судинні, легеневі, ниркові хвороби, цукровий діабет; 2) наявність ускладнень ІЕ: серцева недостатність, періанулярні ускладнення, септичний шок, інсульт, ниркова недостатність; 3) мікроорганізми: золотистий стафілокок, гриби, грамнегативні бактерії; 4) ехоКГ ознаки: великі вегетації, дисфункція протезованого клапана, періанулярні ускладнення, низька фракція викиду, виражена лівобічна клапанна регургітація, підвищений діастолічний тиск, легенева гіпертензія.
Глава 34. Хвороби міокарда
Міокардити
Міокардит — це запальне захворювання серцевого м’яза, зумовлене безпосереднім або опосередкованим через імунні механізми впливом інфекції, паразитарної або протозойної інвазії, хімічних і фізичних факторів, а також яке виникає при алергічних, аутоімунних захворюваннях і трансплантації серця.
Епідеміологія
Поширеність міокардиту, за даними різних авторів, становить 4–11% захворювань серцево-судинної системи взагалі і 20% некоронарогенних захворювань серця. У багатоцентрових дослідженнях встановлено, що поширеність міокардиту значно варіює. У дослідженні Myocarditis Treatment Trial (MTT) ознаки запалення при біопсії міокарда визначили тільки у 10% із 2233 обстежених пацієнтів. В епідеміологічному дослідженні ESETCID (European Study of Epidemiology and Treatment of Cardiac Inflammatory Diseases) відмічали позитивні результати біопсії у 17,2% із перших 3055 хворих. Докази міокардиту виявляють за результатами 1–9% рутинних аутопсій і до 20% випадків нез’ясованої раптової смерті в молодому віці. Реальну поширеність легких форм міокардиту складно встановити, оскільки більшість випадків минає без тривалої клінічної картини.
Класифікація
В Україні застосовується класифікація міокардиту, яка прийнята на IХ Конгресі кардіологів України (2008), що включає наступні рубрики;
Стадії:
- блискавичний
- гострий (І40)
- підгострий (І40.10)
- хронічний (І51.4)
- міокардіофіброз (І51.4)
Етіологія:
- зі встановленою етіологією І40, І41 (інфекційний — I40, бактеріальний — I41.0, вірусний —I41.1, паразитарний — I41.2, при інших захворюваннях — I41.8)
- неуточнений — I40.9
Морфологічні особливості інфільтрату (за даними ендоміокардіальної біопсії — ЕМБ):
- лімфоцитарний
- еозинофільний
- гігантоклітинний
- гранулематозний
Поширеність:
- вогнищевий — I40.1
- дифузний — I40.8
Характер перебігу:
- легкий
- середньої тяжкості
- тяжкий
Ускладнення: порушення ритму серця та провідності, тромбоемболії та ін.
Серцева недостатність: 0–III стадії, I–IV ФК.
Етіологія
Нині вважається загальновизнаним, що запальне ураження міокарда може виникати при будь-яких інфекційних захворюваннях. Етіологічними факторами можуть бути віруси, бактерії, гриби, найпростіші, гельмінти, рикетсії та спірохети. Провідна роль у розвитку міокардиту належить вірусам (50%), серед яких найбільш важливе значення мають ентеровіруси, зокрема віруси Коксакі групи В, а також аденовіруси, віруси грипу A та B, гепатиту C, парвовірус В19. Нещодавно в кардіоміоцитах виявлені специфічні так звані Коксакі аденовірусні рецептори (CAR), які служать мішенню для вірусів Коксакі групи В та 2- і 5-серотипів із сімейства аденовірусів. Серед збудників невірусних інфекційних захворювань в останні роки важливе значення має дифтерійна паличка, підвищується частота міокардиту грибкової етіології. Міокардит також може виникати при бруцельозі, лептоспірозі, сальмонельозі, трипаносомозі, скарлатині, висипному тифі.
Із неінфекційних етіологічних факторів міокардиту відомі різні імунопатогенні агенти: деякі лікарські засоби (антибіотики (доксорубіцин), сульфаніламіди, НПЗП (метамізол натрій), трициклічні антидепресанти, добутамін та ін.), лікувальні сироватки і вакцини, токсичні хімічні речовини, а також голодування, інтоксикація при опіковій хворобі, деякі фізичні фактори (перегрівання, вплив високих доз іонізуючого випромінювання).
Міокардит може розвиватися після укусів павуків і змій. Міокардит може супроводжувати системні запальні захворювання: саркоїдоз, хворобу Крона, тиреотоксикоз, системний червоний вовчак, склеродермію, ревматоїдний артрит.
Запальні зміни міокарда можуть виникати після трансплантації серця.
Патологічна анатомія
Макроскопічно виявляють локальну дилатацію або гіпертрофію, порожнина лівого шлуночка може мати нормальні розміри. Відзначається в’ялість міокарда, строкатість малюнка на розрізі внаслідок запального інфільтрату. При хронічному перебігу міокардиту характерні замісний інтерстиціальний фіброз, гіпертрофія із ділянками деструкції м’язових волокон, вогнищами інтерстиціальної інфільтрації мононуклеарними клітинами, наявність тяжів грануляційної та фіброзної тканини.
Мікроскопічно виявляють порушення структури і взаємного розташування кардіоміоцитів, характерна наявність лімфогістіоцитарних інфільтратів із невеликою кількістю плазматичних клітин в інтерстиціальній тканині, деструкція м’язових волокон різного ступеня та інтерстиціальний набряк, відкладення імуноглобулінів і комплементу в сарколемі та інтерстиції із ушкодженням ендотелію капілярів. Відзначають повнокров’я судин мікроциркуляторного русла, спазм артеріол, парез капілярів і вен, еритроцитарні стази і фібринові мікротромби, порушується судинна проникність і розвивається набряк строми.
Електронно-мікроскопічні та гістохімічні дослідження виявляють порушення ультраструктури міокарда, пошкодження мітохондрій, зменшення кількості гранул глікогену в м’язових клітинах, порушення процесів утилізації глюкози та β-окиснення жирних кислот.
Морфологічний варіант міокардиту пов̓ язаний з етіологічним фактором, що його спричинив. Переважним типом клітин у запальному інфільтраті при вірусному міокардиті є лімфоцити, при бактеріальних інфекціях — нейтрофільні гранулоцити, при алергічному медикаментозному міокардиті, паразитарній інвазії — еозинофільні гранулоцити. При гігантоклітинному або гранулематозному міокардиті в серцевому м’язі виявляють багатоядерні гігантські клітини, що локалізуються головним чином по краях великих ділянок некрозу і складаються переважно із макрофагів. У вогнищі некрозу виявляється запальний інфільтрат, що складається з еозинофільних гранулоцитів, гістіоцитів та інших клітин. Фіброз відсутній. При некротизуючому еозинофільному міокардиті визначається дифузний запальний інфільтрат із превалюванням еозинофільних гранулоцитів, поширений некроз кардіоміоцитів. Характерні патологічні зміни в міокарді, пов’язані з вірусом гепатиту, — невеликі вогнища некрозу ізольованих м’язових пучків, оточені лімфоцитами та дифузним серозним запаленням. Шлуночки можуть бути дилатовані із петехіальними крововиливами в міжшлуночкову перегородку, включаючи ділянку провідної системи серця. При дифтерійному міокардиті при патоморфологічному дослідженні виявляють дилатоване серце зі значною внутрішньоклітинною жировою інфільтрацією кардіоміоцитів, часто з інтерстиціальним запальним інфільтратом, міоцитолізом та гіаліновим некрозом м’язових волокон, виснаження депо глікогену. З часом розвиваються фіброз і гіпертрофія клітин міокарда, що залишилися. Часто уражується провідна система серця. При менінгококовому міокардиті патоморфологічні знахідки включають геморагічні ураження міокарда, часто відзначають інтерстиціальний міокардит з інфільтрацією, яка складається з лімфоцитів, плазматичних клітин і поліморфноядерних лейкоцитів, зрідка з некрозом міокарда. При менінгококцемії, яка розвивається раптово і швидко, можливі фокальний некроз міокарда, виражене жирове переродження і зерниста дистрофія кардіоміоцитів. При лептоспірозі відзначають петехіальні або більш великі вогнища геморагій, часто локалізовані в епікарді. Можлива інтерстиціальна міокардіальна інфільтрація в субендокардіальних шарах із залученням сосочкових м’язів. При грибковому міокардиті при посмертних дослідженнях визначали дифузні міокардіальні абсцеси, а також оклюзію коронарних судин грибковим міцелієм і тромбами.
Патогенез
Ураження міокарда при інфекційному міокардиті може бути наслідком прямої інфільтрації міокарда інфекційним агентом, впливу токсинів, які виділяються збудниками безпосередньо в серці або досягають його гематогенним шляхом, ураження ендотелію дрібних вінцевих артерій із розвитком коронариту, а також результатом розвитку імунопатологічних реакцій. Специфічність етіологічного фактора має значення лише в гострій стадії захворювання, надалі перебіг міокардиту значно більшою мірою зумовлений імунними та аутоімунними реакціями і безпосередньо процесами запалення/загоєння міокарда.
Особливості міокардиту вірусної етіології зумовлені можливим прямим проникненням вірусу в кардіоміоцити з наступною його реплікацією і цитотоксичним ефектом аж до лізису клітини або опосередкованою дією через гуморальні та клітинні імунні реакції в міокарді. Досягаючи поверхні міокардіоциту, віруси проникають всередину нього шляхом зв’язування зі специфічними молекулами САR (Coxackie adenovirus receptor) і DAF (decay accelerating factor) і взаємодіють зі структурними елементами, ушкоджують генетичний апарат клітини, глибоко порушують внутрішньоклітинний метаболізм. Ще одним патогенетичним механізмом є вплив ентеровірусної протеази на деградацію дистрофіну й саркогліканового комплексу, що призводить до зміни архітектоніки клітин міокарда, порушення координації скорочувальної функції серця, некрозу або індукції апоптозу кардіоміоцитів, розвитку імунних та аутоімунних реакцій. Утворення нових вірусних часток відбувається за рахунок використання білкових молекул клітини, яка при цьому може загинути. При руйнуванні м’язової клітини вірусні частки, що утворилися (віріони) вивільняються та можуть проникати в сусідні кардіоміоцити. У клінічних дослідженнях установлено, що кардіотропний вірус як тригерний фактор індукує активацію клітинного і гуморального імунітету і запальне ушкодження тканини міокарда, опосередковане реакціями імунітету у вигляді реакції гіперчутливості уповільненого типу. Більшість вірусів фагоцитується та виводиться з організму протягом 10–14 днів. При цьому в ушкоджених клітинах міокарда зберігаються порушення нуклеїнового обміну, що призводить до утворення антитіл класу IgG, які вступають у реакцію з неушкодженими кардіоміоцитами, запускаючи аутоімунні процеси. Після первинної інфекції міокардит розвивається через кілька тижнів, а ушкодження міокарда носить інфекційно-алергічний характер. Вважається, що у випадках блискавичного міокардиту прямий вірусопосередкований вплив може бути первинним механізмом ушкодження міокарда. При гострому та хронічному активному міокардиті вірусна фаза може бути під контролем імунних реакцій, при персистувальній реакції внаслідок глибокого порушення вірусами внутрішньоклітинного білкового обміну клони імунних клітин активуються проти власних протеїнів. Швидкому видужанню, ймовірно, сприяє генетично наслідуваний фактор, що забезпечує швидку відповідь у вигляді утворення віруснейтралізуючих антитіл класів IgM й IgG, які перешкоджають реплікації вірусів та сприяють їх елімінації. Якщо не відбувається елімінації вірусу з організму, ушкодження міокарда переходить у другу (аутоімунну) стадію внаслідок порушення структури знову синтезованих поліпептидів як результат вірусозалежних змін у генетичному апараті клітини. У патогенезі міокардиту також мають значення активація ферментних систем і вивільнення біологічно активних субстанцій (гістаміну, серотоніну, брадикініну, ацетилхоліну та ін.), що викликає ушкодження судин мікроциркуляторного русла серцевого м’яза, розвиток гіпоксії та ушкодження кардіоміоцитів із утворенням мікронекрозів. При переході захворювання в хронічну стадію ознаки запалення в міокарді можуть не виявлятися, при цьому зберігаються міокардіальна дисфункція та кардіомегалія.
Бактерії можуть безпосередньо викликати розвиток запалення в міокарді, а також бути інфекційно-токсичним і сенсибілізувальним фактором. При виникненні міокардиту на тлі туберкульозу, сифілісу, черевного тифу імунний компонент запалення особливо виражений. Міокардит на фоні дифтерії, скарлатини відрізняється особливою тяжкістю перебігу, що зумовлено прямим впливом мікробних токсинів, які ушкоджують ферментні системи м’язової клітини, інгібують синтез білка, а також викликають аутоімунне ушкодження різних відділів міокарда. При дифтерії дисфункція міокарда, ймовірно, зумовлена порушенням метаболізму жирів, оскільки дифтерійний токсин викликає значне виснаження резервів міокардіального карнітину — кофактора необхідного для їхнього бетаокиснення.
Клінічна картина
Кардіальна патологія у більшості випадків проявляється наприкінці 1-го або на 2-й тиждень від початку інфекційного захворювання. Скарги різноманітні і неспецифічні. Класична маніфестація міокардиту — тахікардія, біль у грудях або задишка на фоні гострої фебрильної лихоманки. Біль в ділянці серця є найбільш частим й одним із ранніх симптомів міокардиту, може бути різної інтенсивності і тривалості, не пов’язаний із фізичним навантаженням. Задишка нерідко передує больовому синдрому, характерне відчуття серцебиття і перебоїв у роботі серця. Перебіг міокардиту з незначною або без дисфункції лівого шлуночка може бути взагалі без кардіальних симптомів. Іноді першими проявами міокардиту бувають швидка втомлюваність, підвищене потовиділення, артралгія, астенізація. При гігантоклітинному міокардиті і некротизуючому еозинофільному міокардиті початок захворювання може бути гострим зі швидким прогресуванням гемодинамічних порушень. При еозинофільному міокардиті захворювання зазвичай починається з бівентрикулярної серцевої недостатності, виникнення аритмій може призводити до раптової смерті. Як правило, гіпереозинофілія передує або збігається з появою симптомів ураження серця, але може з’являтися пізніше. У пацієнтів з ознаками дисфункції лівого шлуночка найбільш частими симптомами є прояви застійної (частіше лівошлуночкової) серцевої недостатності (задишка, втома, дискомфорт в ділянці серця). Підвищення венозного тиску, периферичні набряки свідчать про ураження правого шлуночка. Клінічна картина хронічного міокардиту складається з послідовного ряду загострень, які настають через невизначені проміжки часу.
При фізикальному обстеженні можуть визначатися приглушеність (глухість) тонів серця, поява III і IV тонів серця, систолічний шум на верхівці, не пов’язаний з I тоном, його інтенсивність не змінюється при зміні положення тіла.
Діагностика
Суттєве значення у встановленні діагнозу має ЕКГ-дослідження. Найчастіше на фоні синусової тахікардії відзначаються неспецифічні зміни зубця Т і сегмента ST, а також зниження амплітуди всіх зубців, зміщення інтервалу S–T вниз або вгору від ізолінії в одному або декількох відведеннях, збільшення інтервалу P–Q. Можуть реєструватися різні порушення ритму і провідності серця: передсердні та шлуночкові екстрасистоли, фібриляція та тріпотіння передсердь, AВ-блокади різного ступеня, аж до повної, блокади ніжок пучка Гіса. Зміни ЕКГ у гострий період характеризуються швидкою зміною патологічних ознак, часто їх сукупністю та повною нормалізацією картини ЕКГ при видужанні. Холтерівське моніторування ЕКГ дозволяє виявити передсердні та шлуночкові аритмії, які не виявляють при звичайній реєстрації ЕКГ.
При ехоКГ-дослідженні можна визначити збільшення розмірів порожнин серця, зниження скорочувальної функції міокарда лівого шлуночка (правого шлуночка), гіпо- або акінез різних ділянок міокарда, що супроводжує ексудативний перикардит, внутрішньошлуночкові тромби (15%).
При катетеризації порожнин серця у хворих на міокардит пік систолічного тиску зміщений у бік фази скороченого вигнання крові з шлуночка, що вказує на зниження швидкості скорочення міофібрил у період вигнання, фаза напруження зменшена в основному за рахунок асинхронного скорочення, у той час як фаза ізометричного скорочення зменшена, різко подовжений час ізометричного розслаблення міокарда, характеризуючи порушення діастолічної піддатливості міокарда.
Рентгенологічне дослідження виявляється інформативним лише у хворих із дифузним міокардитом, коли можна виявити дилатацію серця, зміни амплітуди та форми зубців на рентгенокімограмі. Рентгенографія органів грудної клітки іноді виявляє збільшення розмірів серця від незначних до кардіомегалії та/або ознаки застою в легенях.
Істотну допомогу в діагностиці міокардиту (особливо хронічного) надає радіоізотопна сцинтиграфія з використанням технецію-99, цитрату галію-67 (має афінність до клітин, здатних мігрувати до вогнища запалення — моноцитів, нейтрофільних гранулоцитів, активованих Т-лімфоцитів), індій-111-оксима (використовується для маркування моноклональних антитіл до міозину). В останні роки використовують КТ і МРТ як чутливі методи виявлення гострого міокардиту. Однак методи недостатньо специфічні, тому що вони виявляють активне запалення незалежно від його етіології, у тому числі після інфаркту міокарда та при інших патологічних процесах у міокарді. Використання цих методів при проведенні ЕМБ допомагає здійснити прицільне взяття біоптатів та істотно підвищити її діагностичну цінність. У хворих із симптомами гострої фебрильної лихоманки необхідно проводити дослідження сироватки крові на наявність вірусних збудників, які виявляють найчастіше: вірус Коксакі В, грипу А і В, цитомегаловірус та відповідні аденовірусні форми. Для верифікації діагнозу використовують визначення віруснейтралізуючих антитіл (імуноглобулінів класів G і М (IgG, IgМ)) у плазмі крові, а також ПЛР і гібридизацію in situ, які є досить специфічними при вірусному міокардиті та дозволяють швидко підтвердити або виключити наявність вірусної реплікації і диференціювати вірусний та аутоімунний міокардит. На нещодавно перенесену вірусну інфекцію вказує чотириразове підвищення титру противірусного IgG у період видужання порівняно із гострим періодом. При міокардиті з більшою, ніж при іншій патології серця, частотою виявляють антитіла до міозину, актину, міолеми. Органоспецифічним для міокардиту вважають визначення аутоантитіл до аденіннуклеотидного транслокатора — ферменту внутрішньої мембрани мітохондрій. Результати лабораторних досліджень виявляють ознаки імунологічної перебудови, у тому числі підвищення рівня ЦІК, титру антитіл до мембран кардіоміоцитів, білків міокарда, зниження абсолютної та відносної кількості Т-лімфоцитів (у тому числі активних) та зміна співвідношення їх субпопуляцій (хелпери, супресори), зміна стану нейтрофільних гранулоцитів і моноцитів, процентного вмісту дегранульованих форм базофільних гранулоцитів у периферичній крові (в нормі 10%), підвищення експресії маркерів ранньої активації запалення (антигенів CD25-рецепторів для ІЛ-2 і CD71-рецепторів трансферину).
Міокардит зазвичай діагностується як «підозрюване» запалення міокарда. Діагностика міокардиту ґрунтується на динаміці патологічних змін ЕКГ, наявності кардіомегалії, застійної серцевої недостатності, що гостро почалася і прогресує. Серологічний аналіз для хворих із підозрюваним міокардитом повинен включати дослідження рівня кардіоспецифічних ферментів — КФК, МВ-фракції КФК, ЛДГ та кардіальних фракцій із співвідношенням ЛДГ1/ЛДГ2 >1, а також тропонінів. Недавні дослідження показали, що визначення тропоніну I і тропоніну Т є більше чутливим методом діагностики міокардіального ушкодження у хворих з міокардитом порівняно з виявленням у крові кардіоспецифічних ферментів. Підвищення рівня цих показників свідчить про ушкодження серця.
Для встановлення діагнозу «міокардит» загальновизнаних критеріїв не існує. Відповідно до рекомендацій NYHA до великих діагностичних критеріїв відносять перенесену інфекцію та появу протягом 10 днів після неї застійної серцевої недостатності/кардіогенного шоку, повної AВ-блокади із синдромом Морганьї — Адамса — Стокса, патологічних змін ЕКГ, підвищення активності міокардіальних ферментів у сироватці крові. До малих критеріїв відносять лабораторні підтвердження перенесеного вірусного захворювання (позитивні реакції нейтралізації, гальмування гемаглютинації та зв’язування комплементу), тахікардія, послаблення I тону, ритм галопу, результати ЕМБ. Для діагностики легкого перебігу міокардиту досить наявності в анамнезі попередньої інфекції й поєднання двох великих критеріїв або одного з них із двома малими. Наявність же в числі критеріїв одного із трьох перших дозволяє констатувати середньотяжкий або тяжкий перебіг міокардиту. Дані лабораторних досліджень у діагностиці міокардиту неспецифічні та суперечливі. Виявлення в сироватці крові протикардіальних антитіл є серйозним аргументом на користь діагнозу «міокардит», однак ці зміни виявляють переважно при тяжких його формах. Використання ЕМБ при лікуванні хворих після трансплантації підвищило її значення при захворюваннях нативного міокарда, гістологічні докази запалення стали золотим стандартом у діагностиці міокардиту. Однак чутливість ЕМБ історично була обмежена помилками аналізу зразків, і посмертні дослідження хворих, у яких підтверджений міокардит, продемонстрували чутливість ЕМБ у межах 60–70%.
У 1984 р. групою провідних американських морфологів розроблені так звані даллаські діагностичні критерії міокардиту, згідно з якими за результатами первинної ЕМБ виділяли певний міокардит, граничний (імовірний) міокардит або відсутність міокардиту. При повторних біопсіях визначали міокардит, що триває (персистувальний), міокардит, що завершується (загоюється), та міокардит, що завершений (загоївся). Суворим діагностичним критерієм гострого міокардиту визнається наявність запальної клітинної інфільтрації міокарда з некрозом та/або дегенерацією кардіоміоцитів, нетиповою для ішемічного ураження. Термін «граничний міокардит» використовують у разі виявлення клітинної інфільтрації та неушкоджених кардіоміоцитів. Дані повторних біопсій дозволяють визначити динаміку або результат процессу і говорити про персистувальний процес, що завершується або завершений.
У 1998 р. Всесвітньою кардіологічною федерацією прийнятий Консенсус із визначення запальної кардіоміопатії (міокардиту). Гострий міокардит розглядається як активний міокардит, хронічний — як граничний або міокардит, що загоюється. При первинній біопсії можна діагностувати:
1. Гострий (активний) міокардит: наявність інфільтрату (дифузного або локального) з визначенням не менше 14 інфільтруючих лімфоцитів на 1 мм2 (переважно Т-лімфоцитів (CD45ro) або активованих Т-лімфоцитів і до 4 макрофагів). Кількісно інфільтрат має бути визначений імуногістохімічним методом. Виявляють некроз або дегенерацію, необхідно враховувати також фіброз, наявність якого не обов’язкова.
2. Хронічний міокардит: наявність інфільтрату, що містить не менше 14 інфільтруючих лімфоцитів на 1 мм2 (переважно Т-лімфоцитів (CD45ro) або активованих Т-лімфоцитів і до 4 макрофагів). Кількісно інфільтрат слід визначати імуногістохімічним методом. Некроз і дегенерація, як правило, не виражені, слід враховувати фіброз.
3. Відсутність міокардиту: не виявляють інфільтруючі клітини або їхня кількість <14 на 1 мм2. Для оцінки фіброзу розроблені критерії: відсутність фіброзу — 0-й ступінь, початковий фіброз — 1-й ступінь, помірний фіброз — 2-й ступінь, виражений фіброз — 3-й ступінь.
При наступних біопсіях можна діагностувати:
1. Триваючий (персистувальний) міокардит: критерії 1 або 2.
2. Міокардит, що завершується (загоюється): критерії 1 або 2, але імунологічний процес більш млявий, ніж при первинній біопсії.
3. Міокардит завершений (що загоївся), який відповідає «даллаським» критеріям. Усі категорії можуть супроводжуватися або не супроводжуватися фіброзом.
Сьогодні використовують результати гістоморфологічних, імуногістохімічних методів і молекулярно-біологічних технологій (ПЛР, гібридизація in situ), за допомогою яких можна верифікувати діагноз міокардиту. Вірусний міокардит характеризується інтракардіальною наявністю вірусу з ознаками запалення. Хронічний аутоімунний міокардит підтверджується імунологічно активним запаленням, при цьому вірус у молекулярних біологічних середовищах не визначається. Критеріями вилікуваного міокардиту або переходу його в дилатаційну кардіоміопатію (ДКМП) є зникнення ознак запалення в міокарді, а також негативні результати ПЛР і гібридизації in situ. При хронічному міокардиті ЕМБ не дозволяє виявити патогномонічні ознаки вже через 1–2 міс від початку захворювання після проведення адекватної терапії.
Відповідно до об’єднаних рекомендацій АНА, ACR і Європейського товариства кардіологів (2007) ЕМБ показана при наступних клінічних ситуаціях:
- при непоясненому виникненні симптомів серцевої недостатності протягом <2 тиж при нормальному або дилатованому лівому шлуночку і порушеннях гемодинаміки (можуть визначатися лімфоцитарний міокардит, гігантоклітинний міокардит і некротизуючий еозинофільний міокардит);
- при непоясненому виникненні симптомів серцевої недостатності протягом від 2 тиж до 3 міс при дилатованому лівому шлуночку та зі шлуночковими аритміями, які виникли знову, блокадою за типом Мобітц II 2–3-го ступеня або недостатньою ефективністю звичайної терапії протягом 1–2 тиж (може визначатися гігантоклітинний міокардит);
- при непоясненому виникненні симптомів серцевої недостатності протягом >3 міс при дилатованому лівому шлуночку та із вперше виявленими шлуночковими аритміями, блокадою за типом Мобітц II 2–3-го ступеня або недостатньою ефективністю стандартної терапії протягом 1–2 тиж (може визначатися ідіопатичний гранулематозний міокардит);
- при непоясненому виникненні симптомів серцевої недостатності, які пов’язані із ДКМП будь-якої тривалості, що асоціюється з підозрою на алергічну реакцію з еозинофілією (гіперсенситивний міокардит, некротизуючий еозинофільний міокардит);
- у виняткових випадках при непоясненій шлуночковій аритмії (лімфоцитарний міокардит, гранулематозний міокардит).
У більшості пацієнтів із доведеним за даними ЕМБ запаленням виявляють лімфоцитарний міокардит з переважанням лімфоцитів в міокардіальному клітинному інфільтраті. Превалювання еозинофільних гранулоцитів виявляється при міокардиті, пов’язаному із алергічною реакцією або периферичною еозинофілією. Лімфоцитарний міокардит визначають приблизно у 10% пацієнтів, яким була проведена біопсія, а у 1–2% — гігантоклітинний міокардит, що характеризується наявністю багатоядерних гігантських клітин. Гіперсенситивний міокардит виявляють в основному при аутопсії в 2,4–7% випадків. ЕМБ у більшості випадків не рекомендована через відсутність специфічних, що базуються на цьому методі, рекомендацій із терапії. Достовірний діагноз «міокардит» встановити дуже тяжко, оскільки захворювання може бути зовсім безсимптомним або проявлятися різними неспецифічними симптомами. Всі інструментальні і лабораторні методи дослідження дозволяють підтвердити наявність міокардиту, однак негативні результати не є критерієм виключення діагнозу, навіть негативні результати ЕМБ. У гостру фазу міокардиту активність серцевих ферментів зазвичай підвищена, що може спричинити помилкову діагностику інфаркту міокарда. Діагноз «міокардит» майже завжди є певною мірою ймовірним, але стає досить переконливим, якщо до нього приєднуються ознаки супутнього перикардиту.
Лікування
Пацієнти з легким перебігом вогнищевого міокардиту можуть проходити лікування в амбулаторних умовах. Хворі з міокардитом середнього ступеня тяжкості та тяжким перебігом підлягають госпіталізації. Принципи лікування хворих із міокардитом включають застосування немедикаментозних підходів, медикаментозного й хірургічного лікування.
До немедикаментозної терапії можна віднести обмеження фізичної активності, повноцінне раціональне харчування з обмеженням споживання кухонної солі, відмова від паління, обмеження вживання алкоголю.
Медикаментозне лікування міокардиту передбачає кілька напрямків:
- вплив на запальні, аутоімунні та алергічні процеси, у тому числі на зменшення вираженості ушкоджувальної дії антиміокардіальних антитіл;
- скорочення продукції біологічно активних речовин;
- відновлення і підтримка гемодинаміки;
- вплив на метаболізм міокарда;
- активна санація вогнищ інфекції.
Специфічного лікування міокардиту на сьогодні не існує, основними напрямками лікувальної тактики можуть бути:
- етіотропна терапія при встановленому збуднику:
- противірусні засоби,
- антибактеріальні засоби,
- протипаразитарні засоби;
- неспецифічна протизапальна терапія:
- НПЗП,
- ГК;
- вплив на аутоімунний процес;
- симптоматична терапія ускладнень.
Значення специфічної антивірусної терапії не встановлено. При виявленні вірусу гриппу (А і В) застосовують римантадин протягом 7 діб (лікування починають не пізніше 48 год із моменту появи симптомів). При ідентифікації вірусу Varicella zoster, вірусу простого герпеса, вірусу Епштейна — Барр призначають ацикловір, цитомегаловірусу — ганцикловір або фоскарнет натрій, зидовудин. У випадках вірусної етіології міокардиту в перші 2–3 тиж протипоказане призначення НПЗП, ГК, оскільки під їх впливом у гострій фазі можливі значне прискорення реплікації вірусів, збільшення ушкодження міокарда, зменшення продукції інтерферону. При інфікуванні Mycoplasma pneumoniae ефективний еритроміцин. При інфікуванні S. аureus (до визначення чутливості збудника до антибіотиків) призначають ванкоміцин, при зараженні грибами — амфотерицин В. Якщо збудником є Cryptococcus neoformans (найчастіший збудник), застосовують комбінацію амфотерицину В і флуцитозиду. При ідентифікації Toxoplasma gondii комбінують піриметамін + сульфадіазин, а також фолієву кислоту для профілактики мієлосупресії. При дифтерійному міокардиті лікування малоефективне. Для поліпшення ефективності лікування захворювання необхідно негайно ввести протидифтерійний антитоксин, терапія антибіотиками менш невідкладна. НПЗП виявляють активну протизапальну дію. Найчастіше використовують диклофенак або інгібітори ЦОГ-2 — групу препаратів, що впливають на метаболізм арахідонової кислоти і знижують синтез ендогенних простагландинів внаслідок блокади ізоформи ферменту ЦОГ (ЦОГ-2), інгібування якого має велике значення в усуненні запалення. Призначення НПЗП виправдане за наявності ознак супутнього перикардиту. Немає єдиної думки щодо застосування імуносупресивної терапії в пацієнтів із гострим міокардитом вірусної етіології.
У багатоцентровому дослідженні Myocarditis Treatment Trial (MTT) оцінювали застосування преднізолону і циклоспорину в лікуванні доведеного даними біопсії лімфоцитарного міокардиту. Терапевтичного ефекту імуносупресії не було досягнуто, оскільки збільшення фракції викиду лівого шлуночка становило 10% у групі з імуносупресивною терапією і 7% — у контрольній групі. Не отримано різниці в довгостроковому виживанні (p=0,96) при загальній смертності 20% за 1 рік і 56% — за 4,3 року. Призначення імуносупресивної терапії на ранніх стадіях захворювання не виправдано, оскільки пригнічення імунної відповіді може призводити до пролонгації персистування вірусу або його фрагментів в організмі та хронізації запального процесу в міокарді. Імуносупресивна терапія, що включає ГК, може бути ефективною в пацієнтів з аутоімунною фазою міокардиту. При тяжкому перебігу міокардиту — вираженій прогресуючій серцевій недостатності або тяжких порушеннях ритму, рефрактерних до антиаритмічної терапії, призначають ГК у поєднанні з цитостатиками (у разі тяжкого перебігу дифузного міокардиту, рецидивуючого перебігу, що супроводжується алергозами або вираженими ознаками активності запального процесу, що зберігаються при лікуванні НПЗП). Найчастіше застосовують преднізолон у дозі 70–80 мг/добу та азатіоприн у дозі 150–200 мг/добу. Лікування в основному продовжують 6 міс і більше (дозу преднізолону поступово знижують). Призначення імуносупресивної терапії показане у випадку підтвердженого при ЕМБ гігантоклітинного міокардиту.
Сьогодні вивчаються можливості застосування імуномодуляторів, у 2002 р. розпочато Європейське рандомізоване дослідження BICC (Betaferon in Chronic Viral Cardiomyopathy Trial). Ефективність імуноглобуліну для в/в введення у дорослих з міокардитом оцінювали у багатоцентровому дослідженні Intervention in Myocarditis and Acute Cardiomyopathy (IMAC), в якому рандомізовано 62 пацієнти для інфузії імуноглобуліну в дозі 2 г/кг маси тіла або плацебо. Не виявлено ніяких переваг в/в введення імуноглобуліну, оскільки за 6 міс приріст фракції викиду становив 14% в обох групах і через 12 міс — 16% у групі лікування порівняно з 15% — у групі плацебо. Загальна виживаність (події визначені як смерть, проведення трансплантації серця або необхідність у використанні допоміжних пристроїв для підтримки функції лівого шлуночка) становила 91% через 1 рік і 88% за 2 роки; ніяких істотних відмінностей між групами не виявлено.
У комплексній терапії міокардиту можливе використання препаратів системної ензимотерапії.
Ефективність лікування і прогноз при міокардиті значно відрізняються залежно від функції лівого шлуночка. При легкому перебігу міокардиту (підозрі на міокардит) і відсутності дисфункції лівого шлуночка медикаментозна терапія не потрібна, рекомендовано уникати аеробних фізичних навантажень або підйому надмірної ваги протягом як мінімум 6–8 тиж.
Обстеження, що включає проведення ЕКГ та ехоКГ, варто повторити через 6–8 тиж після початку захворювання для оцінки прогресування ураження міокарда. При дисфункції лівого шлуночка пацієнтам із серцевою недостатністю терапія проводиться за загальними правилами. Усім хворим рекомендоване застосування інгібіторів АПФ (при їх непереносимості — антагоністів рецепторів ангіотензину II) і блокаторів β-адренорецепторів. Застій у легенях або симптоми перенавантаження рідиною необхідно усувати шляхом застосування петлевих діуретиків у мінімальній ефективній дозі. Дигоксин додають до схеми лікування у хворих з III–IV функціональним класом NYHA, однак слід враховувати його потенційні проаритмогенні і прозапальні ефекти, призначаючи в мінімальних ефективних підтримувальних дозах або не призначаючи зовсім у хворих з мінімально вираженою симптоматикою (I або II функціональний клас). Навіть на фоні застосування серцевих глікозидів у звичайних дозах існує високий ризик глікозидної інтоксикації. Серцева недостатність при міокардиті відрізняється резистентністю до терапії серцевими глікозидами і діуретиками. Важливо усунути гіпокаліємію та ацидоз. Для лікування рефрактерної серцевої недостатності виправдане застосування периферичних вазодилататорів. Нітрати зменшують венозне повернення крові до серця, тиск у судинах малого кола кровообігу, тобто переднавантаження на лівий шлуночок, що проявляється клінічним поліпшенням. Застосування серцевих глікозидів на фоні периферичних вазодилататорів дозволяє досягти адекватної дигіталізації за відсутності токсичного ефекту. У пацієнтів із блискавичним перебігом міокардиту можуть використовуватися механічні пристрої для підтримання функції шлуночків, у разі якщо їх стан не поліпшується, незважаючи на максимальну інотропну терапію. У хворих, розмір і систолічна функція лівого шлуночка в яких повністю нормалізувалися, терапія інгібіторами АПФ і блокаторами β-адренорецепторів у принципі може бути припинена за умов ретельного контролю функції лівого шлуночка і хронічних захворювань. У багатьох пацієнтів, що одужали, можуть відмічати суб’єктивні обмеження функціональної здатності і об’єктивні обмеження при навантажувальних пробах, можливо, завдяки більш повільному відновленню діастолічної функції лівого шлуночка. Непрямі антикоагулянти призначають хворим із тромбоемболічними ускладненнями (у тому числі в анамнезі), наявністю тромбів у порожнинах серця і фібриляцією передсердь на фоні кардіомегалії. При порушеннях ритму призначають антиаритмічні препарати або імплантують кардіовертер-дефібрилятор, електрокардіостимулятор. Трансплантацію серця слід планувати тільки на пізніх стадіях захворювання, особливо враховуючи ризик високої ранньої післяопераційної смертності в результаті відторгнення трасплантата в пацієнтів, імунна система яких була спочатку активована проти антигенів міоцитів. При відторгненні трансплантата, що частіше розвивається в перші 3 міс після трансплантації серця, призначають ГК у високих дозах (пульс-терапія). У термінальній стадії використовують двокамерну стимуляцію, апаратну підтримку скорочення шлуночків.
Прогноз
Прогноз значною мірою залежить від ступеня відновлення функції лівого шлуночка. У більшості випадків (до 90%) перебіг міокардиту безсимптомний і протягом 1–2 міс завершується повним одужанням, зникають усі суб’єктивні симптоми, нормалізується ЕКГ у спокої та при проведенні велоенергометрії. У 30–50% пацієнтів із дисфункцією лівого шлуночка відбувається істотне її поліпшення протягом першого року після захворювання. У багатьох хворих зберігаються залишкові зміни ЕКГ, що свідчать про розвиток вогнищевого міокардитичного кардіосклерозу (міокардіофіброзу), що підтверджується при ехоКГ-дослідженні. За наявності клінічних проявів дисфункції лівого шлуночка прогноз гірший: у 10–33% хворих розвивається ДКМП. Гігантоклітинний міокардит і некротизуючий еозинофільний міокардит можуть мати блискавичний розвиток симптомів з несприятливим прогнозом. При гігантоклітинному міокардиті тривалість життя без трансплантації серця в середньому становить 5,5 міс, через 4 роки залишаються живими тільки 11%, при лімфоцитарному міокардиті — 44% хворих. Несприятливими чинниками, що обтяжують прогноз міокардиту, вважають підвищення тиску в лівому передсерді, низький серцевий індекс, діастолічну дисфункцію правого шлуночка, розвиток застійної серцевої недостатності, тромбоемболії, порушення провідності та аритмії високих градацій.
Дилатаційна кардіоміопатія
ДКМП — захворювання серцевого м’яза, що характеризується дилатацією і систолічною дисфункцією лівого шлуночка за відсутності порушень наповнення (гіпертензія, клапанні вади) або ішемічної хвороби серця, здатних викликати глобальне погіршення систолічної функції. Може також відзначатися дилатація і дисфункція правого шлуночка, проте це не є обов’язковим для встановлення діагнозу.
Епідеміологія
Частота виявлення ДКМП у різних регіонах неоднакова, тому визначити її справжню поширеність складно. Ідіопатичну ДКМП відзначають у 0,4 випадку на 1 тис. населення, щорічно виявляють 0,08 нового випадку на 1 тис. населення, що становить близько 25% всіх випадків кардіоміопатії і є причиною щорічної смерті 10 тис. хворих. Чоловіки хворіють у середньому в 3 рази частіше, ніж жінки.
Етіологія
Етіологія ДКМП остаточно не встановлена. Багато дослідників дотримуються поліетіологічної гіпотези походження захворювання — достатньо описано випадків розвитку ДКМП, яка є кінцевим результатом різних патологічних процесів. Виділяють ідіопатичну, сімейну (або генетичну), вірусну (та/або імунну) й асоційовану з відомими серцево-судинними захворюваннями ДКМП. Ідіопатична ДКМП, що є первинним захворюванням міокарда невідомої етіології, розвивається у 40% хворих. Сімейна ДКМП головним чином асоціюється з мутаціями в генах цитоскелета й екстрацелюлярного матриксу. Останнім часом досягнутий значний прогрес у сфері молекулярної генетики ДКМП. У 20% хворих захворювання успадковується або виявляється в сімейному анамнезі. При спадкових формах виявлено аутосомно-домінантний, аутосомно- рецесивний, а також пов’язаний з Х-хромосомою гена або мітохондріальною трансмісією тип успадкування. Мутації виявлені в генах кардіоміоцитів, які кодують контрактильні білки або їх регулювальні елементи, включаючи компоненти саркомера, цитоскелета, а також різні механізми, що забезпечують узгодження процесів збудження — скорочення, бета-адренергічні шляхи проведення і процеси, що призводять до дефіциту енергетичних механізмів, у тому числі мутації мітохондрій, метаболізм глікогену, обміну кальцію, а також транскрипторну регуляцію. Виникнення ДКМП пов’язують із варіантами мутацій гена актину, важливе значення також приділяється патології гена білка дистрофіну, що входить до складу комплексу, який зв’язує м’язовий цитоскелет кардіоміоциту з екстрацелюлярним матриксом.
Результати дослідження CARDIGENE (1999) свідчать, що при ДКМП мають значення генетичні порушення шляхів ендотеліну та поліморфізм гена ендотелінових рецепторів типу А — перший ідентифікований генетичний чинник ризику розвитку хвороби. Існує вірусно-імунологічна теорія виникнення ДКМП. При ДКМП виявлено ряд порушень імунної регуляції, включаючи гуморальну і клітинну аутоімунну реактивність по відношенню до міоцитів, зниження вмісту і клітинної активності природних кілерів і відхилення у діяльності супресорних клітин. Наявність порушень імунної регуляції і великої кількості антиміокардіальних антитіл при ДКМП узгоджується з цією гіпотезою: при імунологічному дослідженні підвищення титрів антитіл до вірусу Коксакі В3 виявляли у 40% хворих з ДКМП і тільки у 2% — в групі здорових осіб, при цьому в ендоміокардіальних біоптатах не визначали ознак міокардиту. У 50% хворих на ДКМП виявляють антитіла до міокарда, у 49% — антиінтерфібрилярні антитіла, у 30% — сенсибілізацію до серцевого антигену. Активація аутоімунних процесів призводить до утворення антитіл до міозину і β1- адренорецепторів. Причиною пригнічення активності природних кілерів може бути первинне порушення їх дозрівання, детерміноване антигенами системи HLA, у хворих з ДКМП найчастіше виявляють гаплотипи HLA B27, HLA A2, HLA DQ4 і HLA DR4, що вказує на спадкову схильність до захворювання та свідчить про можливу його імунну основу. Незважаючи на виявлення порушень гуморального імунітету, в цілому вірусно-інфекційно-аутоімунна гіпотеза на сьогодні залишається не доведеною.
Патогенез
Патогенез ДКМП треба насамперед розглядати на молекулярному рівні, оскільки порушується експресія генів, що призводить до зміни фенотипу. Виділяють кілька механізмів, які лежать в основі розвитку захворювання: одиничний генний дефект; поліморфні зміни генів-модифікаторів (у гені АПФ та інших компонентах ренін-ангіотензин-альдостеронової системи (РААС) і β2-адренергічних рецепторах); порушення експресії нормальних генів, що кодують білки, які регулюють скорочувальну функцію серця або формують структуру його порожнин. Мутації генів, що кодують білки позаклітинного матриксу, можуть бути причиною послаблення механічного взаємозв’язку між матриксом і кардіоміоцитами, можливим наслідком чого є дилатація серця, що прогресує. В кардіоміоцитах, що залишилися, відбувається ушкодження внутрішньоклітинних органел, у тому числі відповідальних за енергетику клітини. Внаслідок цього відбувається швидке розщеплення АТФ, що веде не тільки до порушення процесу скорочення, але й до контрактури міокарда в результаті нестачі енергії й кальцієвого перевантаження. Певна роль у розвитку фіброзу приділяється деградації нормального колагенового матриксу металопротеїназами, активація яких відбувається внаслідок активації прозапальних цитокінів та експозиції вільних кисневих радикалів (оксидантного стресу). При ушкодженні міокарда і дилатації серця відбувається активація різних компенсаторних механізмів, спрямованих на нормалізацію серцевої діяльності. Довгострокові компенсаторні механізми включають розвиток гіпертрофії життєздатних кардіоміоцитів, що залишились, і зміну геометрії камер серця, що становить суть ремоделювання лівого шлуночка. Гіпертрофія міокарда не досягає адекватного ступеня і не відповідає потребам дилатованого серця, оскільки скорочувальна активність гіпертрофованого міокарда на одиницю маси нижча, ніж у здоровому серці. Поступово відбувається зміна геометрії шлуночка від нормальної еліпсоїдної до сферичної форми (ремоделювання) і поступове переважання дилатації над гіпертрофією його стінки. Серцевий викид підтримується тахікардією і більшим об’ємом діастолічного наповнення, що, в свою чергу, підвищує внутрішньоміокардіальне напруження як у систолу, так і в діастолу та збільшує потребу міокарда в кисні. Ці процеси є пусковим механізмом активації РААС, стимулюють виділення норадреналіну симпатичними терміналями, а також секрецію натрійуретичного пептиду (НУП). Тривала надмірна активація РААС спричиняє ушкоджувальну дію, наслідком якої є потенціювання активності інших нейрогормональних систем, коронарна та системна вазоконстрикція, безпосередня токсична, ушкоджувальна дія ангіотензину II на кардіоміоцити, що призводить до їх дисфункції та загибелі. Тривала активація симпато-адреналової системи чинить ряд негативних ефектів на серцево-судинну систему, включаючи підвищення потреби в кисні, зменшення сили скорочення (внаслідок підвищення ЧСС), підвищення концентрації катехоламінів у плазмі крові, зниження чутливості β-адренорецепторів до катехоламінів, пригнічення функції мітохондрій, порушення окиснювально-відновної рівноваги, що реалізується шляхом дії через β-рецептори серця і цАМФ. У результаті порушень з боку β-адренергічних шляхів проведення, що значною мірою модулюють функцію серця на рецепторному та клітинному рівнях, відбувається значне зменшення кількості та щільності β1-рецепторів в ураженому серці, тоді як щільність β2-рецепторів залишається практично без змін. У сукупності з підвищенням концентрації блокуючих G-протеїнів і посиленням процесів β-рецепторного фосфорилування ці зміни посилюють порушення скорочувальної функції ураженого міокарда.
Активація нейрогормонів, цитокінів, перебудова біомеханіки міокарда зумовлюють зміну генної експресії, загибель кардіоміоцитів, клітинне ремоделювання, що, в свою чергу, є причиною дисфункції міокарда та його ремоделювання. Можливо, певний генотип первинно призводить до активації нейрогормонів і цитокінів. Велику роль відіграють ендотелін-1 і ФНП-α. Порушення насосної функції серця при ДКМП може бути зумовлено зменшенням кількості самих кардіоміоцитів. Активація процесів, пов’язаних з механічним перерозтягненням стінок ураженого серця, з ефектами системи нейрогуморальної регуляції і цитокінами відображається у прогресуючій втраті контрактильних елементів шляхом апоптозу та некрозу.
Патологічна анатомія
Захворювання характеризується різкою дилатацією всіх порожнин серця, маса серця може досягати 1000 г і більше. У порожнинах передсердь і шлуночків часто (за даними розтину >50%) виявляють пристінкові тромби. AВ-отвір значно розтягнутий, стулки клапана злегка білуваті, подовжені, як і сухожильні нитки. Сосочкові м’язи гіпертрофовані, часто різко склерозовані. Специфічних змін, що виявляються мікроскопічно, немає, визначають нерівномірну гіпертрофію кардіоміоцитів з великими неправильної форми ядрами, також помітна вогнищева жирова дистрофія м’язових волокон, дезорганізація міофібрил, втрата міофіламентів, мікроцитоліз, колапс строми, інтерстиціальний фіброз. Новоутворені колагенові структури характеризуються спотвореним співвідношенням між типами колагену і порушенням архітектоніки взаємного розташування волокон. При ДКМП переважає колаген I+III типу, помірний вміст колагену VI типу та знижений вміст колагену IV типу. Відзначають виражені дегенеративні та некробіотичні зміни кардіоміоцитів (зникнення поперечної смугастості, вакуолізація ядер, некроз, помірний вміст глікогену). Нерідко визначають запальні інфільтрати, при гістологічному дослідженні біоптатів міокарда в інтерстиціальній тканині міокарда налічують від 5 до 10 лімфоцитів при збільшенні в 400 або 200 разів відповідно.
Клінічна картина
Домінуючим клінічним симптомом є наростаюча серцева недостатність. Як правило, ранні стадії захворювання виявляють випадково при профілактичному рентгенологічному або ехоКГ-дослідженні. Період від появи перших симптомів до виникнення розгорнутої клінічної картини становить зазвичай 2 роки. Клінічні симптоми зумовлені дисфункцією лівого, правого або обох шлуночків. У ⅓ хворих можливий біль за грудниною, нерідко — різні порушення ритму, характерними є епізоди тромбоемболії судин великого і малого кола кровообігу. При прогресуванні захворювання та появі декомпенсації симптоми загалом характерні для серцевої недостатності.
Діагностика
На ЕКГ специфічних ознак немає, можуть виявляти порушення внутрішньошлуночкової провідності, блокаду лівої ніжки пучка Гіса, зниження вольтажу зубця R, можливі порушення ритму серця, в тому числі екстрасистолічну аритмію, пароксизмальну тахікардію, фібриляцію передсердь. За даними добового моніторування ЕКГ, часто виявляють тяжкі шлуночкові порушення ритму, які не відзначають при звичайній реєстрації ЕКГ.
Гіпертрофічна кардіоміопатія
ГКМП — первинне ураження міокарда, зумовлене генетичною неповноцінністю скорочувальних білків, характеризується гіпертрофією лівого шлуночка за відсутності серцевої або системної причини.
Епідеміологія
Значно поширена серед багатьох расових груп у країнах Європи, США, Канаді, Ізраїлі, Південній Америці і Далекому Сході. У загальній популяції її поширеність становить 0,2% і відзначається переважно у чоловіків.
Етіологія
На сьогодні, за даними численних досліджень, підтверджена роль генетичних порушень у розвитку ГКМП. Причиною хвороби є різні мутації генів, які кодують протеїни саркомерів, що дозволяє визначити захворювання як порушення контрактильного апарату міоциту. У більшості пацієнтів захворювання успадковується за аутосомно-домінантним типом, приблизно у 50–60% хворих виявляють мутації в одному з восьми генів, які кодують різні компоненти серцевого саркомера та асоційованих білків: важкого ланцюга β-міозину (14-та хромосома), серцевого тропоніну-Т (1-ша хромосома), серцевого тропоніну-І (19-та хромосома), α-тропоміозину (15-та хромосома), серцевого протеїну С, що зв’язує міозин (11-та хромосома), легких ланцюгів есенціального та регуляторного міозину (3-тя і 12-та хромосоми відповідно), а також серцевого актину (15-та хромосома). За останній час також виявлені мутації трьох інших генів протеїнів саркомера: титіну, тропоніну С і важкого ланцюга α-міозину.
При виникненні захворювання може також мати значення порушення взаємодії серця плода, що розвивається, з катехоламінами, тиреоїдними гормонами, соматотропіном, аденозином. Розповсюдженість і локалізація гіпертрофії значно варіюють, навіть серед родичів. Однакова мутація може призвести до тяжкої гіпертрофії шлуночка у одного члена родини і помірної гіпертрофії — у іншого. Причини цих фенотипових розбіжностей в осіб з ідентичною генетичною мутацією не зовсім зрозумілі, але можуть бути наслідком інших генетичних чинників, які відіграють роль в експресії гіпертрофії серця, таких як DD-генотип АПФ, НУП та інші детермінанти росту міоцитів.
Патогенез
До основних патогенетичних чинників ГКМП належать зниження еластичності і скорочувальної здатності гіпертрофованого міокарда лівого шлуночка з погіршенням його діастолічного наповнення, в результаті чого в перерахунку на одиницю маси міокарда робота серця істотно зменшується, коронарний кровотік не відповідає ступеню гіпертрофії міокарда. Порушується швидкість проведення збудження в шлуночках з асинхронним скороченням різних відділів міокарда, що знижує пропульсивну здатність лівого шлуночка. У результаті діастолічної дисфункції виникає хронічне підвищення кінцево-діастолічного тиску лівого шлуночка, тиску заклинювання капілярів легеневої артерії, застій у легенях, гіпертрофія передсердь, що прогресує. Систолічна функція не порушена або навіть посилена внаслідок гіпердинамічності лівого шлуночка (фракція викиду досягає 80–90%), при цьому кінцевий діастолічний об’єм зменшений (нерідко <100 мл, іноді навіть <70 мл).
Патологічна анатомія
Захворювання характеризується гіпертрофією міокарда, найчастіше в ділянці міжшлуночкової перегородки, дезорганізацією кардіоміоцитів і міофібрил, фіброзом міокарда та ураженням дрібних судин. Макроскопічно розрізняють три варіанти ГКМП: асиметрична (60–95%) — ізольована гіпертрофія міжшлуночкової перегородки (ізольований гіпертрофічний субаортальний стеноз); гіпертрофія різних відділів лівого шлуночка, частіше апікальної частини; симетрична — тотальна концентрична гіпертрофія (5%). У 25% хворих визначають обструкцію виносного тракту лівого шлуночка у стані спокою. Рівномірна концентрична гіпертрофія міокарда супроводжується значним збільшенням маси серця, але обструкція шлуночків не відзначається. Правий шлуночок залучається до патологічного процесу приблизно в 50% випадків, що значно погіршує перебіг захворювання. Залежно від вираженості потовщення міокарда виділяють три ступені гіпертрофії: помірна (15–20 мм), середнього ступеня (21–25 мм), виражена гіпертрофія (>25 мм). Характерною анатомічною ознакою ГКМП є структурна зміна мітрального клапана, передняя стулка якого розміщена під кутом до площини клапана, потовщена і «випадає» в просвіт виносного тракту лівого шлуночка, що утворює додаткову перешкоду кровотоку. Порожнина лівого шлуночка невеликих розмірів, ліве передсердя часто гіпертрофоване і дилатоване.
Типові патогістологічні зміни включають гіпертрофію кардіоміоцитів і порушення взаємної орієнтації м’язових волокон (як найбільш частий результат мутацій саркомерів), а також вогнища фіброзу і рубцеві зміни внаслідок некрозу міокарда. Волокна розташовуються короткими рядами, мають схильність до закручування за відсутності змін інтрамуральних судин. Ядра клітин змінені, мають потворну форму, часто оточені світлою зоною («перинуклеарним німбом»), у якій відзначається накопичення глікогену.
Класифікація
Залежно від ступеня вираженості перешкоди відтоку крові виділяють дві основні форми ГКМП: обструктивна (рубрика І42.1 за МКХ-10), характеризується наявністю градієнта тиску між порожниною лівого шлуночка й аортою, і необструктивна (рубрика І42.2 за МКХ-10) — без градієнта тиску.
Клінічна картина
Ступінь обструкції і гіпертрофії лівого шлуночка не корелює з наявністю клінічних симптомів. Захворювання може бути повністю безсимптомним або маніфестувати в будь-якому віці, найчастіше симптоми проявляються у пацієнтів віком 40–50 років. Класична тріада симптомів включає біль у серці, задишку при навантаженні та синкопальні стани. Біль у ділянці грудної клітки відзначають 75% хворих, класичну стенокардію напруження — у 25%. У багатьох хворих виявляють післяобідню стенокардію. Непритомність найчастіше виникає у хворих молодого віку. У деяких випадках раптова смерть може бути першим проявом захворювання, наступає з частотою близько 1% на рік. При необструктивній формі фізикальне обстеження відхилень від норми може не виявити, проте іноді визначають збільшення тривалості верхівкового поштовху, аускультативно IV тон серця. При обструктивній формі при фізикальному обстеженні виявляється пульсація сонних артерій, швидкий «уривчастий» пульс при пальпації сонних артерій; посилений тривалий верхівковий поштовх, що займає всю систолу аж до II тону. Аускультативно тони серця глухі, визначається IV тон, а також систолічний шум (crescendo diminuendo), що не проводиться або слабко проводиться на сонні артерії і ділянку спини, посилюється при зменшенні наповнення серця і зниженні загального периферичного судинного опору (ЗПСО) (підйом із положення сидячи навпочіпки, натужування, прийом нітрогліцерину) і слабшає при збільшенні наповнення серця, підвищенні ЗПСО (у положенні лежачи, сидячи навпочіпки, при стисканні кулаків). ГКМП має повільно прогресуючий перебіг, тяжкість залежить від локалізації та ступеня гіпертрофії міокарда в зоні потовщення, а також від обструкції виносного тракту лівого шлуночка. Погіршують перебіг захворювання серйозні ускладнення. Раптову смерть реєструють у 2–4% дорослих і 4–6% дітей. Іншими ускладненнями є передсердні аритмії, тромбоемболії, ІЕ (5–9%) і застійна серцева недостатність (10–15%).
Діагностика
Зміни на ЕКГ виявляють у 95% хворих. Найбільш частими з них є збільшення лівого передсердя, порушення реполяризації лівого шлуночка у вигляді депресії сегмента ST та інверсії зубця Т і патологічні зубці Q (25–30%), ознаки гіпертрофії лівого шлуночка. При апікальній гіпертрофії можливі глибокі «гігантські» (до 4 см) симетричні негативні зубці Т. Добове моніторування ЕКГ дозволяє виявити порушення ритму серця: шлуночкові екстрасистоли (88%), пароксизми шлуночкової тахікардії (25–30%), суправентрикулярні тахіаритмії (30–40%), а також порушення провідності. Рентгенологічну картину багато в чому визначає ступінь вираженості захворювання. Можливі наступні рентгенологічні зміни: в першій косій проекції з’являється вибухання лівого шлуночка, зумовлене гіпертрофією шляхів відтоку, відсутність талії серця та заокруглення дуг, розширення лівого передсердя.
ЕхоКГ є методом вибору і дозволяє виявити гіпертрофію лівого шлуночка — потовщення стінки >1,5 мм в період діастоли без збільшення його порожнини, збільшення лівого передсердя, порушення діастолічної функції при допплерівській ехоКГ. За даними ехоКГ-обстеження в М-режимі найчастіше можна виявити асиметричний характер гіпертрофії перегородки, систолічний передній рух мітрального клапана, невеликий розмір порожнини лівого шлуночка, зменшення рухливості перегородки і передчасне закриття аортального клапана. При двомірному зображенні визначаються різні варіанти локалізації гіпертрофії міокарда. Систолічна функція зазвичай не порушена, фракція викиду збільшена (часто >80%). Приблизно у 25% хворих відзначають градієнт тиску між порожниною і виносним трактом лівого шлуночка у стані спокою. Хоча клінічне значення градієнта виносного тракту у хворих з ГКМП інтенсивно обговорюється багато років, нині градієнт розглядається як показник справжньої обструкції викиду лівого шлуночка. Прийнято угоду, за якою обструкція виносного тракту лівого шлуночка визначається за наявності градієнта не менше 30 мм рт. ст. (Maron B.J. et al., 2003). Обструкція клінічно важлива (від середнього ступеня до тяжкого) тільки у випадках, якщо градієнт виносного тракту становить >50 мм рт. ст. Залежно від величини градієнта тиску відповідно до класифікації NYHA виділяють наступні стадії захворювання:
I стадія — градієнт тиску до 25 мм рт. ст., як правило, скарг хворі не пред’являють;
II стадія — градієнт тиску до 36 мм рт. ст., самопочуття погіршується при фізичному навантаженні;
III стадія — градієнт тиску до 44 мм рт.ст., виражені клінічні симптоми — стенокардія, задишка та порушення гемодинаміки;
IV стадія — градієнт тиску 80 мм рт. ст. і вище, є значні порушення гемодинаміки.
При проксимальній формі ГКМП (субаортальному стенозі) найбільш характерними ехоКГ-ознаками є потовщення міжшлуночкової перегородки і зниження її екскурсії в базальному сегменті, збільшення співвідношення товщини міжшлуночкової перегородки і задньої стінки >1,3 (1,5–2), наявність градієнта тиску при допплерівській ехоКГ. Для дистальної форми (апікальної ГКМП) при ехоКГ-дослідженні найбільш характерне потовщення та зменшення амплітуди руху міжшлуночкової перегородки у верхівковому сегменті по довгій осі у двовимірному зображенні, порожнина лівого шлуночка у лівій апікальній чотирикамерній позиції пікоподібної форми за рахунок гіпертрофії дистальних відділів міжшлуночкової перегородки і відділів задньої стінки лівого шлуночка, що прилягають до неї. Концентрична (симетрична) форма характеризується потовщенням міжшлуночкової перегородки і задньої стінки лівого шлуночка у діастолу при значному збільшенні загальної маси міокарда, зменшенням систолічного і діастолічного об’ ємів лівого шлуночка, підвищенням індексу співвідношення розмірів лівого передсердя й устя аорти. Ізольована гіпертрофія правого шлуночка при ехоКГ-дослідженні має наступні ознаки: збільшення діастолічної товщини і зменшення амплітуди руху міжшлуночкової перегородки у верхівковому сегменті, потовщення передньої стінки і зменшення діастолічного розміру правого шлуночка.
При біопсії міокарда виявляють хаотичне розташування та вкорочення волокон міокарда, дегенеративні зміни із зникненням міофібрил, деформацію ядер клітин, фіброзне заміщення міокарда. Катетеризацію порожнин серця зазвичай проводять при клінічно вираженій мітральній регургітації для оцінки можливості хірургічного лікування. Внутрішньошлуночкові градієнти тиску виявляють у лівому і рідше в правому шлуночку. Градієнт підвищується після екстрасистол, під час проби Вальсальви і після інгаляції амілнітриту. Кінцево-діастолічний тиск підвищений внаслідок зниженої піддатливості шлуночка. За даними вентрикулографії виявляють характерну деформованість камери, що залежить від форми ГКМП і також іноді підтверджують мітральну регургітацію. Коронарні артерії зазвичай широкі з адекватним кровотоком. За допомогою МРТ можна найбільш точно оцінити морфологічні зміни, поширеність і вираженість гіпертрофії міокарда, особливо при діагностиці верхівкової форми і гіпертрофії нижньої частини міжшлуночкової перегородки і правого шлуночка, визначити систолічну і діастолічну функцію лівого шлуночка. При підозрі на ГКМП діагноз встановлюють за даними генетичного дослідження (аналіз ДНК), що дозволяє виявити характерні мутації генів, відповідальних за синтез скорочувальних білків кардіоміоцитів.
Лікування
Має бути спрямоване на зменшення діастолічної дисфункції, гіпердинамічної функції лівого шлуночка й усунення порушень ритму серця. Загальні заходи включають обмеження фізичних навантажень, які збільшують гіпертрофію міокарда, підвищують внутрішньошлуночковий градієнт тиску і ризик раптової смерті. Блокатори β-адренорецепторів є препаратами першої лінії у хворих незалежно від наявності або вираженості градієнта внутрішньошлуночкового тиску, що мають симптоми задишки або непереносимості фізичних навантажень, зі зниженою скоротністю лівого шлуночка, обмеженим латентним градієнтом виносного тракту, зниженим споживанням кисню міокардом та ішемією. Блокатори β-адренорецепторів зменшують вираженість симптомів у 70% хворих, знижуючи ЧСС і у такий спосіб покращуючи пасивне наповнення шлуночків і зменшуючи потребу міокарда в кисні. Альтернативою може бути застосування верапамілу, який у дозі до 480 мг/добу у хворих як з необструктивною, так і обструктивною ГКМП зменшує вираженість симптоматики, особливо біль у ділянці серця, покращує розслаблення і наповнення шлуночків, зменшує ішемію міокарда і скоротність лівого шлуночка. При застосуванні верапамілу може виникати погіршення гемодинаміки, збільшення обструкції виносного тракту, підвищення тиску в легеневій артерії. З обережністю слід давати навантаження хворим з обструкцією виносного тракту лівого шлуночка у стані покою. За наявності порушень серцевого ритму доцільно призначати блокатори β-адренорецепторів й антиаритмічні засоби, проте слід зазначити, що застосування останніх не знижує ризик раптової смерті. У симптоматичних пацієнтів з обструкцією дизопірамід діє як антиаритмічний засіб (по відношенню як до суправентрикулярних, так і шлуночкових аритмій), і як засіб з негативною інотропною дією викликає зменшення вираженості симптомів. У дозах 300–600 мг/добу може зменшувати обструкцію виносного тракту й обсяг мітральної регургітації. Для зменшення вираженості побічних ефектів можна застосовувати в комбінації з блокаторами β-адренорецепторів у низьких дозах. Не слід застосовувати дизопірамід з соталолом/аміодароном внаслідок ризику проаритмогенної дії. Наявність фібриляції передсердь зазвичай добре переноситься, проте у хворих з тяжкою діастолічною дисфункцією втрата передсердного «внеску» внаслідок аритмії може мати незворотні гемодинамічні наслідки, що вимагає невідкладного відновлення синусового ритму шляхом електричної або медикаментозної кардіоверсії за допомогою аміодарону. Останній ефективний для попередження пароксизмів фібриляції передсердь. Контроль ритму за допомогою блокаторів β-адренорецепторів або верапамілу покращує клінічний статус пацієнтів. Застосування варфарину показано як при пароксизмальній, так і при постійній формі фібриляції передсердь. При лікуванні серцевої недостатності у хворих з ГКМП терапевтична стратегія повинна бути спрямована на стимуляцію регресії гіпертрофії лівого шлуночка і усунення симптомів серцевої недостатності шляхом зниження тиску наповнення лівого шлуночка без зменшення величини серцевого викиду. У цих випадках препаратами вибору можуть бути інгібітори АПФ і антагоністи рецепторів ангіотензину II у зв’язку з їх здатністю блокувати РААС і викликати зворотний розвиток гіпертрофії лівого шлуночка. Клінічні дослідження, проведені за останні роки, продемонстрували сприятливу дію інгібіторів АПФ на ряд важливих показників діастолічної функції, включаючи діастолічне наповнення, ізоволюмічне розслаблення і взаємозв’язок тиск — об’єм лівого шлуночка і можливість зворотного розвитку процесів ремоделювання міокарда. При цьому покращання діастолічної функції (діастолічної розтяжності і здатності до розслаблення міокарда, зниження кінцево-діастолічного тиску наповнення лівого шлуночка) більш виражене у хворих з попередньо тяжчим ступенем дисфункції. Лікувальні заходи при ГКМП і серцевій недостатності певною мірою мають парадоксальний характер. Діуретики слід застосовувати з обережністю, переважно за відсутності значної обструкції виносного тракту. Засоби з інотропним ефектом, спрямовані на стимуляцію систолічного викиду (серцеві глікозиди і пресорні аміни), можуть спричинити несприятливий гемодинамічний ефект — вони посилюють обструкцію виносного тракту і не знижують підвищений кінцево-діастолічний тиск, можуть викликати розвиток асистолії. При збереженій систолічній функції може виникнути негативний ефект у зв’язку з посиленням скоротності шляхом підвищення внутрішньоклітинної концентрації іонів кальцію.
Таким чином, при ГКМП «чистий» ефект від інотропних засобів, що мають позитивний ефект (як збільшення жорсткості міокарда, так і підвищення тиску наповнення лівого шлуночка), призводить до погіршення діастолічної функції. Протее дигоксин можна застосовувати у хворих з діастолічною дисфункцією і фібриляцією передсердь для зниження ЧСС і/ або для відновлення синусового ритму.
Показання до проведення немедикаментозної терапії:
- значна обструкція виносного тракту лівого шлуночка (максимальний градієнт ≥50 мм рт. ст.) і симптоми вираженої задишки;
- біль у ділянці серця і пресинкопальні або синкопальні стани;
- рефрактерність до максимальної медикаментозної терапії.
Останнім часом успішно апробований новий метод для зменшення обструкції виносного тракту у хворих, рефрактерних до медикаментозної терапії — перкутанна алкогольна абляція міжшлуночкової перегородки. Успішна алкогольна абляція супроводжується прогресивним зменшенням градієнта в період від 6 до 12 міс у 80% хворих, що супроводжується покращанням клінічного статусу, зменшенням вираженості симптомів і діастолічної дисфункції та збільшенням переносимості фізичних навантажень. Метою хірургічного втручання при обструкції виносного тракту є усунення систолічного переднього руху мітрального клапана і септально-мітрального контакту шляхом розширення виносного тракту лівого шлуночка. Найчастіше виконується септальна міотомія-міектомія, в результаті якої у 95% хворих визначають значне зменшення градієнта виносного тракту, мітральної регургітації, у 70% поліпшуються клінічні симптоми. Приблизно у 5% пацієнтів операція ускладнюється аортальною регургітацією, що зазвичай гемодинамічно незначима.
Попередження раптової смерті включає ідентифікацію маркерів ризику (включаючи синкопальні стани, раптову смерть родичів, наявність градієнта виносного тракту). Наявність множинних клінічних факторів ризику підвищує ризик раптової смерті; таким хворим необхідна імплантація кардіовертера-дефібрилятора.
Прогноз
Несприятливий; найчастіше хворі вмирають раптово, на фоні важкого фізичного навантаження, у тому числі при безсимптомному перебігу захворювання. Хронічна серцева недостатність розвивається не так часто. Встановлені чинники ризику раптової смерті при ГКМП: маніфестація захворювання в молодому віці (до 16 років), наявність в сімейному анамнезі епізодів раптової смерті, часті синкопальні стани, нетривалі епізоди шлуночкової тахікардії, виявлені при 24-годинному моніторуванні ЕКГ, патологічна зміна рівня артеріального тиску при фізичному навантаженні. Ступінь гіпертрофії лівого шлуночка або наявність обструкції виносного тракту лівого шлуночка прогностичного значення не мають.
Рестриктивна кардіоміопатія
Рестриктивна кардіоміопатія — інфільтративне або фіброзне ураження міокарда, що характеризується ригідними, непіддатливими стінками шлуночків, зменшенням наповнення і діастолічного об’єму одного або обох шлуночків з нормальною або майже незміненою систолічною функцією і товщиною стінок. В основі захворювання лежить розповсюджений інтерстиціальний фіброз.
Етіологія
Безліч вроджених або набутих захворювань можуть призводити до рестриктивної кардіоміопатії із залученням міокарда або ендокарда. Міокардіальні форми рестриктивної кардіоміопатії можуть бути інфільтративними та неінфільтративними. Хвороба може бути ідіопатичною або пов’язаною з іншими інфільтративними, системними захворюваннями (амілоїдоз, гемохроматоз, саркоїдоз та ін.). До менш розповсюджених причин належать міокардит, трансплантація серця. Однією з ідентифікованих причин рестриктивної кардіоміопатії є гіпереозинофілія. У хворих з рестриктивною кардіоміопатією описані міссенс-мутації в гені тропоніну I.
Патогенез
Провідна роль у розвитку рестриктивної кардіоміопатії належить порушенням імунітету з гіпереозинофільним синдромом, на фоні якого внаслідок інфільтрації дегранульованих форм еозинофільних гранулоцитів в ендоміокарді формуються грубі морфологічні порушення структури серця. У патогенезі має значення дефіцит Т-супресорів, що призводить до гіпереозинофілії, дегрануляції еозинофільних гранулоцитів, що супроводжується виділенням катіонних білків, які проявляють токсичну дію на клітинні мембрани і ферменти, що беруть участь у диханні мітохондрій, а також тромбогенна дія на уражений міокард. Пошкоджувальні фактори гранул еозинофільних гранулоцитів частково ідентифіковані, включають нейротоксини, еозинофільний білок, що викликає основну реакцію, яка, очевидно, ушкоджує клітини. Нейтрофільні гранулоцити також можуть чинити пошкоджувальну дію на міоцити. Нещодавно встановлено, що асоційовані з рестриктивною кардіоміопатією мутації в гені тропоніну I викликають надмірне накопичення іонів Ca2+ у міофіламентах і недостатнє інгібуванняCa2+АТФази, що призводить до погіршення діастолічної функції.
Патологічна анатомія
Розміри серця невеликі, маса його становить 170–500 г. Характерно стовщення, що прогресує (може досягати 1 см), пристіночного ендокарда, дуже щільного на дотик, що складається з фіброзної тканини. Фіброз локалізується переважно в ендокарді верхівок обох шлуночків, задній стінці лівого шлуночка і задній стулці мітрального клапана, фіброз також може захоплювати сосочкові м’язи і хорди, що призводить до порушення функції клапанного апарату та формування вади серця. У передсердях і шлуночках виявляють пристіночні тромби. Перикард не змінений.
У розвитку рестриктивної кардіоміопатії виділяють три морфологічні стадії:
- некротичну — триває близько 5 тиж, характеризується гіпереозинофілією, масивною дегенерацією та загибеллю еозинофільних гранулоцитів, вираженою інфільтрацією ендокарда дегранульованими еозинофільними гранулоцитами, наявністю міокардиту з артеріїтом;
- тромботичну — триває до 10 міс, характеризується пристіночним внутрішньопорожнинним тромбоутворенням і порушеннями мікроциркуляції в коронарному руслі, зворотним розвитком інфільтрації міокарда та стовщенням ендокарда;
- стадію фіброзу — триває роками, характеризується поряд з ураженням парієтального ендокарда ураженням сосочкових м’язів і клапанного апарату, виникненням мітральної та трикуспідальної недостатності.
Фіброз може бути значно виражений у внутрішніх шарах міокарда з наступним розвитком гіпертрофії і дилатації серця, може носити вогнищевий або дифузний характер. Характерна гістологічна картина: виявляють шари стовщеного ендокарда, під тромбами відзначаються шари колагенової тканини, під якою розміщується грануляційна тканина з більшою кількістю кровоносних судин і запальною інфільтрацією, переважно еозинофільною. В ендокарді й міокарді можуть бути вогнища некрозу, пізніше — склеротичне стовщення ендокарда. Волокна міокарда орієнтовані нормально, іноді гіпертрофовані. При інфільтративних захворюваннях відбувається накопичення амілоїду (амілоїдоз), гранульоми (саркоїдоз), заліза, переважно в саркоплазматичному ретикулумі (гемохроматоз) або глікогену в міокарді або інтерстиції.
Патологічна фізіологія
Незалежно від етіологічних факторів при рестриктивній кардіоміопатії внаслідок інфільтрації та фіброзу ендокарда або міокарда збільшується твердість (або знижується піддатливість) міокарда одного або обох шлуночків, порушується їх діастолічне розслаблення, що викликає різке підвищення тиску в шлуночку при невеликій зміні об’єму, що змінює кровонаповнення серця. Звичайний тиск вище в лівому порівняно з правим шлуночком, що може відображати знижену піддатливість першого порівняно з останнім. Товщина стінок шлуночків незначно або помірно збільшена. Заповнення ригідного лівого шлуночка відбувається в основному у фазу швидкого наповнення, у той час як у фазу повільного наповнення і у систолу передсердя об’єм істотно не змінюється. Стовщення ендокарда або інфільтрація міокарда із втратою міоцитів, компенсаторною гіпертрофією і фіброзом можуть зумовити порушення функції AВ-клапана, які призводять до мітральної або трикуспідальної регургітації. Залучення вузлів і провідних шляхів викликає дисфункцію синоатріального вузла та блокади серця різного ступеня. Основним гемодинамічним результатом наявності ригідних, непіддатливих камер серця є високий тиск наповнення правого і лівого шлуночка, часто виникає легенева гіпертензія. Порушення систолічної функції нехарактерне, однак може виникнути, якщо компенсаторна гіпертрофія стає неадекватною для підтримки скорочувальної функції інфільтрованих або фіброзних камер.
Класифікація
Нині у рамках ідіопатичної рестриктивної кардіоміопатії виділяють наступні захворювання:
- первинна ідіопатична рестриктивна кардіоміопатія;
- гіпереозинофільний синдром:
- ендоміокардіальний фіброз;
- ендокардит Леффлера;
- ендокардіальний фіброеластоз (у немовлят і дітей молодшого віку).
Обговорюється, що, можливо, ці захворювання мають подібний патогенез або являють собою різні стадії одного процесу.
Клінічна картина
Клінічні симптоми тісно пов’язані з величиною тиску в лівому передсерді, необхідному для компенсації недостатнього наповнення шлуночка. Це призводить до типового порушення толерантності до фізичного навантаження в ранніх стадіях, задишки в спокої та симптомів низького серцевого викиду (підвищена слабкість) при подальшому розвитку. Підвищення ЧСС зменшує час діастоли та адекватне наповнення шлуночків. Клінічні симптоми залежать від типу ураження серця: право-, лівошлуночкового або змішаного право- і лівошлуночкового. Кардіалгічний синдром і синкопальні стани нехарактерні. До найбільш характерних клінічних симптомів рестриктивної кардіоміопатії належать:
1. При ураженні правого шлуночка:
- збільшення серця з перевагою гіпертрофії та дилатації правих відділів, тяжка правошлуночкова недостатність;
- тричленний ритм (ритм галопу), систолічний шум недостатності тристулкового клапана;
- підвищення венозного тиску, набухання яремних вен;
- збільшення печінки;
- виражені периферичні набряки, асцит, анасарка.
2. При ураженні лівого шлуночка:
- систолічний шум;
- ознаки лівошлуночкової недостатності;
- мітральна недостатність;
- збільшення розмірів лівого передсердя;
- застій у легенях;
- гіпертензія малого кола кровообігу.
3. При ураженні обох шлуночків:
- тотальна серцева недостатність.
4. Випіт у перикард, плевру.
5. Тяжкі порушення ритму (фібриляція передсердь, ектопічні аритмії).
6. Тромбоемболічний синдром.
Діагностика
На ЕКГ при ураженні правого шлуночка виявляють:
- синусову тахікардію, аритмію, найчастіше — фібриляцію передсердь;
- блокаду правої ніжки пучка Гіса;
- зниження вольтажу комплексу QRS;
- патологічний зубець Q у відведеннях V1–2;
- зниження сегмента ST, інверсію зубця Т;
- високий зубець Р.
На ЕКГ при ураженні лівого шлуночка виявляють:
- синусову тахікардію;
- ознаки гіпертрофії лівого передсердя і лівого шлуночка;
- блокаду лівої ніжки пучка Гіса;
- надшлуночкові аритмії.
Фібриляція передсердь часто виникає внаслідок дилатації передсердя. Шлуночкові аритмії або поперечна блокада часто розвиваються у тяжких випадках і є причиною смерті.
При рентгенологічному дослідженні хворих з ураженням правого шлуночка виявляють значне збільшення розмірів правого передсердя, малу амплітуду зубців рентгенокімограми правого шлуночка на фоні відсутності ознак застою в легенях. При ураженні лівого шлуночка збільшене ліве передсердя, поблизу верхівки серця і в ділянці шляху відтоку визначається лінійна тінь кальцифікації, в легенях — характерна картина змішаного застою.
ЕхоКГ-обстеження є основним у діагностиці захворювання, можуть виявляти наступні ознаки:
- систолічна функція в початковій стадії не порушена;
- дилатація передсердь;
- парадоксальний рух міжшлуночкової перегородки;
- гіпертрофія міокарда частіше відсутня;
- стовщення ендокарда;
- зменшення порожнини ураженого шлуночка (або обох шлуночків);
- пансистолічне пролабування задньої стулки мітрального клапана;
- функціональна мітральна регургітація внаслідок інфільтрації міокарда і сосочкових м’язів або стовщення ендокарда;
- збільшення швидкості потоку трикуспідальної регургітації;
- скорочення часу ізоволюмічного розслаблення;
- підвищення піку раннього наповнення;
- зниження піку пізнього передсердного наповнення шлуночків;
- розширення шляху відтоку з правого шлуночка і збільшення правого передсердя при ураженні правого шлуночка;
- зміна кровотоку в легеневій вені.
Можуть візуалізуватися внутрішньопорожнинні тромби. У кінцевій стадії захворювання можливе також порушення систолічної функції серця. За допомогою допплерографії можливе проведення деталізованого ехоКГ-аналізу діастолічної функції. Імпульсно-хвильовий режим допплерівського дослідження трансмітрального потоку показує рестриктивний тип діастолічної дисфункції, збільшення співвідношення максимальної швидкості потоку раннього наповнення (Е) до пізнього (А) >2, скорочення часу уповільнення раннього діастолічного потоку на мітральному клапані <150 мс (у нормі 150–220 мс), а також зменшення часу ізоволюметричного розслаблення <60 мс (в нормі 60–100 мс). При диференційному діагнозі з констриктивним перикардитом центральну роль відіграє тканинна допплерографія. Значне зниження швидкості руху медіального мітрального кільця в ранню (Еа) і пізню (Аа) діастолу є ознакою тяжкої рестрикції. При величині Еа ≥8,0 см/с можна з високою точністю (чутливість 95% і специфічність 96%) встановити діагноз констриктивного перикардиту. За допомогою методів МРТ і КТ можна виявити порушення наповнення в ранні та пізні стадії рестриктивної кардіоміопатії, що збігається з даними ехоКГ. Для верифікації діагнозу золотим стандартом є підтвердження рестриктивного наповнення за допомогою комбінованої катетеризації лівого і правого шлуночка. Крива тиску в шлуночках характерно змінена: на початку діастоли тиск у шлуночку знижується, а потім швидко підвищується і досягає плато, залишаючись незмінним у середині і кінці діастоли, має вигляд «діастолічного западіння і плато» (вид квадратного кореня), кінцево-діастолічний тиск у лівому шлуночку зазвичай на кілька міліметрів ртутного стовпчика вищий, ніж у правому, визначається легенева гіпертензія. Рестриктивна гемодинаміка є абсолютним критерієм діагнозу рестриктивної кардіоміопатії.
Верифікуючим методом діагностики є ЕМБ. На ранній стадії захворювання виявляють характерні еозинофільні інфільтрації, далі — інтерстиціальний фіброз і стовщення ендокарда, фіброз міокарда. При диференційній діагностиці рестриктивної кардіоміопатії з констриктивним перикардитом певне діагностичне значення має визначення рівня мозкового НУП, що у середньому підвищується до 756 пг/мл при рестриктивній кардіоміопатії проти 143 пг/мл при констриктивному перикардиті (у нормі <100 пг/мл). Діагностика рестриктивної кардіоміопатії утруднюється через розмаїтість клінічних варіантів застійного симптомокомплексу і відсутності кардіомегалії. Слід зазначити, що через відсутність патогномонічних ознак, у тому числі морфологічних, діагноз «ідіопатична рестриктивна кардіоміопатія» встановлюють тільки після виключення інфільтративних і системних захворювань міокарда.
Лікування
Терапія для більшості пацієнтів не розроблена, в основному симптоматична. Для деяких форм рестриктивної кардіоміопатії (амілоїдоз, гемохроматоз) існує специфічна терапія. На ранніх стадіях захворювання призначають ГК. На більш пізніх стадіях лікування в основному зводиться до усунення симптомів застійної серцевої недостатності, що є провідним проявом рестриктивної кардіоміопатії. Діуретики зменшують вираженість симптомів застою, однак їх призначають з обережністю через здатність знижувати переднавантаження, за допомогою якого ригідні шлуночки підтримують серцевий викид. Звичайне лікування починають із тіазидних діуретиків 1–2 рази на тиждень, потім застосовують петлеві діуретики. Інгібітори АПФ можуть поліпшувати функцію лівого шлуночка. Вазодилататори, що знижують постнавантаження, можуть викликати артеріальну гіпотензію і їх зазвичай не застосовують. Симптоми ішемії є провісниками раптової смерті, необхідне застосування блокаторів β-адренорецепторів. Серцеві глікозиди допомагають за наявності фібриляції передсердь знизити ЧСС і подовжити діастолічне наповнення. Препарати наперстянки допомагають трохи зменшити порушення гемодинаміки, однак за відсутності дилатації і збереженої систолічної функції лівого шлуночка при синусовому ритмі застосування серцевих глікозидів невиправдане. β1-Адреноміметики й інгібітори фосфодіестерази підвищують інотропну функцію серця і поліпшують його діастолічну функцію, однак препарати не рекомендується застосовувати довгостроково (підвищують смертність), їх призначають лише в тяжких випадках для короткострокового лікування. У деяких ситуаціях при значно вираженій компенсаторній гіпертрофії міокарда можуть бути застосовані блокатори кальцієвих каналів. Для лікування тромбоемболічних ускладнень використовують непрямі антикоагулянти. Єдиним радикальним методом лікування рестриктивної кардіоміопатії є трансплантація серця. Іноді ефективна резекція ендокарда з протезуванням або пластикою мітрального і тристулкового клапанів, однак це пов’язано з високою летальністю (15–25%).
Прогноз
Прогноз несприятливий, захворювання з прогредієнтним перебігом, смертність становить 70% протягом перших 5 років. Збільшення товщини стінки лівого шлуночка і зменшення амплітуди комплексу QRS є несприятливими прогностичними ознаками.
Первинна ідіопатична рестриктивна кардіоміопатія
Рідкісна форма неінфільтративного захворювання міокарда з діастолічною дисфункцією за рестриктивним типом. Характерна відсутність гіпертрофії лівого шлуночка і систолічної дисфункції. Часто успадковується й асоціюється зі скелетною міопатією. Зазвичай хвороба виникає спорадично, але може успадковуватися за аутосомно-домінантним типом. Виникнення ригідного шлуночка може бути наслідком патології міоцитів, включаючи порушення кальцієвого обміну, акумуляцію десміну (компонента цитоскелета), порушення структури міофібрил, а також патологію екстрацелюлярного матриксу, включаючи проліферацію колагенових волокон та еластичних елементів. Серед клінічних проявів найбільш часті симптоми застійної серцевої недостатності, підвищення венозного тиску, ознаки мітральної і трикуспідальної регургітації.
Для встановлення діагнозу проводять катетеризацію порожнин серця та/або ехоКГ-обстеження й ЕМБ. При ехоКГ визначають збільшення передсердь і відсутність дилатації шлуночків з майже нормальною систолічною функцією та рестриктивний тип діастолічної дисфункції при допплерівській ехоКГ. Для встановлення діагнозу важлива наявність зниження тиску наповнення при катетеризації серця. ЕМБ проводять для оцінки гіпертрофії міоцитів та інтерстиціального фіброзу. У хворих з прогресуючою серцевою недостатністю, що погано піддається медикаментозному лікуванню, і повною блокадою серця необхідне встановлення кардіостимулятора. Предикторами поганого прогнозу є старший вік, чоловіча стать, збільшення лівого передсердя, високий функціональний клас хвороби. У пацієнтів з тяжким перебігом і фракцією викиду у межах 40–55% необхідна трансплантація серця, при цьому на прогноз впливає наявність супутньої скелетної міопатії.
Гіпереозинофільний синдром
Має дві форми: ендоміокардіальний фіброз та ендокардит Леффлера. Загальним є патологічна еозинофілія в крові й ендокарді, що викликає рестриктивну кардіоміопатію з фіброзним потовщенням ендокарда в ділянці верхівки і підклапанних структур, викликаючи порушення наповнення шлуночка. Причина гіпереозинофілії не встановлена, може бути пов’язана з паразитарними і протозойними інфекціями, малігнізацією (лейкемія), алергічними або аутоімунними реакціями. Раніше вважалося, що це різні форми одного захворювання, які виникають внаслідок ушкодження тканин через токсичний ефект еозинофілів і мають подібні патологічні зміни в пізніх стадіях. Проте існують важливі розходження.
Ендоміокардіальний фіброз
Це ендемічна для екваторіальних країн хвороба (Уганда, Нігерія, Бразилія), яка рідко розвивається в нетропічних країнах. Захворювання виникає, зазвичай, у молодому віці однаково часто у чоловіків і жінок і призводить до смерті протягом 1–4 років з моменту появи перших симптомів. Ендоміокардіальний фіброз характеризується значущим потовщенням і рубцюванням ендокарда, що призводить до облітерації порожнини шлуночка. Пристінковий тромбоз і системні емболи можуть викликати ще більше зменшення об’єму порожнини шлуночків. Часто до процесу залучається й субендокардіальний міокард. Ендоміокардіальний фіброз уражує обидва шлуночки у 50% випадків, лівий — у 40%, правий — в 10%, його відзначають в трьох різних ділянках: верхівці лівого або правого шлуночка і субклапанному просторі. Зазвичай правий шлуночок буває більш облітерованим, ніж лівий. Мікроскопічно в ендокарді виявляють товстий колагеновий шар поверх сполучної тканини з грануляціями, що досягають міокарда. Внаслідок фіброзування мітрального і тристулкового клапанів виникає клапанна регургітація. Клініка ендоміокардіального фіброзу залежить від того, який шлуночок залучений у патологічний процес. При лівобічному ураженні переважає застій у легенях, при правобічному — симптоми правошлуночкової серцевої недостатності. Часто виникають мітральна та трикуспідальна регургітація, в 25% — фібриляція передсердь, особливо при ураженні правого шлуночка. У хворих часто виявляють емболії, випіт у плевральну та перикардіальну порожнину. При ехоКГ-обстеженні визначаються облітерація верхівок шлуночків і гіперкінез базальних відділів, дилатація передсердь, при лівобічному ураженні — зниження рухливості задньої стулки мітрального клапана; при допплерівській ехоКГ — ознаки мітральної і трикуспідальної регургітації, рестриктивний тип наповнення шлуночків. Катетеризація серця підтверджує рестриктивний тип гемодинаміки. При ЕМБ виявляють еозинофільні інфільтрати, що підтверджує діагноз. Загальний прогноз несприятливий, показане паліативне лікування — хірургічна резекція фіброзного ендокарда та заміна клапана, що може поліпшити симптоми, хоча інтраопераційна смертність становить 15–25%. У хворих з тяжким перебігом 2-річна смертність становить 35–50%.
Ендокардит Леффлера
Хвороба жарких країн, спорадично виникає в усьому світі. Частіше хворіють чоловіки віком до 50 років. Захворювання починається як гострий артеріїт з еозинофілією, формуванням тромбів на ендокарді, хордах і прогресуванням фіброзу AВ-клапана. Причина еозинофілії не встановлена, може бути викликана лейкемією або виникати вторинно внаслідок паразитарної інвазії. Еозинофільні гранулоцити накопичуються в міокарді, викликаючи його ушкодження. Пристіночний тромбоз і системні емболи можуть викликати ще більше зменшення об’єму порожнини шлуночків. Клінічні прояви включають зменшення маси тіла, лихоманку, кашель, шкірні висипання, ціаноз, тяжку право- і лівошлуночкову серцеву недостатність, системну тромбоемболію, підвищення тиску в правому передсерді. У крові виявляють гіпереозинофілію. На ЕКГ — неспецифічні зміни, інверсія зубця Т. На рентгенограмі визначається збільшення розмірів серця. При ехоКГ часто визначаються муральний тромбоз, облітерація верхівки і нерухомість задньої стулки мітрального клапана з ознаками мітральної регургітації. Систолічна функція лівого шлуночка часто збережена. У хворих у фіброзній фазі при допплерографії реєструється рестриктивний тип наповнення шлуночків. Трансезофагеальну ехоКГ зазвичай проводять для оцінки діастолічної функції. Катетеризація серця виявляє зниження піддатливості лівого шлуночка внаслідок щільного рубця, мітральну і трикуспідальну регургітацію. ЕМБ необхідна для підтвердження діагнозу, хоча важко отримати адекватний зразок тканини.
Лікування залежить від стадії хвороби. На ранній стадії застосовують ГК для лікування міокардиту, асоційованого з гіпереозинофілією. Інтерферон використовували у обмеженої кількості хворих з багатообіцяючими результатами. Стандартна підтримувальна терапія серцевої недостатності включає дигоксин, діуретики, зниження постнавантажень, антикоагулянти. Хірургічне втручання проводять у стадії фіброзу, воно включає видалення фіброзних бляшок з поверхні ендокарда, заміну клапана, імплантацію кардіостимулятора.
Глава 35. Вроджені вади серця
Вроджені вади серця являють собою структурні аномалії та деформації клапанів, отворів або перегородок між камерами серця або судин, які відходять від нього, порушують внутрішньосерцеву та системну гемодинаміку, що призводить до розвитку гострої або хронічної недостатності кровообігу.
Епідеміологія
Частота вроджених вад серця становить до 1% всіх захворювань серця.
Етіологія
Виникнення вад ембріонального розвитку може бути зумовлене генними мутаціями, різними інфекційними процесами та інтоксикаціями (ендогенними і екзогенними) у період вагітності. Із вродженими вадами серця пов’язані певні хромосомні порушення, найчастіше — трисомія 21 (синдром Дауна), а також трисомії 13, 14, 15 і 18. У більше ніж 50% пацієнтів із синдромом Дауна відзначають вроджені вади серця (найбільш часто АВ- або шлуночкові септальні дефекти). Мутації в декількох специфічних генах ідентифіковані в деяких випадках вроджених вад серця. Мутації в TBX5 виявляють у більшості пацієнтів із синдромом Хольта — Орама, аутосомним захворюванням із септальними дефектами. Мутації в гені еластину ідентифіковані як причина надклапанного аортального стенозу, мутації в NKX2.5 пов’язані з аутосомним домінантним фенотипом дефекту передсердної перегородки при тетраді Фалло.
Патогенез
Вроджена вада серця є наслідком порушення нормального процесу розвитку первинної та вторинної міжпередсердних перегородок та ендокардіальних подушок у період їх ембріонального формування. При порушенні розвитку м’язової частини міжшлуночкової перегородки в ній утворюються поодинокі або множинні отвори, частіше в мембранозній її частині, при цьому можуть утворитися високі або низькі її дефекти, які нерідко захоплюють розташовані нижче частини м’язової перегородки. Порушення розвитку аортолегеневої перегородки може бути локальним, на невеликій ділянці, тоді формується вада за типом аортолегеневої нориці. Іноді поділу артеріального стовбура на аорту й легеневу артерію взагалі не відбувається та формується вада, яка називається загальним артеріальним стовбуром. У процесі формування цієї перегородки напрямок її росту може порушитися і йти не по спіралі, як звичайно, а прямо — у таких випадках формується вада, яка називається транспозицією аорти й легеневої артерії. У ряді випадків порушення розвитку перегородки артеріального конуса призводять до її відхилення в той чи інший бік, внаслідок чого виникає звуження аорти або легеневої артерії. До звуження останньої часто приєднується порушення розвитку складок конуса в місці, де вони беруть участь у формуванні мембранозної частини перегородки, — в ній утворюється дефект, розширена аорта зміщується вправо та розташовується прямо над дефектом — розвивається вада, яка отримала назву «тетрада Фалло». Виникнення ряду вроджених вад серця та магістральних судин пов’язане із порушеннями в постнатальний період. Порушення в процесі нормального закриття артеріальної протоки призводить до формування відкритої артеріальної протоки. При поєднанні незарощення овального отвору з недорозвиненням вторинної перегородки формується дефект міжпередсердної перегородки в ділянці овального вікна.
Класифікація
За етіологією вроджені вади поділяють на дві групи:
1. Вади, зумовлені порушенням формоутворення, що є патологією ембріонального розвитку серцево-судинної системи.
2. Вади, що розвинулися внаслідок хвороб ендокарда, перенесених у внутрішньоутробний період.
Із урахуванням морфології ураження серед вроджених вад серця виділяють:
- аномалії розташування серця;
- аномалії передсердної, міжшлуночкової перегородки;
- аномалії легеневої артерії, артеріальної протоки, аорти;
- переміщення великих судин;
- аномалії клапанного апарата серця.
Систематизація вроджених вад серця
Загальні:
- вроджена коригована транспозиція магістральних артерій;
- аномалії розташування серця;
- вроджена повна блокада серця.
Вади серця «білі» з шунтуванням крові зліва направо
1. Шунт крові на рівні передсердь:
1) дефект міжпередсердної перегородки;
2) дефект міжпередсердної перегородки в поєднанні з мітральним стенозом (синдром Лютамбаше);
3) часткове аномальне приєднання легеневих вен.
2. Шунт крові на рівні шлуночків:
1) дефект міжшлуночкової перегородки;
2) дефект міжшлуночкової перегородки в поєднанні з недостатністю клапана аорти;
3) дефект міжшлуночкової перегородки зі скиданням крові з лівого шлуночка у праве передсердя.
3. Шунт крові з устя аорти в праву частину серця:
1) розрив аневризми синуса Вальсальви;
2) коронарна артеріовенозна фістула;
3) аномальне відгалуження виходу лівої коронарної артерії зі стовбура легеневої артерії.
4. Шунт крові між аортою та легеневою артерією:
1) аортопульмональне вікно;
2) відкрита артеріальна протока.
5. Багаторівневі шунти крові:
1) повний загальний передсердно-шлуночковий канал;
2) поєднання дефекту міжшлуночкової перегородки з дефектом міжпередсердної перегородки;
3) поєднання дефекту міжшлуночкової перегородки з відкритою артеріальною протокою.
Вади серця «білі» без шунтування крові
1. Вади лівих відділів серця:
1) вроджена обструкція притоку крові в ліве передсердя;
2) мітральна недостатність;
3) первинний фіброеластоз ендокарда, що дилатує;
4) стеноз устя аорти;
5) недостатність аортального клапана;
6) коарктація аорти.
2. Вади правих відділів серця:
1) аномалія тристулкового клапана (Ебштейна) без ціанозу;
2) стеноз легеневої артерії;
3) уроджена недостатність клапана легеневої артерії;
4) ідіопатичне розширення стовбура легеневої артерії.
Вади серця «сині»
Із посиленим легеневим кровотоком:
1. Повна транспозиція магістральних артерій.
2. Синдром Тауссіг — Бінга.
3. Артеріальний стовбур.
4. Повна аномалія приєднання легеневих вен.
5. Єдиний шлуночок без стенозу легеневого стовбура.
6. Загальне передсердя.
7. Тетрада Фалло з атрезією легеневого стовбура з посиленням колатерального артеріального кровотоку.
8. Атрезія тристулкового клапана з великим дефектом міжшлуночкової перегородки без стенозу легеневого стовбура.
9. Гіпоплазія лівих відділів серця (атрезія аорти й мітрального клапана).
З нормальним або зменшеним легеневим кровотоком:
1. Атрезія тристулкового клапана.
2. Аномалія Ебштейна з шунтуванням крові з правого передсердя в ліве.
3. Атрезія легеневого стовбура з інтактною міжшлуночковою перегородкою.
4. Стеноз або атрезія легеневого стовбура з дефектом міжшлуночкової перегородки (тетрада Фалло).
5. Стеноз легеневого стовбура із шунтуванням крові з правого передсердя в ліве.
6. Повна транспозиція магістральних судин у поєднанні зі стенозом легеневого стовбура.
7. Відходження обох судин, які виносяться з правого шлуночка у поєднанні зі стенозом легеневого стовбура.
8. Єдиний шлуночок зі стенозом легеневого стовбура.
9. Артеріовенозні нориці легень.
10. Сполучення між порожнистими венами та лівим передсердям.
Лікування
Відповідно до протоколу надання медичної допомоги пацієнтам із вродженими вадами серця лікувальна програма передбачає перелік та обсяг обов’язкових медичних послуг, в які включене оперативне лікування вад серця й лікування серцевої недостатності та її ускладнень (наказ МОЗ України № 436 від 03.07.2006 р.).
Аномалії положення серця
До аномалій внутрішьогрудного положення серця відносять порушення розташування верхівки серця, її відповідності характеру положення органів черевної порожнини та випадки неправильного формування серця, такі як зворотнє або невизначене розташування ембріональних закладок правого й лівого передсердь.
Епідеміологія
Частота цих аномалій становить 1,5% серед усіх вроджених вад серця, найбільш частими серед них є правосформоване праворозташоване серце — ізольована декстрокардія і лівосформоване праворозташоване серце— «дзеркальна декстрокардія» (відповідно у 54 і 33% хворих із правобічним положенням серця). У першому випадку розташування інших органів грудної клітки та топографія органів черевної порожнини відповідають фізіологічній нормі, у другому — відзначають повне зворотне розташування внутрішніх органів.
Клінічна картина й діагностика
Якщо будову серця і судин не порушено, воно в цілому функціонує нормально, порушення кровообігу не відбувається. При пальпації серцевий поштовх розташовується в п’ятому міжребер’ї праворуч по середньоключичній лінії. Аускультативно тони серця найкраще прослуховуються праворуч відповідно до загальноприйнятих аускультативних точок, симетрично зміщеним на праву половину грудної клітини. На ЕКГ при ізольованій декстрокардії характерний позитивний зубець Р у I, aVL, aVF, V1–4 відведеннях. У грудних відведеннях вольтаж комплексів QRS прогресивно підвищується від V6R до V1–2 і далі знижується до V6, вказуючи на правобічне положення серця. При цьому потенціали правого шлуночка у формі комплексів r або RS реєструють у відведеннях від V6R до V2–3, лівого — у вигляді q або qRS — у V3–6. На ЕКГ при істинній декстрокардії характерний негативний зубець Р у I, aVL, V5–6 відведеннях, позитивний Р у II, III, V1 відведеннях, негативні комплекси QRS у I, aVR, підвищення їх вольтажу від V1 до V6R і зниження — від V1 до V6, негативні Т у I, aVL, V5–6. У I стандартному відведенні реєструють зубці, що зазвичай мають протилежний напрямок: II стандартне відведення відповідає нормальному III і, навпаки, III стандартне відведення — II, відведення aVL відбиває відведення aVR й навпаки, aVR–aVL, відведення V1 рівноцінно V2R, V3–V3R, V4–V4R, V5–V5R, V6–V6R. Відведення aVF не змінюється. Якщо поміняти місцями електроди правої й лівої руки й накласти грудні електроди дзеркально до нормального серця, то ЕКГ можна аналізувати як при нормальному положенні серця. Рентгенологічне дослідження виявляє тінь серця переважно в правій половині грудної клітки, верхівка серця орієнтована вправо. Аномальне положення серця підтверджується при ехоКГ-обстеженні.
Лікування
Аномальне розташування серця саме по собі не супроводжується розладами гемодинаміки. Хірургічному лікуванню підлягають лише хворі із супутніми вадами серця.
Аномалії міжпередсердної перегородки
Дефект міжпередсердної перегородки
Дефект міжпередсердної перегородки — вроджена вада серця, при якій є сполучення між двома передсердями, що розвивається в результаті аномального розвитку первинної й вторинної міжпередсердних перегородок та ендокардіальних подушок. Дефект міжпередсердної перегородки в сполученні зі стенозом лівого АВ-отвору називається синдромом Лютамбаше.
Епідеміологія
Поширеність дефекту міжпередсердної перегородки становить 5–10% усіх вроджених вад, є найбільш частою вадою серця у дорослих (30%), у жінок відзначають частіше, ніж у чоловіків (2:1).
Патологічна анатомія
Існує кілька морфологічних типів дефекту міжпередсердної перегородки. Найбільш частий варіант — високий дефект міжпередсердної перегородки за типом ostium secundum (75% випадків), що виникає внаслідок порушення розвитку вторинної перегородки, локалізується в центральній частині міжпередсердної перегородки в ділянці овальної ямки. Дефект типу ostium primum (15%) являє собою варіант дефекту ендокарда, виникає внаслідок неповного розвитку первинної перегородки, розташовується в нижньому відділі перегородки безпосередньо над рівнем передсердношлуночкових отворів, поєднується з розщепленням стулок мітрального й рідше тристулкового клапанів. Поєднання порушення розвитку перегородок передсердя з неправильним розташуванням венозного синуса призводить до утворення складних дефектів. Дефекти венозного синусу (10%) частіше відзначають високо в міжпередсердній перегородці поблизу від впадання верхньої порожнистої вени, пов’язані з атиповим впаданням легеневих вен у праве передсердя або верхню порожнисту вену. Набагато рідше дефекти венозного синусу можуть локалізуватися в нижній частині перегородки над устям нижньої порожнистої вени. У деяких випадках відзначають загальне передсердя — відсутність більшої частини міжпередсердної перегородки або наявність тільки рудиментарних її елементів, часто поєднується з розщепленням АВ-клапанів. Відкритий овальний отвір, що не закривається у 20% дорослих, не слід розглядати як різновид дефекту міжпередсердної перегородки, оскільки при істинному дефекті є недостатність тканини, а при відкритому овальному вікні сполучення здійснюється за рахунок клапана, що відкривається при особливих обставинах. Синдром Лютамбаше морфологічно характеризується наявністю дефекту міжпередсердної перегородки (частіше вторинного) і звуженням лівого АВ-отвору (вродженого або набутого). Характерним є розширення легеневої артерії, що іноді вдвічі перевищує розмір аорти.
Порушення гемодинаміки
Наявність дефекту міжпередсердної перегородки призводить до скидання артеріальної крові з лівого передсердя в праве внаслідок наявності градієнта тиску між ними. У результаті виникають перевантаження об’ємом правої половини серця, дилатація правого шлуночка і збільшення об’єму циркулюючої крові у малому колі. При більших дефектах це може призводити до легеневої гіпертензії, однак виражена гіпертензія в малому колі в перші 20 років виникає у близько 2% хворих. Основну роль у компенсації порушення кровообігу грає правий шлуночок, робота якого збільшується в кілька разів. Недостатність правого шлуночка розвивається після 10 років існування вади, у більш старшому віці приєднується недостатність лівого. Остання, викликаючи зменшення його піддатливості, може призводити до збільшення обсягу шунта зліва направо. При синдромі Лютамбаше величина шунта збільшується пропорційно зростанню стенозувального дефекту мітрального отвору, крім цього, відбувається порушення відтоку крові з малого кола та виникнення легеневої гіпертензії.
Клінічна картина
Клінічні прояви вади залежать від ступеня порушення гемодинаміки та змінюються з віком. При відносно невеликому дефекті в молодому віці хворі можуть не мати скарг і ваду виявляють при випадковому обстеженні. Скарги на задишку та напади серцебиття при фізичному навантаженні виникають у віці старше 40 років, потім збільшується вираженість слабкості і стомлюваності, з’являються різні аритмії, серцева недостатність, що зумовлена вираженою легеневою гіпертензією. При більших дефектах міжпередсердної перегородки задишка є одним із симптомів захворювання вже у молодому віці. У хворих часто виникають напади серцебиття. При перкусії відзначають розширення границь серця переважно вправо, а при більших дефектах — вліво. В окремих випадках описано наявність серцевого горба (за рахунок збільшення правих відділів серця), а також систолічне тремтіння з лівого краю груднини. Характерна аускультативна картина: над легеневою артерією ліворуч біля груднини вислуховується систолічний шум помірної інтенсивності, що виникає внаслідок збільшеного кровотоку через клапан легеневої артерії. Другий тон над легеневою артерією посилений та роздвоєний. Рідше визначають діастолічний шум над нижньою частиною груднини, пов’язаний із відносним стенозом отвору тристулкового клапана при збільшеній кількості крові, що проходить через нього. Після того як збільшений легеневий судинний опір призводить до зниження величини скидання крові зліва направо, інтенсивність шумів знижується, з’являється діастолічний шум, викликаний недостатністю клапана легеневої артерії.
Діагностика
На ЕКГ електрична вісь серця у хворих із вторинним дефектом відхилена вправо, з первинним — вліво. При кожному з дефектів виявляють різний ступінь перевантаження і гіпертрофії правого шлуночка і правого передсердя, вираженість якої залежить від величини тиску в легеневому стовбурі. Виявляють ознаки часткової блокади правої ніжки пучка Гіса (феномен rSR або rsR у правих грудних відведеннях), можуть визначати передсердну екстрасистолічну аритмію, пароксизмальну суправентрикулярну тахікардію. У хворих із синдромом Лютамбаше нерідко виявляють фібриляцію передсердь. На рентгенограмі серце збільшене в поперечнику, в косих проекціях визначається збільшення правих відділів серця. Дуга легеневої артерії вибухає, дуга аорти зменшена, посилений судинний малюнок легень, корені легень розширені, характерна їх пульсація. Діагноз встановлюють методом трансторакальної та допплерівської ехоКГ, за допомогою якої можна встановити локалізацію, розмір дефекту (добре візуалізується оstium secundum та primum), а також напрямок скидання крові. При ехоКГ-обстеженні виявляють наступні ознаки:
- збільшення порожнини правого передсердя й правого шлуночка;
- парадоксальний рух міжшлуночкової перегородки;
- гіпердинамічний рух стінок лівого передсердя;
- ознаки легеневої гіпертензії;
- ознаки розщеплення мітрального та тристулкового клапанів з проявами їх недостатності при первинному дефекті;
- пролапс мітрального клапана при вторинному дефекті;
- стеноз мітрального отвору при синдромі Лютамбаше;
- шунтування крові між передсердями зліва направо або cправа наліво.
Дефекти венозного синуса вимагають додаткових діагностичних підходів — черезстравохідна ехоКГ забезпечує найкращу візуалізацію міжпередсердної перегородки, легеневих вен, особливо у дорослих пацієнтів з недостатніми «акустичними вікнами».
Катетеризації порожнин серця зазвичай не потрібно, крім випадків супутньої легеневої гіпертензії або коли неінвазивне дослідження неповноцінне. Наявність дефекту міжпередсердної перегородки підтверджується проведенням катетера із правого передсердя через перегородку в ліве й підвищенням насичення крові киснем у порожнині правого передсердя порівняно із пробами крові, взятої біля устя порожнистих вен. Різниця в 2 об.% і більше (або 8–10%) розглядається як абсолютна ознака шунтування крові. Коронароангіографію зазвичай проводять пацієнтам віком старше 40 років перед запланованою хірургічною корекцією вади. Дефект міжпередсердної перегородки також може бути діагностовано за допомогою КТ або МРТ, що є методом вибору для оцінки позасерцевої анатомії, у тому числі великих судин, гілок легеневої артерії, а також системних та легеневих венозних з’єднань.
Лікування
Хірургічне закриття дефекту рекомендоване, якщо співвідношення легеневого до системного кровотоку більше ніж 1,5:1 і відношення легеневого до системного судинного опору менше 0,7. Немає єдиної думки щодо хірургічного лікування асимптомних хворих віком 25–40 років, однак воно виправдано для попередження прогресування симптомів. Унаслідок можливості збільшення скидання крові зліва направо, появи фібриляції передсердь і розвитку легеневої гіпертензії з віком бажане виконання хірургічної корекції вади до появи ознак погіршення функції серця. У симптомних хворих віком старше 40 років хірургічне закриття дефекту поліпшує толерантність до фізичного навантаження й виживаність порівняно з медикаментозною терапією та запобігає подальшому погіршенню функціонального стану, хоча не знижує ризик розвитку суправентрикулярної аритмії, серцевої недостатності і цереброваскулярних подій. Хірургічне втручання у 80% хворих віком старше 60 років зі значним скиданням крові приводить до зменшення вираженості симптомів. У 70% хворих старшого віку після операції зберігаються порушення ритму, у 10–25% вони виникають уперше, підвищується ризик системної артеріальної гіпертензії невідомої етіології.
Показання до хірургічного лікування
- неефективна медикаментозна терапія серцевої недостатності;
- значне артеріовенозне скидання;
- відставання у фізичному розвитку;
- підвищення тиску в малому колі кровообігу.
Протипоказання до оперативного втручання
1. Веноартеріальне (справа наліво) скидання, оскільки це ознака вираженої гіпертензії й часто незворотніх змін у малому колі кровообігу.
2. Виражена лівошлуночкова недостатність.
Хірургічне лікування полягає в ушиванні або пластиці латкою дефекту міжпередсердної перегородки. Незначні дефекти зашивають, більші закривають гомотрансплантатами або протезами з пластмасової губки. При первинному дефекті зі значною мітральною недостатністю додатково ушивають розщеплену стулку або проводять протезування мітрального клапана. У результаті хірургічного лікування поліпшується стан хворого: зменшуються задишка, серцебиття, розміри серця. В останні роки зростає роль пристроїв для закриття дефекту (вперше застосовані в 1976 р.), вибір типу пристрою залежить від локалізації дефекту. На сьогодні немає порівняльних досліджень щодо впровадження пристроїв і хірургічних методів операції, не існує консенсусу відносно тривалого спостереження після установки пристроїв і довгострокові результати невідомі, включаючи ризик розвитку передсердної аритмії, серцевої недостатності та інсульту; передбачається, що їх результати будуть порівнянними. При синдромі Лютамбаше хірургічне лікування полягає в одномоментній корекції — усуненні дефекту міжпередсердної перегородки та мітральної комісуротомії. Протипоказаннями до операції є лише тяжка стадія легеневої гіпертензії та різко виражена дистрофія міокарда, що зумовлює термінальну стадію серцевої недостатності. Медикаментозна терапія включає симптоматичні засоби: антиаритмічні препарати при фібриляції передсердь і пароксизмальній суправентрикулярній тахікардії, лікування серцевої недостатності.
Прогноз
Вторинний дефект має сприятливий природний перебіг у перші 20–30 років життя. Причиною смерті за відсутності оперативної корекції є правошлуночкова серцева недостатність, рідше — тромбоз легеневої артерії, аритмія. При первинному дефекті перебіг менш сприятливий, прогноз гірший, раніше виникають клінічна симптоматика та ускладнення, основним обтяжувальним чинником є легенева гіпертензія.
Аномалії міжшлуночкової перегородки
Дефект міжшлуночкової перегородки
Дефект міжшлуночкової перегородки — вроджена вада серця, при якій є патологічне сполучення між правим та лівим шлуночком серця.
Епідеміологія
Виявляється в 25–30% випадків усіх вроджених вад серця, однаково часто в чоловіків і жінок.
Патологічна анатомія
Дефекти можуть розташовуватися вище або нижче надшлуночкового гребеня, в мембранозній або м’язовій частині міжшлуночкової перегородки, найбільш часті перимембранозні дефекти (75–80%). м’язові дефекти або дефекти трабекулярної перегородки відзначають у 20% усіх дефектів міжшлуночкової перегородки. Приблизно половина дефектів невеликого розміру, але вони можуть варіювати від 1 до 30 мм і більше, мати різну форму: круглу, еліпсовидну, з м’якими або фіброзно зміненими краями. При ваді також виявляють гіпертрофію міокарда і дилатацію порожнин обох шлуночків, передсердь, розширення стовбура легеневої артерії, іноді значне.
Порушення гемодинаміки
Функціональні розлади залежать насамперед від розмірів отвору та стану легеневого судинного русла. При невеликих дефектах (до 10 мм) виникає значний градієнт тиску в правому і лівому шлуночку, а в систолу відбувається незначне АВ-скидання крові через дефект. Через низький опір крові в малому колі кровообігу тиск у правому шлуночку і легеневій артерії або незначно підвищується, або залишається нормальним. У діастолу в результаті підвищення кінцевого діастолічного тиску в правому шлуночку частина крові з його порожнини може повертатися в ліві відділи, викликаючи об’ємне перевантаження лівого передсердя, а особливо лівого шлуночка. Помірні або великі дефекти викликають застій в легенях та перевантаження об’ємом лівого шлуночка, що може призводити до виникнення легеневої гіпертензії. Великі дефекти міжшлуночкової перегородки не створюють перешкоди скиданню крові зліва направо, обидва шлуночки функціонують як єдина насосна камера з двома виходами, призводячи до вирівнювання тиску в системному і легеневому колі кровообігу. Величина скидання крові зворотно пропорційна відношенню легеневого та системного судинного опору. Якщо загальнолегеневий опір нормальний або підвищений, але становить менше половини від опору у великому колі кровообігу, відбувається велике скидання крові, легеневий кровотік у 2 рази і більше перевищує системний, відзначаються значне підвищення тиску в малому колі кровообігу, об’ємні та систолічні перевантаження лівого і правого шлуночка, що зумовлює розвиток вираженої декомпенсації кровообігу. У цих хворих дуже рано відзначають розвиток структурних змін у легенях, а також вторинної легеневої гіпертензії. Якщо загальнолегеневий опір становить половину та більше ЗПСО, то обсяг скидання зменшується.
Клінічна картина
Клінічна картина залежить від віку хворого, розмірів дефекту, величини судинного опору легень. При невеликих дефектах клінічні прояви вади відсутні, задишка при фізичному напруженні найчастіше є першим проявом декомпенсації. При дефектах більших розмірів (діаметром >10 мм або більше половини діаметра устя аорти) хворі скаржаться на задишку за типом тахіпное за участю допоміжних м’язів, відчуття серцебиття, біль в ділянці серця, стійкий кашель, що підсилюється при зміні положення тіла. При пальпації грудної клітки часто визначають систолічне тремтіння в четвертому міжребер’ї ліворуч та в ділянці мечоподібного відростка. Основною клінічною ознакою вади є характерний гучний, пов’язаний з I тоном, голосистолічний шум Роже, який вислуховується у третьому– четвертому міжребер’ї ліворуч від груднини, та проводиться до верхівки серця.
Діагностика
При невеликих дефектах ЕКГ у межах фізіологічної норми. При більших дефектах виявляють неспецифічні ознаки комбінованої гіпертрофії обох шлуночків та передсердь, зміни ST–T, фібриляцію передсердь, порушення внутрішньошлуночкової провідності. На рентгенограмі при невеликих дефектах серце нормальних розмірів, при більших — кардіомегалія, посилення легеневого малюнка за рахунок переповнення артеріального русла. При вираженій легеневій гіпертензії посилені прикореневі зони, а судинний малюнок периферичних відділів легень виглядає «збідненим». Дуга легеневого стовбура вибухає по лівому контуру, при рентгеноскопії відзначають посилення її пульсації. ЕхоКГ-обстеження з колірним допплерівським картуванням дозволяє верифікувати діагноз — безпосередньо визначити розміри й розташування дефекту, наявність і напрямок скидання крові. Градієнт тиску між лівим і правим шлуночком можна оцінити за допомогою постійнохвильової допплерографії. При ехоКГ-обстеженні можна виявити:
- збільшення розмірів усіх камер серця;
- гіперкінез стінок лівого шлуночка;
- візуалізуючий дефект міжшлуночкової перегородки (>10 мм);
- турбулентний потік через перегородку зліванаправо.
При катетеризації правих відділів серця відзначають значне підвищення тиску в правому шлуночку та легеневій артерії, а також підвищення насичення крові киснем, що починається на рівні правого шлуночка і збільшується в легеневому стовбурі. Селективну ангіокардіографію зазвичай проводять у пацієнтів віком старше 40 років при планованому хірургічному втручанні, метод дозволяє виначати локалізацію дефекту, його розміри, а також виключити супутню патологію.
Лікування
Хворим із симптомами серцевої недостатності призначають медикаментозну терапію для стабілізації стану перед проведенням хірургічної корекції. Абсолютними показаннями до операції є критичний стан або серцева недостатність, що не піддається консервативній терапії, а також підозра на незворотні зміни, що розвиваються в судинах легень. Відносними показаннями до хірургічного втручання є великий дефект з ознаками значного скидання крові, часті респіраторні захворювання, відставання у фізичному розвитку. Хірургічне лікування протипоказане, якщо систолічний тиск у легеневій артерії дорівнює системному, артеріовенозне скидання крові становить менше 40% хвилинного обсягу малого кола кровообігу і є шунт справа наліво.
Прогноз
Спонтанне закриття дефекту відбувається у 15–60% випадків. Необхідне диспансерне спостереження хворих через можливість виникнення надалі різних ускладнень (ураження провідної системи серця, недостатність тристулкового клапана, фібриляція передсердь). У цілому 25-річна виживаність для всіх пацієнтів становить 87%, смертність підвищується з розміром дефекту. У неоперованих хворих з ізольованим невеликим дефектом і нормальним тиском у правому шлуночку прогноз сприятливий, хоча в них зберігається високий ризик розвитку ІЕ. При помірних і більших дефектах високий ризик розвитку різних ускладнень, включаючи ІЕ, аортальну недостатність, порушення ритму й провідності, дисфункцію лівого шлуночка, раптову смерть.
Комплекс Ейзенменгера
Комплекс Ейзенменгера — високий дефект міжшлуночкової перегородки, зсув аорти вправо та відходження її одночасно від обох шлуночків серця («сидяча верхи аорта»), розширення легеневої артерії.
Епідеміологія
Поширеність вади становить 4% серед вроджених вад серця у дорослих і визначається у 10% хворих з дефектами міжшлуночкової перегородки.
Порушення гемодинаміки
При цій ваді відбувається скидання крові з лівого шлуночка у правий (через дефект міжшлуночкової перегородки) і викид крові із правого шлуночка одночасно в аорту та легеневу артерію. Патологічні зміни в легеневому судинному басейні корелюють зі станом гемодинаміки і можуть бути розділені за ступенем тяжкості. Перший ступінь характеризується збільшенням легеневого кровотоку з нормальним середнім тиском у легеневій артерії. Другий ступінь характеризується підвищенням середнього тиску в легеневій артерії, помірну гіпертрофію виявляють у проксимальних судинах як результат гіпертрофії та проліферації гладких м’язів, а також збільшення елементів сполучної тканини. Третій ступінь характеризується підвищенням легеневого судинного опору, що корелює зі зменшеною кількістю дистальних легеневих судин.
Клінічна картина
Клінічна картина багато в чому подібна до дефекту міжшлуночкової перегородки, однак більш чітко проявляються порушення гемодинаміки. Найбільш частими клінічними симптомами є синюшність шкірних покривів внаслідок змішування артеріальної й венозної крові, задишка, серцебиття при фізичному навантаженні, кровохаркання. Аускультативно ліворуч біля груднини в місці прикріплення III–IV ребра, а також у проекції аорти вислуховується грубий систолічний шум. Діастолічний шум зумовлений аортальною недостатністю.
Діагностика
На ЕКГ нерідко відзначають ознаки внутрішньошлуночкової блокади, гіпертрофії правого шлуночка, фібриляцію та тріпотіння передсердь. При холтерівському моніторингу ЕКГ у 36% хворих виявляють суправентрикулярну аритмію, що є провісником клінічного погіршення та смерті. ЕхоКГ та допплерівська ехоКГ у режимі колірного картування та постійнохвильовому режимі дозволяють визначити наявність дефекту, напрямок скидання крові, підвищення тиску в правому шлуночку і легеневій артерії. На рентгенограмі в передньозадній проекції різко виступає дуга легеневої артерії. Судинний пучок посилений, корінь легень сильно пульсує («танець коренів»). Розміри серця збільшені за рахунок обох шлуночків. При катетеризації серця відзначають підвищення тиску в правому шлуночку і легеневій артерії. Насичення венозної крові киснем може бути рівномірно знижене в правих відділах серця та легеневій артерії або незначно підвищене в правому шлуночку. При введенні контрастної речовини в порожнину лівого шлуночка виявляють її перехід у порожнину правого шлуночка у систолу та подальше одночасне надходження в аорту і легеневу артерію.
Лікування
Медикаментозне лікування симптоматичне, включаючи терапію серцевої недостатності. Терапія не повинна зменшувати післянавантаження через збільшення скидання крові справа наліво. Варто призначати антигіпертензивні препарати, оскільки некоригована артеріальна гіпертензія може підвищувати ризик розвитку легеневих кровотеч. Зазвичай пацієнти добре переносять блокатори β-адренорецепторів. Призначення антагоністів ендотеліну зменшує функціональний клас серцевої недостатності і поліпшує переносимість фізичних навантажень. На відміну від первинної легеневої гіпертензії, де доведена ефективність антикоагулянтів для запобігання тромбозу легеневої артерії, немає аргументів для їх застосування в хворих з комплексом Ейзенменгера і їх не слід призначати через підвищений ризик виникнення кровотеч. Антикоагулянти показані за наявності фібриляції та тріпотіння передсердь, рецидивуючих тромбоемболій, механічних протезів клапанів. Необхідно виключити потенційно нефротоксичні препарати (включаючи НПЗП). Хірургічне лікування — операція накладення анастомозу — протипоказано, оскільки підвищений тиск у системі легеневої артерії унеможливило би правильне функціонування штучної протоки. Немає єдиної думки про роль трансплантації серця у цих хворих.
Прогноз
Пацієнти можуть доживати до середнього віку. До ускладнень комплексу Ейзенменгера відносять легеневі кровотечі й емболії, зумовлені аневризматичними розширеннями легеневої артерії в їх дрібніших відгалуженнях.
Ізольований стеноз легеневої артерії
Ізольований стеноз легеневої артерії характеризується наявністю перешкоди на шляху надходження крові на рівні клапана легеневого стовбура.
Епідеміологія
Досить розповсюджений (9% усіх уроджених вад серця).
Патологічна анатомія
Морфологічно вада неоднорідна, обструкція викиду крові з правого шлуночка може локалізуватися на надклапанному, клапанному та/або підклапанному рівнях, виділяють декілька її форм:
- клапанний стеноз легеневої артерії — найбільш часта форма, вада утворюється в результаті зрощення стулок по комісурам, і клапан отримує вид діафрагми з отвором округлої або злегка овальної форми діаметром від 1 до 10 мм і більше;
- інфундибулярний (підклапанний) стеноз утворюється за рахунок фіброзно-м’язової смуги у місці з’єднання порожнини правого шлуночка та артеріального конуса або за рахунок гіпертрофованих м’язів, що формують звужений вихід з правого шлуночка та розташовуються під клапанами легеневої артерії або нижче у вивідному тракті;
- найрідша форма — звуження просвіту легеневої артерії, зумовлене нерівномірним розподілом артеріального стовбура.
Морфологічно визначають концентричну гіпертрофію м’язів вихідного відділу, ендокард товщає і у вихідному відділі нерідко відзначають формування вираженого фіброзу. У стінці легеневої артерії відбуваються дегенеративні зміни, вона стає тоншою, виникає характерне постстенотичне розширення легеневої артерії, що нерідко поширюється на ліву гілку. Тристулковий клапан часто має ознаки дисплазії.
Порушення гемодинаміки
Перешкода відтоку крові створює перевантаження правого шлуночка, систолічний тиск у ньому значно підвищується (до 200 мм рт. ст. і більше), у результаті чого утворюється систолічний градієнт тиску між правим шлуночком і легеневою артерією, виникає гіпертрофія правого шлуночка. Систолічний тиск у легеневій артерії в більшості випадків у межах норми або незначно знижений. При розвитку гіпертрофії міокарда підвищується діастолічний тиск у правому шлуночку, що призводить до підвищення тиску в правому передсерді, його гіпертрофії та дилатації. Ступінь стенозу з віком підвищується, оскільки змінений потік крові через звужений отвір збільшує клапанну деформацію.
Клінічна картина
Клінічні ознаки цілком залежать від ступеня стенозу, часто виявляють фізичну недорозвиненість. Скарги на виражену задишку, особливо після фізичного напруження у зв’язку з недостатньою артеріалізацією легень, біль у серці, перебої в роботі серця, запаморочення, непритомність. При огляді відзначають наявність серцевого горба, посилення серцевого поштовху, систолічне тремтіння передньої частини грудної стінки в ІІ–ІІІ міжребер’ї ліворуч від груднини, перкуторно розширення границь серця вправо. Аускультативно визначається інтенсивний грубий систолічний шум над легеневою артерією, що добре проводиться до верхньої частини лівої половини грудної клітки та на ділянку спини, підсилюється на вдиху, II тон над легеневою артерією при клапанному та підклапанному (інфундибулярному) стенозі ослаблений або відсутній, при надклапанному стенозі може бути посилений.
Діагностика
На ЕКГ електрична вісь відхилена вправо, часто визначають ознаки гіпертрофії та перевантаження правого шлуночка і правого передсердя, які корелюють зі ступенем стенозу. Зсув сегмента SТ униз і негативний зубець Т у правих грудних відведеннях свідчать про крайній ступінь перевантаження. На рентгенограмі в передньозадньому положенні виявляється розширення контуру правого шлуночка. Легенева артерія при підклапанному ураженні зменшена, при надклапанному звуженні — розширена з виступаючою дугою. Характерне розходження між пульсуючим основним стовбуром легеневої артерії і нерухомими артеріями середнього калібру в ділянці коренів легень. Аорта часто буває недорозвинена, лівий шлуночок невеликих розмірів, легені підвищеної прозорості внаслідок зниженого кровопостачання. ЕхоКГ-обстеження дозволяє виявити ваду і деталізувати її анатомічну будову, хоча іноді можуть виникати ускладнення з візуалізацією клапана легеневої артерії. Визначають потовщення стулок клапана з неповним відкриттям їх у систолу, зменшенням отвору через зрощення стулок по комісурам, а також гіпоплазію клапанного кільця. За допомогою допплерівського дослідження виявляють турбулентний систолічний потік у легеневому стовбурі, можна розрахувати градієнт тиску в місці перешкоди між правим шлуночком і стовбуром легеневої артерії. Катетеризація є верифікуючим методом діагностики в незрозумілих випадках: патогномонічною ознакою служить градієнт систолічного артеріального тиску між правим шлуночком та легеневою артерією. Тиск у порожнинах серця характерно змінений: у правому шлуночку підвищується до 100–200 мм рт. ст. і вище, у легеневій артерії залишається нормальним або трохи зниженим, при вираженому стенозі це зниження значне (<8 мм рт. ст.). За характером зміни тиску від легеневої артерії до правого шлуночка можна судити про локалізацію стенозу, що важливо для вибору тактики хірургічного лікування: при клапанному стенозі (сприятливому для операції) перепад тиску відбувається різко, у момент проходження клапана може реєструватися негативний тиск — ефект Вентури; при інфундибулярному (підклапанному) стенозі тиск змінюється більш поступово, в порожнині лівого шлуночка тиск незмінений.
Лікування
Єдиним ефективним методом лікування є хірургічна корекція вади, показаннями до якої є виражена клінічна картина вади й наявність градієнта тиску між правим шлуночком і легеневою артерією >40 мм рт. ст. Хірургічне лікування протипоказане при вираженій правошлуночковій недостатності. Летальність після хірургічного усунення вади не перевищує 3%, однак різко підвищується при вираженій кардіомегалії, зумовленій гіпертрофією та дилатацією правих відділів серця. Віддалені результати лікування хороші і прямо залежать від віку хворого, якому зроблена операція, і початкової тяжкості стенозу.
Прогноз
Залежить від ступеня стенозу та вираженості клінічних проявів. При незначному стенозі легеневої артерії пацієнти доживають до старості. При більш вираженому стенозі і прогресуванні захворювання може розвиватися недостатність правого шлуночка, що є основною причиною раптової смерті в молодому віці. Частими ускладненнями вади є туберкульоз легень та ІЕ.
Тетрада Фалло
У комплекс тетради Фалло входять чотири аномалії: високий дефект міжшлуночкової перегородки, декстрапозиція аорти (одночасне відходження аорти від правого і лівого шлуночка), стеноз або атрезія легеневої артерії з обструкцією виносного тракту правого шлуночка, гіпертрофія правого шлуночка.
Епідеміологія
Тетрада Фалло відноситься до найпоширенішої «синьої» вади серця, частота її становить у старшому віці 12–14% усіх уроджених вад серця, трохи частіше в чоловіків. У 15% хворих з тетрадою Фалло виявляють делецію хромосоми 22q11.
Патологічна анатомія
Основу вади становить недорозвиненість вихідного відділу правого шлуночка і зміщення конусної перегородки вперед та вліво, що зумовлює стеноз вихідного відділу правого шлуночка, як правило, з порушенням розвитку фіброзного кільця легеневого стовбура, клапанного апарата та дуже часто стовбура і гілок. Нерідко відзначають двостулковий клапан легеневої артерії. Характерний розвиток колатерального кровообігу в судинах малого кола кровообігу, зміни в судинах легень, які піддаються значній перебудові: деякі судини склерозуються, облітеруються, у просвіті іноді утворюються багатостовбурові судини.
Порушення гемодинаміки
Звуження легеневої артерії приводить до перевантаження правого шлуночка тиском, крім того, збільшення повернення крові до правого шлуночка викликає одночасно і перевантаження об’ємом. Скидання крові з лівого в правий шлуночок не має великого значення внаслідок значного підвищення тиску в правому шлуночку, тому градієнт тиску між шлуночками виявляється незначним. У випадках помірного стенозу легеневої артерії, коли опір викиду крові в легені нижче, ніж в аорту, скидання крові через дефект міжшлуночкової перегородки відбувається зліва направо і легеневий кровотік виявляється збільшеним, що клінічно проявляється так званою блідою (аціанотичною) формою тетради Фалло. При збільшенні вираженості стенозу виникає перехресне, а потім стабільне скидання крові справа наліво (веноартеріальне), що клінічно позначається переходом у ціанотичну («синю») форму вади.
Клінічна картина
Клінічні прояви залежать від ступеня звуження вихідного відділу правого шлуночка і легеневої артерії та ступеня гіпоксемії. Уже з раннього дитинства у хворих відзначають дифузійний ціаноз (в аорту одночасно надходить кров з правого і лівого шлуночка, відбувається змішування артеріальної й венозної крові, що викликає синюшність шкірних покривів), поліцитемію та згущення крові. Можуть виникати часті легеневі кровотечі. Найбільш характерним симптомом є напади задишки з появою ціанозу, які виникають внаслідок спазму м’язів у вихідному відділі правого шлуночка, у результаті чого кров з нього надходить в аорту, при цьому збільшується кисневе голодування та може настати втрата свідомості (гіпоксична кома). Напад починається раптово, з посилення задишки, ціанозу, можливі апное, судоми з наступною появою геміпарезу, може закінчитися летально. Хворі не в змозі переносити фізичне навантаження, оскільки при навантаженні збільшується скидання венозної крові, підсилюється гіпоксемія, що веде до збільшення гіпоксії тканин. Типова задишка при невеликому навантаженні, різка слабкість після навантаження, запаморочення, тахікардія, посилення ціанозу. Найбільш частими клінічними симптомами є:
- зміна форми нігтів («годинникове скло»);
- деформація пальців у вигляді барабанних паличок (як реакція на тривалу гіпоксемію);
- відставання у фізичному розвитку;
- діти віддають перевагу положенню «навпочіпках», при якому створюються особливо сприятливі умови для кровообігу в легенях (зменшується обсяг венозного скидання крові в аорту);
- судомний синдром внаслідок гіпоксії мозку;
- видимі слизові оболонки та шкірні покриви ціанотичні;
- артеріальний тиск зазвичай знижений;
- систолічне тремтіння в ІІ–ІІІ міжребер’ї, викликане стенозом устя легеневої артерії.
При аускультації характерними ознаками є ослаблення II тону над легеневою артерією, грубий «сухий» систолічний шум у ІІ–ІІІ міжребер’ї ліворуч біля груднини.
Діагностика
На ЕКГ виявляють значне відхилення електричної осі вправо, ознаки вираженої гіпертрофії правого шлуночка і правого передсердя, може реєструватися порушення провідності по правій ніжці пучка Гіса. На рентгенограмі в передньозадній проекції в більшості випадків відзначають нерізке випинання нижньої частини правого контуру за рахунок гіпертрофії правого шлуночка. Аорта частіше зміщена вправо, стравохід відхилений вліво. Легеневі поля підвищеної прозорості, у більш пізніх стадіях може бути посилення малюнка коренів легень внаслідок розвинених колатералей. При ехоКГ-обстеженні добре виявляються всі ознаки вади, можливе безпосереднє визначення величини зсуву аорти, дефекту міжшлуночкової перегородки, ступеня легеневого стенозу і гіпертрофії правого шлуночка. Найбільш характерними ехоКГ-ознаками є:
- розрив між міжшлуночковою перегородкою та передньою стінкою аорти;
- локалізація аорти над міжшлуночковою перегородкою;
- розширення устя аорти;
- гіпертрофія правого шлуночка;
- клапанний і субклапанний стеноз легеневої артерії;
- гіпоплазія кільця клапана легеневої артерії, легеневого стовбура та проксимальних відділів легеневої артерії;
- закидання контрастної речовини з правого шлуночка у вихідний відділ лівого шлуночка та аорту;
- при допплерівському дослідженні турбулентний систолічний потік у правий шлуночок (шунт зліва направо), можливий турбулентний потік у вихідний відділ лівого шлуночка (шунт справа наліво);
- додатковий турбулентний діастолічний потік у разі функціонуючого аортолегеневого анастомозу в легеневому стовбурі;
- наявність градієнта тиску між правим шлуночком і легеневою артерією.
Катетеризація порожнин серця і ангіокардіографія — найбільш інформативні методи діагностики тетради Фалло. При катетеризації порожнин виявляють значне підвищення тиску в правому шлуночку, рівне системному градієнту тиску між правим шлуночком і легеневою артерією. Характерною гемодинамічною ознакою вади є однаковий систолічний тиск у правому шлуночку та аорті, куди нерідко вдається провести катетер через дефект міжшлуночкової перегородки. Тиск у правому передсерді частіше нормальний, у легеневій артерії помірно знижений. При введенні контрастної речовини в порожнину правого шлуночка відзначається одночасне його надходження в легеневу артерію й аорту. Визначається також дефект наповнення інфундибулярної частини правого шлуночка або клапана легеневої артерії, що дозволяє визначити ступінь її стенозу.
Лікування
Консервативна терапія неефективна. Хірургічне лікування показане всім хворим із тетрадою Фалло і може бути радикальним або паліативним. Радикальна корекція вади включає усунення легеневого стенозу та закриття дефекту міжшлуночкової перегородки в умовах штучного кровообігу. При тяжких формах ціанотичні напади, високий рівень гемоглобіну (>200 г/л), стан хворого, який швидко погіршується, можуть бути показаннями до проведення паліативної операції — накладання аортолегеневих анастомозів з метою збільшення легеневого кровотоку та зменшення гіпоксії. Ці операції дозволяють пацієнтам дожити до 5–6-річного віку, коли можливе проведення радикальної корекції вади з меншим ступенем ризику.
Прогноз
Віддалені результати радикального хірургічного лікування неускладнених форм тетради Фалло, як правило, хороші. Погіршення стану у віддалений термін може бути зумовлене тією чи іншою мірою стенозом легеневої артерії, що залишився, легеневою недостатністю, реканалізацією міжшлуночкової перегородки, порушеннями ритму серця. Останні зазвичай зумовлені травмуванням провідних шляхів під час оперативного втручання, залишковим високим тиском у правому шлуночку, вираженою кардіомегалією через реканалізацію міжшлуночкової перегородки або розвиток аневризми правого шлуночка. Саме аритмія є причиною раптової смерті хворих у різний термін після оперативної корекції вади.
Відкрита артеріальна протока
Відкрита артеріальна протока (ductus arteriosus, боталова протока) — незарощення артеріальної протоки плода, що з’єднує аорту й легеневу артерію.
Епідеміологія
Відзначають у 5–10% уроджених вад серця. У жінок ваду виявляють частіше, ніж у чоловіків (3:1).
Патологічна анатомія
Залежно від форми протоки існують різні анатомічні його типи: циліндричний, воронкоподібний, вікончатий, аневризматичний. При тривалому існуванні протоки виникає її кальциноз, що захоплює і аорту. Стовбур і розгалуження легеневої артерії розширені. У дрібних легеневих артеріях та артеріолах у міру розвитку легеневої гіпертензії відбуваються характерні морфологічні зміни — м’язово-фіброзне переродження стінок і зменшення їх просвіту.
Порушення гемодинаміки
При незарощенні артеріальної протоки внаслідок різниці тиску між аортою та легеневою артерією відбувається скидання оксигенованої крові в легеневу артерію й далі в легені, потім через судини малого кола кровообігу вона повертається назад у ліву половину серця й аорту, тобто збільшується кровотік у малому колі кровообігу та гіпертензія в системі легеневої артерії, кровонаповнення лівого передсердя і лівого шлуночка збільшене. При малих розмірах протоки обсяг шунта невеликий і тиск в легеневій артерії залишається нормальним. При великому діаметрі протоки значна кількість крові надходить у легеневу артерію, потім у ліві відділи, викликаючи їхнє об’ємне перевантаження. Тиск в аорті передається безпосередньо через протоку в легеневу артерію, що зумовлює ранній розвиток легеневої гіпертензії, при цьому ступінь останньої може бути досить високим.
Клінічна картина
Клінічна симптоматика і перебіг захворювання варіюють залежно від ступеня порушення гемодинаміки. При невеликих і середніх розмірах протоки перебіг вади довгостроково може бути безсимптомним і ваду виявляють випадково. У більшості випадків протягом тривалого часу хворі не пред’являють скарг; у фазі декомпенсації на перший план виступають задишка та серцебиття. Зазвичай відзначається блідість шкірних покривів. Ціаноз, що з’являється, не є прямим наслідком вади, а виникає тільки при значному підвищенні тиску в легеневій артерії, зумовленому застоєм в легенях. Пізньому ціанозу, як правило, передує ціаноз при навантаженні (збільшення споживання кисню периферичними тканинами). При більшій вираженості шунтів у хворих виявляють відставання у фізичному розвитку, швидку стомлюваність, задишку та серцебиття при фізичному навантаженні. Систолічний артеріальний тиск нормальний або незначно підвищений, діастолічний — різко знижений і при фізичному навантаженні може знижуватися до нуля, що зумовлює типовий високий пульсовий тиск. При пальпації визначається посилення верхівкового поштовху, парастернальний серцевий горб, збільшення печінки й селезінки. Важливою діагностичною ознакою є шум над легеневою артерією — грубий протяжливий систолодіастолічний «машинний шум», «шум поїзда в тунелі», що супроводжується систолодіастолічним або систолічним тремтінням («котяче муркотіння») у проекції основи серця. Із розвитком легеневої гіпертензії діастолічний компонент шуму зменшується, а потім зовсім зникає. При вирівнюванні тиску у великому й малому колі кровообігу, коли тиск крові в легеневій артерії стає вищим, ніж в аорті, напрямок кровотоку через шунт змінюється і вада стає практично «афонічною».
Діагностика
На ЕКГ при вираженій ваді виявляють відхилення електричної осі вправо (при вираженій легеневій гіпертензії) або вліво, ознаки гіпертрофії обох шлуночків, іноді реєструють неповну блокаду лівої ніжки пучка Гіса, передсердні аритмії. При рентгенологічному дослідженні відзначають посилення судинного малюнка, що відповідає величині артеріовенозного скидання крові, розширення або вибухання стовбура легеневої артерії зі збільшенням діаметру часткових та сегментарних судин легень (кардіоторакальний індекс становить 55–60%) за рахунок спочатку лівого, а потім обох шлуночків і лівого передсердя. З віком відзначається розширення висхідної частини аорти. За відсутності легеневої гіпертензії зміни на рентгенограмі можуть бути відсутні. На ехоКГ специфічних ознак немає, однак характерне збільшення порожнини лівого шлуночка і зміна відношення розміру лівого передсердя до діаметру аорти (1:2 і більше). Як прояв об’ємного перевантаження лівого шлуночка є збільшення швидкості руху передньої стулки мітрального клапана в діастолу. При проведенні допплерівського дослідження часто вдається візуалізувати протоку, визначити постійний потік крові з аорти в легеневу артерію та виміряти градієнт тиску. При контрастній ехоКГ визначається закидання контрастної речовини з легеневої артерії в аорту. Діагноз може бути встановлений та/або підтверджений катетеризацією порожнин серця за наявності сатурації кисню на рівні легеневої артерії і візуалізацією протоки при аортографії. При катетеризації правих відділів серця виявляється підвищення тиску в правому шлуночку (близько 45 мм рт. ст.) і в легеневій артерії (до 100 мм рт. ст.). При введенні контрастної речовини в порожнину шлуночка визначають його подальше надходження не тільки в аорту, але й в систему легеневої артерії. Найбільш достовірною ознакою є проведення катетера з легеневої артерії в аорту, коли він описує своєрідну характерну криву. Під час видалення катетера реєструється крива аортального, потім легеневого тиску.
Лікування
Показання до хірургічного лікування залежать від розмірів протоки, ступеня легеневої гіпертензії та клінічної картини серцевої декомпенсації. Віддалені результати хірургічної корекції вади показують, що своєчасне оперативне втручання дозволяє досягти повного видужання. У хворих із вираженою легеневою гіпертензією результат оперативного втручання залежить від вихідного стану й зворотності структурних і функціональних змін легеневих судин і міокарда. Найчастішими ускладненнями після оперативного втручання є виникнення легеневої гіпертензії, дисфункції лівого шлуночка, аритмії. Найбільш складним є питання про оперативне втручання у хворих з високою легеневою гіпертензією, оскільки відомо, що в них протока часто склерозована, спроби її перев’язки можуть призвести до тяжкої кровотечі внаслідок розриву протоки, прорізування лігатури і т.д. Абсолютно протипоказане хірургічне лікування хворим зі скиданням крові справа наліво.
Прогноз
Відкрита артеріальна протока навіть невеликих розмірів веде до передчасної смерті, що зумовлено зниженням компенсаторних можливостей міокарда та судин малого кола кровообігу, приєднанням різних ускладнень (пневмонія, легенева гіпертензія, ІЕ, серцева недостатність, розрив аневризми та ін.), частіше у осіб віком старше 40 років. При великому діаметрі протоки розвивається типова картина з ціанозом і задишкою. При неускладненому перебігу середня тривалість життя — 50–60 років, хоча описані окремі випадки, коли хворі доживали до 70–79 років (діаметр протоки у них не перевищував 3 мм).
Коарктація аорти
Коарктація аорти — вроджене сегментарне звуження аорти, що розташовується в ділянці її перешийка.
Епідеміологія
Є однією з найпоширеніших — до 15% усіх вроджених вад серця, у чоловіків виявляють в 2–2,5 раза частіше, ніж у жінок.
Патологічна анатомія
Коарктація частіше локалізується в ділянці дуги аорти дистально від місця відхождення лівої підключичної артерії поблизу артеріальної протоки або відповідної їй зв’язки. За анатомічними особливостями виділяють три варіанти вади:
1) ізольована коарктація аорти;
2) коарктація в поєднанні з відкритою артеріальною протокою:
- постдуктальна (розташована нижче відгалуження відкритої артеріальної протоки);
- юкстадуктальна (протока відкривається на рівні звуження);
- предуктальна (артеріальна протока відходить нижче рівня коарктації);
3) коарктація аорти в поєднанні з іншими вродженими вадами серця (дефектом міжшлуночкової, міжпередсердної перегородки, стенозом аорти, аневризмою синуса Вальсальви, транспозицією магістральних судин).
У стінці аорти в ділянці коарктації наростає склеротичний процес, що призводить до потовщення інтими, її значних змін. Дистально від коарктації стінка тоншає, просвіт аорти розширюється, іноді аневризматично (внаслідок впливу турбулентного потоку крові після проходу місця звуження). Характерні склеротичні зміни в судинах верхньої половини тіла, посилений розвиток колатералей, по яких кров переходить з верхньої частини аорти в постстенотичну її ділянку, дистрофічні зміни міокарда. Особливо значно розширені підключичні артерії, гілки пахвової артерії. Лівий шлуночок серця значно гіпертрофований, у тому числі його м’язовий і трабекулярний апарат, що може викликати звуження шляху відтоку.
Порушення гемодинаміки
Механічна перешкода на шляху кровотоку в аорті викликає перевантаження лівого шлуночка і веде до розвитку двох режимів кровообігу: гіпертонічного (верхня половина тулуба) і гіпотонічного (черевна порожнина, нижні кінцівки). Проксимальніше місця звуження артеріальний тиск підвищений, що супроводжується збільшенням хвилинного об’єму крові і роботи лівого шлуночка. Дистальніше перешкоди артеріальний тиск (особливо пульсовий) знижений, кровопостачання частково, а іноді й повністю здійснюється за рахунок колатералей. При постдуктальній коарктації кров з аорти під високим тиском скидається через відкриту артеріальну протоку в легеневу артерію, при цьому може рано розвинутися легенева гіпертензія. При предуктальному варіанті напрямок скидання через артеріальну протоку буде визначатися різницею тисків між легеневою артерією та низхідною аортою нижче місця коарктації, скидання може бути артеріовенозним і веноартеріальним. Останній пояснює диференційований ціаноз (є на ногах і відсутній на руках) як клінічну ознаку предуктальної коарктації. Патогенез артеріальної гіпертензії при коарктації аорти складний і до кінця не з’ясований. Передбачається, що механічна обструкція, активація РААС на фоні недостатньої перфузії нирок та органічні зміни в судинній стінці призводять до збільшення ЗПСО.
Клінічна картина
Клінічна картина вади визначається віком, анатомічними змінами, рівнем артеріального тиску. У дітей старшого віку та дорослих у разі відсутності скарг підвищений артеріальний тиск виявляють випадково. Пацієнти скаржаться на головний біль, запаморочення, підвищену стомлюваність, слабкість і біль у ногах, судоми м’язів ніг, мерзлякуватість стоп, носові кровотечі. При огляді відзначають:
- диспропорційний розвиток скелетних м’язів: м’язи верхньої половини тіла гіпертрофовані при відносній гіпотрофії м’язів таза та нижніх кінцівок;
- підвищену пульсацію при пальпації міжреберних артерій (при нахилі вперед з опущеними руками);
- посилену пульсацію сонних і підключичних артерій, артерій верхніх кінцівок;
- пульсацію аорти в яремній ямці;
- пульсацію в міжлопатковому просторі, у пахвовій западині;
- різко ослаблену пульсацію на стегновій артерії та судинах нижніх кінцівок, артеріальний тиск на останній нерідко знижений або взагалі не визначається;
- виражену артеріальну гіпертензію (переважно систолічну — до 220 мм рт. ст.);
- невеликий пульсовий тиск на ногах: систолічний артеріальний тиск на ногах на 50–60 мм рт. ст. нижчий, ніж на руках, при нормальному діастолічному тиску;
- верхівковий поштовх посилений;
- границі серцевої тупості зазвичай розширені вліво, аорта розширена;
- шкіра верхньої половини тіла тепла, нижньої — більш холодна, бліда.
Дані аускультації неспецифічні, I тон приглушений, акцент II тону на аорті внаслідок підвищення артеріального тиску у початковому відділі аорти, систолічний шум середньої інтенсивності в другому міжребер’ї ліворуч, добре проводиться в міжлопатковий простір.
Діагностика
На ЕКГ часто виявляють відхилення електричної осі вліво, визначають ознаки вираженої гіпертрофії міокарда лівого шлуночка. При рентгенографічному дослідженні в передньозадній проекції визначається збільшення лівого шлуночка, розширення висхідної аорти. Визначається узурація нижніх країв ребер внаслідок тиску різко розширених та звитих міжреберних артерій. У передній косій проекції чітко помітна різниця в діаметрі висхідної та нисхідної частин аорти (тінь аорти має вигляд числа «3»). Легеневий малюнок зазвичай виражений, судинний пучок вибухає праворуч за рахунок розширення висхідної частини аорти, лівий контур його згладжений. Посилено пульсацію лівого шлуночка висхідної частини аорти та плечоголовних судин. За допомогою ехоКГ можлива візуалізація місця звуження аорти, його діаметру і довжини, а також співвідношення з гілками дуги аорти та відкритою артеріальною протокою. За допомогою допплерівської ехоКГ можна визначити систолічний турбулентний потік та градієнт тиску в місці коарктації, точно оцінити ступінь обструкції.
Проведення МРТ має переваги в дорослій популяції для візуалізації анатомічних особливостей аорти, особливо показано в тому випадку, якщо ехоКГ неможливе. При катетеризації аорти діагноз вади підтверджується при визначенні величини градієнта систолічного тиску між висхідною та низхідною аортою, місця звуження, вираженості аневризматичних змін аорти. У порожнині лівого шлуночка й аорті виявляють значне підвищення систолічного тиску. При предуктальному варіанті катетер з легеневої артерії безперешкодно проходить через відкриту артеріальну протоку в низхідну аорту, де визначається зниження насичення крові киснем.
Лікування
Наявність коарктації аорти є абсолютним показанням до хірургічного втручання, ризик якого неоднаковий у різні вікові періоди та залежить від тяжкості стану хворих, анатомії вади та порушень кровообігу. Показання до оперативної корекції вади у дорослих включають застійну серцеву недостатність, гіпертензію в судинах верхніх кінцівок та/або градієнт >20 мм рт. ст. Консервативне лікування ускладнень (серцева недостатність, артеріальна гіпертензія), як правило, малоефективне. У хворих віком старше 20 років питання про показання до оперативного втручання варто вирішувати індивідуально: операція необхідна пацієнтам, у яких відсутні склеротична форма легеневої гіпертензії та тяжкий кальциноз аорти. За наявності вираженої декомпенсації кровообігу хірургічне лікування пов’язане з високим ризиком, тому показання до операції визначають із обережністю, оцінивши скорочувальну здатність міокарда та причину серцевої недостатності. У дорослих хворих відновлення прохідності аорти зазвичай легко здійснюється шляхом резекції звуженої ділянки аорти та накладення анастомозу кінець у кінець або ж заміщення звуженої ділянки судинним протезом. Рідше використовують операцію прямої або непрямої істмопластики аорти. Із постопераційних ускладнень слід зазначити артеріальну гіпотензію, що може розвинутися відразу ж після відновлення кровотоку по аорті, абдомінальний синдром і кровотечу, що виникають у найближчий післяопераційний період. Специфічним ускладненням після успішного усунення коарктації аорти є розвиток післяопераційної або так званої парадоксальної гіпертензії, що відзначають у 50–80% хворих, частіше в осіб старшого віку. Наявність артеріальної гіпертензії є ризиком передчасного розвитку ішемічної хвороби серця, дисфункції лівого шлуночка, розриву аневризм аорти або мозкових судин, раптової смерті. Незважаючи на відсутність рандомізованих досліджень, блокатори β-адренорецепторів зазвичай рекомендують як препарати першого вибору.
Прогноз
Вада у разі відсутності лікування характеризується вкрай несприятливим перебігом, середня тривалість життя без хірургічного лікування — 35 років. Більшість пацієнтів помирають від прогресуючої серцевої недостатності, ІЕ, іноді від розриву аорти або її аневризми, інсульту. Летальність у віддалений термін у хворих, прооперованих у віці старше 25 років, досягає 35%, у 20% хворих нормалізації рівня артеріального тиску не відбувається, можливе утворення аневризми аорти та рестенозу. Загальна 30-річна виживаність після хірургічної корекції вади становить усього 72%. Основні причини смерті у віддалений післяопераційний період — серцева недостатність та інфаркт міокарда.
Аномалія Ебштейна
Аномалія розвитку тристулкового клапана, що характеризується різним ступенем дисплазії та зміщенням стулок клапана в порожнину правого шлуночка.
Епідеміологія
Поширеність вади становить менше 1% всіх вроджених вад серця, в 1,5 раза частіше відмічають у дівчаток.
Патологічна анатомія
Основна анатомічна особливість вади полягає у зсуві тристулкового клапана в порожнину правого шлуночка у напрямку до верхівки серця, зазвичай до місця з’єднання припливної і трабекулярної його частин. Ступінь дисплазії, деформації стулок, їх структур варіюють в широких межах. У всіх випадках в порожнину шлуночка зміщені задня стулка та досить часто — перегородкова, місцем найбільшого зсуву є комісура між ними. Зміщені стулки часто різко деформовані, стоншені, хорди їх укорочені, сосочкові м’язи гіпоплазовані. До фіброзного кільця прикріплюється тільки малозмінена передня стулка, що найчастіше є єдиною функціонуючою стулкою тристулкового клапана, вона значно збільшена в розмірах, нерідко є парусовидною, іноді вільний її край прикріплюється у вивідному відділі правого шлуночка і викликає стенозування шляхів відтоку. Патологія тристулкового клапана супроводжується розширенням фіброзного кільця, що призводить до вираженої недостатності клапана.
Зміщені стулки ділять порожнину правого шлуночка на дві функціональні частини: велика (верхня) частина, розташована над зміщеним клапаном, є «атріалізованою» частиною правого шлуночка та утворює з правим передсердям загальну, велику за об’ємом порожнину. Менша (нижня) частина розташовується під зміщеним клапаном і разом із трабекулярним та вихідним відділом функціонує як правий шлуночок. Стінка правого передсердя гіпертрофована, у той час як стінка передсердної частини правого шлуночка стоншена, аневризматично вибухає, товщина її становить 1–3 мм, міокард дистальної камери нормальний або трохи стовщений.
Порушення гемодинаміки
Зміни гемодинаміки визначаються ступенем зсуву і дисплазії тристулкового клапана, наявністю або відсутністю міжпередсердного сполучення. Анатомічні зміни призводять до дефіциту легеневого кровотоку, недостатності тристулкового клапана, скидання крові справа наліво через міжпередсердне сполучення. Зменшення легеневого кровотоку зумовлене меншим, ніж у нормі, ударний об’єм крові правого шлуночка. Крім цього, відзначають обмеження припливу крові в дистальний відділ правого шлуночка і в діастолу: у систолу правого передсердя «атріалізована» камера правого шлуночка перебуває у фазі діастоли, через що просування крові в його дистальну камеру затримується, а ефективність систоли передсердя знижується. У результаті тиск у правому передсерді підвищується, що зумовлює його дилатацію й гіпертрофію. При виражених змінах виникає нездатність правого передсердя до подальшого розширення, що створює перешкоду відтоку крові з порожнистих вен.
Клінічна картина
Клінічні симптоми різноманітні і залежать від вираженості анатомічних порушень, серед них найчастіше відзначають задишку, низьку толерантність до фізичних навантажень, напади пароксизмальної тахікардії, що призводять до втрати свідомості. При огляді виявляють різний ступінь ціанозу, набухання шийних вен, границі серцевої тупості значно розширені вліво і вправо, визначається «серцевий горб», зумовлений гігантськими розмірами правого передсердя та верхньою частиною правого шлуночка. Аускультативно тони серця ослаблені, роздвоєння I тону, розщеплення, ослаблення II тону, наявність III і IV тонів, що створюють ритм галопу, систолічний шум недостатності тристулкового клапана і діастолічний шум трикуспідального стенозу в проекції мечоподібного відростка справа біля груднини.
Діагностика
На ЕКГ електрична вісь частіше відхилена вправо. Зубці Р у I, II і в правих грудних відведеннях високі та гострі (гіпертрофія і дилатація правого передсердя), амплітуда шлуночкових зубців низька. Нерідко відзначають WPW-синдром (25%) із проявами пароксизмальної шлуночкової і передсердної тахікардії, екстрасистолію, тріпотіння та фібриляцію передсердь. Часто виявляють повну або неповну блокаду правої ніжки пучка Гіса, перший ступінь АВ-блокади (40–50%), збільшення інтервалу P–Q. На рентгенограмі в передньозадній проекції відзначають підвищену прозорість легеневих полів (за рахунок збідніння легеневого малюнка), різке збільшення розмірів серця за рахунок розширених правих відділів. Тінь серця може мати форму кулі, судинний пучок залишається вузьким. Ліві відділи не змінені. ЕхоКГ дозволяє встановити правильний діагноз у більшості пацієнтів. При ехоКГ виявляють наступні найбільш характерні ознаки:
- зсув нижче фіброзного кільця однієї, двох або всіх стулок тристулкового клапана (більше ніж на 8 мм);
- запізнення закриття тристулкового клапана порівняно з мітральним (0,065 с);
- збільшення амплітуди відкриття та зниження швидкості раннього діастолічного закриття передньої стулки тристулкового клапана;
- деформація ехосигналу від стулок;
- об’ємне збільшення розмірів правого передсердя, ідентифікація «атріалізованої» частини правого шлуночка.
За допомогою допплерівської ехоКГ можлива оцінка ступеня недостатності тристулкового клапана, шунтуючого потоку справа наліво через дефект міжпередсердної перегородки. Катетеризацію порожнин серця необхідно проводити з великою обережністю, оскільки нерідко розвиваються небезпечні для життя порушення серцевого ритму. При катетеризації визначається підвищення тиску в правому передсерді, при проведенні катетера у вихідний відділ правого шлуночка нерідко реєструють діастолічний градієнт, що зв’язаний із зсувом і дисплазією тристулкового клапана або зумовлений відносним стенозуванням правого АВ-отвору порівняно з різко розширеним правим передсердям. Систолічний тиск в правому шлуночку і легеневій артерії зазвичай нормальний або трохи знижений. Введення контрастної речовини в порожнину правого шлуночка допомагає виявити різке розширення шляху припливу та зниження шляху відтоку. Важливі діагностичні ознаки можна отримати при одночасній реєстрації тиску та проведенні внутрішньопорожнинної ЕКГ.
Лікування
При безсимптомному перебігу захворювання хірургічне лікування не проводять. Оперативне втручання показане за наявності ціанозу, ознак недостатності кровообігу та тяжких порушень ритму серця, рефрактерних до медикаментозного лікування. Радикальна операція полягає в пластичній реконструкції тристулкового клапана, за неможливості її виконання роблять протезування клапана. При поєднанні аномалії Ебштейна з WPW-синдромом одномоментно виконують пластику тристулкового клапана і деструкцію додаткових шляхів проведення імпульсу. Абляція додаткових шляхів часто супроводжується ускладненнями та менш результативна (75%), ніж у хворих без вад (95%). Не встановлено впливів оперативного лікування на виникнення раптової смерті. Медикаментозна терапія спрямована на лікування серцевої недостатності та усунення порушень ритму серця.
Прогноз
Захворювання характеризується прогресуючим перебігом. У неоперованих хворих старшого віку прогностично несприятливими факторами та причиною смерті зазвичай є прогресуюча кардіомегалія, серцева недостатність і порушення ритму, які призводять до раптової смерті (3–4%).
Тести для самоконтролю
Ревматоїдний артрит
- Вкажiть найбiльш типову локалiзацiю уражень суглобiв при РА:
- проксимальні міжфалангові суглоби кистей,стоп
- гомiлковостопнi
- променезап’ястковi, лiктьовi
- скронево-щелепнi, реберногрудниннi з’єднання
- кульшовi суглоби
- Вкажіть основні варіанти ураження легень при РА:
- хронічний iнтерстицiальний пневмонiт
- плеврит
- негоспітальна пневмонія
- ревматоїдні вузлики
- легеневий васкуліт
- Який найбiльш ефективний препарат в лiкуваннi iнтерстицiального пневмонiту при РА?
- ГК
- НПЗП
- антибiотик
- сульфасалазин
- метотрексат
- Вкажіть основні гістологічні типи ураження судин при РА
- дигітальний артеріїт з пролiферацiєю ендотелiю та тромбозами
- некроз середньої оболонки судини
- продуктивний васкулiт шкіри з мононуклеарною інфільтрацією
- панваскулiт за типом ВП
- аортит зi склерозом аорти
- Предикторами агресивного перебігу РА вважають:
- значне підвищення титру РФ, антитіл до циклічного цитрулінового пептиду
- значне збільшення СРБ
- системні прояви в дебюті
- поліартрит з ураженням понад 17 суглобів
- жіноча стать, молодий вік
- все перераховане
- Які ураження серця відмічають при РА?
- перикардит
- мiокардит
- ендокардит пристiнковий
- мiокардiодистрофiю
- формування недостатностi клапана аорти
- Які рентгенологічні зміни суглобів типові для РА?
- навколосуглобовий ОП
- анкілози
- звуження міжсуглобових щілин
- узури суглобових поверхонь
- усе перераховане
- Дайте рентген-характеристику I стадiї РА за Штейнброкером
- навколосуглобовий ОП
- звуження суглобової щiлини, кистоподiбнi просвiтлення
- узурацiя хрящiв
- узури на суглобових поверхнях
- анкiлоз
- Дайте рентген-характеристику II стадiї РА за Штейнброкером
- навколосуглобовий ОП
- звуження суглобової щiлини
- поодинокі узури суглобових поверхонь
- анкiлоз
- кистоподiбнi просвiтлення, узурацiя хрящiв
- Дайте рентген-характеристику III стадiї РА за Штейнброкером
- ОП
- множинні узури суглобових поверхонь
- кистоподiбнi просвiтлення, узурацiя хрящiв
- звуження суглобової щiлини
- Дайте рентген-характеристику IV стадiї РА за Штейнброкером
- ОП
- множинні узури суглобових поверхонь
- звуження суглобової щiлини
- кістковий анкілоз
- усе перераховане
- Що таке серопозитивний РА?
- наявнiсть антинуклеарного фактора в сироватцi кровi
- високий вміст СРБ
- титр РФ 1:16
- вiдсутнiсть РФ в сироватцi кровi
- титр РФ бiльше 1:32
- Синовiальна рiдина при РА
- в’язкість знижена
- виявляють РФ та імунні комплекси
- лейкоцитоз
- муциновий згусток рихлий
- переважають моноцити
- Що таке синдром Фелтi?
- лiмфоаденопатiя
- спленомегалiя
- панцитопенiя
- варіант РА
- РА з наявнiстю силікозу
- Лабораторнi критерiї неактивного РА
- α-2 глоб. до 10%, СРБ (–), ШОЕ до 12 мм/год
- α-2 глоб. до 10%
- СРБ (–), ШОЕ до 12 мм/год
- ШОЕ до 12 мм/год
- ШОЕ 20 мм/год
- Лабораторнi критерiї РА I ступеня активностi
- α-2 глоб. до 12%, СРБ I(+), ШОЕ до 20 мм/год
- α-2 глоб. 10%, СРБ (–)
- СРБ I(+++)
- ШОЕ 10 мм/год
- α-2 глоб. 12%
- Лабораторнi критерiї РА II ступеня активностi
- α-2 глоб. 15%, СРБ 2(+), ШОЕ 40 мм/год
- СРБ 2(+)
- α-2 глоб. 12%
- ШОЕ 40 мм/год
- ШОЕ 20 мм/год, СРБ I(+)
- Лабораторнi критерiї РА III ступеня активностi
- α-2 глоб. бiльше 15%, СРБ 3(+), ШОЕ бiльше 40 мм/год
- ШОЕ 20 мм/год
- СРБ 4(+)
- α-2 глоб. 17%
- СРБ I(+)
- Клiнiчнi критерiї ремісії РА
- припухлість періартикулярних м’яких тканин відсутня
- ШОЕ не перевищує у жінок 30 мм/год, у чоловіків 20 мм/год
- біль в суглобах при пальпації та рухах відсутній
- скарги на біль в суглобах відсутні
- ранішня скутiсть до 15 хв
- усе перераховане
- Клiнiчнi критерiї РА II ступеня активностi
- ранкова скутiсть до 12:00
- біль помiрний
- гiпертермiя помiрна
- скутiсть упродовж дня
- ексудативнi явища помiрнi
- Який з перерахованих препаратів є блокатором рецепторів до ІЛ-6?
- тоцилізумаб
- адалімумаб
- ритуксимаб
- інфліксимаб
- Який з базисних біологічних препаратів був синтезований першим для лікування РА?
- тоцилізумаб
- адалімумаб
- ритуксимаб
- інфліксимаб
- Який з базисних біологічних препаратів може застосовуватись для лікування РА як монотерапія?
- тоцилізумаб
- адалімумаб
- ритуксимаб
- інфліксимаб
- Який з базисних біологічних препаратів може застосовуватись підшкірно?
- тоцилізумаб
- адалімумаб
- ритуксимаб
- інфліксимаб
- Який з базисних біологічних препаратів застосовується як препарат ІІ лінії?
- тоцилізумаб
- адалімумаб
- ритуксимаб
- інфліксимаб
- Які з базисних біологічних препаратів, перерахованих нижче, відносяться до блокаторів ФНП-α?
- тоцилізумаб
- адалімумаб
- ритуксимаб
- інфліксимаб
- Які скринінгові тести обов’язкові при застосуванні базисних біологічних препаратів?
- туберкулінові проби
- маркери до вірусного гепатиту
- рентгенографія органів грудної клітки
- усі перераховані
- жоден з перерахованих
Еталони відповідей: 1) 1; 2) 1, 2, 4, 5; 3) 1; 4) 1, 2, 3, 4; 5) 6; 6) 1, 2, 4, 5; 7) 5; 8) 1; 9) 1, 2, 3; 10) 1, 2, 4, 5; 11) 5; 12) 5; 13) 1, 2, 3, 4; 14) 4; 15) 1; 16) 1; 17) 1; 18) 1; 19) 6; 20) 1, 2, 3, 5; 21) 1; 22) 5; 23) 1; 24) 2; 25) 3; 26) 2,4; 27) 4
Серонегативні спондилоартропатії
- Важливий дiагностичний критерiй ПсА
- ураження проксимальних суглобiв
- ознаки двобiчного сакроiлеїту
- симетричне ураження крупних суглобiв
- наявнiсть РФ
- бiль та припухість всiх трьох суглобiв одного пальця руки
- Базисні препарати при ПсА
- метотрексат
- селективні ЦОГ-2 НПЗП
- сульфасалазин
- солі золота
- блокатори ФНП
- Хвороба Бехтєрева характеризується ушкодженням:
- крижово-здухвинних з’єднань
- суглобiв щелепи
- легень
- моноартритом
- анкiлозом колiнних суглобiв
- Анкiлозуючий спондилоартрит частiше виявляють у чоловiкiв вiком:
- 10–15 рокiв
- 20–40 рокiв
- 60–70 рокiв
- 35–45 рокiв
- 1–3 роки
- Поширення анкiлозуючого спондилоартриту серед населення:
- до 2%
- до 10%
- до 20%
- до 90%
- до 30%
- Як часто виявляють антиген гiстосумiсностi HLA-B27 при хворобi Бехтєрева?
- 90–97% випадкiв
- 30–40% випадкiв
- 10–20% випадкiв
- 15–30% випадкiв
- не виявляється
- Патанатомiчна картина анкiлозуючого спондилоартриту:
- анкiлозуючий сакроiлеїт
- однобічнi остеофiти
- кальциноз м’язiв
- остеолiз хребцiв
- Найбiльш типова локалiзацiя ураження суглобiв при хворобi Бехтєрева?
- кульшовi
- лiктьовi
- плечовi
- суглоби ступнi
- суглоби кистi
- Для хвороби Бехтєрева характерні:
- вiдсутнiсть РФ
- гіпергаммаглобулінемія
- наявність HLA-B27
- збільшення вмісту СРБ
- тромбоцитопенiя
- Фактори, якi сприяють легеневiй патологii при хворобi Бехтєрева?
- обмеження рухливостi грудної клiтки
- схильність до респіраторних захворювань
- немає причин для розвитку легеневої патологiї
- розвиток сакроiлеїту
- ураження шийного вiддiлу хребта
- Чим зумовлений розвиток легеневого серця при хворобi Бехтєрева?
- обмеженням рухомостi грудної клiтки
- первинним ураженням альвеол
- гiпотонiєю у малому колі кровообiгу
- гіпертензією
- вадою серця
- Початковi прояви хвороби Бехтєрева:
- люмбалгiчний синдром
- суглобовий синдром
- ранкова скутiсть при рухах
- біль у сiдницях
- біль у груднiй клiтцi
- Характернi клiнiчнi прояви хвороби Бехтєрева:
- біль вiд перебування в одному положенні
- пiдвищена втомлюванiсть
- зменшення маси тіла
- обмеження дихальної екскурсії грудної клітки
- гiпертермiя
- Клініко-рентгенологічні критерії АС:
- біль в нижній частині спини, протягом 3 міс
- обмеження рухів в поперековому відділі хребта в сагітальній і фронтальній площині
- зменшення дихальної екскурсії грудної клітки
- рентгенологічно двобічний сакроілеїт (ІІ–ІV стадія)
- відсутність сакроілеїту
- Типовi ураження нервової системи при хворобi Бехтєрева:
- iшiалгiї
- радикулiти
- мiжребернi невралгiї
- біль у черевнiй порожнинi
- вiдсутнiсть шийного синдрому
- Ураження очей при хворобi Бехтєрева:
- iрит
- iридоциклiт
- увеїт
- блефарит
- кон’юнктивiт
- Ураження нирок при хворобi Бехтєрева:
- амiлоїдоз
- нирково-кам’яна хвороба
- гiпернефрома
- медикаментозна нефропатiя
- киста нирки
Еталони відповідей: 1) 5; 2) 1, 3, 4, 5; 3) 1; 4) 1, 2; 5) 1; 6) 1; 7) 1; 8) 1, 3; 9) 1,2,3,4; 10) 1, 2; 11) 1; 12) 1, 3, 4, 5; 13) 1, 2, 3, 4; 14) 1, 2, 3, 4; 15) 1, 2, 3; 16) 1, 2, 3; 17) 1, 2, 4
Кристалічні артропатії
- Що викликає пiдвищення рiвня сечової кислоти в кровi?
- прийом діуретиків
- променева терапія
- надмiрне фiзичне навантаження
- вживання великої кiлькостi жиру
- вживання великої кiлькостi клітковини
- При яких захворюваннях виникає вторинна подагра?
- лейкоз
- хвороба Бехчета
- псорiаз
- пернiцiозна анемiя
- хронiчна гемолiтична анемiя
- метаболічний синдром
- Затримка виведення сечової кислоти нирками вiдбувається при:
- мікседемі
- цукровому діабеті
- у період вагітності
- хронiчнiй нирковiй недостатностi
- анальгетичній нефропатiї
- прийомi дiуретикiв
- Вкажіть нормальну концентрацiю сечової кислоти в кровi у чоловiкiв:
- 0,120 ммоль/л
- 0,42 ммоль/л
- 0,62 ммоль/л
- 0,540 ммоль/л
- Якою є нормальна екскрецiя сечової кислоти з сечею?
- 3,8 ммоль/л
- 1,2 ммоль/л
- 5,4 ммоль/л
- 7,55 ммоль/л
- 0,9 ммоль/л
- Якi фази видiляють в патогенезi подагри?
- вiдкладення уратiв в тканинах
- гiперурикемiя
- гостре подагричне запалення
- ураження органiв травлення
- порушення обмiну електролiтiв
- В яких органах i тканинах вiдкладаються урати?
- суглобах i сухожиллях
- нирках
- суглобових сумках
- шкiрi
- еритроцитах
- Якi твердження вірні відносно гострого подагричного артриту?
- раптовий початок вночі
- ураження ілеосакральних з’єднань
- симетрія уражень
- характерні припухлість, яскрава гіперемія
- різкий біль в I плеснофаланговому суглобі
- Який з наведених органiв уражується при подагрi найчастiше?
- серце
- легенi
- печiнка
- нирки
- кишечник
- Вкажіть рентгенологiчнi ознаки подагри на початку захворювання:
- симптом «пробiйника»
- потовщення періартикулярних тканин
- вiдсутнiсть Ro-змiн кісткової тканини
- ОП
- змiни в тазових кiстках
- Які рентгенологiчні ознаки типові для подагри:
- округлi дефекти кiсткової тканини — «пробiйники»
- великi ерозiї кiсток
- анкілози
- ущiльнення м’яких навколосуглобових тканин
- ОП
- Характерними змiнами в синовiальнiй рiдинi при подагричному артритi є:
- помiрна в’язкiсть
- лейкоцитоз з переважанням нейтрофiлiв
- виявлення РФ
- виявлення кристалiв урату натрiю
- рiдина мутна з грудками бiлого кольору
- Вкажiть головний симптом, що дозволяє вiдрiзнити подагру вiд остеоартриту:
- вузлики Бушара
- тофуси
- звуження суглобової щiлини
- гiперурикемiя
- ущiльнення м’яких тканин
- Які вiдмiнності мiж анкілозуючим спондилоартритом i подагрою?
- гіперурикемія
- стiйкi артрити периферичних суглобiв
- порушена толерантність до глюкози
- вiдсутнiсть кристалiв сечової кислоти в синовiальнiй рiдинi
- рентген-ознаки двобiчного сакроiлеїту
- наявнiсть тофусiв
- Назвiть ознаки, якi вiдрiзняють подагру вiд псорiатичної атропатiї:
- одночасна поява шкiрних i суглобових змiн
- ураження дистальних суглобiв з почервонiнням шкiри над ними
- гіперурикемія
- лейкопенія
- відсутність змiн в сечi
- Які методи лiкування подагри є патогенетичними?
- антибiотикотерапiя
- дiєтотерапiя, урикостатичнi,урикозуричнi препарати
- фiзiотерапiя
- протизапальна терапiя
- хiрургiчне лiкування
- Який симптом є показником тривалостi й тяжкостi порушення сечокислого обмiну?
- наявнiсть гiперурикемiї
- деформацiя суглобiв
- наявнiсть тофусiв
- пiдвищення температури тiла
- змiни в сечi Які ураження нирок можливі при гiперурикемiї?
- сечокам’яна хвороба
- iнтерстицiальний нефрит
- полікістоз
- нефросклероз
- нирковий діабет
- Назвiть механiзм дiї алопуринолу:
- протизапальна
- сечогiнна
- урикозурична
- урикодепресивна
- антиалергiчна
- Якi побiчнi явища можуть з’являтися при прийомi алопуринолу?
- глюкозурія
- набряк Квiнке
- висипка на шкiрi
- диспепсія
- погiршення функцiї печінки
- Якi твердження відносно обміну пуринів вірні?
- сечова кислота — кінцевий продукт обміну пуринів в організмі людини
- при подагрі урати відкладаються в тканинах
- урати фільтруються в ниркових клубочках
- сечова кислота виводиться через кишечник
- джерелом пуринів є обмін нуклеотидів
Еталони відповідей: 1) 1, 2, 3, 4; 2) 1, 3, 4, 5, 6; 3) 1, 2, 4, 5, 6; 4) 2, 4; 5) 1; 6) 1, 2, 3; 7) 1, 2, 3, 4; 8) 1, 4, 5; 9) 4; 10) 3; 11) 1, 2, 4; 12) 4, 5; 13) 2; 14) 1, 2, 4, 5, 6; 15) 1, 2; 16) 2; 17) 3; 18) 1, 2, 4; 19) 4; 20) 2, 3, 4, 5; 21) 1, 2, 3, 5
Артрити, пов’язані з інфекцією
- Екзоферменти стрептококів:
- стрептолізин О
- стрептолізин S
- стрептокіназа
- урокіназа
- стрептогіалуронідаза
- протеаза
- Основним етiологiчним фактором ГРЛ є:
- вiрусна iнфекцiя
- вiрусно-бактерiальна iнфекцiя
- змiшана iнфекцiя
- стафiлококова iнфекцiя
- iнфекцiя В-гемолiтичним стрептококом групи А
- Успадкування яких антигенiв системи HLA сприяє розвитку ГРЛ?
- HLA-B27
- HLA-D35
- HLA-B40
- HLA-А4
- HLA-A11, -B5, -DR2
- Генетично детермiнована перехресна реактивнiсть сприяє розвитку ГРЛ за наявностi її мiж:
- Т-лiмфоцитами й антигенами легень
- В-лiмфоцитами й антигенами селезiнки
- макрофагами й антигенами стрептококу
- еозинофiлами i тканинами серця
- В-лiмфоцитами, тканинами серця й антигенами стрептококу
- Який вiдсоток осiб, що перенесли гостру стрептококову iнфекцiю, хворiє на ГРЛ?
- 90–93%
- 50–54,8%
- 40–41,7%
- 20–26,3%
- 0,3–3%
- Наявнiсть яких антитiл у високих титрах свiдчить про стрептококову iнфекцiю при ГРЛ?
- противiрусних Коксакi В3 i В4
- iмуноглобулiнiв А
- iмуноглобулiнiв Е
- iмуноглобулiнiв G
- АСЛ-0, АСГ, АСК, анти-ДНКази В
- На якiй стадiї морфологiчного розвитку ГРЛ можливi зворотнiсть або обмеженiсть процесу?
- мукоїдного набухання
- фiбриноїдних змiн
- фiбриноїдного некрозу
- утворення Ашоф-Талалаєвських гранульом
- склерозування
- Якi вiковi групи населення найчастiше хворіють на ГРЛ?
- дитяча
- пiдлiткова
- похилого вiку
- середнього вiку
- В осіб якої статi найчастiше виявляють ГРЛ?
- чоловiки i жiнки однаковою мiрою
- чоловiки в 1,7 раза частiше
- жiнки в 4–5 разiв частiше
- чоловiки в 2,1 раза частiше
- жiнки в 2,5–3 рази частiше
- Назвiть характернi ознаки ГРЛ:
- гострий початок, лихоманка та полiсиндромнiсть
- швидке зростання та швидкий зворотний розвиток симптомiв хвороби
- цикл розвитку клiнiчних проявiв атаки триває 2–3 мiс
- високi лабораторнi показники активностi
- летючі поліартралгії
- Склад ревматичної гранульоми:
- базофільні клітини гістіоцитарного походження
- гігантські багатоядерні клітини
- лімфоїдні, плазматичні й гладкі клітини
- нейтрофіли
- лейкоцити
- Якi ферменти стрептококу А мають кардiотоксичний ефект?
- стрептолiзин S
- стрептокiназа
- гiалуронiдаза
- стрептолiзин S, стрептокiназа
- стрептолiзин О
- Вкажiть великi дiагностичнi критерiї ГРЛ?
- кардит, дерматит, енцефалiт
- полiартрит, полiсерозит, ендокардит
- хорея, полiсерозит, пiдвищення температури тіла, потовиділення
- пiдвищення температури тіла, адинамiя, астенiчний синдром, поява СРБ
- кардит, полiартрит, хорея, кiльцеподiбна еритема, пiдшкiрнi вузлики
- Розвиток якої вади серця найбiльш характерний для ГРЛ?
- недостатнiсть мiтрального клапана
- стеноз лiвого АВ-отвору
- стеноз гирла легеневої артерiї
- стеноз правого АВ-отвору
- недостатнiсть аортального клапана
- Яка симптоматика найбільш характерна для ревматичного мiокардиту?
- послаблення звучностi I тону, подовження iнтервалу P–Q
- систолiчний шум на верхiвцi, скорочення iнтервалу P–Q
- систолiчний шум на аортi, скорочення iнтервалу P–Q
- акцент II тону на аортi, подовження iнтервалу P–Q
- шум тертя перикарду, скорочення iнтервалу P–Q
- Визначте характернi шкiрнi прояви ГРЛ:
- вузлувата еритема
- еритема на щоках
- періорбiтальний набряк з пурпурною еритемою
- депiгментацiя
- кiльцеподiбна еритема, пiдшкiрнi вузлики
- Визначіть характернi ознаки ревматичної хореї:
- гiперкiнези, якi посилюються при хвилюванні
- мiопатiя
- гемiпарез
- гемiплегiя
- ознаки фунікулярного мiєлозу
- Яким антибактерiальним препаратам надається перевага при ГРЛ та ХРХС у разі непереносимостi пенiцилiну?
- сульфанiламiди
- нiтроксолiн
- тетрациклiн
- макроліди
- стрептомiцин
- Механiзм передачi iнфекцiї при хворобi Лайма:
- повiтряно-крапельний
- статевий
- фекально-оральний
- укуси комарiв
- укуси клiщiв
- Дiагностичними критерiями хвороби Лайма є:
- артрити мiжфалангових суглобiв ступней з дифузним їх припуханням у виглядi «сосисок»
- негативний тест на РФ
- рецидивуючий моно- або олiгоартрит i мiгруюча еритема
- полiартрит з вираженим ексудативним компонентом запалення, сильним болем
- Дiагностичнi лабораторнi тести при хворобi Лайма:
- дослiдження загального аналiзу кровi
- бiохiмiчне дослiдження кровi
- бактерiологiчне дослiдження кровi, сечi, калу
- серологiчне дослiдження кровi
- Для хвороби Лайма не характерно:
- наявнiсть мiгруючої еритеми
- загальнi симптоми (озноб, слабкiсть, лихоманка, головний бiль, мiалгiї, лiмфаденопатiя)
- рецидивуючий моно- або олiгоартрит
- прогресуюче ураження дрібних суглобів кистей та стоп
- неврит черепних нервiв, менiнгiт, мiєлiт
- Для хвороби Уїпла характерно:
- поєднання рецидивуючого моно- або олiгоартриту з мiгруючою еритемою
- поєднання мiгруючого артриту та прогресуючого ураження тонкої кишки (дiарея, стеаторея)
- поєднання моно- або олiгоартриту з дисбiозом
- виникнення несиметричного артриту та кон’юнктивiту пiсля сечостатевої інфекцiї
- виникнення артриту та кон’юнктивiту пiсля кишкової iнфекцiї
- Пiд «ревматизмом Понсе» розумiють:
- асиметричний моно- або олiгоартрит, або «летючий полiартрит»
- реактивний полiартрит у хворих на вiсцеральний туберкульоз
- ОА з реактивним синовiтом
- хондроматоз суглобiв
- iнтермiтуючий гiдроартроз
- Для «ревматизму Понсе» не характерні наступнi дiагностичнi ознаки:
- стiйкий артрит без деформацiй
- вiдсутнiсть ерозивних змiн на рентгенограмi суглобiв
- туберкульоз в анамнезi або в теперiшний час
- наявнiсть переважно лейкоцитiв в синовiальнiй рiдинi
- ефективнiсть протитуберкульозної терапiї
- Вiдмiнними ознаками вiрусних артритiв є:
- артрит асиметричний з летючим характером
- повна зворотнiсть артриту через 1–6 мiс
- зв’язок з вiрусною iнфекцiєю
- схильнiсть до розвитку однобічного сакроiлеїту
- періартрити (тендосиновiти, синдром карпального каналу)
- Для артритiв при паразитарних iнвазiях не характерно:
- позитивний тест на РФ
- пiдвищення вмiсту еозинофiлiв у кровi
- пiдвищення рiвня IgE в синовiальнiй рiдинi
- виявлення паразитiв в калi
- артралгiї
- Найбiльш частими етiологiчними факторами гострих iнфекцiйних артритiв є:
- гриби
- вiруси
- стафiлококова iнфекцiя
- стрептококова iнфекцiя
- ентеробактерiї
- Фактори, якi сприяють розвитку iнфекцiйного артриту:
- похилий i старечий вiк
- зловживання алкоголем
- злоякiснi пухлини
- наявнiсть первинного або вторинного iмунодефiциту
- хвороби кровi
- дитячий вік
- Назвiть характерний клiнiчний варiант iнфекцiйного артриту:
- поступовий початок, помiрнi мiсцевi прояви
- гострий початок, припухлiсть, гiперемiя i гiпертермiя на фонi загальнотоксичних явищ
- гострий початок, слабо вираженi мiсцевi ознаки запалення
- гострий початок без суглобового синдрому
- Вкажiть необхiднi специфiчнi лабораторнi дослiдження при дiагностицi iнфекцiйного артриту:
- загальний аналiз кровi
- бiохiмiчне дослiдження кровi
- iмунологiчне дослiдження кровi
- бактерiологiчне дослiдження кровi i синовiальної рiдини
- виявлення HLA-B27
- Дiагноз iнфекцiйного артриту пiдтверджується:
- асиметричним моноолiгоартритом
- швидким розвитком кiсткової деструкцiї
- позитивними серологiчними реакцiями
- ефектом антибактерiальної терапiї
- спонтанним одужанням
- Високий лейкоцитоз в синовiальнiй рiдинi є характерним для:
- хвороби Бехтєрева
- ПсА
- подагричного артриту
- септичного артриту
- хондрокальцинозу
- Мутна синовiальна рiдина визначається при:
- хондрокальцинозi
- СЧВ
- ПсА
- септичному артритi
- остеоартритi
Еталони відповідей: 1) 1, 2, 3, 4, 5; 2) 5; 3) 5; 4) 5; 5) 5; 6) 5; 7) 1; 8) 1, 2; 9) 5; 10) 1, 2, 3, 4; 11) 1, 2, 3; 12) 5; 13) 5; 14) 2; 15) 1; 16) 1, 5; 17) 1; 18) 4; 19) 5; 20) 3; 21) 4; 22) 4; 23) 2; 24) 2; 25) 4; 26) 1, 2, 3, 4; 27) 1; 28) 3, 4; 29) 1, 2, 3, 4, 5; 30) 2; 31) 4; 32) 1, 2, 3, 4; 33) 4; 34) 4
Системний червоний вовчак
- Пік захворюваності на СЧВ припадає на вік:
- 0–20 років
- 20–40 років
- 40–60 років
- 60–80 років
- Серед хворих на СЧВ у віці 20–40 років кількість жінок становить:
- 50%
- 60%
- 70%
- 80%
- 90%
- Провокуючі фактори розвитку СЧВ:
- інсоляція
- гормональні перебудови
- стреси
- ангіна
- вакцинація
- Основнi фактори, якi вважаються етiологiчними для розвитку СЧВ:
- спадкова схильнiсть i вплив перенесеної вiрусної iнфекцiї
- iнсоляцiя, ультрафiолетове опромiнення пiд час фiзiотерапевтичних процедур
- введення сироваток i вакцин
- рентгенiвське опромiнення i введення радiоактивних iзотопiв
- перенесенi бактерiальнi iнфекцiї
- Назвiть препарати, якi сприяють виникненню медикаментозного СЧВ:
- преднiзолон, полькортолон, дексаметазон, допегіт, метилдопа
- апресин, прокаїнамiд, хлорпромазин
- диклофенак натрiю
- інгібітори АПФ
- Про роль вiрусної iнфекцiї в етiологiї СЧВ свiдчать:
- наявнiсть лiмфоцитотоксичних антитiл в сироватцi кровi
- тромбоцитопенiя
- виявлення LE-клiтин
- лiмфопенiя
- пiдвищення рiвня ЦIК в сироватцi кровi
- Пiдвищення яких антигенiв гiстосумiсностi відмічають при СЧВ?
- HLA-B27
- HLA-В5
- HLA-А11, -В7, -В35, -С4, -DR3
- DR1
- DР7
- Що лежить в основi iмунорегулюючих порушень при СЧВ?
- дефiцит супресорної функцiї Т-лiмфоцитiв та антиiдеотипiв i посилення утворення аутоантитiл проти ядер клiтин i їх компонентiв
- дисбаланс Т-хелперiв i Т-супресорiв
- накопичення ЦIК
- порушення фагоцитозу
- змiни в системi комплементу
- Розвиток iмунного запалення при СЧВ пов’язаний з дiєю:
- антитiл до ДНК
- iмунних комплексiв
- iнтерлейкiну-10
- комплементу
- iмуноглобуліну Е
- Який тип антитіл із перерахованих є маркером медикаментозного СЧВ?
- до базальної мембрани судин
- до РФ
- до рибонуклеопротеїду
- до гiстону
- АНФ
- Патогенетична роль iмунних комплексiв при СЧВ пiдтверджується:
- загальною гiперкоплементемiєю
- зниженням окремих компонентiв комплементу-С1, -С3, -С4
- збiльшення рiвня великодисперсних ЦІК у сироватцi кровi
- пригнiчення Т-клiтинного iмунiтету
- гiперфункцiя В-клiтин
- Особливостями патоморфологiчних змiн при СЧВ є:
- системна дезорганiзацiя сполучної тканини
- генералiзоване ураження мiкроциркуляторного русла
- патологiя ядер клiтин — деформацiя ядер i збіднiння їх хроматином, утворення гематоксилiнових тiлець
- плазмоклiтинна та макрофагальна реакцiя
- явища диспротеїнозу
- Найбiльш частий варiант перебiгу СЧВ:
- гострий
- пiдгострий
- хронiчний
- синдром дискоїдного червоного вовчака
- синдром Верльгофа
- Класичною дiагностичною трiадою при СЧВ є:
- артрит, астеновегетативний синдром, пiдвищення температури тіла
- шкiрний висип, трофiчнi розлади, швидке схуднення
- дерматит, артрит, полiсерозит
- синдром Рейно, артрит, пiдвищене випадiння волосся
- ураження нервової системи, схуднення, прискорення ШОЕ
- При лiкуваннi суглобового синдрому у хворих на СЧВ перевага надається:
- диклофенаку
- фенілбутазону
- ібупрофену
- німесуліду
- мiсцевiй терапiї гелями НПЗП
- При СЧВ частiше відмічають наступні ураження серця:
- перикардит зі спайками та невеликою кiлькiстю рiдини
- iнфаркт мiокарда
- розшаровуюча аневризма аорти
- iдiопатичний гiпертрофiчний субаортальний стеноз
- мiксома лiвого передсердя
- Ураження ЦНС при СЧВ:
- відмічають у бiльше нiж у половини хворих
- у всiх хворих
- не виявляють
- Назвiть характернi ознаки ендокардиту Лiбмана — Сакса:
- ураження трикуспiдального клапана
- злоякiсний перебiг
- ураження аортального клапана
- абактерiальний варукозний ендокардит, який частiше локалiзується на мітральному клапанi
- Якi особливостi синдрому Рейно при СЧВ?
- одна iз самих раннiх i частих ознак хвороби
- стiйкий синдром Рейно є прогностично несприятливою ознакою
- нестiйкий синдром Рейно може бути показником доброякiсного перебiгу процесу
- не характерний для СЧВ
- Якi ознаки можуть вказувати на люпус-нефрит?
- нефритичний або нефротичний синдром у хворих із суглобовим синдромом, лихоманкою, стiйким збiльшенням ШОЕ
- ураження нирок за типом нефропатiї вагiтних
- розвиток гострого нефротичного синдрому
- наявнiсть сечового синдрому та злоякiсної гiпертензiї
- наявнiсть ознак пiєлонефриту жiнок молодого вiку зi стiйкою артерiальною гiпертензiєю
- Якi особливостi перебiгу СЧВ у чоловiкiв?
- наявнiсть сакроiлеїту при негативному HLA-В27
- характерна велика частота гострого початку хвороби та ураження життєво важливих органiв
- частiше виявляють синдром Шегрена
- велика частота анемiї
- велика частота збiльшення ШОЕ
- З чим пов’язаний розвиток синдрому Верльгофа при СЧВ?
- наявнiсть iнтоксикацiї
- дiя антитромбоцитарних антитiл
- розвиток васкулiтiв
- порушення дозрiвання тромбоцитiв
- порушення ретракцiї кров’яного згустку
- Якi препарати є засобами першої лінії в терапії СЧВ при гострому перебiгу?
- НПЗП
- ГК
- амiнохiнолiновi препарати
- iмуносупресори
- iмуностимулятори
- Добова доза ГК та термiн застосування, необхiднi для лiкування хворих на СЧВ з клiнiкою люпус-нефриту та/або нейролюпусу:
- 1–2 мг/кг/добу протягом 3–6 мiс
- 0,5 мг/кг/добу протягом 2 мiс
- 1–2 мг/кг/добу протягом 2 тиж
- 3 мг/кг/добу протягом 3 мiс
- 1–2 мг/кг/добу протягом 1 мiс
- Якi препарати з групи iмуносупресорів є препаратами вибору при СЧВ з ураженням нирок?
- 6-меркаптопурин
- циклофосфамiд
- метотрексат
- хлорбутин
- мофетилу мікофенолат
- Маркером хронiчного перебiгу СЧВ є:
- полiсерозит
- дискоїдний червоний вовчак, синдром Шегрена, синдром Рейно
- пульмонiт
- синдром Шегрена
- синдром Рейно
- Найбiльше значення у дiагностицi вовчакового гломерулонефриту має:
- iзотопна ренографiя
- бiопсiя нирок
- екскреторна урографiя
- проба Зимницького
- аналiз сечi за Нечипоренком
- Найбiльш несприятливим у прогностичному вiдношеннi серед клiнiчних проявiв СЧВ є:
- ендокардит Лiбмана — Сакса
- люпус-нефрит
- ексудативний плеврит
- нейролюпус
- гематологiчний криз
Еталони відповідей: 1) 2; 2) 5; 3) 1, 2, 3, 5; 4) 1; 5) 2; 6) 1; 7) 3; 8) 1; 9) 1, 2, 3, 4; 10) 4; 11) 2, 4, 5; 12) 1, 2, 3, 4; 13) 2; 14) 3; 15) 1; 16) 1; 17) 1; 18) 4; 19) 1, 2, 3; 20) 1; 21) 1, 2, 5; 22) 2; 23) 2; 24) 1; 25) 2, 5; 26) 2; 27) 2; 28) 5
Системна склеродермія
- Якi вiддiли ШКТ найбiльш часто уражаються при ССД:
- стравохiд
- товстий кишечник
- печiнка
- пiдшлункова залоза
- жовчовивiднi шляхи
- Яка з ознак характерна для вогнищевої склеродермiї?
- склеродактилiя
- синдром Рейно
- дисфагiя
- телеангiектазiя
- змiни на шкiрi у виглядi «удару шаблею»
- Ураження легень, які найчастіше відмічають при ССД:
- базальний пневмосклероз
- вогнищева пневмонiя
- крупозна пневмонiя
- фiброзуючий альвеолiт
- абсцедуюча пневмонiя
- Клінічні форми ССД:
- лімітована
- склеродермія без склеродерми
- перехресний синдром
- дифузна
- ювенільна склеродермія
- все перераховане
- Симптоми характерні для склеродермії без склеродерми:
- відсутнє ущільнення шкіри
- феномен Рейно
- ознаки легеневого фіброзу, гостра склеродермічна нирка, ураження серця, ШКТ
- акросклероз
- наявність антинуклеарних антитіл (Scl-70, антицентромірних, нуклеолярних)
- Ознаки, характерні для дифузної форми склеродермії:
- генералізоване ураження шкіри кінцівок, тулуба, обличчя протягом року
- ранній розвиток вісцеральної патології
- відсутність ураження шкіри
- значна редукція капілярів з формуванням аваскулярних зон
- наявність антитіл до моноізомерази-1 (Scl-70)
- Симптоми, характерні для лімітованої форми склеродермії:
- тривалий період ізольованого феномену Рейно
- обмежене ураження шкіри обличчя, кисті, стоп
- CREST-синдром, розширення капілярів без аваскулярних зон
- наявність антицентромірних антитіл
- раннє ураження нирок
- Який найбiльш характерний синдром облiтеруючої ангiопатiї відмічають при хронiчному перебiгу ССД:
- синдром Рейно
- склеродермiчна нефропатiя
- ураження легеневих судин
- ураження коронарних артерiй
- ураження церебральних судин
- Ураження обличчя при склеродермiї:
- амiмiя
- загострення носа
- мікростомія
- блискуча, напружена шкiра
- симптом «окулярів»
- Якi фактори можуть бути iндукторами у виникненнi ССД?
- тривале переохолодження
- хлорвiнiл, вiнiлхлорид
- дiя кремнiю
- інсоляція
- вiбрацiя
- Найчастiше ураження серця при ССД:
- ендокардит
- мiокардит
- перикардит
- панкардит
- Який тип пневмосклерозу найчастiше виявляють при ССД?
- кiстовидний базальний
- компактний базальний
- кiстовидний дифузний
- компактний дифузний
- Раннi ознаки ССД:
- синдром Рейно
- дисфагiя
- пневмосклероз
- артралгiї
- набряк та ущiльнення шкiри
- Якi характернi змiни для ССД відмічають у пiдшкiрнiй клiтковинi?
- вузлики Гебердена
- тофуси
- вузлики Бушара
- синдром Тiб’єржа — Вейссенбаха
- ревматоїднi вузлики
- Препаратами базової терапiї при ССД є:
- D-пеніциламін
- ГК
- каптоприл
- дезагреганти
- ацетилсалiцилова кислота
- Найбiльш характернi ураження ШКТпри ССД:
- езофагiт
- атонiя кишечнику
- дисбактерiоз кишечнику
- зниження секреторної функцiї шлунка
- зниження секреторної функцiї пiдшлункової залози
- Які препарати найбільш ефективні для лікування злоякiсної гiпертензiї при ССД?
- преднiзолон
- інгібітори АПФ
- блокатори β-адренорецепторів
- дигідропіридинові похідні
- клонідин
- Ураження суглобів та кісток при ССД виявляють у вигляді:
- артритів
- переломів хребців
- поліартралгії, симптому тертя сухожиль
- згинальних контрактур
- акроостеолізу
- Синдром Рейно при ССД характеризується наступними фазами:
- побiлiння та синюшнiсть шкiри
- синюшнiсть шкiри
- мармуровість
- почервонiння та синюшнiсть шкiри
- побiлiння, синюшнiсть та почервонiння шкiри
- Лiкування D-пеніциламіном може ускладнюватися:
- лейкоцитозом
- тромбоцитопенiєю
- лейкопенiєю, тромбоцитопенiєю
- протеїнурiєю
- лейкопенiєю, протеїнурiєю, тромбоцитопенiєю
- Найбільш характерні ураження ШКТ при ССД:
- гіпотонія,стриктура стравоходу, ерозії та виразки
- гіпотонія шлунка
- шлункова кровотеча
- синдром мальабсорбції
- ураження товстої кишки
Еталони відповідей: 1) 1; 2) 5; 3) 1; 4) 6; 5) 1, 2, 3, 5; 6) 1, 2, 4, 5; 7) 1, 2, 3, 4; 8) 1; 9) 1, 2, 3, 4; 10) 1, 2, 3, 5; 11) 3; 12) 1; 13) 1; 14) 4; 15) 1; 16) 1; 17) 2; 18) 1,3,4,5; 19) 5; 20) 5; 21) 1
Антифосфоліпідний синдром
- При АФС у кровi визначаються:
- антитiла до кардiолiпiну
- вовчаковий антикоагулянт
- LE-клітини
- реакцiя Вассермана
- Симптомами АФС є:
- розповсюджений тромбоз
- сiтчате лiведо
- інфаркт печінки
- внутрiшньоутробна загибель плода
- ураження ЦНС
- асептичні некрози кісток
Еталони відповідей: 1) 1,2,4; 2) 1,2,4,5,6
Синдром Шегрена
- Синдром Шегрена супроводжує такі захворювання:
- біліарний цироз печінки
- гепатити
- системні захворювання сполучної тканини
- РА
- хвороба Рейтера
- тиреоїдит Хашимото
- При якому iз перерахованих захворювань найчастiше виявляють синдром Шегрена:
- ССД
- РА
- СЧВ
- полiмiозит
- ОА
- До системних проявiв хвороби Шегрена вiдносяться:
- рецидивуючий неерозивний артрит
- панкреатит
- паротит
- ксеростомiя
- сухiсть бронхiв
- Схема лiкування хвороби Шегрена за наявностi ураження нирок (iмунокомплексний гломерулонефрит):
- преднiзолон 30–40 мг, хлорбутин 4–6 мг
- преднiзолон 30–40 мг
- хлорбутин 4–6 мг
- пульс-терапiя
- гідроксихлорохін
- Якi органи та системи частiше уражуються при хворобi Шегрена?
- надниркова залоза
- слиннi залози
- пiдшлункова залоза
- печiнка
- шкiра
- Найбiльш характернi клiнiчнi ознаки хвороби Шегрена:
- карiєс зубiв
- ксеростомiя, карієс зубів
- спрага
- дисфагiї
- лiмфоаденопатiя
- За наявностi якого клiнiчного синдрому застосовується пульс-терапiя при хворобi Шегрена:
- помiрний мiозит
- легеневий фiброз
- гломерулонефрит
- виразково-некротичний васкулiт
- жовтяниця
- Схема лiкування хвороби Шегрена при ураженнi легень:
- преднiзолон 30–60 мг
- пульс-терапiя
- цитостатики
- преднiзолон 60–90 мг
- преднiзолон 10–15 мг
- Яке ускладнення при синдромi (хворобi) Шегрена є найбiльш небезпечним для життя?
- гнiйний паротит
- нитковий кератит
- злоякiсна лiмфома
- анемiя
- абсцес сльозного мiшка
- Яка клiнiчна ознака частiше відмічається при синдромi (хворобi) Шегрена?
- гiпергаммаглобулiнемiчна пурпура
- синдром Рейно
- гiперпiгментацiя шкiри
- бронхiт
- полiмiозит
- Характернi змiни з боку кровi при синдромi (хворобi) Шегрена:
- анемiя
- збiльшення ШОЕ
- лейкоцитоз
- гiпопротеїнемiя
- тромбоцитопенiя
- Препарати вибору при неускладненому синдромi Шегрена:
- ГК
- НПЗП
- iмунодепресанти
- амiнохiнолiновi
- Схема лiкування хвороби Шегрена за наявностi гiпергаммакрiоглобулiнемiчної пурпури:
- преднiзолон 20–30 мг, циклофосфамiд 200 мг в/м 1 раз на тиждень, пульс-терапiя
- преднiзолон 20–30 мг
- пульс-терапiя
- циклофосфамiд 200 мг в/м 1 раз на тиждень
- iбупрофен 200 мг
- Схема лiкування хвороби Шегрена за наявностi цереброваскулiту:
- преднiзолон 60–100 мг, циклофосфамiд 200 мг в/м 1 раз на тиждень, пульс-терапiя
- преднiзолон 60–100 мг
- пульс-терапiя
- циклофосфамiд 200 мг в/м 1 раз на тиждень
- iндометацин 75 мг
- Схема лiкування хвороби Шегрена за наявностi виразково-некротичного васкулiту:
- преднiзолон 60–100 мг, циклофосфамiд 200 мг в/м 1 раз на тиждень, пульс-терапiя
- преднiзолон 60–100 мг
- пульс-терапiя
- циклофосфамiд 200 мг в/м 1 раз на тиждень
- мелоксикам 7,5 мг
- Для якого iз перелічених захворювань є характерним розвиток карiєсу?
- хвороба Шегрена
- хвороба Рейно
- СЧВ
- дерматомiозит
- рецидивуючий полiхондрит
- Характерна лабораторна ознака при хворобi Шегрена:
- підвищення вмісту СРБ
- виявлення антинуклерних антитіл до SS-A
- LE-клiтини
- лейкоцитоз
- нормальна ШОЕ
- Можливi варiанти перебiгу хвороби Шегрена:
- хронiчний
- гострий
- пiдгострий
- пiдгострий, хронiчний
- гострий, пiдгострий
- Препарати вибору для лiкування пiдгострого перебiгу хвороби Шегрена:
- НПЗП
- цитостатики
- ГКС
- препарати золота
- амiнохiнолiновi препарати
- Який iз системних проявiв частiше відмічають при хворобi Шегрена:
- рецидивуючий неерозивний артрит
- мiозит
- лiмфаденопатiю
- синдром Рейно
- гепатоспленомегалiю
- Найбiльш характерне ураження легень при хворобi Шегрена
- альвеолярний легеневий фiброз
- інтерстиціальна пневмонія
- абсцедуюча пневмонiя
- кандидоз легень
- ексудативний плеврит
- Характерна лабораторна ознака для хвороби Шегрена:
- гiперпротеїнемiя
- лейкоцитоз
- тромбоцитопенiя
- СРБ
- зниження рiвня гаммаглобулiнiв
- З якими з перелічених захворювань частiше проводиться диференцiальна дiагностика хвороби Шегрена?
- РА
- ССД
- лiмфолейкоз
- цукровий дiабет
- хвороба Микулича
Еталони відповідей: 1) 1, 2, 3, 4, 6; 2) 1; 3) 1; 4) 1; 5) 2; 6) 2; 7) 4; 8) 1; 9) 3; 10) 1; 11) 2; 12) 1; 13) 1; 14) 1; 15) 1; 16) 1; 17) 2; 18) 4; 19) 3; 20) 1; 21) 1,2; 22) 1; 23) 2
Ідіопатичні запальні міопатії (ревматична поліміалгія)
- Для хворих на РПМ характерним є:
- бiль при активних рухах в плечових i кульшових суглобах
- висока ШОЕ
- висока ефективнiсть преднiзолону
- біль в дистальних відділах кінцівок
- Для РПМ характерно:
- ураження серця
- вiк хворого старше 50 рокiв
- наявнiсть болю у шиї, плечовому та тазовому поясi
- двобiчна локалiзацiя болю, висока ШОЕ
- втомлюванiсть, слабкiсть, лихоманка, анемiя
- Вкажiть ознаки, загальнi для РПМ та полiмiозиту:
- лейкоцитоз
- змiни на електромiограмi
- наявнiсть мiастенiї
- пiдвищення вмiсту КФК у сироватцi кровi
- наявнiсть креатинурiї
- При диференцiальнiй дiагностицi РПМ необхiдно виключати:
- гострий панкреатит
- плечелопатковий періартрит
- полiмiозит
- мієломну хворобу
- гiпотиреоз
- В лiкуваннi РПМ головна роль належить:
- ГК у середнiх дозах
- ГК у високих дозах
- НПЗП
- анальгетикам
- iмуносупресантам
- Яку патологію часто супроводжує РПМ:
- гігантоклітинний артерiїт
- артерiальну гiпертензiю
- СЧВ
- ССД
- дерматополіміозит
- РПМ — це:
- захворювання, що належить до системного ураженням сполучної тканини
- неспецифiчна реакцiя м’язiв на iнтоксикацiю
- вiковi змiни в м’язах
- варiант перебiгу ревматизму
- запальні зміни в скелетних м’язах
- Яка лабораторна ознака є найбiльш типовою для РПМ:
- пiдвищення ШОЕ
- анемiя
- лейкоцитоз
- еозiнофiлiя
- тромбоцитопенiя
- НА РПМ найчастіше хворiють:
- жінки віком старше 55 років
- жiнки дiтородного вiку
- пiдлiтки
- чоловiки середнього вiку
- люди всiх вiкових груп
Еталони відповідей: 1) 1,3; 2) 2,3,4,5; 3) 1; 4) 2,3; 5) 1; 6) 1; 7) 1; 8) 1; 9) 1
Змішане захворювання сполучної тканини
- Яке iз захворювань печiнки близьке за своєю природою до системних захворювань сполучної тканини?
- аутоімунний гепатит
- гепатит як прояв гемохроматозу
- мiкронодулярний цироз печiнки
- ураження печінки при хворобі Вільсона
- До системних уражень сполучної тканини вiдносяться такі нозологiчнi одиницi, крiм:
- ОА
- дерматомiозиту
- ССД
- СЧВ
- дифузного еозинофiльного фасциту
- Яке захворювання було вперше описане Шарпом та спiвавторами?
- дерматомiозит
- СЧВ
- ССД
- РА
- ЗЗСТ
- Хто частiше хворiє на ЗЗСТ?
- чоловiки
- особи похилого вiку
- дiвчата
- хлопчики
- жiнки середнього вiку
- Яка iз перерахованих ознак найчастiше відмічають при хворобi Шарпа?
- ураження кiсток
- синдром Рейно
- алопецiю
- ураження серозних оболонок
- лихоманку
- Який iз перерахованих органiв найрідше уражується при хворобi Шарпа?
- стравохiд
- суглоби
- нирки
- легенi
- м’язи
- Найбiльш характерна лабораторна ознака при хворобi Шарпа
- гiпохромна анемiя
- лейкопенiя
- реакцiя Вассермана
- антитiла до рибонуклеопротеїду
- протеїнурiя
- Найбiльш характерне ураження кистей при хворобi Шарпа
- iшемiчнi некрози
- «сосископодiбний» набряк пальцiв
- петехiальний висип
- пiгментацiя шкiри
- остеолiз кiнцевих фаланг
- Найбiльш характернi ознаки ураження м’язiв при хворобi Шарпа
- біль i м’язова слабкiсть
- ущiльнення м’язiв
- набряк м’язiв
- атрофiя м’язiв
- кальциноз м’язiв
- Найбiльш ефективна група препаратiв при ураженнi м’язiв у пацієнтів із хворобою Шарпа
- НПЗП
- амiнохiнолiновi препарати
- антибiотики
- ГК
- антигiстамiннi препарати
- Наявність яких із перелічених ознак при хворобi Шарпа є критерієм несприятливого перебiгу захворювання?
- ураження м’язiв
- синдром Рейно
- ураження стравоходу
- фіброзуючий альвеоліт
- ураження нирок
- Який iз синдромiв є несприятливим для прогнозу життя при хворобi Шарпа?
- синдром Шегрена
- полiнейропатiї
- суглобовий синдром
- ущiльнення шкiри
- фiброзуючий альвеолiт з легеневою гiпертензiєю
- Критерiї синдрому Шарпа:
- гломерулонефрит
- набряк кистi, синдром Рейно
- мiозит
- синовiт, набряк кистi
- високi титри антитiл до рибонуклеопротеїду
- При якому iз перерахованих захворювань розвивається симптом гiперв’язкостi:
- хвороба Шегрена
- РА
- СЧВ
- ССД
- рецидивуючий полiхондрит
- Для дифузного еозінофільного фасциту характерно:
- переважне ураження фасцiй
- судиннi розлади
- порушення трофiки
- синдром Рейно
- вiсцеропатiї
- З яким захворюванням найбiльш часто необхiдно диференцiювати еозинофiльний фасцит:
- ССД
- дерматомiозит
- тендовагiнiт
- РА
- РПМ
- Основний лiкарський засiб при лiкуванні дифузного еозинофiльного фасциту:
- ГК
- НПЗП
- D-пеніциламін
- диметилсульфоксид
- Для дифузного еозинофiльного фасциту характерно:
- еозинофiлiя i гiпергаммаглобулiнемiя
- лейкоцитоз
- еозинофiлiя
- гiпербiлiрубiнемiя
- пiдвищенння ШОЕ
- Найбiльш раннiм проявом дифузного еозинофiльного фасциту є:
- вiдчуття стягування шкiри переважно нижнiх кiнцiвок
- вiдчуття свербежу
- болюче затвердiння шкiри стоп
- артралгiї
- вiсцеропатiї
- Основними провокуючими факторами дифузного еозинофільного фасциту є:
- гострi iнфекцiї
- алергiчнi реакції
- травми
- фізичне перенапруження
- перегрiвання
- Для якого захворювання з групи системних захворювань сполучної тканини характерна лімфоїдно-макрофагальна iнфiльтрацiя з плазмоцитозом та еозинофiлiєю в фасцiї?
- ССД
- СЧВ
- дерматомiозит
- рецидивуючий полiхондрит
- дифузний еозинофільний фасцит
- Яка з наведених ознак рідко відмічається при дифузному еозинофільному фасциті?
- артралгія
- міалгія
- гіперпігментація шкіри
- синдром запястного каналу
- синдром Рейно
- Змiни шкiри за типом «апельсинової шкiрки» характерні для:
- еозинофiльного фасциту
- склеродерми Бушке
- вузлуватої еритеми
- ССД
- вогнищевої склеродермiї
Еталони відповідей: 1) 1; 2) 1; 3) 5; 4) 5; 5) 2; 6) 3; 7) 4; 8) 2; 9) 1; 10) 4; 11) 4,5; 12) 5; 13) 2, 3, 4, 5; 14) 1; 15) 1; 16) 1; 17) 1; 18) 1; 19) 1; 20) 1, 2, 3, 4; 21) 5; 22) 5; 23) 1
Системні васкуліти
- До системних васкулiтiв вiдносяться захворювання:
- хвороба Бюргера
- хвороба Шенляйна — Геноха
- хвороба Такаясу
- хвороба Педжета
- хвороба Хортона
- Що лежить в основi розвитку клiнiчних симптомiв системних васкулiтiв?
- ураження елементiв судинної стiнки
- пошкодження ендотелiю
- порушення мiкроциркуляцiї
- iшемiчне пошкодження тканин
- синовіти
- До АНЦА-залежних васкулiтiв належать:
- геморагiчний васкулiт
- синдром Черджа — Стросс
- гранулематоз Вегенера
- мікроскопічний поліангіїт
- хвороба Кавасакі
- Для облiтеруючого тромбангiїту типовим є ураження:
- середнiх, дрiбних артерiй та вен
- великих артерiй
- капiлярiв
- аорти
- Для облiтеруючого тромбангiїту типовими є наступнi морфологiчнi змiни в судинах:
- продуктивнi ендоваскулiти i деструктивно-продуктивнi тромбоваскулiти
- периваскулiт
- некротизуючий васкулiт
- гранулематозно-некротизуючий васкулiт
- панваскулiт
- Клінічними ознаками у бiльшостi хворих на облiтеруючий тромбангiїт є:
- перемiжна кульгавість
- бiль у суглобах
- бiль у попереку
- цiаноз нижнiх кiнцiвок
- оніміння та мерзлякуватість пальців
- Для вiсцерального варiанту облiтеруючого тромбангiїту типовим є часте ураження судин:
- вiнцевих
- пiдшлункової залози
- церебральних
- ниркових
- печiнкових
- Для хвороби Хортона характерним є ураження:
- вен
- капiлярiв
- судин басейну сонної артерiї
- стегнових артерiй
- артерiол
- Для хвороби Хортона характерними є такі ознаки:
- послаблення пульсації скроневих артерій
- слiпота
- синдром дуги аорти
- гангрена кiнцiвок
- мiалгiі
- Ознаками активностi запального процесу при хворобi Хортона є:
- лихоманка
- збiльшення ШОЕ
- диспротеїнемiя з альфа-2-гiперглобулiнемiєю
- наявнiсть тяжких клiнiчних проявiв
- початок хвороби у віці старше 50 років
- Вкажіть, які твердження вірні для неспецифiчного аортоартерiїту:
- починається у віці до 50 років
- виявляють антиаортальні антитіла
- відмічають сегментарні стенози й оклюзії судин
- можлива роль мікобактерій туберкульозу
- патогенез пов’язаний з АНЦА
- Для пурпури при геморагiчному васкулiтi характерні наступні ознаки:
- зв’язок із тромбоцитопенiєю
- ортостатизм
- рецидивування
- симетричнiсть
- висип визначається при пальпацiї
- Якi твердження вiдносно гранулематозу Вегенера є вiрними?
- деструкцiя тканин верхнiх та нижнiх дихальних шляхiв
- гранулематозний васкулiт
- цитоплазматичнi антинейтрофiльнi антитiла
- перебіг гострий, підгострий, хронічний
- уражуються артерії м’язового типу та мікроциркуляторне русло
- частіше уражується аорта
- Якi ознаки характерні для гранулематозу Вегенера?
- протеїнурія
- гнiйний синусит
- iнфiльтрати в легенях, схильні до розпаду
- виявлення антитiл до цитоплазми нейтрофiлiв
- виявлення АНФ
- Для гранулематозу Вегенера характерним є некротизуючий гранулематозний васкулiт:
- судин верхнiх дихальних шляхiв
- судин нирок
- судин нижнiх кiнцiвок
- судин легень
- судин серця
- Дiагностичними критерiями гранулематозу Вегенера є наступнi ознаки:
- виразково-некротичний процес слизової оболонки дихальних шляхiв
- стiйкий нежить із серозно-гнiйним видiленням
- інфільтрати при рентгенографії легень
- полiартрит
- сечовий синдром
- Який показник найбiльш достовiрно пiдтверджує дiагноз мiкроскопiчного полiангiїту?
- залiзодефiцитна анемiя
- лейкоцитоз
- антитiла до нейтрофiлiв,що реагують з протеїназою-3
- прискоренна ШОЕ
- антитiла до нейтрофiлiв,що реагують з мiєлопероксидазою
- Для мiкроскопiчного полiангiїту характерним є ураження:
- артерiй середнього калiбру
- артерiол, капiлярiв шкiри, легень та нирок
- аорти та гiлок, якi вiд неї вiдходять
- капiлярiв ЦНС
- артерiй верхнiх дихальних шляхiв
- Для хвороби Кавасакi характерні:
- двобiчний кон’юнктивiт
- гострий негнiйний лiмфаденiт
- ураження слизових оболонок порожнини рота
- ураження шкiри кiнцiвок
- двoбiчний отит
- Які твердження відносно хвороби Кавасакi вірні:
- зазвичай хворіють діти
- типовим є ураження судин коронарних, ниркових
- це системний васкулiт
- характерно виникнення аневризм
- відмічається генералізована лімфаденопатія
- При гранулематозі Вегенера мофетилу мікофенолат використовують:
- у дозі 2 г/добу
- для підтримки ремісії
- не використовують
- при швидкому прогресуванні хвороби
- при полінейропатії
- Клiнiчною трiадою синдрому Бехчета є:
- рецидивуючий афтозний стоматит, гломерулонефрит, кон’юнктивiт
- гломерулонефрит, артрит, кардит
- рецидивуючий афтозний стоматит, виразково-некротичнi змiни на статевих органах, увеїт
- кардит, артрит, гепатит
- увеїт, артрит, полiсерозит
- Вкажіть найбільш типове ураження суглобів для синдрому Бехчета:
- моноартрит
- поліартрит
- сакроiлеїт
- ерозивно-деструктивнi змiни тканин суглобiв
- артрити кульшових суглобів
- Які твердження відносно хвороби Бехчета є вірними:
- доведена роль інфекції в розвитку хвороби
- частіше відмічають за наявності HLA-B51
- в біоптатах шкіри виявляють васкуліт венул, капілярів, артеріол
- при енцефаліті відмічають пролiферацiю мiкроглiї, астроцитiв
- венозних тромбозів не виявлено
- Який дiагностичний критерiй хвороби Бехчета є обов’язковим:
- афтознi виразки в ділянцi статевих органiв
- шкiрний васкулiт
- рецидивуючий афтозний стоматит
- увеїт
- синовiт
- До дiагностичних критерiїв еозинофiльного гранулематозного васкулiту належать:
- гiпереозинофiлiя
- мононейропатiя
- еозинофiльнi iнфiльтрати в легенях
- синусити
- рецидивуючий афтозний стоматит
- З яким гепатотропним вірусом пов’язують розвиток есенціального крiоглобулiнемiчного васкулiту?
- вiрус гепатиту В
- вiрус гепатиту С
- вiрус гепатиту А
- вiрус гепатиту D
- Для ВП характерним є ураження:
- артерiй середнього та рідше дрiбного калiбру
- капiлярiв
- венул
- великих артерiй
- аорти
- Які ураження серця типові для ВП?
- коронарит
- безбольовий інфаркт міокарда
- аритмії
- ендокардит
- вада серця
- Бронхоспастичний синдром є типовим для дебюту:
- синдрому Черджа — Стросс
- хвороби Хортона
- хвороби Такаясу
- ВП
- мiкроскопiчного полiангiїту
- Яка ознака з перелічених є найбiльш характерною для ВП?
- полiнейропатiя
- iнфаркт мiокарда
- iнтерстицiальний пневмонiт
- артрити
- порушення функцiї печiнки
- Вкажiть типовi ознаки при поєднаннi ВП з хронiчним гепатитом В:
- ураження шкiри, печiнки, гiпокомплементемiя
- фiброзуючий альвеолiт
- персистування HBsAg
- лихоманка
- ураження суглобів
- Яке ураження шкiри при ВП вiднесено до класифiкацiйних критерiїв?
- пурпура
- iндурацiя
- виразки
- сiтчасте лiведо
- Які ураження нирок типові для ВП:
- васкуліт міждолевих артерій
- інфаркт нирки
- розрив аневризм ниркових судин
- гломерулонефрит
- пієлонефрит
- Для синдрому Черджа — Стросс характерним є:
- астма
- еозинофiлiя
- ураження артерiй дрібного калібру
- рецидивуючі синусити
- еозинофільний гастроентерит
- ураження великих судин
- Вкажіть, які ураження серця відмічають при синдромi Черджа — Стросс:
- порушення ритму
- ваду серця
- бiлатеральну блокаду нiжок пучка Гiса
- недостатнiсть кровообiгу
- перикардит
- Якi твердження вiдносно синдрому Черджа — Стросс вiрнi?
- характерна пурпура
- наявнiсть проявiв алергiї в анамнезi
- часте ураження нирок
- характерна полінейропатія
- типові легеневі інфільтрати
- Хворим на ВП при виявленні маркерів реплікації HBV протипоказано:
- преднiзолон
- плазмаферез
- вiдарабiн
- циклофосфамiд
- iнтерферон
Еталони відповідей: 1) 1, 2, 3, 4, 5; 2) 1, 2, 3, 4; 3) 2, 3, 4; 4) 1; 5) 1; 6) 1, 3, 4; 7) 1, 3; 8) 3; 9) 1, 2, 3, 5; 10) 1, 2, 3, 4; 11) 3; 12) 2, 3, 4, 5; 13) 1, 2, 3, 4, 5; 14) 1, 2, 3, 4; 15) 1, 2, 4, 5; 16) 1, 2, 3, 5; 17) 5; 18) 2; 19) 1, 2, 3, 4; 20) 1, 2, 3, 4; 21) 1, 2; 22) 3; 23) 2; 24) 1, 2, 3, 4; 25) 3; 26) 1, 2, 3, 4; 27) 2; 28) 1; 29) 1, 2, 3; 30) 1; 31) 1, 2, 4, 5; 32) 1, 3; 33) 4; 34) 1, 2, 3; 35) 1, 2, 3, 4, 5; 36) 1, 3, 4, 5; 37) 1, 2, 4, 5; 38) 4
Остеоартроз
- Складовi частини суглобового хряща:
- протеоглiкани
- Т-лімфоцити
- колаген
- гіалуронова кислота
- хондроцити
- Основні структурні складові матриксу хряща:
- протеоглікани
- гіалуронова кислота
- ліпіди
- колаген
- агрекан
- Якi цитокіни сприяють деструкції хряща при ОА?
- інтерлейкін-1B
- туморозний некротичний фактор-L
- інтерлейкін-17
- інтерлейкін-18
- трансформуючий фактор росту-B
- Основними етiологiчними факторами ОА є всі перелічені, за винятком:
- дисплазiї
- порушення статики
- травми суглоба
- функцiонального перевантаження суглоба
- порушення вуглеводного обмiну
- Якi рентгенологiчнi ознаки характерні для ОА?
- остеосклероз й остеофiтоз
- звуження суглобової щiлини
- остеолiз епiфiзiв
- крупнi кiсти в субхондральнiй кiстцi
- ОП, багаточисленнi узури
- Основними причинами розвитку ОА є:
- механiчне перевантаження суглоба
- посилене заняття спортом
- ожирiння
- порушення нормальної конгруентностi суглобових поверхонь хряща
- перенесене інфекційне захворювання
- Якi суглоби частiше уражуються при ОА?
- кульшовi, колiннi
- мiжхребцевi суглоби
- проксимальнi мiжфаланговi суглоби
- лiктьовi, плечовi суглоби
- груднино-ключичний суглоб
- Симптом «блокади суглоба» при ОА проявляється:
- зменшенням вираженості болю в суглобах
- раптовим сильним болем, який обмежує рухи
- поступовим наростаючим болем
- нiчним болем
- крепiтацiєю при рухах в суглобах
- Больовий синдром при артрозах характеризується:
- посиленням болю у спокої
- посиленням болю в нiчний час
- зменшенням вираженості болю при фiзичному навантаженнi
- появою болю при навантаженнi на суглоб
- немає чiтких взаємозв’язаних причин
- Стартовий біль у хворих на ОА можуть вiдмiчати:
- за наявностi реактивного синовiту
- при венознiй гiперемiї i стазi в субхондральнiй частинi кiстки
- при наявностi «суглобової мишi»
- при фiброзi капсули суглобу
- при утвореннi сухожильно-м’язових контрактур
- Симптом «блокади суглоба» при ОА зумовлений:
- защемленням «суглобової мишi»
- наявнiстю реактивного синовiту
- наявнiстю тендобурситу
- стазом в субхондральнiй частинi кiстки
- набряком періартикулярних тканин
- Kлiнiчнi ознаки ОА:
- хруст в суглобах i незначний біль
- швидка втомлюванiсть навколосуглобових м’язiв
- нетривала тугорухливiсть в суглобi
- вранішня скутість більше години
- припухлість суглобів
- Для хворих на ОА найбiльш тяжкими є:
- хода по рiвнiй мiсцевостi
- спуск i пiдйом по сходах
- положення лежачи на спинi
- положення лежачи на боку
- положення лежачи на животi
- Яку клiнiчну форму ОА відмічають найчастіше?
- ОА колiнного суглоба
- ОА гомiлкового суглоба
- артроз дистальних мiжфалангових суглобiв кистей
- артроз першого плеснофалангового суглоба
- Якi рентгенологiчнi ознаки ОА є найбiльш характерними:
- кальцифiкацiя суглобового гiалiнового хряща
- звуження суглобової щiлини, крайовi остеофiти
- великі кiсти в субхондральнiй частинi кiстки
- епiфiзарний ОП
- ерозування суглобових поверхонь
- У хворих на ОА може вiдмiчатися збiльшення ШОЕ:
- при «блокадi» суглоба
- при артрознiй хворобi
- при вторинному синовiтi
- при вираженiй деформацiї суглобiв
- при атрофiї регiонарних м’язiв
- Якi особливостi синовiальної рiдини відмічають при ОА:
- незапальний характер із задовільною в’язкiстю i невеликою кiлькiстю клiтин
- високий цитоз, кристали урату натрiю
- низька в’язкiсть, фагоцити
- мутна, високий цитоз
- висока в’язкiсть, LE-клiтини
- Для ОА характерним є:
- швидкий прогресуючий перебiг
- тривалий, хронiчний перебiг, без рiзко виражених загострень
- хронiчний з рiдкісними загостреннями
- можлива генералiзацiя процесу з кiстково-суглобовою деструкцiєю
- сприятливий перебiг
- Перебiг ОА є найбiльш сприятливим:
- у молодих жiнок
- у чоловiкiв
- у дiтей
- у пiдлiткiв
- у жiнок в перiод менопаузи
- Вiдмiнностями артрозу вiд артриту є:
- стiйка деформацiя суглобу, яка зумовлена кiстковими змiнами
- залучення у процес періартикулярних тканин
- перiодична припухлiсть суглоба
- пiдвищення температури шкiри
- обмеженiсть рухiв
- Раннiми симптомами коксартрозу є:
- біль у колiнних суглобах
- біль механiчного характеру в ділянці кульшового суглоба
- біль у попереку
- біль в ділянці гомiлки
- біль у ступнi
- Назвiть основнi причини гонартрозу:
- часта травматизацiя колiнного суглоба
- порушення осанки
- порушення перемiщення надколiнка
- ожирiння
- заняття спортом
- Яке ускладнення гонартрозу є найбiльш частим:
- вторинний реактивний синовiт
- блокада суглоба
- спонтанний гемартроз
- зовнiшнiй пiдвивих надколiнка
- Якi ознаки вторинного реактивного синовiту відмічають при гонартрозi?
- посилення болю
- поява випоту в порожнинi суглоба
- пiдвищення температури шкiри
- пальпаторна болючiсть по ходу суглобової щiлини
- деформація
- Якою є типова локалiзацiя вузликiв Гебердена?
- проксимальнi мiжфаланговi суглоби
- дистальнi мiжфаланговi суглоби
- лiктьовi суглоби
- перший плеснофаланговий суглоб
- мiжхребцевi суглоби
- Особливостями реактивного синовiту при ОА дистальних мiжфалангових суглобiв є:
- частi рецидиви синовiту
- вiдсутнiсть чiткої видимої причини
- гiперемiя, болючiсть, припухлiсть м’яких тканин
- обмеження рухiв
- всi переліченi ознаки
- Де локалiзуються вузлики Бушара?
- дистальнi мiжфаланговi суглоби
- проксимальнi мiжфаланговi суглоби
- колiннi суглоби
- мiжхребцевi суглоби
- перший плеснофаланговий суглоб
- Вкажiть на особливостi ОА зап’ястно-п’ясткового суглоба великого пальця:
- процес двобiчний
- характерний біль по внутрiшньому краю зап’ястка
- обмеження рухiв у великому пальцi та хруст
- деформацiя кистi
- припухлість та почервоніння
- Якi суглоби найчастіше уражуються при поліостеоартрозі?
- груднино-ключичнi суглоби
- колiннi, кульшовi, дистальнi мiжфаланговi суглоби
- гомiлковостопнi суглоби
- суглоби великого пальця ступнi
- плечовi суглоби
- Дiагностичними критерiями ОА є наступні, за винятком:
- рiзкого обмеження рухливостi суглобу
- болю в суглобах «механiчного» характеру
- симптому «блокади суглоба»
- рентгенологiчного звуження суглобової щiлини, субхондрального остеосклерозу
- повiльного непомiтного початку хвороби
- Дiагноз ОА встановлюють на основi наступних критерiїв:
- переважне ураження суглобiв нiг та дистальних мiжфалангових суглобiв
- симптом «блокади суглоба»
- біль у суглобах при навантаженнi
- нормальнi показники кровi
- ОП на рентгенограмі
- Якими є головнi етiологiчнi фактори ОА?
- порушення нормальної конгруентностi суглобових поверхонь хряща
- функцiональне перевантаження суглоба
- метаболiчнi порушення
- неспецифiчне запалення суглоба
- перенесені ангіни
- Найбiльш часто ОА необхiдно диференцiювати з:
- артритом рiзного походження
- палiндромним ревматизмом
- синдромом Бехчета
- iнтермiтуючим гiдроартрозом
- саркоїдозом
- Головними диференцiйно-дiагностичними ознаками ОА i РА є:
- гострота дебюту захворювання
- вираженiсть мiсцевої запальної активностi
- вираженiсть ранкової скутостi
- наявнiсть ревматоїдних вузликiв
- ураження міокарда
- До «базисних» засобiв для лiкування ОА вiдносяться:
- цитостатики
- НПЗП
- орготеїн
- хондропротектори
- препарати золота
- Розвантаження ураженого суглоба у хворих на ОА здiйснюється:
- тривалою ходьбою
- тривалим стоянням
- пересуванням хворого з палицею, милицями
- спусканням по сходах
Еталони відповідей: 1) 1, 3, 4, 5; 2) 1, 2, 4, 5; 3) 1, 2, 3, 4; 4) 5; 5) 1; 6) 1, 2, 3, 4; 7) 1; 8) 2; 9) 4; 10) 1, 2, 3; 11) 1; 12) 1, 2, 3; 13) 2; 14) 1; 15) 2; 16) 3; 17) 1; 18) 2, 3, 4, 5; 19) 2; 20) 1, 5; 21) 1, 2, 3; 22) 1, 2, 3, 4; 23) 1, 2; 24) 1, 2, 3, 4; 25) 2; 26) 5; 27) 2; 28) 1, 2, 3, 4; 29) 2, 3, 4; 30) 1; 31) 1; 32) 1, 2, 3, 4; 33) 1; 34) 1, 2, 3, 4; 35) 4; 36) 3
Остеопороз
- Який із ГК-препаратiв найменше викликає ОП, мiопатiю та затримку рiдини:
- гiдрокортизон
- тріамцинолон
- дексаметазон
- беклометазон
- метилпреднiзолон
- Діагностичні критерії ГК-ОП:
- швидке зниження МЩКТ, особливо в перші 6–12 міс терапії ГК
- після припинення прийому ГК мінеральна щільність може відновлюватися
- переломи кісток скелету частіше відмічають у хребті та кульшовій кістці
- у двох третин пацієнтів із ГК-ОП переломи хребців безсимптомні
- після відміни ГК мінеральна щільність не відновлюється
- Профілактика ГК-ОП:
- оптимізація ГК-терапії (відміна або максимально можливе зниження дози ГК)
- зміна стилю життя (відмова від тютюнопаління, вживання алкоголю, регулярні фізичні вправи, їжа з високим вмістом кальцію та вітаміну D)
- масаж та мануальна терапія
- уникати падінь, прийому снодійних та седативних препаратів, лікування артеріальної гіпертензії та аритмій
- застосування комбінації кальцію та вітаміну D у всіх хворих, які приймають ГК більше 3 міс
- Який антиостеопоротичний препарат, показаний для лікування ГК-індукованого ОП, вводиться в/в?
- алендронат
- ризедронова кислота
- золедронова кислота
- стронцію ренелат
- ібандронат
- Який з перелічених антиостеопоротичних препаратів має подвійний ефект — антирезорбтивний та кістковоутворювальний?
- алендронат
- ризедронова кислота
- золедронова кислота
- стронцію ренелат
- ібандронат
- ГК сприяють:
- процесам неоглюкогенезу
- пригнiченню синтезу бiлка
- затримці регенераторних процесів
- посиленню утилiзацiї глюкози клiтинами
- гiперкалiємiї
Еталони відповідей: 1) 5; 2) 1, 2, 3, 4; 3) 1, 2, 4, 5; 4) 3; 5) 4; 6) 1, 2, 3
Фіброміалгії
- Дайте визначення активної мiофасцiальної тригерної точки:
- локальний бiль у м’язi, пов’язаний із запаленням нервового волокна, що його iнервує
- фокус вiдбитого болю в зонi Захар’їна — Геда
- фокус збiльшеної подразливостi м’яза або його фасцiї, який виявляється у виглядi болю
- локальний бiль у м’язi, пов’язаний із його запаленням
- локальний бiль у фасцiї, пов’язаний з її алергiчним ураженням
- Назвiть синонiм асоцiативної мiофасцiальної тригерної точки:
- зона локалiзацiї вiдбитого вiд м’язiв болю
- гiпоподразнена дiлянка шкiри
- сателiтна тригерна точка
- гiперподразнена дiлянка, яка виникає у м’язi чи його фасцiї
- фокус вiдбитого болю в зонi Захар’їна — Геда
Еталони відповідей: 1) 3; 2) 3
Хвороба Бехчета
- Для хвороби Бехчета характерним є наявнiсть наступних iмуногенетичних маркерiв:
- HLA-B51
- HLA-A9, -B5, HLA-DR4
- всiх перелiчених
- HLA-B7, -B8, -DR2
- Клiнiчною трiадою синдрому Бехчета є:
- рецидивуючий афтозний стоматит, гломерулонефрит, кон’юнктивiт
- гломерулонефрит, артрит, кардит
- рецидивуючий афтозний стоматит, виразково-некротичнi змiни на статевих органах, увеїт
- кардит, артрит, гепатит
- увеїт, артрит, полiсерозит
- Вкажіть найбільш типове для синдрому Бехчета ураження суглобів:
- моноартрит
- поліартрит
- сакроiлеїт
- ерозивно-деструктивнi змiни тканин суглобiв
- артрити кульшових суглобів
- Які твердження відносно хвороби Бехчета є вірними:
- доведена роль інфекції в розвитку хвороби
- частіше відмічають за наявності HLA-B51
- у біоптатах шкіри відмічають васкуліт венул, капілярів, артеріол
- при енцефаліті відмічають пролiферацiю мiкроглiї, астроцитiв
- венозні тромбози не виявляють
- Який дiагностичний критерiй хвороби Бехчета є обов’язковим:
- афтознi виразки в ділянцi статевих органiв
- шкiрний васкулiт
- рецидивуючий афтозний стоматит
- увеїт
- синовiт
- При лiкуваннi менiнгоенцефалiту при хворобі Бехчета препаратом вибору є:
- преднізолон у високих дозах
- 6-меркаптопурин
- циклофосфамiд
- левамiзол
- купренiл
Еталони відповідей: 1) 1; 2) 3; 3) 2; 4) 1, 2, 3, 4; 5) 3; 6) 1
Ювенільний артрит
- Що таке ювенільний артрит?
- гетерогенне захворювання
- клінічна форма РА в осіб похилого віку
- РА з вісцеритами
- РА високого ступеня активності
- моно-, олігоартрит
- Рентгенологічні ознаки при ювенільному хронічному артриті:
- помірна деструкція кістки та хряща
- анкілозування (рідко)
- анкілоз великих суглобів
- немає рентгенологічних змін
- остеоліз
- Яка етіологія ювенільного хронічного артриту?
- сукупність причин
- мікоплазма і вірус
- селективний дефіцит імуноглобуліну А
- гіпогаммаглобулінемія
- Згідно із сучасною концепцією патологічні процеси у хворих на ювенільний хронічний артрит мають фази:
- ексудативна
- фаза хронічного запалення
- ексудативна і фаза хронічного запалення
- Які суглоби не уражуються при ювенільному хронічному артриті?
- дистальні міжфалангові суглоби
- ліктьові
- колінні
- акроміально-ключичний, груднинно-ключичний
- Які рентгенологічні ознаки характерні для ранніх стадій ювенільного хронічного артриту?
- набряк періартикулярних тканин
- локальний ОП
- розширення суглобової щілини
- ОП
- Системний варіант ювенільного хронічного артриту включає ознаки:
- лихоманка
- висип
- лімфоаденопатія
- гепатолієнальний синдром
- артралгії
- синусит
Еталони відповідей: 1) 1; 3) 1; 4) 1; 5) 3; 6) 4; 8) 1, 2, 3; 9) 1, 2, 3, 4, 5