
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ДОКЛІНІЧНІ ДОСЛІДЖЕННЯ БЕЗПЕКИ ЯК ПІДҐРУНТЯ КЛІНІЧНИХ ВИПРОБУВАНЬ ЗА УЧАСТЮ ЛЮДИНИ
ТА РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ
(ICH M3(R2))
СТ-Н МОЗУ 42-6.0:2014
Видання офіційне
Київ
Міністерство охорони здоров’я України
2014
Передмова
1 РОЗРОБЛЕНО: ДП «Державний експертний центр МОЗ України»
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: О. Нагорна, канд. мед. наук; Т. Бухтіарова, д-р мед. наук, чл.-кор. НАМН України; Т. Талаєва, д-р мед. наук, професор; В. Коваленко, д-р біол. наук, професор; Л. Ковтун, канд. мед. наук; М. Козлов, канд. мед. наук; Л. Бондаренко, д-р біол. наук; С. Распутняк; О. Тур
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Міністерство охорони здоров’я України
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 19 вересня 2014 р. № 661
3 Ця настанова відповідає документу EMA/CPMP/ICH/286/1995 (ICH M3(R2)) «Guideline on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals — December 2009» (Доклінічні дослідження безпеки як підґрунтя клінічних випробувань за участю людини та реєстрації лікарських засобів — грудень 2009)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2014
© Державний експертний центр МОЗ України
Національний вступ
В Україні доклінічні дослідження проводяться відповідно до статті 6 Закону України «Про лікарські засоби» [1], «Порядку проведення доклінічного вивчення лікарських засобів та експертизи матеріалів доклінічного вивчення лікарських засобів» [2] та Настанови з належної лабораторної практики [3]. Дослідження рекомендовано планувати таким чином, щоб забезпечити якомога швидший, надійний і економічний перехід від доклінічного вивчення до клінічних випробувань та запровадження його в медичну практику. Одночасно для адекватної оцінки ризиків використання нового лікарського засобу для організму людини рекомендується розробити належну стратегію доклінічних досліджень, яка б, наскільки це можливо, враховувала необхідність дотримання принципу максимального обмеження ризиків від самого початку застосування у людини, стосовно кожної фази клінічних випробувань, а також під час терапевтичного застосування.
Для створення сучасних положень щодо стандартизації планування та проведення доклінічного вивчення у Європейському Союзі (ЄС) введено настанову EMA/CPMP/ICH/286/1995 (ICH M3(R2)) «Non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals» [4]. Цією настановою рекомендується впровадження міжнародних стандартів проведення доклінічних досліджень, які слід розглядати не як послідовність певних окремих тестів, а вони мають бути тісно пов’язані з програмою запланованих клінічних випробувань і введені в процес розробки лікарського засобу в цілому. Описана в даній настанові схема послідовних етапів полегшує розробку стратегії досліджень, порядок доклінічного вивчення і клінічних випробувань, їх тривалість і взаємну узгодженість у часі залежно від певного лікарського засобу, його складу та особливостей застосування та схеми призначення людині.
З огляду на вищевикладене, актуальною проблемою є впровадження в Україні цієї настанови, яка містить рекомендації щодо проведення доклінічних досліджень з огляду на їх вид, тривалість та узгодженість у часі залежно від відповідної фази клінічних випробувань. Дана настанова сприяє безпеці, вдосконаленню принципів етики (включаючи зменшення використання лабораторних тварин) та прискоренню впровадження нових лікарських засобів у медичну практику. Такі рекомендації повинні бути гармонізовані з положеннями відповідної настанови ЄС [4].
Ця настанова розроблена на підставі настанови з доклінічного дослідження безпеки лікарських засобів: EMA/CPMP/ICH/286/1995 (ICH M3(R2)) «Guideline on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals — December 2009» (Доклінічні дослідження безпеки як підґрунтя клінічних випробувань за участю людини та реєстрації лікарських засобів — грудень 2009) [4].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, що відповідають чинному законодавству України.
До цієї настанови внесено окремі зміни, зумовлені правовими положеннями і прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо до пунктів, яких вони стосуються.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до положень ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [5];
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Перелік редакційних змін та доповнень», а також національний додаток «Бібліографія», які оформлені згідно з положеннями ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладення, оформлення та вимоги до змісту нормативних документів» [5] та ДСТУ 1.7-2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [6]. «Зміст» цієї настанови подано з урахуванням додаткових структурних елементів;
- основні положення викладені у розділі «Доклінічні дослідження безпеки як підґрунтя клінічних випробувань за участю людини та реєстрації лікарських засобів»; при цьому кожний структурний елемент та його номер у даній настанові відповідають таким у настанові EMA/CPMP/ICH/286/1995 [4];
- у розділі «Нормативні посилання» додатково наведено бібліографічний опис нормативних документів, що згадуються у даній настанові;
- у національному додатку «Бібліографія» додатково наведено бібліографічний опис нормативних документів, посилання на які наведено у даній настанові;
- перелік скорочень, що використовуються у цій настанові, наведено в розділі «Познаки та скорочення»;
- у цій настанові слова «дозвіл на продаж» («marketing authorisation») замінено на «реєстрація»;
- по всьому тексту внесено редакційні зміни у посилання на структурні елементи цієї настанови, наприклад, замість «(see section 2.5)» вказано «(див. розділ 2.5 цієї настанови)»;
- додатково до посилань на настанови ICH зроблено посилання на відповідні гармонізовані документи, затверджені в Україні.
Ця настанова застосовна як методичні рекомендації для проведення доклінічних досліджень безпеки лікарських засобів як підґрунтя для подальшого проведення їх клінічних випробувань та реєстрації.
Правовий статус цієї настанови відповідає правовому статусу відповідної настанови у ЄС та інших країнах ІСН, з якими гармонізовано розроблену настанову. Цю настанову слід розглядати як технічний документ для надання консультацій заявникам та власникам реєстраційних посвідчень, компетентним уповноваженим органам та/або іншим зацікавленим особам щодо найкращого та найбільш прийнятного способу дотримання положень, встановлених законодавством України у фармацевтичній сфері. Ця наукова настанова пов’язана зі специфічними науковими питаннями щодо проведення дослідження безпеки лікарських засобів як підґрунтя їх клінічних випробувань та реєстрації. Положення цієї настанови відображують гармонізований (у рамках ЄС та ІСН) підхід, вони базуються на останніх наукових досягненнях у цій галузі знань.
У рамках чинного законодавства ця настанова носить рекомендаційний характер. Дотримання її положень зацікавленими сторонами (такими як заявники, власники реєстраційних посвідчень, розробники та виробники лікарських препаратів, експертні та регуляторні органи) підвищить безпеку проведення клінічних випробувань, сприятиме вдосконаленню принципів етики та зменшенню використання лабораторних тварин, прискоренню впровадження в медичну практику нових лікарських засобів. Однак, можуть бути застосовані альтернативні підходи за умови їх відповідного наукового обґрунтування.
Такий підхід до правового статусу більшості наукових настанов викладено у документі Європейського агентства з лікарських засобів (EMA) [7]. Вказаний підхід відповідає позиції ВТО щодо застосування стандартів.
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Доклінічні дослідження безпеки як підґрунтя клінічних випробувань за участю людини та реєстрації лікарських засобів
(ICH M3 (R2))
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Доклинические исследования безопасности для обоснования клинических испытаний с участием человека и регистрации лекарственых средств
(ICH М3 (R2))
MEDICINAL PRODUCTS
Non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals
(ICH М3 (R2))
Чинна від 2014-09-19
Сфера застосування
Ця настанова визначає положення (рекомендації) щодо проведення доклінічних досліджень безпеки лікарських засобів як підґрунтя клінічних випробувань за участю людини з метою їх подальшої реєстрації.
Ця настанова застосовується до лікарських препаратів, що розробляються, реєструються та виробляються в Україні для медичного застосування в Україні та з метою експорту або імпортуються в Україну.
Ця настанова поширюється на планування та проведення доклінічних досліджень безпеки лікарських препаратів відповідно до фази клінічних випробувань та реєстрації лікарських засобів, а також експертизи матеріалів реєстраційного досьє.
Ця настанова рекомендується для суб’єктів господарювання (далі — організацій), які займаються розробкою, доклінічним та клінічним вивченням, поданням заявок на реєстрацію лікарських засобів на території України незалежно від відомчого підпорядкування та форми власності, для відповідних заявників та підприємств-виробників, продукція яких реєструється та імпортується в Україну, для науково-експертних організацій, експертів, що проводять експертизу при реєстрації (перереєстрації) лікарських засобів, а також для аудиторів та інспекторів.
Нормативні посилання
ICH Topic S6 Document «Safety Studies for Biotechnological Products» (Дослідження з безпеки біотехнологічних препаратів).
ICH Topic E8 Document «General Considerations for Clinical Trials» (Загальні принципи клінічних випробувань).
ICH Topic S5(R2) Document «Detection of Toxicity to Reproduction for Medicinal Products and Toxicity to Male Fertility» (Визначення репродуктивної токсичності лікарських препаратів та їх токсичності щодо чоловічої фертильності).
ICH Topic S1C(R2) Document «Dose Selection for Carcinogenicity Studies of Pharmaceuticals» (Вибір дози для дослідження канцерогенності лікарських засобів).
ICH Topic S7A Document «Safety Pharmacology Studies for Human Pharmaceuticals» (Вивчення фармакології безпеки лікарських засобів для людини).
ICH Topic S7B Document «Nonclinical Evaluation of QT Interval Prolongation» (Доклінічна оцінка подовження інтервалу Q–T).
ICH Topic S3A Document «Note for Guidance on Toxicokinetics-The Assessment of Systemic Exposure in Toxicity Studies» (Примітка до настанови з токсикокінетики — оцінка системної експозиції в токсикологічних дослідженнях).
ICH Topic S2B Document «Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals» (Генотоксичність: стандартний набір тестів генотоксичності лікарських засобів).
ICH Topic S1A Document «Guideline on the Need for Carcinogenicity Studies for Pharmaceuticals» (Рекомендація щодо необхідності дослідження канцерогенності лікарських засобів).
ICH Topic Q3A(R2) Document «Impurities in New Drug Substances» (Домішки в субстанціях нових лікарських препаратів).
ICH Topic Q3B(R2) Document «Impurities in New Drug Products» (Домішки в нових лікарських препаратах).
ICH Topic S8 Document «Immunotoxicity Studies for Human Pharmaceuticals» (Вивчення імунотоксичності лікарських засобів для людини).
Познаки та скорочення
|
AUC |
— |
area under the concentration curve (площа під кривою концентрації) |
|
Cmax |
— |
максимальна концентрація у плазмі крові |
|
CPMP або СНМР |
— |
Committee for Medicinal Products for Human Use (Комітет з лікарських препаратів для людини) |
|
EMA |
— |
European Medicines Agency (Європейське агентство з лікарських засобів) |
|
GLP |
— |
Good laboratory practice (Належна лабораторна практика) |
|
ICH |
— |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини) |
|
MFD |
— |
maximum feasible dose (максимальна доза, яку можна ввести з урахуванням технічних можливостей) |
|
MTD |
— |
maximum tolerated dose (максимальна толерантна доза) |
|
NOAEL |
— |
no observed adverse effect level (максимально безпечний рівень дози) |
|
siRNA |
— |
small interfering RNA (малі інтерферуючі РНК) |
|
в/в |
— |
внутрішньовенно |
|
ВІЛ |
— |
вірус імунодефіциту людини |
|
ГХЛ |
— |
гонадотропін хоріону людини |
|
ЄС |
— |
Європейський Союз |
|
ЖЗНД |
— |
жінки, здатні народжувати дітей |
|
ЗСА |
— |
зв’язок «структура — активність» |
|
ПЕТ |
— |
позитронно-емісійна томографія |
|
ФД |
— |
фармакодинаміка |
|
ФК |
— |
фармакокінетика |
1. Вступ
1.1. Мета
Метою даної настанови є рекомендації стосовно обсягу і тривалості доклінічних досліджень безпеки, що рекомендуються як підґрунтя клінічних випробувань, а також реєстрації лікарських засобів та сприяння їх гармонізації з міжнародними стандартами.
Узгодження настанови з доклінічного вивчення безпеки лікарських засобів сприятиме окресленню діючих рекомендацій і зниженню ймовірності існування істотних відмінностей між регіонами.
Дана настанова має сприяти своєчасному проведенню клінічних випробувань, зменшенню кількості використаних у доклінічних дослідженнях тварин відповідно до принципів 3R (reduce/refine/replace — зменшити/удосконалити/замінити) та зменшенню використання інших ресурсів у процесі розробки лікарських засобів. Слід звернути увагу на використання нових in vitro альтернативних методів оцінки безпеки, хоча це й не є предметом даної настанови. У разі їх валідації та затвердження усіма регуляторними органами ці методи можуть бути використані замість поточних стандартних методів досліджень.
Дана настанова сприяє безпеці, вдосконаленню принципів етики та доступності лікарських засобів.
1.2. Передумова
Рекомендації даної переглянутої настанови сприяють гармонізації доклінічних досліджень безпеки, що проводяться як підґрунтя різних стадій клінічних випробувань у країнах ЄС, Японії та США. Дана настанова є консенсусом, що діє з огляду на вид та тривалість доклінічних досліджень безпеки і їх координацію в часі відносно клінічних випробувань і реєстрації лікарських засобів.
1.3. Сфера застосування
Доклінічна оцінка безпеки лікарського засобу, що впроваджується, зазвичай включає фармакологічні, загальнотоксикологічні, токсикокінетичні та фармакокінетичні дослідження, вивчення репродуктивної токсичності, генотоксичності і, для препаратів спеціального застосування або призначених для тривалого використання, — оцінку канцерогенного потенціалу. Інші доклінічні дослідження щодо фототоксичності, імунотоксичності, токсичності для ювенільних тварин і потенціалу лікарської залежності слід проводити у разі необхідності. Даною настановою окреслені обсяг доклінічних досліджень безпеки і їх зв’язок з клінічними випробуваннями на людях.
Цей документ стосується ситуацій, що зазвичай виникають у ході розробки лікарських засобів, і має розглядатися як загальноприйнята настанова з розробки лікарських засобів.
Доклінічні дослідження безпеки та клінічні випробування на людях повинні плануватися і проводитися з дотриманням відповідних наукових і етичних положень. Для біотехнологічних препаратів відповідні доклінічні дослідження безпеки мають проводитися згідно з настановою ICH S6 «Safety Studies for Biotechnological Products»*. Для зазначених препаратів дана настанова лише обумовлює узгодження у часі доклінічних досліджень відносно клінічних випробувань.
Стосовно лікарських засобів, що розробляються для призначення за життєвими показаннями або при тяжких захворюваннях (наприклад, останні стадії раку, резистентна ВІЛ-інфекція і спадкові захворювання дефіциту ферментів), для яких не існує ефективної терапії, необхідно розглядати кожний випадок окремо щодо токсикологічної оцінки та клінічних випробувань з метою оптимізації та прискорення впровадження лікарського засобу. У цих випадках, а також для препаратів, створених з використанням інноваційних методів терапії (наприклад, малих інтерферуючих РНК (siRNA)), так само, як і для ад’ювантів вакцин, окремі дослідження можуть бути скорочені, відтерміновані, вилучені або додатково заплановані. Мають враховуватися діючі настанови стосовно конкретних груп препаратів.
*Настанова видана в Україні у вигляді методичних рекомендацій «Доклінічне вивчення безпеки лікарських засобів біотехнологічного походження» [8].
1.4. Загальні принципи
Розробка лікарського засобу є поетапним процесом, що включає отримання даних щодо його безпеки та оцінку ефективності препарату на тваринах і людях. Завдання доклінічної оцінки безпеки зазвичай включають характеристику токсичного впливу на органи-мішені, дозозалежність, взаємозв’язок з експозицією і, за необхідності, потенціал зворотності дії. Ці дані використовують для встановлення безпечної стартової (початкової) дози, діапазону доз для клінічних випробувань, а також визначення параметрів клінічного моніторингу потенційних побічних ефектів. Незважаючи на те що на початку клінічних випробувань доклінічні дослідження безпеки, як правило, обмежені, проте вони мають бути достатніми для встановлення потенційного негативного впливу, що може виникати при проведенні клінічних випробувань.
Клінічні випробування проводять для вивчення ефективності й безпеки препарату, починаючи з відносно низького рівня системної дії на невеликій кількості осіб. В подальших клінічних випробуваннях експозиція лікарського засобу, як правило, підвищується шляхом збільшення її тривалості та/або розміру популяції пацієнтів. Клінічні випробування можуть бути розширені на основі даних про безпеку, отриманих у попередньому(-іх) клінічному(-их) випробуванні(-ях), а також додаткових даних доклінічного вивчення безпеки, що стали доступними в процесі проведення поточних клінічних випробувань.
Виявлення серйозного негативного ефекту препарату за результатами клінічних або доклінічних досліджень може впливати на тривалість клінічних випробувань. У межах загального клінічного вивчення ці дані можуть бути використані для визначення доцільності та особливостей дизайну додаткових доклінічних та/або клінічних досліджень.
У цій настанові загалом використовується термінологія, визначена настановою ICH Е8 «General Considerations for Clinical Trials». Але, оскільки існує тенденція до об’єднання фаз клінічного випробування, у деяких випадках цей документ також стосується доклінічних досліджень стосовно тривалості та обсягу клінічних випробувань, а також характеристик включених у випробування суб’єктів.
1.5. Вибір високої дози для загальнотоксикологічних досліджень
Як правило, в токсикологічних дослідженнях ефекти, що мають потенційну значимість для клініки, можуть бути адекватно охарактеризовані шляхом використання доз, які досягають рівня максимальної толерантної дози (MTD). Не обов’язково у кожному дослідженні використовувати MTD. До інших цілком прийнятних граничних доз належать підходи, що передбачають або повторні уведення вищих доз, або насичувальні експозиції, або використання максимальних доз, які можна ввести з урахуванням технічних можливостей (MFD). Такі граничні дози (рисунок) запобігають використанню для тварин доз, що не додають значимості до оцінки прогнозування клінічної безпеки. Дані рекомендації узгоджуються з дизайном досліджень репродуктивної токсичності і канцерогенності, для яких вже визначені граничні дози та/або експозиції [9, 10].
Гранична доза при вивченні гострої, субхронічної та хронічної токсичності становить 1000 мг/кг/добу для гризунів та негризунів і вважається прийнятною для всіх випадків, за винятком тих, що обговорюються нижче. В окремих випадках, коли доза 1000 мг/кг/добу не забезпечує середнє значення в межах 10-кратної клінічної дози, а клінічна доза перевищує 1 г/добу, дози в токсикологічних дослідженнях повинні обмежуватися або 10-кратним збільшенням експозиції, або дозою 2000 мг/кг/добу, або MFD, якою б низькою вона не була.
У таких виняткових випадках, коли доза 2000 мг/кг/добу забезпечує меншу експозицію, ніж клінічна експозиція, має розглядатися можливість використання MFD.
Дози, що забезпечують 50-кратну межу експозиції (як правило, ґрунтуються на середніх значеннях площі під кривою концентрації (AUC) (див. примітку 1 розділу 19) для вихідного лікарського засобу або фармацевтично активного інгредієнта проліків) стосовно клінічної системної експозиції, зазвичай, також розглядаються як прийнятні максимальні дози для досліджень гострої токсичності та токсичності при повторних введеннях тваринам незалежно від виду.
Так, наприклад, для обґрунтування фази III клінічних випробувань у США гранична токсична доза, як правило, визначається щонайменше на одному виді тварин, якщо як граничну дозу використовують 50-кратну межу експозиції. В іншому разі рекомендується проведення доклінічного дослідження протягом одного місяця або довше на одному виді тварин з використанням граничної дози 1000 мг/добу, MTD або MFD, якою б низькою вона не була. Однак в окремих випадках невиправданим є в дослідженнях коротшої тривалості визначати дозозалежну токсичність, використовуючи дозу вищу, ніж та, що призводить до 50-кратного перевищення діапазону експозиції.
Якщо загальнотоксикологічні дослідження містять кінцеві точки генотоксичності, відповідні максимальні дози повинні бути обрані з урахуванням MFD, MTD чи граничної дози 1000 мг/кг/добу.
2. Фармакологічні дослідження
Дослідження фармакології безпеки та фармакодинаміки (ФД) визначаються ICH S7A «Safety Pharmacology Studies for Human Pharmaceuticals». Основний комплекс досліджень фармакології безпеки включає оцінку впливу на серцево-судинну, центральну нервову і дихальну системи і, як правило, здійснюється до початку випробувань на людях відповідно до ICH S7A і S7B «Nonclinical Evaluation of QT Interval Prolongation».
За відповідних обґрунтувань додаткові і наступні дослідження з фармакології безпеки можуть проводитися протягом подальшого клінічного випробування. Для зменшення використання тварин слід розглянути можливість додатково до загальнотоксикологічних досліджень включити всі дані досліджень in vivo.
Крім того, для встановлення механізму дії та/або впливу речовини на задану терапевтичну мішень проводяться попередні дослідження ФД (in vivo та/або in vitro), які, як правило, проводяться на етапі фармацевтичної розробки, а, отже, зазвичай, не вимагають дотримання положень належної лабораторної практики (GLP). Ці дослідження можуть сприяти вибору доз для доклінічних і клінічних випробувань.
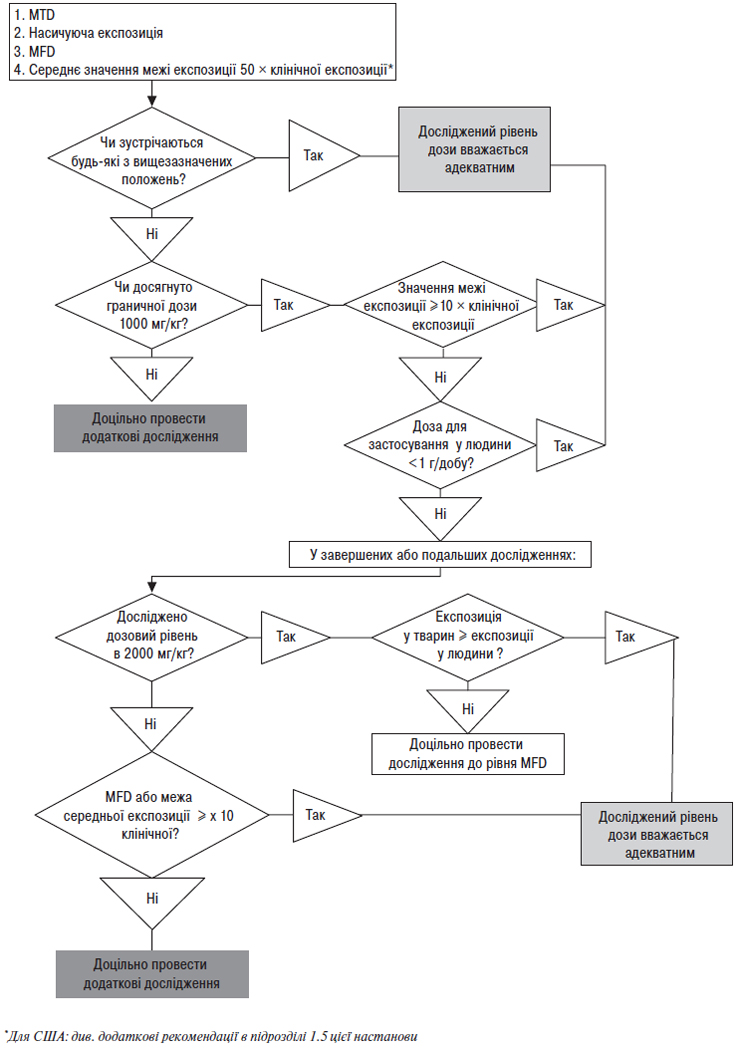
Рисунок. Рекомендації щодо вибору високої дози для дослідження загальної токсичності
3. Токсикокінетичні та фармакокінетичні дослідження
Перед початком клінічних випробувань на людях, як правило, мають бути проаналізовані дані in vitro щодо метаболізму та зв’язування з білками плазми крові у тварин і людини, а також дані системної експозиції (ICH S3A «Note for Guidance on Toxicokinetics. The Assessment of Systemic Exposure in Toxicity Studies») для тварин, види яких використовуються в дослідженнях токсичності за повторних введень. Перед введенням досліджуваного препарату великій кількості людей або протягом тривалого часу (як правило, перед початком фази III) мають бути надані додаткові дані досліджень фармакокінетики (ФК) (наприклад, абсорбції, розподілу, метаболізму і виведення) на тваринах відповідного виду і результати біохімічних досліджень in vitro стосовно потенційної взаємодії ліків. Ці дані можуть бути використані для порівняння метаболітів людини і тварин та визначення доцільності проведення додаткових досліджень.
Доклінічне вивчення метаболітів людини вимагається лише в разі, коли вони становлять понад 10% введеної речовини і їх рівень у людини значно перевищує максимальну експозицію у токсикологічних дослідженнях. Такі дослідження слід проводити як підґрунтя фази ІІІ клінічних випробувань. Якщо речовина вводиться щодня в дозі <10 мг, доцільно дослідити найбільші фракції її метаболітів. Деякі метаболіти не викликають занепокоєння щодо їх токсичності (наприклад, більшість кон’югатів з глутатіоном) і не потребують дослідження. Доцільність доклінічного вивчення властивостей метаболітів та визначення причин для занепокоєння (наприклад, метаболіти, притаманні лише людині) має розглядатися у кожному конкретному випадку.
4. Дослідження гострої токсичності
З історичної точки зору, дані гострої токсичності отримували при вивченні токсичності за одноразового введення ссавцям двох видів з використанням клінічного і парентерального шляхів введення. Однак така інформація може бути отримана при проведенні досліджень, що передбачають підвищення доз або короткострокове вивчення дозозалежності з метою встановлення МTD для загальнотоксикологічних досліджень на тваринах [11, 12]. Якщо у будь-якому дослідженні одержані дані стосовно гострої токсичності, окреме тестування з одноразовим введенням не рекомендується. Дослідження, що передбачають отримання інформації стосовно гострої токсичності, можуть бути обмежені лише шляхом введення, який передбачається застосовувати в клініці; такі дослідження можуть проводитися без дотримання положень GLP за умови, що шлях введення ґрунтується на дослідженнях токсичності за повторних введень відповідно до GLP. Летальність не слід розглядати як кінцеву точку визначення гострої токсичності.
В окремих випадках (наприклад, клінічні випробування з використанням мікродоз, див. розділ 7 цієї настанови) дослідження гострої токсичності або токсичності за одноразового введення може бути основним підґрунтям клінічних випробувань. У цих випадках вибір високої дози може відрізнятися від описаного у підрозділі 1.5 розділу 1, але має достатньою мірою обґрунтовувати очікувані дозу і шлях введення в клініці. Ці дослідження слід проводити відповідно до правил GLP.
Дані гострої токсичності активних фармацевтичних інгредієнтів можуть бути використані для прогнозування наслідків передозування у людини і мають бути надані до початку фази ІІІ клінічних випробувань. Попередня оцінка гострої токсичності є важливою при визначенні терапевтичних показань стосовно популяції з групи високого ризику щодо передозування (наприклад, депресія, біль, деменція) у амбулаторних пацієнтів — учасників клінічних випробувань.
5. Дослідження токсичності за повторних введень
Як правило, тривалість досліджень токсичності за повторних введень рекомендується встановлювати залежно від тривалості, терапевтичних показань та обсягу запланованих клінічних випробувань. В принципі, тривалість досліджень токсичності на тваринах, яка проводиться на ссавцях двох видів (один вид негризуни), повинна бути однаковою або перевищувати тривалість клінічних випробувань, сягаючи максимально рекомендованої тривалості для досліджень токсичності за повторних уведень (табл. 1). Граничні дози/експозиції, що вважаються прийнятними в дослідженнях токсичності за повторних введень, описані в підрозділі 5.1 цього розділу.
За певних умов, коли виявлено значний терапевтичний ефект, клінічне випробування може бути продовжене за межі терміну досліджень токсичності за повторних введень лікарського засобу, що проводилися як підґрунтя клінічних випробувань.
5.1. Клінічні випробування на етапі розробки лікарського засобу
Дослідження токсичності за повторних введень лікарського засобу тваринам двох видів (один вид негризуни) з мінімальною тривалістю 2 тиж (див. табл. 1), як правило, має бути підґрунтям для будь-якого клінічного випробування при тривалості застосування препарату не більше 2 тиж у подальшому. Більш тривале клінічне випробування повинне бути підтверджене токсикологічними дослідженнями за повторних введень щонайменше еквівалентної тривалості. Тривалість досліджень 6 міс для гризунів і 9 міс для негризунів вважається достатньою основою клінічного застосування понад 6 міс (винятки наведені у примітках до табл. 1).
Таблиця 1. Рекомендована тривалість досліджень токсичності за повторних уведень як підґрунтя клінічних випробувань
|
Максимальна тривалість клінічного випробування |
Рекомендована мінімальна тривалість дослідження токсичності за повторних введень як підґрунтя клінічних випробувань |
|
| Гризуни | Негризуни | |
|
До 2 тиж |
2 тиж |
2 тижа |
|
Від 2 тиж до 6 міс |
Аналогічно клінічній |
Аналогічно клінічній |
|
Більше 6 міс |
6 місб, в |
9 місб, в, г |
Примітки.
аУ США для обґрунтування випробувань, що передбачають одноразове введення людям, як альтернативу доклінічним дослідженням протягом 2 тиж можуть проводити розширені дослідження токсичності за одноразового введення (див. примітку в до табл. 3). Клінічні випробування, що тривають менше ніж 14 днів, можуть проводитися після токсикологічних досліджень протягом терміну, аналогічного клінічному.
бЗа певних обставин клінічні випробування тривалістю понад 3 міс можуть бути розпочаті за наявності даних доклінічних випробувань як на гризунах, так і негризунах протягом 3 міс. Перед розширенням дозування при проведенні клінічних випробувань понад 3 міс відповідно до місцевих регуляторних положень щодо проведення клінічних випробувань мають бути надані результати завершених досліджень хронічної токсичності на гризунах і негризунах.
В окремих індивідуальних серйозних випадках, а також при загрозі для життя таке розширення може бути обґрунтоване даними завершених досліджень хронічної токсичності на гризунах та прижиттєвими даними і результатами розтину в дослідженнях на негризунах. Повністю дані гістопатологічних досліджень на негризунах повинні бути надані протягом наступних 3 міс.
вЯкщо основною популяцією є педіатрична, попередньо мають бути надані результати досліджень на тваринах (фармакологічні або токсикологічні), якими визначається наявність потенційних ризиків щодо розвитку органів-мішеней. У таких випадках може бути доцільним проведення довготривалого дослідження токсичності на ювенільних тваринах (див. розділ 12 цієї настанови).
гУ ЄС прийнятними є дослідження на негризунах протягом 6 міс. Але, якщо проводилися дослідження більшої тривалості, проведення додаткових досліджень протягом менше ніж 6 міс є недоцільним. Нижче наведено приклади досліджень на негризунах тривалістю до 6 міс, які також можуть бути застосовані в Японії і США:
- якщо імуногенність або непереносимість не дозволяють проведення більш тривалих досліджень;
- повторна короткострокова експозиція препарату, навіть якщо тривалість клінічних випробувань перевищує 6 міс, наприклад, за періодичного лікування при мігрені, еректильній дисфункції або простому герпесі;
- препарати, що вводяться довгостроково для зниження ризику рецидиву раку;
- препарати, що, як очікується, застосовуються протягом короткого терміну.
5.2. Реєстрація
За наявності ризиків для численної популяції та за відносно менш контрольованих умов при проведенні клінічних випробувань може бути доцільним проведення доклінічних досліджень протягом більш тривалого часу. Тривалість доклінічних досліджень за умов повторних уведень перед процедурою реєстрації залежно від терміну лікування наведена в табл. 2. Проте, за певних обставин, коли препарат застосовується від 2 тиж до 3 міс, але існує великий клінічний досвід, який підтверджений широким і тривалим застосуванням, крім показань, що рекомендуються (наприклад, тривога, сезонний алергічний риніт, біль), тривалість досліджень має бути відповідно еквівалентною рекомендованій для лікування понад 3 міс.
Таблиця 2. Рекомендована тривалість досліджень токсичності за повторних уведень як підґрунтя реєстрації лікарського засобу
|
Тривалість показання для лікування |
Гризуни, міс |
Негризуни, міс |
|
До 2 тиж |
1 |
1 |
|
Від 2 тиж до 1 міс |
3 |
3 |
|
1–3 міс |
6 |
6 |
|
Понад 3 міс |
6в |
9в,г |
Див. примітки в і г до табл. 1.
6. Визначення першої дози для людини
Встановлення першої дози для людини є важливим елементом захисту осіб, що беруть участь у першому клінічному випробуванні лікарського засобу. При визначенні рекомендацій щодо першої дози у людини мають бути враховані всі суттєві дані доклінічних досліджень, включаючи фармакологічну дозозалежність, фармакологічний/токсикологічний профіль та фармакокінетику (ФК).
Загалом найбільш важлива інформація — встановлення максимально безпечного рівня дози (NOAEL) — ґрунтується на результатах доклінічних досліджень безпеки, проведених на найбільш придатних видах тварин. Запропонована для клініки початкова доза також залежить від різних факторів, включаючи ФД, особливості будови молекули та дизайну клінічного випробування.
Пілотні клінічні випробування (див. розділ 7 цієї настанови) на людях можуть бути розпочаті за наявності меншого або відмінного від загальноприйнятого об’єму доклінічного вивчення (див. підрозділ 5.1 цієї настанови), тому встановлення стартової (початкової) та максимальної дози для клінічних випробувань може відрізнятися. В табл. 3 описані рекомендації щодо критеріїв визначення стартових (початкових) доз для різних дизайнів пілотних клінічних випробувань.
7. Пілотні клінічні випробування
Загальновизнано, що в ряді випадків попередні дані, отримані на людях, можуть забезпечити більш глибоке розуміння фізіології/фармакології людини, знання властивостей потенційного лікарського засобу та визначення терапевтичної мішені хвороби. Цього можна досягти завдяки добре налагодженим раннім пілотним випробуванням. Пілотні клінічні випробування відповідно до завдань даної настанови проводяться на початку фази І і передбачають обмежену експозицію на людину, що не має терапевтичного значення і не призначена для вивчення клінічної переносимості, а використовуються для встановлення ряду таких параметрів, як ФК, ФД та інших біомаркерів, включаючи взаємодію з рецепторами згідно з позитронно-емісійною томографією (ПЕТ) або іншими діагностичними вимірюваннями. Особи, включені до таких випробувань, можуть бути здоровими добровольцями або пацієнтами обраних популяцій з тяжкими захворюваннями.
Кількість і види доклінічних досліджень визначаються в кожному конкретному випадку окремо і залежать від рівня експозиції, що пропонується для людини, максимальної дози для клініки і тривалості застосування. Нижче, в табл. 3, підсумовані і більш детально викладені 5 різних підходів у якості прикладів до пілотних клінічних випробувань і відповідних програм доклінічних досліджень, що можуть бути рекомендовані для кожного з цих конкретних підходів.
Однак можуть бути застосовані й інші альтернативні підходи, не описані в даній настанові, що включають стратегію вивчення продуктів біотехнологічного походження. Рекомендується попередньо обговорювати та узгоджувати такі альтернативні підходи з відповідним регуляторним органом. Застосування будь-якого з цих принципів може сприяти зменшенню загальної кількості тварин, що використовуються на етапі доклінічного вивчення при розробці ліків.
В табл. 3 наведено 5 підходів для встановлення стартової (початкової) дози та максимальної дози для клінічних випробувань. У всіх випадках важливою є характеристика ФД та фармакології за даними моделей in vivo та/або in vitro, як зазначено у табл. 3 та розділі 2, що має бути використана для обґрунтування дози для людини.
7.1. Випробування з використанням мікродоз
Нижче описані й детально наведені два підходи до випробувань з використанням мікродоз (див. табл. 3). Перший підхід передбачає використання дози, що сумарно не перевищує 100 мкг, яка може бути введена як одноразово, так і розподілена на кілька введень для кожного суб’єкта. Це може бути корисним для вивчення зв’язування з рецептором мішені або розподілу в тканинах за методом ПЕТ. Друга сфера застосування полягає у визначенні ФК з використанням міченої або не міченої ізотопом речовини.
Другий підхід щодо застосування мікродози включає менше 5 уведень, максимум 100 мкг на введення (сумарно 500 мкг на особу). Це може бути корисним для введень, подібних до першого підходу використання мікродоз, як описано вище, але для менш активних лігандів ПЕТ.
У ряді випадків може бути доцільним проведення клінічних випробувань мікродоз з використанням внутрішньовенного (в/в) шляху введення препарату, призначеного для перорального застосування, для якого вже є достатній обсяг доклінічних токсикологічних досліджень для перорального введення. За цих умов в/в введена мікродоза може бути визначена на основі наявних даних дослідження токсичності при пероральному введенні, як зазначено в табл. 1 або табл. 3 (підхід 3), завдяки яким встановлені адекватні межі експозиції. За даних умов не рекомендується досліджувати місцеву переносимість при в/в введенні, оскільки доза, що застосовується, дуже низька (максимум 100 мкг). Якщо застосовується новий в/в розчинник, має бути визначена його місцева переносимість.
7.2. Випробування за умов одноразового введення субтерапевтичних доз або дози в межах заявленого терапевтичного діапазону
Третій підхід передбачає клінічне випробування при введенні однієї дози, що зазвичай починається з субтерапевтичних доз з можливим підвищенням до фармакологічної або очікуваної терапевтичної дії (див. табл. 3). Максимально допустима доза має ґрунтуватися на доклінічних даних, але в подальшому може бути знижена на основі клінічних даних, отриманих у ході випробувань. Цей підхід дозволяє, наприклад, визначати фармакокінетичні параметри з радіоактивно неміченим лікарським засобом або з дозою, наближеною до активної фармакодинамічної. Іншим прикладом може бути оцінка впливу лікарського засобу на мішень або вивчення фармакології після одноразового введення. Цей підхід не призначений для визначення максимальної переносимої клінічної дози (див. винятки, примітка а до табл. 1).
7.3. Випробування за багаторазових уведень
Два різні доклінічні підходи (4 і 5) для обґрунтування клінічних випробувань за багаторазових уведень представлені в табл. 3. Вони передбачають введення препарату до 14 днів для визначення ФК та ФД у людини в терапевтичному діапазоні доз, але не призначені для встановлення максимально допустимої клінічної дози.
Підхід 4 включає дослідження токсичності за повторних уведень протягом 2 тиж на гризунах та негризунах, при цьому вибір дози для тварин ґрунтується на даних очікуваної AUC за повторних уведень максимальної клінічної дози.
Підхід 5 включає 2-тижневі дослідження токсичності на гризунах з наступними дослідженнями на негризунах для підтвердження, що NOAEL на гризунах є нетоксичною дозою і для негризунів. Якщо при введенні препарату негризунам виявлено токсичність дози, яка не викликає проявів токсичності у гризунів, уведення в клініці має бути відтерміноване до проведення додаткових доклінічних досліджень на цих видах тварин (як правило, це стандартні дослідження токсичності (див. розділ 5 цієї настанови)).
8. Вивчення місцевої переносимості
Місцева переносимість оцінюється переважно залежно від очікуваного терапевтичного шляху введення як частина загальнотоксикологічних досліджень; окремі дослідження зазвичай не рекомендовані.
Для обґрунтування обмеженого за часом введення препарату людині шляхом, який не використовується з метою терапії (наприклад, одноразове в/в введення для визначення абсолютної біодоступності засобу для перорального введення), вважається достатнім вивчення місцевої переносимості при одноразовому введенні тваринам одного виду. Якщо внаслідок шляху введення, який не є терапевтичним, очікується системна експозиція (AUC і максимальна концентрація у плазмі крові (Сmax)), то мають бути проведені токсикологічні дослідження в необхідному обсязі, у межах якого кінцеві точки вивчення місцевої переносимості можуть обмежуватися клінічними симптомами і макро- та мікроскопічним аналізом в ділянці аплікації. Лікарська форма, що призначена для вивчення місцевої переносимості, може бути неідентичною, але має бути подібною за складом до призначеної для клінічних випробувань.
При вивченні в/в введення мікродоз, що базуються на даних комплексу токсикологічних досліджень при пероральному введенні (див. розділ 7 цієї настанови), недоцільно аналізувати місцеву дію активної фармацевтичної субстанції. Якщо ж застосовується новий носій або розчинник, то попередньо має досліджуватися його місцева переносимість.
Для препаратів, призначених для парентерального введення, оцінка місцевої дії на ділянках, не передбачених для введення, за необхідності, має здійснюватися до початку застосування препаратів у великої кількості пацієнтів (наприклад, фаза ІІІ клінічних випробувань). Підхід до таких досліджень у різних країнах неоднаковий. Такі дослідження, як правило, не проводяться у США (наприклад, виключенням буде інтратекальний шлях за умов епідурального введення). В Японії та ЄС рекомендується одноразове паравенозне введення для в/в шляху. Інші парентеральні шляхи введення оцінюють у кожному випадку окремо.
Таблиця 3. Доклінічні дослідження, що рекомендуються перед проведенням пілотних клінічних випробувань
|
Клінічні випробування |
Доклінічні дослідження |
|||
|
Доза, що вводиться |
Стартова (початкова) і максимальна дози |
Фармакологія |
Загальні токсикологічні дослідженняа |
Генотоксичністьб/інші |
|
Підхід 1: Загальна доза ≤100 мкг; (максимум 5 введень) (інтервали між дозами не обмежуються) ТАКОЖ Загальна доза ≤1/100 та NOAEL ≤1/100 фармакологічно активної дози (розрахованої в мг/кг маси тіла для в/в введення та в мг/м2 для перорального введення) |
Максимальна та стартова (початкова) дози можуть бути однаковими, але не перевищувати сумарну дозу 100 мкг |
Аналіз профілю мішені/рецептора іn vitro. Для обґрунтування вибору доз для людини мають бути надані дані первинної фармакології (механізм дії та/або ефекти) на основі досліджень з використанням фармакодинамічно обґрунтованих моделей |
Розширене дослідження токсичності за одноразового введення (див. примітки в і г до табл. 3) тваринам одного виду, зазвичай гризунам, за очікуваного шляху введення з токсикокінетичними даними або за в/в введення. Може бути використана доза, яка в 1000 разів перевищує клінічну дозу з розрахунку мг/кг маси тіла для в/в введення та в мг/м2 для перорального введення |
Генотоксичні дослідження не рекомендуються, але будь-які дослідження або встановлення зв’язку «структура — активність» (ЗСА) мають бути включені до заявки на клінічні випробування. Для високорадіоактивних агентів (наприклад, агентів для ПЕТ) мають бути одержані дані ФК та дозиметрії |
|
Підхід 2: Сумарна накопичена доза ≤500 мкг; максимум 5 введень із вимиванням між введеннями (6 або більше фактичних або очікуваних періодів напіввиведення) ТАКОЖ Кожна доза ≤100 мкг ТАКОЖ Кожна доза ≤1/100 NOAEL та ≤1/100 фармакологічно активної дози |
Максимальна щоденна та стартова (початкова) дози можуть бути однаковими, але не перевищувати 100 мкг |
Аналіз профілю мішені/рецептора іn vitro. Для обґрунтування вибору доз для людини мають бути надані дані первинної фармакології (механізм дії та/або ефекти) на основі досліджень з використанням фармакодинамічно обґрунтованих моделей |
Токсикологічні дослідження за повторного очікуваного шляху введення протягом 7 днів тваринам одного виду, що мають включати дані токсикокінетики, або за в/в введення. Мають бути наведені дані розтину, гематологічних, біохімічних, гістопатологічних досліджень. Може бути застосована максимальна доза, що є 1000-кратною стосовно клінічної дози, розрахована у мг/кг маси тіла для в/в та мг/м2 для перорального введення |
Генотоксичні дослідження не рекомендуються, але будь-які дослідження або встановлення ЗСА мають бути включені до заявки на клінічні випробування. Для високорадіоактивних агентів (наприклад, агентів для ПЕТ) мають бути одержані дані ФК та дозиметрії |
|
Підхід 3: Випробування при введенні однієї субтерапевтичної дози або дози в межах очікуваного терапевтичного діапазону |
Стартова (початкова) доза має ґрунтуватися на даних токсичності для найбільш чутливих видів тварин та з урахуванням фармакологічно активної дози. Стосовно інших міркувань щодо визначення початкової дози для людини слід звертатися до відповідних методичних рекомендацій. Максимальна доза може сягати ½ NOAEL для тварин найбільш чутливих видів, якщо у тварин виявлено будь-які суттєві прояви токсичності, передбачається здійснення їх моніторингу і зворотності у людини |
Аналіз профілю мішені/рецептора іn vitro. Для обґрунтування вибору доз для людини мають бути надані дані первинної фармакології (механізм дії та/або ефекти) на основі досліджень з використанням фармакодинамічно обґрунтованих моделей. Базовий набір досліджень з фармакології безпеки (див. розділ 2 цієї настанови) |
Розширене вивчення токсичності за одноразового введення гризунам і негризунам шляхом, передбаченим для клініки (див. примітку в до табл. 3), включаючи дослідження з токсикокінетики, гематології, біохімії, аналіз розтину і гістопатології. В даному випадку вищою дозою мають бути MTD, MFD або гранична доза (див. розділ 1.5 цієї настанови) |
Тест Еймса (або альтернативні методи, якщо тест Еймса не може бути використаний, наприклад, для антибактеріальних препаратів) |
|
Підхід 4: Введення до 14 днів у діапазоні терапевтичних доз, але не призначених для визначення клінічної МTD |
Стартова (початкова) доза визначається за даними токсичності на двох видах тварин та згідно з діючими регіональними настановами. Якщо токсичність не виявлена на обох видах тварин (тобто NOAEL є найвищими дослідженими дозами, а також використані дози не були обмежені іншим чином, наприклад, не були MFD) або показана у тварин лише одного виду, клінічна стартова (початкова) доза має визначатись як така, що забезпечує очікуване значення AUC в клініці (на основі або міжвидових даних моделювання ФК, або перерахунку в мг/м2), що становить близько 1/50 AUC при NOAEL для виду тварин за нижчої експозиції. Щодо інших засад визначення початкових доз для людини, наприклад, очікувана ФД активність, слід враховувати положення регіональних методичних настанов За відсутності токсичності для обох видів тварин рекомендується, щоб максимальна клінічна доза не перевищувала 1/10 нижчої експозиції (AUC) для обох видів тварин при найвищій дозі у тварин. Якщо токсичність виявлена лише у тварин одного виду, максимальна клінічна доза або не повинна перевищувати NOAEL виду тварин, для яких показана токсична дія, або відповідати ½ AUC за введення найвищої дози тваринам виду, для яких не виявлено ознак токсичної дії, залежно від того, яка з них нижча. Якщо показано токсичну дію для обох видів тварин, максимальна клінічна доза має ґрунтуватися на стандартних підходах для оцінки ризику і в цьому конкретному випадку має бути проаналізована клінічна MTD |
Аналіз профілю мішені/рецептора іn vitro. Для обґрунтування вибору доз для людини мають бути надані дані первинної фармакології (механізм дії та/або ефекти) на основі досліджень з використанням фармакодинамічно обґрунтованих моделей. Базовий набір досліджень з фармакології безпеки (див. розділ 2 цієї настанови) з використанням доз, подібних до тих, що використані при проведенні токсикологічних досліджень |
Вивчення токсичності за повторних уведень протягом 2 тиж на гризунах і негризунах з оцінкою стандартних параметрів та вибором доз для тварин на основі багаторазової експозиції очікуваної в клініці AUC за максимальної дози |
Тест Еймса (або альтернативні методи, якщо тест Еймса не може бути використаний, наприклад, для антибактеріальних засобів) та дослідження (in vitro або in vivo), здатні виявити хромосомні пошкодження у ссавців |
|
Підхід 5: Введення до 14 днів, не перевищуючи тривалість введення для негризунів; у діапазоні терапевтичних доз, але не призначених для визначення клінічної МTD |
Передбачається, що експозиції стартових (початкових) доз не повинні перевищувати 1/50 NOAEL у тварин більш чутливих видів в розрахунку мг/м2. Стосовно інших підходів щодо визначення початкових доз для людини слід враховувати діючі регіональні методичні рекомендації. Максимальна експозиція для людини не повинна перевищувати AUC за NOAEL для негризунів або ½ AUC за NOAEL для гризунів, залежно від того, яка з них нижчад |
Аналіз профілю мішені/рецептора іn vitro. Для обґрунтування вибору доз для людини мають бути надані дані первинної фармакології (механізм дії та/або ефекти) на основі досліджень з використанням фармакодинамічно обґрунтованих моделей. Базовий набір досліджень з фармакології безпеки (див. розділ 2) з використанням доз, подібних до тих, що використані при проведенні токсикологічних досліджень |
Стандартне вивчення токсичності на гризунах за повторних уведень протягом 2 тиж (з обґрунтуванням вибору гризунів як виду, придатного для цих досліджень). Найвищою дозою має бути МTD, MFD або гранична доза (див. підрозділ 1.5 цієї настанови). Дослідження на негризунах (n=3), якими підтверджується експозиція NOAEL, попередньо встановлена на гризунах, тривалістю щонайменше 3 дні і не менше, ніж тривалість клінічних випробувань. Як альтернатива проводяться дослідження доз, що підвищуються, на негризунах тривалістю щонайменше 3 дні, але не менше, ніж тривалість запланованих клінічних випробувань за експозиції NOAEL, попередньо встановленої для гризунів |
Тест Еймса (або альтернативні методи, якщо тест Еймса не може бути використаний, наприклад, для антибактеріальних засобів) та дослідження (in vitro або in vivo), здатні виявити хромосомні пошкодження у ссавців. Якщо проводяться дослідження in vivo, вони можуть бути фрагментом вивчення токсичності на гризунах |
Примітки.
аЗагальнотоксикологічні дослідження мають бути проведені відповідно до положень GLP.
бДив. посилання 13 стосовно дизайну досліджень генотоксичності та вибору доз.
вЯк правило, план розширених досліджень токсичності за одноразового введення має включати оцінку гематологічних, клінічних біохімічних даних, аналіз розтину та гістопатології (якщо за введення високої дози не виявлено патологічних проявів, зумовлених її дією, аналіз обмежується групами контролю та високої дози) після одноразового введення з додатковими дослідженнями понад 2 тиж для встановлення віддаленої токсичності та/або її зворотності. Зазвичай план для гризунів включає 10 тварин у групі для кожної статі на день введення та 5 тварин кожної статі для кожної з обраних доз для оцінки на 14-й день після введення. Дизайн для негризунів, як правило, передбачає використання в групі 3 тварин кожної статі на день введення та по 2 тварини кожної статі для кожної дози на 14-й день.
гРівень одноразової дози для оцінки зворотності або віддаленої токсичної дії на 14-й день після введення може бути підґрунтям випробувань з використанням мікродоз. Ця доза не має бути високою, але щонайменше в 100 разів перевищувати клінічну.
дЯкщо за даними клінічних випробувань побічна дія відсутня, може бути доцільним підвищення дози до рівня, що перевищує дану AUC, за умов, що результати досліджень токсичності передбачають можливість здійснення моніторингу, зворотність дії та низький ступінь тяжкості стосовно організму людини.
9. Вивчення генотоксичності
Тест генної мутації, як правило, є достатнім підґрунтям для початку всіх клінічних випробувань за умов одноразового введення. За багаторазових введень препарату при проведенні клінічних випробувань додатково оцінюється його здатність викликати хромосомні пошкодження у ссавців [13]. Весь стандартний набір тестів генотоксичності має бути повністю завершений до початку фази ІІ клінічних випробувань [13].
У разі отримання позитивних результатів розглядається можливість проведення додаткових досліджень [13] для визначення доцільності продовження введення препарату людям.
Дослідження генотоксичності, що рекомендуються до початку пілотних клінічних випробувань, описані в розділі 7.
10. Вивчення канцерогенності
Належні умови проведення досліджень канцерогенності викладені в настанові ICH S1A «Guideline on the Need for Carcinogenicity Studies for Pharmaceuticals». Якщо вивчення канцерогенності рекомендоване за клінічними показаннями, воно має проводитися до отримання дозволу на реєстрацію лікарського засобу. Лише за наявності обставин, згідно з якими існують серйозні причини для занепокоєння щодо канцерогенного ризику, перед початком клінічних випробувань мають бути надані результати доклінічних досліджень канцерогенності. Довга тривалість клінічних випробувань не вважається достатньою причиною для занепокоєння.
Для лікарських засобів, розроблених для лікування деяких тяжких захворювань дорослих або дітей, дослідження канцерогенності, якщо такі рекомендовані, можуть бути завершені після реєстрації лікарського засобу.
11. Дослідження репродуктивної токсичності
Дослідження репродуктивної токсичності [9] повинні бути проведені відповідно до популяції, у якої передбачається застосування препарату.
11.1. Чоловіки
Чоловіки можуть бути включені до фази І і фази ІІ клінічних випробувань до проведення досліджень фертильності на самцях, оскільки оцінка стану репродуктивних органів самців здійснюється в ході вивчення токсичності за повторних введень (примітка 2 розділу 19).
Дослідження фертильної функції у самців [9] мають бути завершені до початку розширених або тривалих клінічних випробувань (наприклад, фази ІІІ).
11.2. Жінки, не здатні народжувати дітей
Жінки, що потенційно не можуть народжувати дітей (наприклад, перманентно стерилізовані, період постменопаузи), можуть бути включені до участі в клінічних випробуваннях без попереднього проведення доклінічних досліджень репродуктивної токсичності, якщо були проведені відповідні дослідження токсичності за повторних введень (що включають оцінку стану жіночих репродуктивних органів). Постменопауза визначається як 12 міс відсутності менструації без альтернативної медичної причини.
11.3. Жінки, здатні народжувати дітей
Для жінок, які потенційно можуть народити дитину (ЖЗНД), існує високий рівень небезпеки непередбаченого впливу на ембріон або плід до надходження даних про потенційні переваги порівняно з можливими ризиками. Рекомендації стосовно координації доклінічних досліджень репродуктивної токсичності для обґрунтування участі ЖЗНД у клінічних випробуваннях однакові для всіх країн ICH.
При включенні ЖЗНД до клінічних випробувань важливо охарактеризувати і звести до мінімуму ризик непередбаченого впливу на ембріон або плід. Одним з підходів для досягнення цієї мети є проведення досліджень репродуктивної токсичності для характеристики очікуваних ризиків застосування лікарського засобу та вжити відповідних заходів при включенні ЖЗНД у клінічні випробування. Другий підхід полягає в обмеженні ризику шляхом проведення заходів безпеки для запобігання вагітності під час клінічних випробувань. Запобіжні заходи для попередження вагітності можуть включати тест на вагітність (наприклад, визначення β-субодиниці гонадотропіну хоріону людини), використання високоефективних методів контролю над народжуваністю (примітка 3 розділу 19) і включення у випробування лише після підтвердження менструального періоду.
Тестування на вагітність під час випробування і навчання пацієнтки повинні бути достатніми для забезпечення дотримання нею заходів попередження вагітності протягом періоду експозиції лікарського засобу (який може перевищувати термін дослідження). Для підтримки цих підходів інформована згода має ґрунтуватися на будь-якій відповідній інформації щодо репродуктивної токсичності, такої як загальна оцінка потенційної токсичності лікарських засобів із подібним складом або фармакологічною дією. За відсутності відповідних даних репродуктивної токсичності слід повідомляти про можливість неочікуваних ризиків для ембріону або плода.
За певних умов в усіх країнах ICH ЖЗНД можуть бути включені на початку клінічних випробувань без даних доклінічного вивчення токсичного впливу на розвиток (наприклад, доклінічні ембріофетальні дослідження). Однією з обставин може бути інтенсивний контроль ризику вагітності при дуже короткострокових клінічних випробуваннях (наприклад, 2 тиж). Іншою обставиною може бути домінування хвороби жінок, і завдання клінічного випробування не може бути ефективно вирішене без включення у нього ЖЗНД. При цьому має бути вжито достатньо заходів для попередження вагітності (див. вище).
Додатково розглядається можливість клінічних випробувань із залученням ЖЗНД без проведення доклінічних досліджень токсичного впливу на розвиток за наявності даних про механізм дії речовини, вид діючої речовини, ступінь впливу на плід, або труднощі проведення досліджень токсичного впливу на розвиток для відповідної моделі тварин. Наприклад, для моноклональних антитіл, для яких ембріофетальний вплив протягом органогенезу у людини згідно з сучасними науковими даними знаходиться на низькому рівні; дослідження токсичного впливу на розвиток можуть бути проведені в ході фази ІІІ. Завершені звіти мають бути надані із заявою на реєстрацію лікарського засобу.
Як правило, за наявності попередніх адекватних даних репродуктивної токсичності для двох видів тварин (див. примітку 4 розділу 19) та застосування засобів попередження вагітності при проведенні клінічних випробувань (див. вище), включення ЖЗНД (до 150 осіб) у відносно короткострокове випробування (до 3 міс) може проводитися до остаточного завершення дослідження репродуктивної токсичності. Підставою для цього є дуже низька частка випадків вагітності в ході контрольованих клінічних випробувань з такою вибіркою і тривалістю (див. примітку 5 розділу 19) та здатністю (належним чином спланованих попередніх досліджень) виявити токсичний вплив на розвиток ембріону та плода, які могли б викликати сумніви щодо залучення ЖЗНД до клінічних випробувань. На кількість ЖЗНД і тривалість їх участі у клінічному випробуванні може вплинути характеристика популяції, що змінює частку випадків вагітності (наприклад, вік, наявність захворювання).
У США дослідження ембріофетального розвитку може бути відтерміноване до фази ІІІ для ЖЗНД, які використовують засоби для попередження вагітності протягом клінічних випробувань. У ЄС і Японії, окрім ситуацій, описаних у попередніх розділах, доклінічні дослідження токсичного впливу на розвиток ембріону та плода повинні бути завершені до включення ЖЗНД у клінічне випробування.
У всіх країнах ICH ЖЗНД можуть бути включені у клінічне випробування фази І і ІІ при повторних введеннях лікарського засобу до проведення досліджень фертильності на самицях, оскільки оцінка стану репродуктивних органів самиць вже здійснювалася в ході досліджень токсичності за повторних введень (примітка 2 розділу 19). Доклінічні дослідження, які конкретно стосуються фертильності самиць [9], повинні бути завершеними до включення ЖЗНД до розширених або тривалих клінічних випробувань (наприклад, фаза ІІІ випробувань).
В усіх країнах ICH для реєстрації лікарських засобів мають бути надані результати досліджень пре- і постнатального розвитку.
Усі дослідження жіночої репродуктивної токсичності [9] і стандартний набір тестів з генотоксичності [13] повинні бути завершені до включення ЖЗНД до будь-яких клінічних випробувань, у ході яких не застосовується високоефективний контроль народжуваності (див. примітку 3 розділу 19) або статус вагітності невідомий.
11.4. Вагітні
Перед включенням вагітних у клінічні випробування повинні бути проведені всі доклінічні дослідження репродуктивної токсичності самиць [9] та стандартний набір тестів з генотоксичності [13]. Крім того, необхідно оцінити безпеку тривалості застосування препарату на основі попередніх даних впливу на людину.
12. Клінічні випробування у педіатричній популяції
При проведенні клінічних випробувань на педіатричній популяції, як правило, мають бути представлені дані про безпеку на основі наявного досвіду застосування у дорослих людей як найбільш значущої інформації, що, зазвичай, має бути доступною до початку педіатричних клінічних випробувань. Відповідність і обсяг даних для дорослих мають визначатися у кожному випадку окремо. До початку проведення клінічних випробувань у педіатричній популяції широкий досвід використання лікарського засобу у дорослих людей може бути й відсутній (наприклад, у разі лише специфічних педіатричних показань).
До початку клінічних випробувань із залученням педіатричної популяції мають бути надані результати дослідження токсичності за повторних уведень лікарського засобу на дорослих тваринах відповідної тривалості (див. табл. 1), основного набору досліджень з фармакології безпеки та стандартного набору тестів з генотоксичності. Вивчення репродуктивної токсичності мають бути релевантними щодо статі та віку педіатричної популяції, залученої до клінічних випробувань. Також важливою інформацією про пряму токсичну дію або ризики стосовно розвитку потомства є доклінічне вивчення репродуктивної токсичності з урахуванням віку та статі педіатричної популяції (наприклад, дослідження фертильності та пре- і постнатального розвитку). Аналіз ембріофетального розвитку не є обов’язковим для початку клінічних досліджень за участю осіб чоловічої статі або жіночої статі препубертатного віку. Проведення токсикологічних досліджень на ювенільних тваринах вважається доцільним лише у разі, коли попередні дані на тваринах і дані щодо безпеки для людини, включаючи дію інших лікарських засобів даного фармакологічного класу, є недостатніми для прийняття рішення про проведення педіатричних випробувань. За наявності обґрунтування для проведення таких досліджень зазвичай достатнім вважається використання тварин одного відповідного виду, переважно гризунів. Дослідження на негризунах мають бути науково обґрунтованими.
Як правило, для педіатричної популяції при проведенні короткострокових досліджень ФК (наприклад, 1–3 введення) не обов’язково попередньо вивчати токсичність на ювенільних тваринах.
До початку короткострокових клінічних випробувань ефективності та безпеки, що передбачають багаторазове введення лікарського засобу, слід розглянути доцільність надання результатів досліджень на ювенільних тваринах залежно від терапевтичних показань, віку педіатричної популяції, даних безпеки, отриманих на дорослих тваринах і при застосуванні у людей.
Вік дітей, які включаються у клінічне випробування, є одним з найбільш важливих чинників, який необхідно враховувати (наприклад, проміжок періоду розвитку, що викликає занепокоєння щодо введення лікарського засобу пацієнтам у ході клінічного випробування). Такий аналіз дозволяє визначити доцільність проведення досліджень на ювенільних тваринах і, в разі підтвердження, узгодити їх тривалість відносно клінічних випробувань.
За наявності рекомендацій щодо вивчення токсичності на ювенільних тваринах як підґрунтя тривалих клінічних випробувань за участю педіатричної популяції, такі доклінічні дослідження мають бути завершені до початку клінічних випробувань.
Іноді педіатрична популяція є основною популяцією, коли дослідження на тваринах можуть виявити ризик виникнення проблем розвитку органів-мішеней (токсикологічних або фармакологічних). У деяких з цих випадків може бути доречним проведення тривалих доклінічних досліджень токсичності на ювенільних тваринах. Може бути доцільним проведення довгострокових досліджень лікарського засобу на тваринах відповідного віку та виду з визначенням кінцевих точок, оскільки це впливає на розвиток їх організму (наприклад, протягом 12 міс на собаках і 6 міс на гризунах). Так, дослідження тривалістю 12 міс може повністю охоплювати період розвитку для собаки. Для інших видів тварин за певних умов даний дизайн досліджень може бути адаптованим для заміни відповідного загальноприйнятого довгострокового вивчення лікарського засобу на окреме дослідження з використанням ювенільних тварин.
Перед початком тривалого застосування лікарського засобу при проведенні педіатричних клінічних випробувань має визначатися доцільність вивчення канцерогенності. За відсутності серйозних причин для занепокоєння (наприклад, підтвердження генотоксичності в ряді тестів, імовірність проканцерогенного ризику, що ґрунтується на аналізі механізму дії або даних загальнотоксикологічних досліджень) не рекомендується вивчення канцерогенності перед проведенням педіатричних клінічних випробувань.
13. Імунотоксичність
Як зазначено в настанові ICH S8 «Immunotoxicity Studies for Human Pharmaceuticals», всі нові лікарські засоби, призначені до застосування у людини, повинні бути оцінені щодо їх імунотоксичної дії на основі аналізу ступеня її вірогідності за даними проявів імунної відповіді у ході стандартних токсикологічних досліджень і додаткових досліджень з імунотоксичності. У разі показань до проведення додаткових досліджень імунотоксичності вони повинні бути завершені до початку проведення клінічних випробувань на великій популяції пацієнтів (наприклад, фаза ІІІ клінічних випробувань).
14. Дослідження фотобезпеки
На доцільність проведення та розрахунок тривалості досліджень фотобезпеки у людини можуть впливати: 1) фотохімічні властивості (наприклад, фотопоглинання і фотостабільність) молекули; 2) інформація щодо фототоксичного потенціалу сполук, подібних за хімічною будовою; 3) розподіл у тканинах; 4) клінічні або доклінічні дані, що свідчать про фототоксичність.
Початкова оцінка потенціалу фототоксичності проводиться на основі фотохімічних властивостей лікарського засобу та належності до відповідного фармакологічного або хімічного класу речовин. Якщо оцінка всіх наявних даних та запропонований дизайн клінічних випробувань вказують на значний потенційний ризик фототоксичності для людини, необхідно вжити належних заходів щодо захисту пацієнтів при проведенні клінічних випробувань. Крім того, для отримання додаткових даних щодо ризику для людини та необхідності подальших випробувань має бути завершений відповідний доклінічний аналіз розподілу лікарського засобу при нанесенні на шкіру та очі. За необхідності експериментального вивчення фототоксичного потенціалу (доклінічного, in vitro або in vivo, клінічного) воно має бути завершене до застосування на великій кількості суб’єктів (фаза ІІІ).
Як альтернатива замість вищезазначеного поетапного підходу може бути здійснена пряма оцінка фототоксичності за даними доклінічних досліджень або клінічних випробувань. У разі негативних результатів цього дослідження немає необхідності у попередньому дослідженні розподілу щодо очей і шкіри та запровадженні заходів захисту при проведенні клінічних випробувань. Якщо оцінка фототоксичності вказує на потенційний ризик фотоканцерогенності, як правило, це необхідно належним чином враховувати при запровадженні заходів захисту пацієнтів, включаючи попередження до інформованої згоди на участь у клінічних випробуваннях та до інформації про препарат, що подається на реєстрацію (примітка 6 розділу 19).
15. Доклінічне вивчення схильності до лікарської залежності
Для засобів, що впливають на діяльність центральної нервової системи, незалежно від терапевтичних показань, необхідно розглянути питання про доцільність визначення потенціалу залежності. Доклінічні дослідження мають сприяти розробці дизайну клінічної оцінки потенціалу залежності, класифікації і внесенню регуляторними органами до відповідних переліків та інформації про препарат. При розробці комплексного плану специфічних досліджень потенціалу залежності слід враховувати діючі регіональні настанови проведення доклінічних досліджень схильності до лікарської залежності.
Для завчасного виявлення показників залежності можуть бути використані дані попередніх доклінічних досліджень, отримані на початку розробки лікарського засобу. Такі попередні показники доклінічних досліджень in vivo, зазвичай, мають бути встановлені до першого введення людині і включати: профіль ФK/ФД для визначення тривалості дії, подібність хімічної структури до структур відомих наркотиків, профіль зв’язування з рецептором, дані поведінки і клінічні симптоми. Якщо за даними ранніх досліджень явна залежність відсутня, розширені доклінічні дослідження щодо схильності до лікарської залежності є недоцільними. Зазвичай, якщо активні речовини виявляють ознаки, що асоціюються з відомими прикладами схильності до залежності, або активна речовина має новий механізм дії на центральну нервову систему, для обґрунтування розгорнутих клінічних випробувань (наприклад, фази ІІІ) рекомендується проведення додаткових доклінічних досліджень.
Якщо профіль метаболітів і мішень дії лікарського засобу у гризунів співпадають з такими у людини, доклінічна оцінка схильності до лікарської залежності може проводитися на гризунах. Примати можуть бути використані лише в окремих обмежених випадках, коли існують переконливі докази, що, на відміну від гризунів, лише даний вид тварин здатний передбачити схильність людини до лікарської залежності.
Оцінка потенціалу схильності до лікарської залежності ґрунтується на трьох видах досліджень: вибіркове відношення до лікарського засобу, самолікування, оцінка відміни.
Як правило, дослідження вибірковості вибору лікарського засобу та самостійного його застосування проводять окремо. Оцінка синдрому відміни іноді може включатися до плану токсикологічних досліджень, що передбачають вивчення зворотності дії. Адекватною дозою для доклінічного дослідження лікарської залежності є максимальна доза, згідно з якою концентрація у плазмі крові у декілька разів перевищує терапевтичну дозу, що використовується в клініці.
16. Інші дослідження токсичності
Якщо попередні доклінічні або клінічні дані щодо препарату або споріднених лікарських засобів вказують на наявність занепокоєння щодо безпеки, то може бути доцільним проведення додаткових доклінічних досліджень (наприклад, з метою визначення потенційних біомаркерів, встановлення механізму дії).
Підходи щодо кваліфікації домішок і продуктів розпаду викладені у настановах ICH Q3A(R2) «Impurities in New Drug Substances» та Q3B(R2) «Impurities in New Drug Products». Якщо показані спеціальні дослідження для кваліфікації домішок і продуктів розпаду, то зазвичай вони не проводяться до початку фази ІІІ клінічних випробувань, за винятком випадків, коли є зміни, що призводять до суттєво нового профілю вмісту домішок (наприклад, новий шлях синтезу, нові продукти розпаду, які утворюються внаслідок взаємодії складових лікарської форми). В даному випадку перед проведенням фази ІІ клінічних випробувань або наступних фаз впровадження мають бути здійснені відповідні дослідження якості.
17. Дослідження токсичності комбінованих лікарських засобів
Даний розділ стосується комбінованих лікарських засобів, призначених для сумісного пакування або введення в одній лікарській формі (фіксована комбінація). Викладені принципи також можуть застосовуватися при розробці препаратів, для яких є рекомендації стосовно одночасного застосування з конкретним лікарським засобом, навіть не у вигляді фіксованої комбінації, та щодо яких існує мінімальна клінічна інформація відносно комбінацій.
Зазначені комбінації можуть включати: (1) два або більше активних фармацевтичних інгредієнтів на більш пізніх стадіях вивчення (визначаються як субстанції, що мають значний досвід застосування в клініці (наприклад, фаза ІІІ клінічних випробувань та/або постмаркетингові дослідження)); (2) один або більше активних фармацевтичних інгредієнтів на пізніх стадіях вивчення і один або більше інгредієнтів на ранніх стадіях досліджень (визначаються як субстанції з обмеженим досвідом клінічного застосування (наприклад, фаза ІІ або більш рання фаза клінічних випробувань); (3) більше одного активного фармацевтичного інгредієнта на ранніх стадіях випробувань.
Для більшості комбінацій, що включають два активні фармацевтичні інгредієнти на пізніх стадіях досліджень і для яких існує достатній клінічний досвід сумісного введення, дослідження токсичності комбінацій як підґрунтя для клінічних випробувань або здійснення реєстрації, як правило, не рекомендується, за винятком випадків, коли існує значна занепокоєність стосовно токсичності (наприклад, подібність токсичного впливу на орган-мішень). Ця занепокоєність може варіювати залежно від коефіцієнту безпеки та можливості моніторингу негативного впливу на людину. Якщо для встановлення причини суттєвого занепокоєння щодо токсичності проводиться доклінічне дослідження, то, як правило, воно має бути завершене до початку клінічних випробувань цієї комбінації.
Якщо є два препарати на пізніх стадіях дослідження, для яких відсутній належний клінічний досвід сумісного введення, але згідно з існуючими даними немає причин для занепокоєння щодо токсичності, як правило, не рекомендується проведення доклінічних досліджень комбінації як підґрунтя обмежених короткострокових клінічних випробувань (наприклад, фаза ІІ випробувань тривалістю до 3 міс). Проте, до початку розширених або довготривалих випробувань комбінацій, а також перед реєстрацією лікарського засобу мають бути проведені доклінічні дослідження комбінацій.
Для комбінацій на ранніх стадіях розробки, що містять активні фармацевтичні інгредієнти, стосовно яких існує клінічний досвід застосування на пізніх стадіях досліджень та для яких відсутні проблеми щодо токсичності, не рекомендується проведення доклінічних досліджень токсичності комбінації як підґрунтя клінічних випробувань тривалістю до 1 міс (доказ правильності концепцій запланованого клінічного випробування). Клінічні випробування комбінації не повинні тривати довше, ніж клінічний досвід застосування окремих активних фармацевтичних інгредієнтів. Пізні стадії досліджень або більш тривалі клінічні випробування повинні бути підкріплені доклінічними дослідженнями токсичності комбінацій.
Для комбінацій, що містять два інгредієнти на ранніх стадіях дослідження, перед початком клінічних випробувань проводять доклінічні дослідження токсичності.
Якщо передбачається проведення доклінічних досліджень окремих активних фармацевтичних інгредієнтів та токсичності комбінації, то повна програма таких досліджень має обґрунтовувати клінічні випробування. Тривалість доклінічного вивчення комбінованих лікарських засобів має бути еквівалентною тривалості клінічних випробувань, сягаючи щонайбільше 90 днів. Дослідження токсичності комбінації протягом 90 днів також має обґрунтувати подальшу реєстрацію лікарського засобу. Залежно від очікуваного клінічного застосування період дослідження токсичності комбінації для обґрунтування реєстрації може бути менш тривалим. Дизайн доклінічних досліджень, що рекомендуються для характеристики комбінації, залежить від фармакологічного, токсикологічного та ФК профілів індивідуальних активних фармацевтичних інгредієнтів, показань до застосування(-нь), популяції пацієнтів та наявних клінічних даних.
Доклінічне вивчення комбінованих лікарських засобів, як правило, має обмежуватися одним адекватним видом тварин. У разі виявлення неочікуваних проявів токсичності може бути доцільним проведення додаткових досліджень.
Якщо повна програма доклінічних досліджень не передбачає вивчення індивідуальних активних фармацевтичних інгредієнтів, допускається проведення повного доклінічного токсикологічного дослідження лише комбінації за умови, що індивідуальні складові призначені виключно для застосування у комбінації.
Як правило, дослідження генотоксичності, фармакології безпеки, канцерогенності комбінацій до початку клінічних випробувань чи етапу реєстрації не проводяться, якщо індивідуальні активні фармацевтичні інгредієнти вивчені відповідно до сучасних стандартів. У тих випадках, коли до популяції пацієнтів включені ЖЗНД, і дані досліджень окремих активних фармацевтичних інгредієнтів показали наявність ембріофетального ризику, доклінічне вивчення комбінації не проводиться, оскільки потенціал небезпеки для розвитку потомства людини вже встановлений. Якщо доклінічним дослідженням ембріофетальної токсичності встановлено, що жодна зі складових не впливає на розвиток потомства людини, дослідження комбінації не рекомендуються за винятком випадків, які базуються на властивостях індивідуальних активних фармацевтичних інгредієнтів, комбінація яких підвищує небезпеку для людини. У разі, коли окремі інгредієнти вивчені стосовно впливу на ембріофетальний розвиток, але обґрунтовано доцільність проведення ембріофетальних досліджень їх комбінації, таке дослідження має бути завершене до подання заяви на реєстрацію лікарського засобу.
18. Подальші заходи з поліпшення гармонізації
Констатується досягнення значних успіхів в гармонізації розрахунку термінів проведення доклінічних досліджень безпеки як підґрунтя для клінічних випробувань фармакологічних засобів, деталізованих даною настановою. Однак залишається ряд відмінностей. Регуляторні органи й виробники продовжать розгляд цих відмінностей і подальше удосконалення процесу створення лікарських засобів.
19. Примітки
Примітка 1. У цьому документі «експозиція» зазвичай означає середнє значення AUC для групи. За деяких обставин (наприклад, коли відомо, що речовина або клас речовин викликають гострі функціональні зміни з боку серцево-судинної системи або клінічні ознаки впливу на центральну нервову систему) може бути доцільним визначення меж експозиції на основі середнього значення для групи Сmax, ніж AUC.
Примітка 2. Фертильність самців та самиць за стандартним гістопатологічним дослідженням сім’яників та яєчників у дослідженні в межах вивчення токсичності за повторних уведень (зазвичай на гризунах) протягом не менше 2 тиж вважається такою ж чутливою, що й у дослідженні фертильності на основі встановлення токсичного впливу на репродуктивні органи самців та самиць [9, 15, 16].
Примітка 3. Високоефективні засоби контролю народжуваності визначаються як такі, що за умов послідовного і правильного застосування окремо чи у комбінації сприяють зменшенню кількості помилок (наприклад, менше 1% на рік). Суб’єктам, що використовують гормональні контрацептивні засоби, має надаватися інформація відносно препарату, що досліджується, та його потенційного впливу на протизаплідний засіб.
Примітка 4. Попереднє ембріофетальне дослідження, як правило, проводиться з використанням адекватних рівнів доз; воно включає аналіз виживання плода, масу тіла, зовнішній та вісцеральний огляд; при цьому для кожної групи використовується щонайменше 6 самиць; включаються самиці, яким препарат уводився протягом періоду органогенезу. Це попереднє доклінічне дослідження має проводитися згідно з високоякісними науковими стандартами із забезпеченням швидкої доступності зібраних зареєстрованих даних або з дотриманням умов GLP.
Примітка 5. Частота настання вагітності у жінок, що вперше намагаються завагітніти, становить близько 17% на менструальний цикл. Частота настання вагітності, очікувана в клінічних випробуваннях фази ІІІ, проведених у ЖЗНД, становила <0,1% на менструальний цикл. У ході цих випробувань пацієнти заохочуються до попередження вагітності та вживають заходів для запобігання вагітності. Дані звіту доклінічних досліджень, проведених до початку фази ІІ, свідчать, що частота настання вагітності була нижчою, ніж у випробуваннях фази ІІІ, але ступінь подальшого зниження не може бути визначений внаслідок обмеженого числа залучених жінок. Виходячи з вищевикладеного досвіду фази ІІІ, оцінка клінічних випробувань фази ІІ із залученням 150 ЖЗНД протягом 3 міс показала значно менше ніж 0,5 випадків вагітності на лікарський засіб, що розробляється.
Примітка 6. Дослідження фотоканцерогенності на гризунах з використанням сучасних доступних моделей (наприклад, безволосих щурів) при розробці лікарського засобу не вважається доцільним і, як правило, не рекомендується. Якщо вивчення фототоксичності свідчить про наявність потенційного ризику фотоканцерогенності і доступних відповідних методів, таке дослідження зазвичай має бути завершене до реєстрації препарату, при цьому мають бути враховані результати оцінки ризику для людини.
Національний додаток (довідковий). Бібліографія
- Закон України «Про лікарські засоби» (із змінами) від 4 квітня 1996 року № 124/96ВР.
- Наказ Міністерства охорони здоров’я України від 14.12.2009 р. № 944 «Про затвердження Порядку проведення доклінічного вивчення лікарських засобів та експертизи матеріалів доклінічного вивчення лікарських засобів», зареєстрований в Міністерстві юстиції України від 19.01.2010 р. за № 53/17348.
- Настанова. Лікарські засоби. Належна лабораторна практика/ О. Стефанов, Т. Бухтіарова, В. Коваленко та ін. — Київ, МОЗ України, 2009.
- EMA/CPMP/ICH/286/1995 (ICH guideline M3(R2)) «Non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals, December 2009» (Доклінічні дослідження безпеки як підґрунтя клінічних випробувань за участю людини та реєстрації лікарських засобів, грудень 2009).
- ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів/І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
- ДСТУ 1.7-2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів/О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
- EMEA/P24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
- Доклінічне вивчення безпеки лікарських засобів біотехнологічного походження. Методичні рекомендації (схвалено Науково-експертною радою ДП «Державний експертний центр МОЗ України»). — Київ, 2011.
- ICH Topic S5(R2) Document «Detection of Toxicity to Reproduction for Medicinal Products and Toxicity to Male Fertility».
- ICH Topic S1C(R2) Document «Dose Selection for Carcinogenicity Studies of Pharmaceuticals».
- National Centre for the Replacement, Refinement and Reduction of Animals in Research «Challenging Requirements for Acute Toxicity Studies: Workshop Report, May 2007» (Проблемні вимоги вивчення гострої токсичності: звіт про семінар, травень 2007).
- A European pharmaceutical company initiative challenging the regulatory requirement for acute toxicity studies in pharmaceutical drug development, April 2008 (Ініціатива європейської фармацевтичної компанії стосовно критичної оцінки нормативних вимог, що регулюють дослідження гострої токсичності при фармацевтичній розробці лікарського препарату, квітень 2008).
- ICH Topic S2B «Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals, 1997» (Генотоксичність: стандартний набір тестів генотоксичності лікарських засобів).
- ICH Topic S1C(R2) «Dose Selection for Carcinogenicity Studies of Pharmaceuticals, March 2008» (Вибір дози для дослідження канцерогенності лікарських засобів, березень 2008).
- Collaborative work to evaluate toxicity on male reproductive organs by 2-week repeated-dose toxicity studies in rats. Overview of the studies, October 2000 (Спільна робота стосовно оцінки токсичності лікарського засобу для чоловічих статевих органів при повторному введенні дози щурам. Огляд досліджень, жовтень 2000).
- Collaborative work on evaluation of ovarian toxicity by repeated-dose and fertility studies in female rats, 2009 (Спільна робота стосовно оцінки токсичності лікарського засобу для яєчників при повторному введенні доз та досліджень народжуваності у самок щурів, 2009).
Ключові слова: лікарські засоби, доклінічні дослідження, клінічні випробування, безпека.