Патогенез остеоартрозу
Содержание
- РОЛЬ БІОМЕХАНІЧНИХ ФАКТОРІВ У ПАТОГЕНЕЗІ ТА ПРОГРЕСУВАННІ ОСТЕОАРТРОЗУ
- ГЕНЕТИЧНІ ТА МЕТАБОЛІЧНІ АСПЕКТИ ПАТОГЕНЕЗУ ОА
- РОЛЬ ЗМІН У СУБХОНДРАЛЬНІЙ КІСТЦІ В ПАТОГЕНЕЗІ ОА
- РОЛЬ ФЕРМЕНТІВ І ЦИТОКІНІВ У ПАТОГЕНЕЗІ ОСТЕОАРТРОЗУ
- РЕПАРАЦІЯ СУГЛОБОВОГО ХРЯЩА І ФАКТОРИ РОСТУ В ПАТОГЕНЕЗІ ОА
- РОЛЬ ВІДКЛАДАННЯ КРИСТАЛІВ У ПАТОГЕНЕЗІ ОА
РОЛЬ БІОМЕХАНІЧНИХ ФАКТОРІВ У ПАТОГЕНЕЗІ ТА ПРОГРЕСУВАННІ ОСТЕОАРТРОЗУ
Результати цілого ряду епідеміологічних досліджень, описаних у главі 1, свідчать, що професії, пов’язані з тривалим повторюваним використанням певних груп суглобів, пов’язані з високим ризиком розвитку ОА. Проте часто важко або навіть неможливо розділити частку механічного фактора в патогенезі ОА і вплив віку, генетичних, гормональних та інших факторів, які можуть сприяти виникненню та прогресуванню хвороби. Так, професії фермера (Thelin A., 1990; Croft P. et al., 1992; Axmacher B., Lindberg H., 1993), балерини (Andersson S. et al., 1989), будівельника (Partridge R.E.H., Duthie O.R., 1968; Andersson J.A.D., 1984; Lindberg H., Montgomery F., 1987; Felson D.T. et al., 1991; Vingard E. et al., 1991), а також професійне заняття футболом (Klunder K.B. et al., 1980; Lindberg H. et al., 1993; Vingard E. et al., 1993), лижним спортом, тенісом (Marti B. et al., 1989; Vingard E. et al., 1993; Kujala U. et al., 1994; Lequesne M.G. et al., 1997) асоційовані з розвитком ОА. Виникає питання, наскільки точно можна пов’язати це захворювання з первинною дегенерацією суглобового хряща, а не із вторинними його змінами після перенесених неминучих при цих видах діяльності травм інших тканин суглоба (менісків, зв’язок, капсули). Травма чи розрив менісків, а також розрив передніх хрестоподібних зв’язок колінного суглоба відносно часто супроводжують професійних гравців у футбол (Roos H., 1994). Результати дослідження кінетики вивільнення протеогліканів суглобового хряща в синовіальну рідину у професійних футболістів свідчать, що їх концентрація була значно підвищена протягом декількох годин після травми й, хоча згодом їх рівень знижувався, він залишався підвищеним протягом декількох років (Lohmander L.S. et al., 1989). Рентгенологічні ознаки ОА у цієї категорії осіб появлялися не менше ніж через 15 років після травми (Roos H., 1994). На меніски колінного суглоба впливає маса тіла людини, вони відіграють важливу механічну роль у нормальній функції суглоба (Shrive N.G. et al., 1978; Seedhom B.B., Hargreaves D.J., 1979; Kurosawa H. et al., 1980), тому їх травмування призводить до того, що суглобові поверхні несуть значно більше навантаження, ніж у нормі, прискорюючи дегенерацію хряща і розвиток ОА (De Haven K.E., 1985).
Вплив фізичних вправ на суглобовий хрящ. Популярність джогінгу (нешвидкий біг підтюпцем — Ред.) серед населення багатьох країн світу останнім часом привернула увагу до бігу на довгі дистанції як до фактора ризику розвитку ОА. Ретроспективні й проспективні дослідження показали, що клінічні та рентгенологічні критерії ОА у бігунів на середні дистанції та марафонців виявляють не частіше, ніж у людей, які не займаються бігом (Puranen J. et al., 1975; Lane N.E. et al., 1986; 1987; Panush R.S. et al., 1986; Sohn R.S., Lyle M.J., 1987; Konradsen L. et al., 1990; Lane N.E., Buckwalter A.J., 1993; Lane N.E. et al., 1993; Lequesne M.G. et al., 1997). Однак у зв’язку з тим, що дизайн більшості цих досліджень має ряд недоліків (некоректний статистичний аналіз, некоректні методи діагностики або оцінки ОА та ін.) їх результати викликають сумніви (Lequesne M.G. et al., 1997). N.E. Lane та співавтори (1986; 1987; 1993) спробували виправити помилки попередніх дослідників. Протягом 9 років вони вивчали рентгенологічні ознаки ОА у бігунів-аматорів похилого віку (середній вік — 65 років). Виявлено, що у цієї категорії осіб захворюваність на ОА (рентгенологічно підтверджена) не перевищувала такої у групі осіб того ж віку, які не займаються бігом. Хоча у групі бігунів-аматорів у жінок частіше реєстрували субхондральний склероз, а у осіб обох статей частіше виявляли остеофіти на рентгенівських знімках, проте автори зробили висновок, що непрофесійне заняття легкою атлетикою не є фактором ризику ОА. Таким чином, наведені дані свідчать про те, що у осіб зі «здоровими» суглобами біг на довгі дистанції не є причиною дегенерації хряща і розвитку ОА.
Дослідження біомеханіки ОА на моделях у тварин підтверджують вищенаведений висновок. P.M. Newton та співавтори (1997) досліджували гончаків, яких тренували бігом зі швидкістю 3,3 км/год по 75 хв/добу протягом 5 днів на тиждень. Кожен собака ніс додаткове «екзогенне» навантаження 11,5 кг (130% маси тіла). Контрольну групу становили дорослі гончаки, яких не тренували і не застосовували додаткове навантаження. Через 52 тиж після початку тренувань було проведене гістологічне дослідження суглобового хряща, менісків і зв’язок. Виявилося, що застосований рівень навантаження не викликав у собак дегенеративних змін у суглобових тканинах. Ніякої різниці між біомеханічними властивостями хряща у тренованих і нетренованих собак не виявлено.
В іншому дослідженні молодих гончаків (з незрілим скелетом) тренували за програмою середньої складності (4 км/год на тредмілі з нахилом 15°) протягом 15 тиж (Kiviranta E. et al., 1987; 1988). Автори виявили потовщення хряща і посилення синтезу протеогліканів порівняно з контрольною (нетренованою) групою тварин. Однак більша частина протеогліканів у хрящі тренованих тварин втратила здатність до агрегації з гіалуроновою кислотою і містила більшу кількість хондроїтин-6-сульфатів (Kiviranta E. et al., 1988). Автори дослідження припустили, що такий рівень навантаження прискорює дозрівання матриксних депозитів у суглобовому хрящі тварин.
У дослідженні, проведеному за участю молодих гончаків, програма тренування була дещо ускладнена: 20 км/добу протягом 15 тиж (Kiviranta E. et al., 1992). Таке навантаження викликало зниження концентрації колагену, підвищення вмісту води, зменшення співвідношення хондроїтин-6- і хондроїтин-4-сульфатів у суглобовому хрящі латеральних виростків стегнових кісток. Збільшення дистанції до 40 км/добу і тривалості тренування до 52 тиж супроводжувалося зниженням вмісту протеогліканів у ПКМ хряща (Arokoski J. et al., 1993; 1994). Найбільш виражену втрату глікозаміногліканів відзначали на верхівках виростків стегнових кісток, особливо в поверхневій зоні хряща (Helminen H.J. et al., 1992; Arokoski J. et al., 1994).
C. Little та співавтори (1997) продемонстрували, що тривалі інтенсивні тренування можуть індукувати зміни метаболізму протеогліканів у суглобах зап’ястка у коней. У рамках цього дослідження автори вивчали вплив помірних або високих тренувальних навантажень на синтез і деградацію великих агрегованих протеогліканів (агрекану) і двох дрібних дерматансульфатвмісних протеогліканів (декорин і біглікан). Експлантати суглобового хряща були взяті з трьох ділянок третьої зап’ясткової кістки, на які доводиться найбільше навантаження і які найчастіше травмуються у спортивних коней. У дослідження було включено 12 коней віком від 3 до 5 років без клінічних або рентгенологічних ознак патології середнього зап’ясткового суглоба. Програма тренувань включала біг зі швидкістю 6 м/с 2000 м 3 дні/тиж зі збільшенням дистанції до 4000 м до кінця 8-го тижня дослідження. Потім усіх тварин розподілили на дві групи — тварини групи А продовжували тренування в попередньому режимі, а у тварин групи В режим тренувань був посилений (біг зі швидкістю 8 м/с на дистанцію 4000 м 4 дні/тиж протягом 17 тиж). Через 16 тиж після закінчення тренувань було зроблено забір матеріалу з певних ділянок третьої зап’ясткової кістки з обох сторін.
У гістологічному дослідженні хряща тварин обох груп виявлено депресію поверхневих його ділянок і руйнування кальцифікованого хряща та «хвилястої границі» лише в ділянці дорзального радіального виступка третьої зап’ясткової кістки. Істотної різниці виявлених гістологічних змін між групами А і В не виявлено. У культурі експлантатів суглобового хряща у тварин групи В вивільнялася більша кількість протеогліканів із хряща дорзального радіального виступка в середовище, ніж у тварин групи А, що свідчить про більш високий рівень катаболізму в групі В. Включення 35S у протеоглікани було менш виражене в експлантатах, отриманих від тварин групи В; водночас у тварин цієї групи спостерігали посилення біосинтезу декорину, змін інтенсивності біосинтезу біглікану не виявлено. Таким чином, отримані результати свідчать про те, що тривалі інтенсивні тренування коней індукують пригнічення синтезу агрекану та посилення синтезу дерматансульфатвмісних протеогліканів.
Функціональна роль декорину в сполучній тканині взагалі та у хрящовій, зокрема, як і раніше, залишається предметом досліджень (див. главу 3). Припускають, що декорин відіграє центральну роль в організації колагенових макромолекул (Brown D.C., Vogel K.G., 1989), проліферації клітин (Kresse H. et al., 1993) і модулюванні активності факторів росту (наприклад ТФР-β) (Yamagouchi Y. et al., 1990). Додавання декорину до колагенового гелю викликало відкладання більш однорідних тонких колагенових фібрил, ніж за його відсутності (Vogel K.G., Trotter J.A., 1987). У тканині шийки матки після пологів руйнування колагенової сітки корелювало з підвищеним рівнем декорину (Rechberger T., Woessner J.F.Jr., 1993). Таким чином, декорин, найімовірніше, відіграє роль «диригента» процесів репарації та ремоделювання сполучної тканини.
Підвищення синтезу декорину хондроцитами суглобового хряща коней на фоні високих динамічних навантажень можна інтерпретувати в такий спосіб: декорин, що вивільняється з ушкоджених хондроцитів у відповідь на механічне перевантаження, виконує роль месенджера. Ця гіпотеза підтверджена дослідженнями in vitro та in vivo, що продемонстрували підвищену продукцію декорину хондроцитами, які піддавали надфізіологічному механічному навантаженню. T.H.V. Korver та співавтори (1992) повідомили, що циклічне навантаження in vitro, що застосовувалося протягом 7 днів, збільшує в 3 рази синтез декорину в експлантатах суглобового хряща. Подібні результати отримані N.A. Vissen та співавторами (1994), які використовували експлантати зрілого й незрілого суглобового хряща. У моделі раннього (гіпертрофічного) ОА, індукованого у собак шляхом перетинання передніх хрестоподібних зв’язок, G.S. Dourado та співавтори (1996) спостерігали підвищення рівня матричної рибонуклеїнової кислоти (мРНК) біглікану, декорину і фібромодуліну в хрящі дестабілізованих суглобів.
Вплив меніскектомії на суглобовий хрящ. Як зазначалося раніше, суглобні меніски відіграють важливу роль у нормальній функції суглоба. Меніски — структури, які збільшують конгруентність суглобових поверхонь стегнової та великогомілкової кістки, підвищують латеральну стабільність і поліпшують розподіл синовіальної рідини, а також обмін поживними речовинами із суглобовим хрящем (Seedhom B.B., Hargreaves D.J., 1979; Kurosawa H. et al., 1980). Тотальна або парціальна меніскектомія призводить до зміни напрямку навантаження на суглобову поверхню великогомілкової кістки (рис. 5.1), внаслідок чого розвивається дегенерація суглобового хряща (Helfet A.J., 1959; Tapper E.M., Hoover N.W., 1969; Appel H., 1970; Johnson R.J. et al., 1974; Cox J.S., Cordell L.D., 1977; Colombo C. et al., 1983).
Вивченню впливу меніскектомії на біомеханіку суглоба, а також індукції дегенеративних процесів у суглобовому хрящі й субхондральній кістці у тварин (зазвичай собак і овець) присвячено багато досліджень. Спочатку дослідники проводили ектомію медіального меніска колінного суглоба (Ghosh P. et al., 1983; 1990; 1993; Armstrong S. et al., 1994), однак згодом виявилося, що ектомія латерального меніска зумовлює більш швидкий розвиток ОА (Ghosh P. et al., 1993; 1997; Little C.B. et al., 1996; 1997).
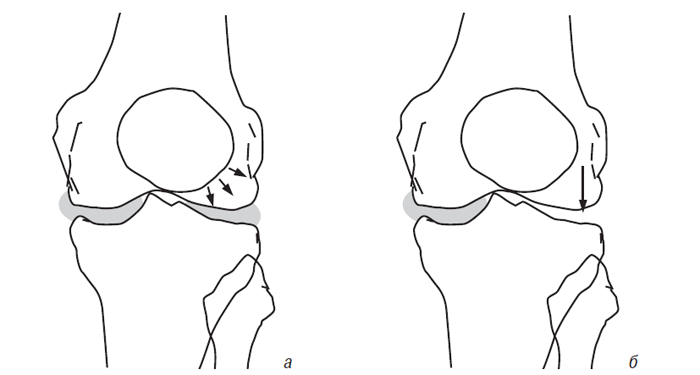
Рис. 5.1. Вектор локального тиску на суглобовий хрящ і субхондральну кістку в колінному суглобі в нормі (а) і після латеральної меніскектомії (б)
Використовуючи латеральну меніскектомію в овець, C. Little та співавтори (1997) досліджували зміни суглобового хряща й субхондральної кістки у декількох ділянках колінного суглоба. Типовими гістологічними знахідками, що ілюструють індуковані зміни в суглобовому хрящі через 6 міс після операції, були розволокнення хряща, зниження концентрації протеогліканів, зменшення кількості хондроцитів. Під місцями зміненого хряща в субхондральній кістці відзначено проростання капілярів у зону кальцифікованого хряща, зміщення назовні «хвилястої границі» і потовщення губчастої речовини субхондральної кістки.
У дослідженні P. Ghosh та співавторів (1998) виявлено, що через 9 міс після латеральної меніскектомії у овець з’являються ознаки ремоделювання субхондральної кістки та підвищення МЩКТ вторинно відносно дегенерації суглобового хряща. У зонах, що потерпають від аномально високого механічного навантаження внаслідок видалення латерального меніска (латеральний виросток стегнової кістки й латеральна пластинка великогомілкової кістки), виявлено підвищений синтез дерматансульфатвмісних протеогліканів (Little C.B. et al., 1996), хоча у хрящі медіальної пластинки також виявлено підвищення синтезу протеогліканів того ж виду. Виявилося, що дерматансульфатвмісні протеоглікани представлені в основному декорином (Little C.B. et al., 1996; Ghosh P. et al., 1997). Найвища його концентрація виявлена в середніх і глибоких зонах суглобового хряща (Poole A.R. et al., 1996; Ghosh P. et al., 1997).
Водночас із підвищенням синтезу дерматансульфатвмісних протеогліканів у зонах хряща, що несуть високе навантаження внаслідок видалення латерального меніска, виявлено підвищений катаболізм агрекану, про що свідчить вивільнення його фрагментів у живильне середовище з експлантатів хряща (Little C.B. et al., 1996), а також висока активність матричної металопротеази (ММП) і агреканаз (Ghosh P. et al., 1998). Внаслідок того, що запальна активність у цій моделі ОА була мінімальною, автори припустили, що джерелом ферментів були хондроцити.
Незважаючи на те що ще залишається багато невирішених питань, в описаних дослідженнях розкривається можлива роль біомеханічних факторів у патогенезі остеоартрозу. Зрозуміло, що хондроцити здатні «відчувати» механічні властивості свого оточення, реагуючи на їх зміни синтезом ПКМ, здатного переносити більше навантаження і у такий спосіб запобігати ушкодженню хряща. У молодих тварин помірні фізичні вправи викликали синтез багатого на агрекан ПКМ. Ця гіпертрофічна (чи адаптивна) фаза відповіді хондроцитів може тривати кілька років, забезпечуючи стабільний рівень механічного навантаження на суглобовий хрящ. Однак порушення цього балансу внаслідок підвищення інтенсивності чи тривалості навантаження, або зміни нормальної біомеханіки суглоба після травми чи хірургічного втручання, або зниження здатності хондроцитів посилювати синтез ПКМ у відповідь на збільшення навантаження (при старінні), дія ендокринних факторів спричиняють значні зміни на клітинному та матриксному рівні: пригнічується синтез протеогліканів і колагену ІІ типу, стимулюється синтез декорину й колагенів І, ІІІ та Х типу. Одночасно зі зміною біосинтезу підвищується катаболізм ПКМ, а також рівень ММП і агреканаз. Не відомо, як механічне навантаження сприяє резорбції хондроцитами оточуючого їх ПКМ, можливо, цей процес опосередкований простаноїдами, цитокінами (такими, як ІЛ-1β або ФНП-α, вільними кисневими радикалами). Тут необхідно згадати про роль синовіта при ОА, тому що найбільш імовірним джерелом вищезазначених медіаторів катаболізму можуть виступати макрофагоподібні синовіцити і лейкоцити, що інфільтрують синовіальну оболонку суглоба.
У дослідженні O.D. Chrisman та співавторів (1981) продемонстровано, що травматичне ушкодження суглоба стимулює продукцію попередника простагландинів (ПГ) — арахідонової кислоти. Джерелом арахідонової кислоти вважають мембрани ушкоджених хондроцитів. Добре відомо, що арахідонова кислота швидко конвертується у ПГ за допомогою ферменту циклооксигенази (ЦОГ). Продемонстровано, що ПГ, зокрема ПГE2, взаємодіють із рецепторами хондроцитів, змінюючи еспресію їх генів. Проте залишається не відомим, стимулює чи пригнічує арахідонова кислота продукцію протеїназ і агреканаз. Більш ранні дослідження показали, що ПГE2 збільшує продукцію ММП і викликає деградацію суглобового хряща. За результатами інших досліджень ПГE2 має анаболічний вплив на ПКМ, а також сприяє цілісності ПКМ, пригнічуючи продукцію цитокінів хондроцитами. Можливо, протилежні дані цих досліджень зумовлені різними концентраціями ПГE2, які в них використовували.
Невелика кількість ІЛ-1β (основний цитокін, що стимулює синтез і вивільнення ММП, а також пригнічує активність їх природних інгібіторів) може утворюватися у відповідь на ушкодження суглобового хряща, що веде до подальшої деградації тканини.
Таким чином, описані в цій главі дослідження показали, що підтримка підпорогового динамічного навантаження на суглоб викликає розмноження хондроцитів, здатних переносити нові механічні умови, що означає настання гіпертрофічної стадії ОА (рис. 5.2). Гіпертрофовані хондроцити — клітини, що перебувають в останній стадії диференціювання, тобто експресія генів основних елементів матриксу в них змінена. Тому синтез агреканових протеогліканів і колагену ІІ типу пригнічений, а синтез декорину, колагенів І, ІІІ та Х типу посилений.
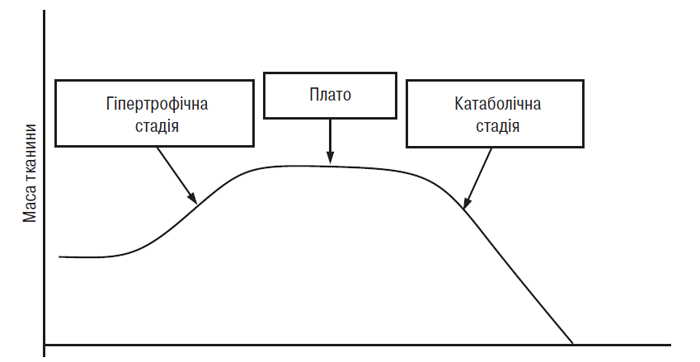
Рис. 5.2. Спрощена схема взаємовідношення між надфізіологічним навантаженням на суглобовий хрящ і зміною метаболізму хондроцитів. Кінцева (катаболічна) стадія відповідає маніфестному ОА (за P. Ghosh, 1999)
Зниження вмісту агрекану і колагену ІІ типу в ПКМ, пов’язане з порушенням балансу між процесами синтезу й деградації, надає суглобовому хрящу властивість неадекватно реагувати на механічне навантаження. Як наслідок хондроцити стають незахищеними, процес переходить у третю, катаболічну, стадію (див. рис. 5.2), що характеризується надмірною протеолітичною активністю і секрецією аутокринних і паракринних факторів регуляції. Морфологічно ця стадія характеризується деструкцією ПКМ суглобового хряща, клінічно — відповідає маніфестному ОА. Ця гіпотеза, безумовно, є спрощеним поясненням всіх складних процесів, що відбуваються при ОА, однак вона узагальнює сучасну концепцію патобіології ОА.
ГЕНЕТИЧНІ ТА МЕТАБОЛІЧНІ АСПЕКТИ ПАТОГЕНЕЗУ ОА
Роль механічних факторів у патогенезі остеоартрозу безсумнівна, однак існують переконливі дані про те, що деякі форми ОА успадковуються за законами Менделя. Спадкові остеоартропатії можна поділити на:
- первинний генералізований ОА;
- кристаласоційовані артропатії;
- передчасний ОА внаслідок спадкової остеохондродисплазії (Williams C.J., Jimenez S.A., 1999).
У 1803 р. W. Heberden описав «злегка щільні вузли розміром, як дрібна горошина» на тильній поверхні дистальних міжфалангових суглобів кистей. Ця ознака, на думку автора, відрізняє ОА від інших захворювань суглобів, включаючи подагру. J. Hayagarth (1805) розширив клінічний опис вузлів Гебердена, відзначивши їх часту асоціацію з артрозом інших локалізацій. Надалі Ch.J. Bouchard описав аналогічні вузли на тильній поверхні проксимальних міжфалангових суглобів кистей. Використовуючи термін «вузлики Гебердена й Бушара», W. Osler розділив «гіпертрофічний артрит» і «деформівний артрит» (1909). У 1953 р. R.M. Stecher і H. Hersh виявили поширення вузлів Гебердена серед членів родини й дійшли висновку, що вони успадковуються за аутосомно-домінантним типом. Дослідження, проведені після відкриття R.M. Stecher і H. Hersh, виявили асоціацію вузлів Гебердена й Бушара з дегенеративним ураженням інших суглобів (Kellgren J.H., Moore R., 1952; Stecher R.M. et al., 1953; Allison A.C., Blumberg B.S., 1958; Crain D.C., 1961; Kellgren J.H. et al., 1963; Buchanan W.W., Park W.M., 1983; Nuki G., 1983). Ґрунтуючись на даних клінічного обстеження та HLA-типування, J.S. Lawrence (1977), J.S. Lawrence та співавтори (1983) припустили наявність полігенного успадкування, а не дефекту одиничного гена.
Фенотипічний спектр спадкового ОА широко варіює від легких форм, які проявляються клінічно лише після досягнення пізнього зрілого віку, до дуже тяжких форм, що маніфестують у дитячому віці. Традиційно всі ці форми класифікували як вторинний ОА. На сьогодні відомо, що причиною деяких із цих фенотипів є мутація генів, які кодують макромолекули ПКМ суглобового хряща, що порушує цілісність хрящового матриксу, а також регуляцію проліферації хондроцитів і експресії генів. Ці спадкові захворювання становлять певну підгрупу ОА, що відрізняється від вторинного ОА (табл. 5.1).
Таблиця 5.1
Відмінності між спадковим і вторинним ОА (за: C.J. Williams і S.A. Jimenez, 1999)
| Показник | Спадковий ОА | Вторинний ОА |
| Етіологія | Мутація генів, що експресують у суглобовому хрящі | Різні спадкові й набуті хвороби |
| Патогенез | Ушкодження структурних або функціональних компонентів суглобового хряща | Вторинні прояви хвороби, яка не завжди уражає лише суглобовий хрящ |
| Лікування | Можлива генна терапія для корекції дефекту гена | Лікування основного захворювання |
Хондродисплазії/остеохондродисплазії — група клінічно гетерогенних хвороб, що характеризуються аномаліями росту й розвитку суглобового хряща і ростової пластинки. Деякі хондродисплазії/остеохондродисплазії призводять до раннього розвитку ОА, що клінічно характеризується тяжким перебігом. Серед них можна виділити такі захворювання (табл. 5.2):
Таблиця 5.2
Спадкові дисплазії, що характеризуються раннім початком ОА (за: C.J. Williams і S.A. Jimenez, 1999)
| Захворювання | Локус | Тип наслідування | Ген, що мутував | Тип мутації |
| Ранній ОА з пізнім початком СЕД (ОАР)* | 12q13.1–q13.2 | АД | COL2A1 | Заміна основи, вставка, делеція |
| Синдром Стиклера (STL1) | 12q13.1–q13.2 | АД | COL2A1 | Заміна основи, вставка |
| Синдром Стиклера (STL2) | 6p21.3 | АД | COL11A2 | Вставка, делеція |
| Синдром Стиклера | 1p21 | АД | COL11A1 | Заміна основи |
| Синдром Вагнера | 12q13.1–q13.2 | АД | COL2A1 | Заміна основи |
| ОСМЕД | 6p21.3 | АР | COL11A2 | Заміна основи |
| Синдром Маршалла | 1p21 | АД | COL11A1 | Вставка |
| Дисплазія Кніста | 12q13.1–q13.2 | АД | COL2A1 | Вставка, делеція |
| МЕД (EDM 1) | 19p13.1 | АД | COMP | Заміна основи |
| МЕД (EDM 2) | 1p32.2–p33 | АД | COL9A2 | Вставка |
| МХД Шміда (MCDS) | 6q21–q22.3 | АД | COL10A1 | Заміна основи, делеція |
| МХД Янсена (MCDJ) | 3p21.2–p21.3 | АД | PTHR1 | Заміна основи |
*У дужках зазначені символи локусу; АД — аутосомно-домінантний; АР — аутосомно-рецисивний; МЕД — множинна епіфізіальна дисплазія; МХД — метафізіальна хондродисплазія; ОСМЕД — отоспондилометаепіфізіальна дисплазія; СЕД — спондилоепіфізіальна дисплазія.
- спондилоепіфізіальні дисплазії;
- синдром Стиклера:
- дисплазія Кніста;
- множинні епіфізіальні дисплазії;
- метафізіальні хондродисплазії;
- деякі отоспондилометаепіфізіальні дисплазії.
Спондилоепіфізіальні дисплазії включають гетерогенну групу хвороб з аутосомно-домінантним типом наслідування, що характеризуються аномальним розвитком осьового скелета й тяжкими змінами епіфізів довгих трубчастих кісток, що часто спричинюють карликовість. Нерідко спондилоепіфізіальні дисплазії мають тяжкий клінічний перебіг, супроводжуються вкороченням тіла і меншою мірою — кінцівок (Spranger J., 1976; Rimoni D.L., Lachman R.S., 1990; Horton W.A., Hecht J.T., 1993; Pyeritz R.E., 1993; Byers P.H., 1994).
При формах спондилоепіфізіальної дисплазії, що маніфестує у більш пізньому віці, фенотип часто мало змінений і може клінічно не проявлятися до підліткового віку, коли розвивається тяжкий ОА. Деформація поперекового відділу хребта може проявлятися звуженням міжхребцевих дисків, платиспондилією і незначним кіфосколіозом. Також виявляють аномалії епіфізів у периферичних суглобах і ранні дегенеративні зміни в них. Найбільш постійною ознакою ураження периферичних суглобів є сплощення суглобових поверхонь гомілковостопних і колінних суглобів, а також сплощення міжвиросткової борозни стегнової кістки. Нерідко виявляють аномалії голівки та шийки стегнової кістки з розвитком ОА кульшового суглоба, що маніфестує в підлітковому віці.
У зв’язку з тим що колаген ІІ типу — головний компонент ПКМ гіалінового хряща, висловлено припущення, що причиною спондилоепіфізіальної дисплазії є кодуючий його ген COL2A1. Перший опис генетичного зв’язку між фенотипом раннього ОА, пов’язаного зі спондилоепіфізіальною дисплазією, що пізно маніфестує, і геном проколагену ІІ типу COL2A1 відноситься до 1989 (A. Palotie та співавтори) і 1990 р. (R.G. Knowlton та співавтори). Перше повідомлення про мутацію COL2A1 у родички з раннім ОА, пов’язаним із спондилоепіфізіальною дисплазією, що пізно маніфестує, стосувалося заміни основи Arg519>Cys (Ala-Kokko L. et al., 1990). На сьогодні виявлені ще чотири родини з аналогічними мутаціями (Bleasel J.F. et al., 1998). У членів іншої родини з раннім ОА та спондилоепіфізіальною дисплазією легкого перебігу виявлено заміну основи Arg75>Cys, хоча фенотип спондилоепіфізіальної дисплазії у членів цієї родини не схожий на фенотип родини із заміною аргініну на цистеїн у положенні 519. У представників родин із спондилоепіфізіальною дисплазією також виявлені й інші мутації COL2A1–gly976>Ser (Williams C.J. et al., 1995), Gly493>Ser (Katzenstein P.L. et al., 1992). J. Spranger та співавтори (1994) застосували термін «тип ІІ колагенопатії» для опису спадкових хвороб хрящової тканини з первинною мутацією гена проколагену II типу COL2A1.
Класична форма синдрому Стиклера була вперше описана у 1965 р. G.B. Stickler та співавторами, які назвали його спадковою артроофтальмопатією. Описаний G.B. Stickler синдром характеризувався ураженням органа зору й тяжким дегенеративним захворюванням суглобів, яке зазвичай розвивається на третьому або четвертому десятилітті життя. Це аутосомно-домінантне захворювання, поширеність якого становить приблизно 1 на 10 тис. немовлят. Клінічна картина хвороби включає міопію, прогресуючу глухоту, щілину піднебіння, гіпоплазію нижньої щелепи (аномалія П’єра — Робена) і гіпоплазію епіфізів. У неонатальний період на рентгенограмах хворих із синдромом Стиклера виявляють збільшені епіфізи, в основному проксимальний стегнової та дистальний великогомілкової кісток. У процесі росту розвивається дисплазія епіфізів, яка проявляється нерегулярністю осифікації епіфізів і подальшими дегенеративними змінами.
У зв’язку з тим, що COL2A1 експресується в суглобовому хрящі і склоподібному тілі очного яблука, з патологією цього гена пов’язували виникнення синдрому Стиклера. Однак обстеження декількох родин із синдромом Стиклера показало, що не у всіх родинах захворювання пов’язане з COL2A1 (Francomano C.A. et al., 1987; Knowlton R.G. et al., 1989; Bonaventure J. et al., 1992). Таку форму хвороби називають І типом синдрому Стиклера (символ локусу STL1).
Спектр клінічних проявів синдрому Стиклера широко варіює, на сьогодні виділено декілька фенотипів. Серед них — синдром Вагнера, який характеризується переважним ураженням очного яблука; ОА при синдромі Вагнера фактично ніколи не розвивається, хоча у хворих виявлена мутація саме гена COL2A1 (заміна основи Gly67>Asp) (Korkko J. et al., 1993). Залишається нез’ясованим, чому така мутація COL2A1 компрометує тільки функцію склоподібного тіла й не впливає на гіаліновий хрящ.
Ще однією формою синдрому Стиклера є так званий голландський варіант; він характеризується всіма класичними проявами синдрому, за винятком ураження органа зору. H.G. Brunner та співавтори (1994) показали, що голландський фенотип синдрому Стиклера пов’язаний із мутацією гена COL11A2: домінуючою мутацією є делеція 54 пари основ з подальшою делецією екзона (Vikkula M. et al., 1995). M. Sirko-Osadsa та співавтори (1998) повідомили про іншу родину, не пов’язану з описаною попередніми авторами, з аналогічним фенотипом і мутацією гена COL11A2 (делеція 27 пари основ), що підтверджує дані H.G. Brunner та співавторів (1994). Цей варіант називають ІІ типом синдрому Стиклера (символ локусу STL2).
Нещодавно був визначений третій локус синдрому Стиклера у членів родини з патологією склоподібного тіла й сітківки ока, які фенотипічно суттєво відрізняються від змін, що спостерігаються при «класичному» варіанті синдрому. У представників цієї родини виявлена мутація гена COL11A1 (заміна основ Gly97>Val) (Richards A.J. et al., 1996). Безперечно, підтвердження знахідки A.J. Richards та співавторів потребує нових описів випадків такого фено- і генотипу синдрому Стиклера.
Тривалий час обговорювалося питання про нозологічні зв’язки синдрому Маршалла і класичного варіанта синдрому Стиклера. На сьогодні синдром Маршалла класифікують як окремий фенотип в основному завдяки більш вираженій деформації лицьового скелета (Williams C.J., Jimenez S.A., 1999), хоча ураження периферичних суглобів схоже з таким при синдромі Стиклера І типу. При синдромі Маршалла ОА колінних суглобів і попереково-крижового відділу хребта починається після 30 років. Причиною синдрому є мутація гена колагену ІХ типу COL11A1 (Griffith A.J. et al., 1998).
Отоспондилометаепіфізіальна дисплазія. Цей фенотип був описаний у голландській родині, у членів якої дегенеративні зміни в суглобах, що нагадують ОА, з’являлися в підлітковому віці та вражали головним чином кульшові, колінні, ліктьові й плечові суглоби; також виявлені своєрідні риси обличчя, посилення поперекового лордозу, збільшення міжфалангових суглобів, туговухість, однак не виявлено ніяких аномалій органа зору (Vikkula M. et al., 1995). Дослідники виявили мутацію гена, що кодує α2-ланцюг колагену ІІ типу COL11A2.
Дисплазія Кніста характеризується вкороченням тулуба й кінцівок, сплощенням обличчя і спинки носу, екзофтальмом і тяжкою аномалією суглобів (Maroteaux P., Spranger J., 1973; Kniest W., Lieber B., 1977). У хворих із синдромом Кніста суглоби, зазвичай великі від народження, продовжують збільшуватися в дитинстві й ранньому підлітковому віці. У них також часто можна виявити міопію, туговухість, щілину піднебіння, клишоногість; у більшості хворих рано розвиваються тяжкі дегенеративні зміни, особливо виражені в колінних і кульшових суглобах. На рентгенограмах хребта виявляють сплощення й значне подовження тіл хребців, платиспондилію. Довгі трубчасті кістки деформовані подібно до гантелі, осифікація епіфізів сповільнена. У суглобах кистей сплощені епіфізи та звужені суглобові щілини. Суглобовий хрящ м’який, його еластичність знижена; гістологічно в ньому виявляють великі кісти (симптом «швейцарського сиру»). Причиною синдрому Кніста є мутація гена проколагену ІІ типу COL2A1.
Множинні епіфізіальні дисплазії — гетерогенна група хвороб, що характеризуються аномалією розвитку ростових пластинок довгих трубчастих кісток, а також раннім (маніфестуючим у дитячому віці) тяжким ОА, що вражають як осьові, так і периферичні суглоби (найчастіше колінні, кульшові, плечові й суглоби кистей). Клінічно множинні епіфізіальні дисплазії проявляються болем і скутістю в суглобах, зміною ходи. У хворих із множинною епіфізіальною дисплазією виявляють також мінімальні зміни з боку хребета (різного ступеня сплощення тіл хребців), іноді хребет може бути інтактним. Також характерний низький зріст хворих, хоча карликовість розвивається рідко. Орган зору не уражується. Множинні епіфізіальні дисплазії включають кілька варіантів, наприклад фенотип Фербенкса (Fairbanks T., 1947) і Риббінга (Ribbing S., 1937).
Множинні епіфізіальні дисплазії успадковуються за аутосомно-домінантним типом з різним ступенем пенетрантності. У зв’язку з тим, що відмінною рисою множинної епіфізіальної дисплазії є аномалія ростової пластинки епіфізів, було висловлено припущення, що причиною цих дисплазій є дефект генів, які кодують макромолекули хряща ростової пластинки. Виявилося, що принаймні три локуси пов’язані з фенотипом множинної епіфізіальної дисплазії. Дослідження E.J. Weaver та співавторів (1993), J.T. Hecht та співавторів (1992) виключили зі списку «винуватців» множинної епіфізіальної дисплазії гени колагенів ІІ і VI типу, стрижньового білка протеогліканів і сполучного білка хряща. J.T. Hecht та співавтори (1993), R. Oehelmann та співавтори (1994) виявили зв’язок між множинною епіфізіальною дисплазією, а також клінічно близьким до неї синдромом псевдоахондроплазії, та перицентромірним регіоном 19-ї хромосоми. Подальші дослідження ідентифікували мутацію гена, що кодує олігомерний матриксний протеїн хряща у 3 хворих із множинними епіфізіальними дисплазіями (символ локусу EDM1) (Briggs M.D. et al., 1995). Оскільки всі 3 мутації відбулися в ділянці гена, що кодує кальцієзв’язувальний домен олігомерного матриксного протеїну хряща, ймовірно, саме кальцієзв’язувальна функція цього білка є невід’ємною для нормального розвитку хряща ростової пластинки.
M.D. Вriggs та співавтори (1994) повідомили про родину з Голландії, фенотип множинної епіфізіальної дисплазії якої був пов’язаний з ділянкою 1-ї хромосоми, що містить один з генів колагену ІХ типу COL9A2 (символ локусу EDM2). Характерно, що виявлена мутація виявилася першим доказом ролі колагену ІХ типу, що локалізується на поверхні фібрил колагену ІІ, у підтримці цілісності гіалінового хряща. M. Deere та співавтори (1995) показали, що фенотип Фербенкса генетично не зв’язаний ні з локусом EDM1, ні з локусом EDM2, що підтвердило гетерогенність множинної епіфізіальної дисплазії.
Метафізіальні хондродисплазії — гетерогенна (описано понад 150 типів) група спадкових захворювань гіалінового хряща, які клінічно проявляються раннім ОА (Sutcliffe J., Stanley P., 1973; Kozlowski K., 1976; Lachman R.S. et al., 1988). Метафізіальні хондродисплазії характеризуються змінами метафізів кісток. Клінічно вони проявляються низьким зростом, укороченням кінцівок, викривленням гомілок, «качиною» ходою. Також у хворих із метафізіальними хондродисплазіями виявляють ознаки ураження інших систем (наприклад імунної та травної). Спостерігають дезорганізацію хряща ростової пластинки, що гістологічно проявляється скупченнями проліферованих і гіпертрофованих хондроцитів, оточених потовщеними перетинками і дезорганізованим матриксом, а також проникненням некальцифікованого хряща в субхондральну кістку.
Синдроми Янсена, Шміда й МакКузика — найбільш вивчені метафізіальні хондродисплазії. Вони подібні за особливостями аномалій скелета, але відрізняються за ступенем тяжкості (синдром Янсена>синдром МакКузика>синдром Шміда). Найчастіше відзначають синдром Шміда (символ локусу MCDS), який успадковується за аутосомно-домінантним типом. Рентгенологічно синдром проявляється coxa vara, укороченням і викривленням трубчастих кісток, чашеподібною деформацією метафізів (більш вираженою у проксимальному, ніж у дистальному відділі стегнової кістки). Найбільш виражені зміни спостерігають у ростових пластинках довгих трубчастих кісток.
Принаймні 17 різних видів мутацій гена колагену Х типу описані у хворих із синдромом Шміда. Колаген Х типу експресується в гіпертрофованих хондроцитах ростових пластинок і, можливо, бере участь у процесах осифікації (Jacenko O. et al., 1993). Таким чином, мутація, що кодує колаген Х типу гена COL10A1 — найбільш імовірна причина синдрому Шміда (Kuivaniemi H. et al., 1997).
У дітей із синдромом Янсена виявляють гіперкальціємію, а також підвищений рівень фосфатів в сечі, зниження рівня паратгормону і паратгормонзв’язаного пептиду (Lenz W., 1969). З аномалією останнього, ймовірно, пов’язане виникнення синдрому Янсена. У 1994 р. A.C. Karaplis та співавтори опублікували результати оригінального дослідження. Після руйнування гена, що кодує паратгормонзв’язаний пептид у стовбурових клітинах ембріонів мишей, миші з дефіцитом за цим алелем помирали одразу після народження. У них виявлені аномалія розвитку субхондральної кістки, порушення росту хряща і зниження проліферації хондроцитів. У 1995 р. E. Schipani та співавтори повідомили про гетерозиготну мутацію гена рецептора паратгормонзв’язаного пептиду у пацієнта із синдромом Янсена. Мутація полягала в заміні основи Gys223>Arg, що призводило до накопичення циклічного аденозинмонофосфату (цАМФ); це означає, що амінокислота гістидин у положенні 223 відіграє вирішальну роль у передачі сигналу. Пізніше E. Schipani та співавтори (1996) повідомили про трьох інших хворих із синдромом Янсена, у двох з яких виявлена аналогічна мутація, а в третього — заміна Try410>Pro.
Найбільш поширеною спадковою формою ОА є первинний генералізований ОА, який був уперше описаний як окрема нозологія J.H. Kellgren і R. Moore у 1952 р. Клінічно для первинного генералізованого ОА характерна поява вузлів Бушара й Гебердена, поліартикулярне ураження. Первинний генералізований ОА характеризується раннім початком маніфестації ОА й швидким його прогресуванням. Рентгенологічно первинний генералізований ОА не відрізняється від неспадкового ОА. Незважаючи на те що питання про етіопатогенез первинного генералізованого ОА все ще дискутується, проведені дослідження демонструють важливу роль спадкової схильності у виникненні та прогресуванні первинного генералізованого ОА (Stecher R.M. et al., 1953; Allison A.C., Blumberg B.S., 1958; Kellgren J.H. et al., 1963; Harper P., Nuki G., 1980; Nuki G., 1983).Так, J.H. Kellgren та співавтори (1963) виявили вузли Бушара й Гебердена у 36% родичів чоловічої статі та у 49% родичів жіночої статі, тоді як у загальній популяції ці цифри становили 17 і 26% відповідно. У осіб із первинним генералізованим ОА частіше виявляють HLA A1B8 гаплотип (Lawrence J.S. et al., 1983; Pattrick M. et al., 1989) і MZ-ізоформу α1-антитрипсину (Pattrick M. et al., 1989). У класичному дослідженні за участю близнюків T.D. Spector та співавтори (1996) виконали рентгенографію колінних суглобів і суглобів кистей у 130 одно- і 120 двояйцевих близнюків жіночої статі з метою виявлення наявності змін, характерних для ОА. З’ясувалося, що конкордантність рентгенологічних ознак ОА всіх локалізацій була в 2 рази вищою у однояйцевих близнюків порівняно з двояйцевими, а внесок генетичних факторів коливався від 40 до 70%. У дослідженні вузликового ОА, проведеного G.D. Wright та співавторами (1997), продемонстровано ранній початок хвороби, високий ступінь тяжкості й негативний кореляційний зв’язок між віком початку хвороби у пацієнтів і віком їх зачаття батьками.
Серед кристаласоційованих артропатій відкладення кристалів сечової кислоти і кальцієвмісних кристалів у порожнині суглоба мають сімейну схильність (табл. 5.3).
Таблиця 5.3
Спадкові кристаласоційовані артропатії (за: Williams C.J. і Jimenez S.A., 1999)
| Захворювання | Локус | Тип успадковування | Мутуючий ген | Тип мутації |
| Подагра (HPRT)* | Xq27 | Пов’язаний із Х-хромосомою | HPRT1 | Заміна основи, делеція |
| Подагра (PRPS) | Xq22–q24 | Пов’язаний із Х-хромосомою | PRPS1 | Заміна основи |
| Первинна пірофосфатна артропатія (CCAL1) | 5p15.1–p15.2 | АД | ? | ? |
| Пірофосфатна артропатія, асоційована з раннім початком ОА (CCAL2) | 8q | АД | ? | ? |
*У дужках зазначені символи локусу; АД — аутосомно-домінантний.
У 1958 р. D. Zintan і S. Sitaj навели клінічні описи патології, яку вони назвали «хондрокальцинозом» у 27 хворих. Більшість пацієнтів належали до п’яти родин, що вказувало на спадковий компонент в етіопатогенезі хвороби. Пізніше D. McCarty і J.L. Hollander (1961) повідомили про 2 хворих, у яких припускали подагру з відкладенням неуратних кристалів у порожнині суглобів. Рентгенологічне дослідження виявило аномальну кальцифікацію гіалінового хряща багатьох суглобів.
Рентгенологічно хвороба відкладання кристалів пірофосфату кальцію дигідрату, або пірофосфатна артропатія, нагадує спорадичний ОА, проте вона частіше уражує суглоби, нетипові для звичайних форм ОА (наприклад п’ястково-фалангові, човноподібно-променевий, пателофеморальний відділи колінного суглоба). При пірофосфатній артропатії частіше формуються кісти субхондральної кістки. Хоча у більшості випадків хондрокальциноз виникає раніше маніфестації вторинного ОА, у деяких осіб захворювання може починатися як ідіопатичний ОА, який супроводжується розладами обміну речовин (гемохроматоз, гіперпаратіреоїдизм, гіпомагніємія та ін.) (Williams C.J., Jimenez S.A., 1999).
Найімовірніше, структурні зміни ПКМ суглобового хряща індукують відкладання кристалів пірофосфату кальцію дигідрату. A.O. Вjelle (1972; 1981) виявив у середній зоні матриксу суглобового хряща членів родини зі Швеції з пірофосфатною артропатією зниження вмісту колагену і фрагментацію колагенових волокон. У цих ділянках не містилося кристалів, тому автори зробили припущення, що описана аномалія матриксу може зумовлювати схильність до їх відкладання і розвитку дегенеративних змін у суглобах. На підставі вивчення спорадичних випадків пірофосфатної артропатії K. Ishikawa та співавтори (1989), I. Masuda та співавтори (1991) дійшли висновку, що причиною хондрокальцинозу є мутація генів, що кодують протеїни ПКМ. C.J. Williams та співавтори (1993), A.J. Reginato та співавтори (1994) виявили гетерозиготну мутацію COL2A1 (заміна основ Arg75>Cys) у членів великої родини із клінічним фенотипом тяжкого раннього ОА з анкілозуванням, пізнім розвитком спондилоепіфізіальної дисплазії та хондрокальцинозом гіалінового та волокнистого хрящів. Однак виявилося, що у членів цієї родини хондрокальциноз носив вторинний характер відносно ОА.
Також було висловлено припущення про те, що утворенню кристалів сприяють неорганічні компоненти ПКМ. Наприклад, гіпомагніємія викликає розвиток хондрокальцинозу шляхом пригнічення ферменту пірофосфатази, що у свою чергу знижує розчинення кристалів (Bennett R.M. et al., 1975). У синовіальній рідині хворих на пірофосфатну артропатію виявлено підвищений вміст неорганічних фосфатів (Silcox D.C., McCarty D., 1974). Результати цього та інших спостережень дозволили висловити припущення, що у хворих на пірофосфатну артропатію виникає локальне порушення метаболізму пірофосфатів (Altman R.D. et al., 1973; Silcox D.C., McCarty D., 1974). Було описано фермент нуклеозид-трифосфат-пірофосфогідролаза, який, можливо, бере участь в утворенні кристалів пірофосфату в зоні їх відкладення у ПКМ (Howell D.S. et al., 1984; Muniz O. et al., 1984; Ryan L.M. et al., 1984; 1985). У спорадичних випадках пірофосфатної артропатії виявлено підвищений вміст цього ферменту, однак у родинних формах хвороби таку аномалію не спостерігали (Ryan L.M. et al., 1986). Проте при культивуванні фібробластів і лімфобластів пацієнтів із родинною пірофосфатною артропатією виявлене підвищення вмісту неорганічних фосфатів (Lust G. et al., 1981), що також підтверджує припущення про роль порушень локального метаболізму пірофосфатів у патогенезі захворювання.
За останні роки розпочато спроби визначення генів, «винних» у виникненні родинних випадків пірофосфатної артропатії. Так, аналіз генетичного матеріалу, отриманого від членів великої родини з пірофосфатною артропатією (штат Мен, США), при якій хондрокальциноз розвивався вторинно стосовно тяжкого швидкопрогресуючого недиспластичного ОА, виключив зв’язок захворювання з локусом COL2A1 (Baldwin C.T. et al., 1995). Однак автори цього дослідження виявили зв’язок між досліджуваним фенотипом пірофосфатної артропатії та локусом, розташованим на довгому плечі 8-ї хромосоми (символ локусу CCAL2). A.G. Hughes та співавтори (1995) виявили зв’язок між фенотипом первинного хондрокальцинозу в родині з Великобританії і локусом CCAL1, який локалізується на короткому плечі 5-ї хромосоми в регіоні 5р15. За даними C.J. Williams та співавторів (1996) локус CCAL1 у членів родини з Аргентини з пірофосфатною артропатією локалізувався дещо проксимальніше, ніж в попередньому випадку — у регіоні 5р15.1. Аналогічний генотип виявлено у членів родини із Франції (Gaucher A. et al., 1977).
Таким чином, дані описаних досліджень свідчать про те, що родинна форма пірофосфатної артропатії становить клінічно й генетично гетерогенне захворювання, причиною якого можуть бути мутації принаймні трьох різних генів.
РОЛЬ ЗМІН У СУБХОНДРАЛЬНІЙ КІСТЦІ В ПАТОГЕНЕЗІ ОА
Поряд із дегенерацією суглобового хряща у патологічний процес при ОА залучається і прилегла кісткова тканина. Припускають, що потовщення субхондральної пластинки спричиняє прогресування ОА (Radin E.L. et al., 1970; Radin E.L., Rose R.M., 1986). З прогресуванням ОА суглобовий хрящ, який є об’єктом для механічного і хімічного стресів, піддається повільній ерозії завдяки дисбалансу процесів катаболізму та репарації хряща. Зокрема, механічний стрес по відношенню до суглобів, що «несуть» масу тіла, спричиняє утворення великої кількості мікропереломів у субхондральній пластинці та хрящі. Внаслідок поступового прогресування ерозування суглобового хряща прогресує склероз субхондральної кістки, підвищується жорсткість кісткової тканини, що у свою чергу зумовлює подальше порушення структури суглобового хряща. Однак питання про первинність або вторинність змін субхондральної кістки при ОА залишається не вирішеним.
Донедавна вважали, що рентгенологічні зміни, які визначаються в губчастій речовині субхондральної кістки, такі як склероз або утворення кіст, у хворих ОА мають вторинний характер. Однак результати клінічних і експериментальних досліджень свідчать про можливу ініціюючу роль субхондральної кістки в патогенезі ОА. Одним із можливих механізмів є різке підвищення градієнта жорсткості субхондральної кістки (Radin E.L. et al., 1970; Radin E.L., Rose R.M., 1986) у зв’язку з тим, що цілісність належної хрящової тканини залежить від механічних властивостей її кісткового «ложа». Дослідження у приматів показали, що зміни в субхондральній кістці можуть передувати змінам у суглобовому хрящі (Carlson C.S. et al., 1994; 1996). Свідчення, що появилися в результаті проведених досліджень на моделях ОА у тварин (Brandt K.D. et al., 1991; Dedrick D.K. et al., 1993; Armstrong S. et al., 1994) і клінічних досліджень (Chai B.F., 1991; Grynpas M.D. et al., 1991; Hulth A., 1993; Shimizu M. et al., 1993) на користь цієї гіпотези й проти неї тільки загострили дискусію. Потовщення трабекул у субхондральній кістці не завжди супроводжується підвищенням мінералізації кісткової тканини, а вірніше, збільшенням обсягу остеоїду (Grynpas M.D. et al., 1991). Ця ознака аномальної мінералізації (Puzas J.E., 1993) свідчить про те, що порушення регуляції ремоделювання кісткової тканини є невід’ємною частиною ОА, а також свідчить на користь концепції про дефект клітин кісткової тканини при ОА. Група J. Dequeker (1989) розглядає останній як «генералізовану метаболічну хворобу кісткової тканини».
Кісткова тканина постійно оновлюється. Цей динамічний процес, який називається ремоделюванням кісткової тканини, становить складну послідовність процесів резорбції та мінералізації. Остеокласти резорбують кісткову тканину, а остеобласти секретують білки, що формують основний органічний компонент для мінералізації (Raisz L.G., 1988). Утворення та резорбція кістки не випадково відбуваються у всьому скелеті, це — запрограмований процес, що відбувається в різних ділянках скелета, названих одиницями кісткового ремоделювання (Parfitt A.M., 1979). На початку циклу остеокласти з’являються на неактивній поверхні; протягом 2 тиж вони утворюють тунель у кортикальному шарі кості або лакуну на поверхні трабекулярної кістки. Частота активації нових одиниць кісткового ремоделювання визначає ступінь відновлення кісткової тканини. У здорової молодої людини процеси формування та резорбції кісткової тканини врівноважені, підтримується нормальна маса кісткової тканини. Внаслідок гормональної регуляції резорбції кісткової тканини, принаймні паратиреоїдний гормон (ПТГ) і ПГE2, активуються не лише остеокласти, а й остеобласти, оскільки під дією цих гормонів вивільняються фактори, що стимулюють резорбцію кістки остеокластами. На сьогодні відомо більше 12 локальних і системних регуляторів росту кісткової тканини, що впливають на її ремоделювання, зокрема ПТГ, 1,25(ОН)2D3, кальцитонін, гормон росту, глюкокортикоїди (ГК), гормони щитовидної залози, інсулін, ІФР-1 і -2, естрогени, ПГE2, андрогени (Simpson E., 1984; Centrella M., Canalis E., 1985).
Кісткові клітини вивільняють ряд білків і цитокінів, які здійснюють ендокринну регуляцію і передачу сигналу. Синтезовані остеобластами білки включають білки кісткового матриксу, такі, як колаген, остеопонтин, остеокальцин, кісткові сіалопротеїни (Whitson S.W. et al., 1984; Price P.A., 1985; Kream B.E. et al., 1986; Lo Y.Y.C. et al., 1996). Крім того, ці клітини вивільняють протеази як в активній, так і в латентній формі, які беруть участь у процесі ремоделювання кісткової тканини — ММП (Heath J.K. et al., 1984; Otsuka K. et al., 1984; Meikle M.C. et al., 1994), компоненти системи активатор плазміногену (АП)/плазмін (Allan E.H. et al., 1991; Hoekman K. et al., 1991; Fawthrop F.W. et al., 1992). У разі ОА остеобласти субхондральної кістки характеризуються надмірною продукцією лужної фосфатази, остеокальцину і, особливо, ТФР-β1, ІФР-1 та урокінази (Mansell J.P., Bailey A.J., 1998; Hilal G. et al., 1999). Крім того, за результатами досліджень останніх років доведено, що зрілі склерозовані остеобласти відрізняються підвищеною продукцією остеокальцину, ІЛ-6, ІЛ-8, С-термінального типу пропетиду проколагену 1, ТФР-β1, остеопонтину та накопиченням остеоїдної речовини. При цьому експресія рецепторів ПТГ є зниженою, що обґрунтовує стійкість субхондральної кістки до стимуляції ПТГ (Hilal G. et al., 2001; Sanchez C. et al., 2005; Massicotte F. et al., 2006; Sanchez C. et al., 2008).
Цитокіни, що вивільняються остеобластами, можуть діяти на місцеві клітини (інші остеобласти, остеокласти) як за допомогою аутокринних механізмів, так і паракринним шляхом (Horowitz M.C., Jilka R.L., 1992; Lowik C.W.G.M., 1992).
Роль субхондральної кістки у патогенезі ОА прийнято розглядати, виходячи з двох позицій. По-перше, відомо, що повторюваний механічний стрес викликає локальну проліферацію кісткових клітин та посилення продукції окремих білків. При цьому передача сигналу механічного стресу може відбуватися за рахунок механочутливих іонних каналів (Yamagouchi D.T. et al., 1987; Moreau R. et al., 1996; 1997). По-друге, підвищення експресії у субхондральній кістці ІФР-1 та ТФР-β1 стимулює процеси ремоделювання кістки, що призводить до розвитку субхондрального склерозу та формування остеофітів (van Beuningen H.M. et al., 1994; Mansell J.P., Bailey A.J., 1998). На кожному з цих патогенетичних напрямків слід зупинитися більш детально.
В умовах in vivo механічне навантаження здатне активувати остеобласти (Pead M.J. et al., 1988), підвищувати рівень циклічних нуклеотидів (Somjen D. et al., 1980; Shimshoni Z. et al., 1984), продукцію ПГ (Somjen D. et al., 1980; Yeh C.K., Rodan G.A., 1984), а також викликати морфологічні зміни, асоційовані з ремоделюванням кісткової тканини (Pead M.J. et al., 1988). В умовах in vitro механічний стрес викликає проліферацію культури остеобластів (Cheng M.Z. et al., 1997), експресію мРНК кісткових білків, що беруть участь в утворенні остеоїда і у процесі мінералізації (Harter L.V. et al., 1995; Cheng M.Z. et al., 1997), вивільнення локальних факторів росту, таких, як ІФР-1 та ІФР-2 (Zaman G. et al., 1997) і молекул адгезії (Keles A.O. et al., 1994). Крім того остеобласти цієї ділянки у відповідь на вплив механічного навантаження виявляються залученими у продукцію продегенеративних факторів, зокрема ММП-1, ММП-13, ПГЕ2, ІЛ-6 з паралельним пригніченням синтезу компонентів матриксу хряща (Massicotte F. et al., 2006). Більше того, зазначені продегенеративні фактори відіграють важливу роль не лише у дегенерації хряща, а також беруть участь у процесах деградації субхондральної кістки (Mansell J.P., Bailey A.J., 1998). Існують непрямі докази порушення функції остеобластів при ОА. G. Gevers і J. Dequeker (1987) продемонстрували підвищення рівня остеокальцину в сироватці крові у жінок із ОА суглобів кистей, а також в експлантатах кортикальної зони кістки, що свідчить про те, що патологія кісткової тканини може виступати частиною ОА. При аутопсії виявлено не тільки потовщення субхондральної кістки, а й аномально низьку мінералізацію голівки стегнової кістки (Grynpas M.D. et al., 1991). У морських свинок із хірургічно індукованим ОА за допомогою комп’ютерної томографії виявлено значне потовщення кісткової фракції в субхондральній зоні (Dedrick D.K. et al., 1991). Дисбаланс між колагеновими і неколагеновими (остеокальцин тощо) білками може призвести до збільшення об’єму кісткової тканини, але не впливає на її мінеральну щільність (Hannan M.T. et al., 1993; Li B., Aspden R.M., 1997). За даними M. Shimizu та співавторів (1993) прогресування дегенеративних змін суглобового хряща асоціюється з більш інтенсивним ремоделюванням субхондральної кістки і підвищенням її жорсткості, що також вказує на дефект клітин кісткової тканини при ОА. Відповідно до запропонованої B. Lee і M. Aspden (1997) гіпотези, проліферація дефектних кісткових клітин може призвести до підвищення жорсткості кісткової тканини, але не викликає підвищення МЩКТ.
C.I. Westacott та співавтори (1997) висунули гіпотезу про те, що аномальні остеобласти безпосередньо впливають на метаболізм хрящової тканини. Культивуючи остеобласти пацієнтів з ОА із хондроцитами людей, у яких не було хвороб суглобів, автори спостерігали значну зміну вивільнення глікозаміногліканів нормальною хрящовою тканиною in vitro, однак рівень вивільнення цитокінів залишався незмінним. G. Hilal та співавтори (1998) показали, що культура остеобластів субхондральної кістки хворих на ОА in vitro має змінений метаболізм — активність системи АП/плазмін і рівень ІФР-1 у цих клітинах підвищені. Спостереження C.I. Westacott та співавторів (1997) можна пояснити підвищенням активності протеаз клітинами субхондральної кістки.
Досі не відомо, чи зміни в субхондральній кістці ініціюють, чи сприяють прогресуванню ОА. D.K. Dedrick та співавтори (1993) продемонстрували, що у собак із хірургічно індукованим ОА потовщення субхондральної кістки не є необхідною умовою для розвитку ОА-подібних змін суглобового хряща, однак це сприяє прогресуванню дегенеративних процесів у хрящі. Результати дослідження А. Saіеd та співавторів (1997) суперечать даним попереднього дослідження. Використовуючи 50 МГц ехографію для оцінки початкових морфологічних змін та їх прогресування в суглобовому хрящі й кістці при експериментальному ОА, індукованому ін’єкціями монойодоцтової кислоти в колінний суглоб щурів, автори продемонстрували одночасний процес змін у кістці та хрящі протягом перших 3 днів після ін’єкції.
Результатами останніх досліджень із використанням гістологічного аналізу та МРТ продемонстровано, що дегенеративні зміни у суглобовому хрящі супроводжуються локальними змінами у субхондральній кістці, зокрема процесами формування кіст, збільшенням товщини трабекул та остеоїду, формування і ремоделювання кістки (Burr D.B. et al., 1997; Matsui H. et al., 1997; Watson P.J. et al., 1998; Huebner et al., 2002).
Остеобласти секретують фактори росту та цитокіни, що беруть участь у локальному ремоделюванні кісткової тканини, що може сприяти ремоделюванню належної хрящової тканини в суглобах, що «несуть масу тіла» після їх проникнення через мікротріщини в кальцифікованому шарі суглобового хряща (Sokoloff L., 1993). Крім того, продукти секреції кісткових клітин виявляють у синовіальній рідині (Sharif M. et al., 1995). Найбільш вірогідними продуктами, що виділяються аномальними остеобластами, здатними запускати процес локального ремоделювання хрящової тканини, є ТФР-β і кісткові морфометричні протеїни (КМП). Обидва представника родини ТФР виділяються і хондроцитами, і остеобластами та обидва здатні модифікувати ремоделювання як кісткової, так і хрящової тканини (Erickson D.M. et al., 1997; Lietman S.A. et al., 1997; Ripamonti U. et al., 1997). J. Martel Pelletier та співавтори (1997) спостерігали підвищення рівня ТФР-β в експлантатах субхондральної кістки хворих на ОА порівняно зі здоровими людьми, що свідчить про імовірну роль цього фактора росту в патогенезі ОА. ІФР також продукується остеобластами. У культурі остеобластоподібних клітин, одержаних від хворих на ОА, виявлене підвищення рівня ІФР (Hilal G. et al., 1998), що змінює метаболізм хряща (Hilal G. et al., 1998).
ТФР-β, ІФР, КМП і цитокіни, що продукуються остеобластами в субхондральній кістці, можуть впливати на продукцію колагенази та інших протеолітичних ферментів у хрящі, що в свою чергу, може сприяти ремоделюванню/деградації хрящового матриксу (Martel-Pelletier J. et al., 1999). Залишається нез’ясованим, чи виробляють остеобласти при ОА менше макрофагального колонієстимулюючого фактора (М-КСФ — стимулятор кісткової резорбції), ніж нормальні клітини. Результати досліджень A.G. Uitterlinden та співавторів (1997) показали, що певну роль в утворенні остеофітів можуть відігравати рецептори вітаміну D, які експресуються остеобластами й регулюють експресію ряду факторів, синтезованих цими клітинами, що частково пояснює роль остеобластів у патогенезі цього захворювання.
Враховуючи результати вищенаведених досліджень, G. Hilal та співавтори (1998), J. Martel-Pelletier та співавтори (1997) запропонували таку робочу гіпотезу взаємин ремоделювання субхондральної кістки й належного суглобового хряща при ОА (рис. 5.3). На ранній або розгорнутій стадії патогенезу ОА інтенсифікується процес ремоделювання кісткової тканини в субхондральній кістці. Водночас повторюване навантаження веде до локальних мікропереломів та/чи появи дисбалансу системи ІФР/ІФР-зв’язувальний білок внаслідок аномальної відповіді остеобластів субхондральної кістки, що спричиняє її склероз. Останнє в свою чергу може сприяти появі мікропереломів прилеглого хряща та ушкодженню його матриксу.
У нормальних умовах це ушкодження усувається шляхом локального синтезу і вивільнення ІФР-1 та ІФР-зв’язувального білка, які стимулюють утворення ПКМ суглобового хряща. Водночас ІФР-система сприяє росту клітин субхондральної кістки й формуванню кісткового матриксу. Анаболічна активність ІФР-системи підвищена в субхондральній кістці хворих на ОА, тоді як локальна активація системи АП/плазмін (місцевий регулятор ІФР-системи) у суглобовому хрящі зумовлює його локальні зміни (Martel-Pelletier J. et al., 1991). В остеобластах при ОА ІФР-1 порушує регуляцію АП плазміном за типом позитивного зворотного зв’язку, отже, може стримувати ремоделювання в кістковій тканині, що в результаті призводить до субхондрального склерозу (Hilal G. et al., 1998). Таким чином, у кістковій та хрящовій тканині локальна індукція ІФР-1 і протеаз веде, з одного боку, до ушкодження хряща, з іншого — до потовщення субхондральної кістки, останнє у свою чергу спричиняє подальше ушкодження хряща. Дисбаланс між ушкодженням хряща, пов’язаним із субхондральним склерозом, і його репаративними здатностями, веде до прогресуючої зміни ПКМ хряща і до розвитку ОА. На думку авторів, ця гіпотеза також пояснює повільне прогресування хвороби.
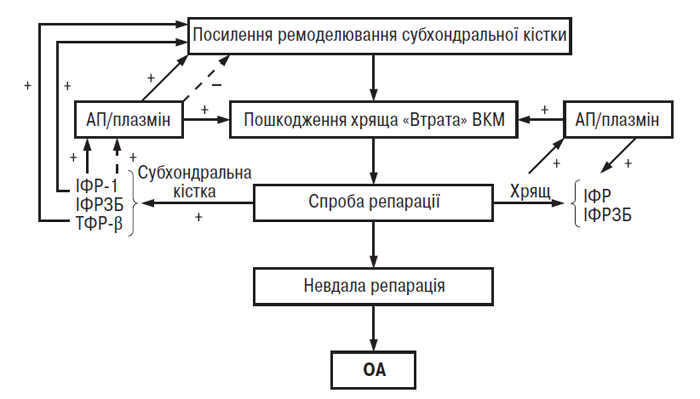
Рис. 5.3. Гіпотетична схема локальних біохімічних процесів у субхондральній кістці й суглобовому хрящі, які ведуть до втрати/ушкодження хряща і розвитку ОА (за J. Martel-Pelletier та співавт., 1999). ІФРЗБ — ІФР-зв’язувальний білок
Необхідно також зазначити, що при ОА субхондральна кістка знаходиться в стані гіперкоагуляції, гіпофібринолізу та тромбозу, пов’язаних зі зниженням кровотоку та підвищенням продукції прокоагулянтних факторів. Все це призводить до венозного стазу і гіпертензії, що спричиняє виникнення тромбозів та вогнищевих ішемічних некрозів кістки (Ghosh P., Cheras P.A., 2001).
M.B. Goldring і S.R. Goldring (2007) описали феномен ангіогенезу у місці з’єднання суглобового гіалінового хряща та суміжної субхондральної кістки у разі ОА. Так судинна інвазія, що виникає, призводить до зменшення товщини субхондральної кістки, при цьому зазначений судинний феномен є частково незалежним від синовіту. Такі зміни у структурі субхондральної кістки в подальшому сприяють розвитку аномального біомеханічного навантаження та посилюють дегенеративні зміни хряща.
РОЛЬ ФЕРМЕНТІВ І ЦИТОКІНІВ У ПАТОГЕНЕЗІ ОСТЕОАРТРОЗУ
В останні роки велика увага дослідників фокусується на ідентифікації протеаз, що відповідають за деградацію ПКМ суглобового хряща при ОА (рис. 5.4). Згідно із сучасним уявленням важливу роль у патогенезі ОА відіграють ММП (Dean D.D., 1991; Pelletier J.P. et al., 1997).
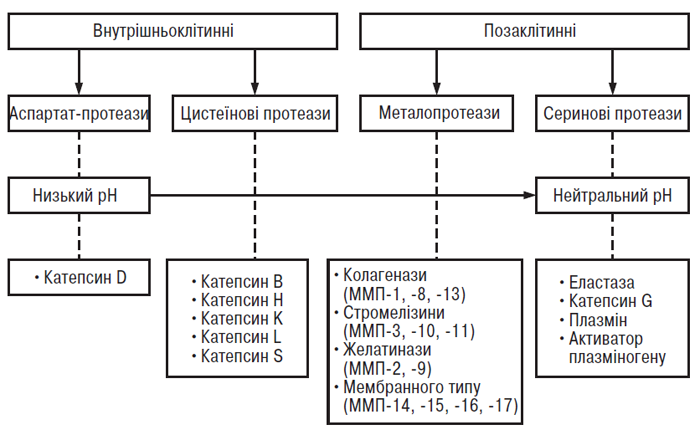
Рис. 5.4. Протеази сполучної тканини. Чотири основні класи протеаз названі згідно з назвою хімічних груп, що беруть участь у каталізі. Цистеїнові й аспартатпротеази фукціонують при низькому рН, здійснюють внутрішньоклітинний (лізосомальний) протеоліз. Ферменти цих груп також можуть вивільнятися в позаклітинне середовище у спеціальні «кишені» з низьким рН поблизу клітинної мембрани, де вони здійснюють деградацію ПКМ. Секретовані серинові та металопротеази функціонують позаклітинно при нейтральних значеннях рН. Деякі з них пов’язані із клітинною мембраною і здійснюють місцевий протеоліз
На сьогодні відомо щонайменше 18 представників родини Zn2+-нейтральних ММП. Зазначену ММП родину прийнято класифікувати на ММП-секреторного типу та ММП-мембранного типу (ММП-МТ). ММП продукуються переважно хондроцитами у відповідь на вплив механічного стресу та стимуляцію цитокінами, але синовіоцити та остеобласти субхондральної кістки також можуть залучатися у процес їх синтезу. Оскільки ММП активуються у нейтральному середовищі, вони здатні впливати на хрящовий матрикс дистантно від хондроцитів.
У хворих на ОА виявляють підвищений рівень трьох представників ММП — колагеназ, стромелізинів і желатиназ. Колагеназа відповідальна за деградацію нативного колагену, стромелізин — колагену IV типу, протеогліканів і ламінину (Fini M. et al., 1987), а желатиназа — за деградацію желатину, колагенів IV, V і XI типу, еластину (Mohtai M. et al., 1993). Іншою субродиною ММП є агреканази, або так звані ADAMTS (дизентегрин та металопротеїнази із тромбосподиновими фрагментами). Їх основне значення полягає у деградації агрекану, зокрема протеоліз хрящових протеогліканових агрегатів.
У суглобовому хрящі людини ідентифіковано три типи колагеназ, рівень яких значно підвищений у хворих на ОА: колагеназа-1 (ММП-1), колагеназа-2 (ММП-8) і колагеназа-3 (ММП-13). Співіснування трьох різних типів колагеназ у суглобовому хрящі свідчить про те, що кожна з них відіграє свою специфічну роль (Martel-Pelletier J., Pelletier J.P., 1996). Дійсно, колагенази-1 і -2 локалізуються головним чином у поверхневій та верхній частині проміжної зони суглобового хряща, тоді як колагеназу-3 виявляють у нижній частині проміжної та у глибокій зонах (Nguyen Q. et al., 1992; Cole A.A. et al., 1996; Martel-Pelletier J., Pelletier J.P., 1996; Moldovan F. et al., 1997; Fernandes J.C. et al., 1998). Більше того, результати імуногістохімічного дослідження продемонстрували, що в процесі прогресування ОА рівень колагенази-3 досягає плато і навіть знижується, тоді як рівень колагенази-1 поступово підвищується (Fernandes J.C. et al., 1998). Є дані про те, що при ОА колагеназа-1 в основному бере участь у запальному процесі в суглобовому хрящі, тоді як колагеназа-3 — у ремоделюванні тканини (Martel-Pelletier J. et al., 1999). Експресована у хрящі хворих на ОА колагеназа-3 здійснює деградацію колагену II типу більш інтенсивно, ніж колагеназа-1 (Mitchell P. et al., 1996).
Із представників другої групи металопротеаз, стромелізинів у людини ідентифіковані також три — стромелізин-1 (ММП-3), стромелізин-2 (ММП-10) і стромелізин-3 (ММП-11). На сьогодні відомо, що лише стромелізин-1 залучений у патологічний процес при ОА (Sirum K.L., Brinckerhoff C.E., 1989; Okada Y. et al., 1992; Hembry R.M. et al., 1995). У синовіальній мембрані хворих на ОА не визначається стромелізин-2, однак він виявлений у дуже малій кількості в синовіальних фібробластах хворих на РА (Sirum K.L., Brinckerhoff C.E., 1989; Hembry R.M. et al., 1995). Стромелізин-3 також виявлено у синовіальній оболонці хворих на РА поблизу фібробластів, особливо в зонах фіброзу (Sirum K.L., Brinckerhoff C.E., 1989).
У групі желатиназ у хрящовій тканині людини ідентифіковано тільки дві — желатиназа 92 кД (желатиназа В, або ММП-9) і желатиназа 72 кД (желатиназа А, або ММП-2); у хворих на ОА визначають підвищення рівня желатинази 92 кД (Mohtai M. et al., 1993).
Згідно з даними S. Ohta та співавторів (1998) при ОА у хрящі також відзначається підвищена експресія матризиліну (ММП-7), який може відігравати важливу роль у деградації компонентів ескстрацелюлярного матриксу, зокрема протеогліканів.
Відносно недавно була ідентифікована ще одна група ММП, які локалізуються на поверхні клітинних мембран і називаються ММП-МТ. До цієї групи належать чотири ферменти — ММП-МТ-1 – ММП-МТ-4 (Martel-Pelletier J. et al., 1999). Експресія ММП-МТ виявлена в суглобовому хрящі людини (Buttner F.H. et al., 1997). Хоча ММП-МТ-1 має властивості колагенази (Ohuchi E. et al., 1997), обидва ферменти ММП-МТ-1 і ММП-МТ-2 здатні активувати желатиназу-72 кД і колагеназу-3 (Takino T. et al., 1995; Imai K. et al., 1996; Buttner F.H. et al., 1997). Роль цієї групи ММП у патогенезі ОА потребує уточнення.
Протеїнази секретуються у формі зимогену, який активується іншими протеїназами чи органічними сполуками ртуті. Каталітична активність ММП залежить від наявності цинку в активній зоні ферменту.
Біологічна активність ММП контролюється специфічними тканинними інгібіторами металопротеаз (ТІМП) (рис. 5.5). Досі ідентифіковано три типи ТІМП, які виявляють у суглобових тканинах людини: ТІМП-1 – ТІМП-3 (Dean D.D. et al., 1989; Apte S.S. et al., 1994; Martel-Pelletier J. et al., 1994). Четвертий тип ТІМП ідентифікований і клонований, однак його досі не виявлено у суглобових тканинах людини (Greene J. et al., 1996). Ці молекули специфічно зв’язуються з активним центром ММП, хоча деякі з них здатні зв’язувати активний центр прожелатинази 72 кД (ТІМП-2, -3, -4) і прожелатинази 92 кД (ТІМП-1 і -3) (Will H. et al., 1996; Bigg H.F. et al., 1997). Дані свідчать про те, що при ОА в суглобовому хрящі існує дисбаланс між ММП і ТІМП, результатом якого є відносний дефіцит інгібіторів (Dean D.D. et al., 1989; Pelletier J.P. et al., 1990), що, можливо, частково пов’язане з підвищенням рівня активних ММП у тканині. ТІМП-1 і -2 виявляють у суглобовому хрящі, вони синтезуються хондроцитами (Dean D.D. et al., 1989; Wolfe G.C. et al., 1993; Martel-Pelletier J. et al., 1994). При ОА в синовіальній оболонці та синовіальній рідині виявлений тільки перший тип ТІМП (Clarck I.M. et al., 1993; Lohmander L.S. et al., 1993; Hembry R.M. et al, 1995). ТІМП-3 виявляють винятково в ПКМ (Gomez D.E. et al., 1997). ТІМП-4 майже на 50% має ідентичну амінокислотну послідовність із ТІМП-2 і -3 і на 38% — із ТІМП-1 (Leco K.J. et al., 1997). В інших клітинах-мішенях ТІМП-4 відповідальний за модуляцію активації прожелатинази 72 кД на поверхні клітин, що свідчить про важливу роль у якості тканиноспецифічного регулятора ремоделювання ПКМ (Bigg H.F. et al., 1997).
Іншим механізмом контролювання біологічної активності ММП є їх фізіологічна активація. Існує думка, що ферменти родини серинових і цистеїнових протеаз, таких, як АП/плазмін і катепсин В відповідно, і є фізіологічними активаторами ММП (Eeckhout Y., Vaes G., 1977; Nagase H. et al., 1990). У суглобовому хрящі хворих на ОА виявлено підвищений рівень урокінази і плазміну (Martel-Pelletier J. et al., 1991).
До патогенезу ОА, крім ММП, залучаються й інші групи протеолітичних ферментів, зокрема цистеїнові, серинові та аспартат протеїнази.
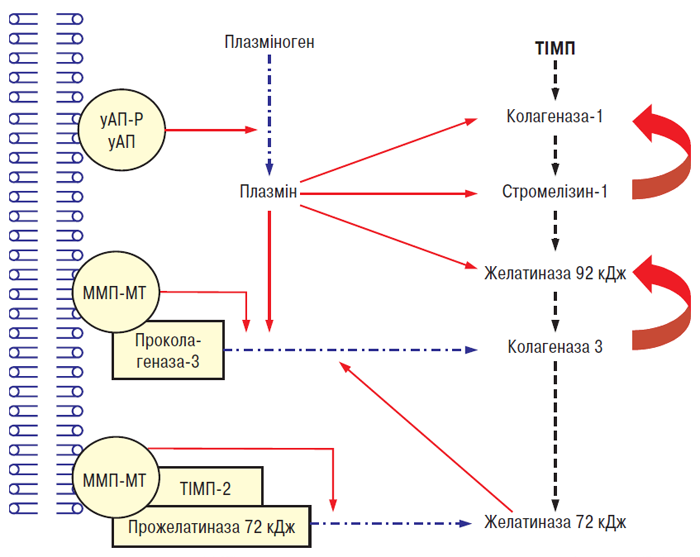
Рис. 5.5. Каскад ММП/ТІМП у суглобовому хрящі людини. уАП — урокіназа, уАП-Р — рецептор урокінази. Суцільні стрілки означають активацію, пунктирні — пригнічення, штрих-пунктирні — перетворення (за: Martel-Pelletier J. et al., 1999)
Цистеїнові протеїнази включають групу катепсинів В, Н, L та S, що беруть участь у деградації матриксу протягом періодів формування, росту, ремоделювання, старіння, а також у разі патологічних процесів. Експресія цистеїнових катепсинів, зокрема катепсин К, значно підвищена у кістках та хрящі (Soderstorm M. et al., 1999).
Так, катепсин В за рахунок як ендо-, так і екзопептидазної активності може призводити до деградації агрекану, подібної до такої, що спостерігається при дії агреканаз (Mort J.S. et al., 1998). На експериментальних моделях ОА на кролях встановлено, що рівень катепсину В значно підвищувався у синовіальній тканині на ранніх стадіях ОА, що при прогресуванні процесу супроводжувалося зростанням його експресії у хрящі. Незважаючи на те що в тканинах суглоба виявляють декілька типів катепсинів, найбільш вірогідним активатором ММП у хрящі вважають катепсин-В (Martel-Pelletier J. et al., 1990; Buttle D.J. et al., 1993).
Цистеїнові протеази родини папаїну розглядаються як потенційні мішені терапевтичної стратегії при захворюваннях кістково-м’язової системи. Найбільший інтерес викликають катепсин К та S, які характеризуються селективною експресією клітинами імунної системи та клітинами, що беруть участь у процесі деградації колагену та інших білків позаклітинного матриксу (Yasuda Y et al., 2005).
Серинові протеази структурно схожі на трипсин, що активуються за умови нейтрального рН. Дія цих ферментів може бути непрямою, через активацію інших протеаз, а також прямою шляхом деградації макромолекул позаклітинного матриксу. Крім того, серинові протеази — важливі активатори попередників ММП. Так, плазмін може активувати ММП шляхом розриву пропептидів латентного проферменту. В свою чергу, плазмін є продуктом перетворення попередника плазміногену із залученням АП двох типів — тканинного та урокіназного АП.
Інші серинові протеїнази, зокрема еластаза поліморфнонуклеарних лейкоцитів (ПМН еластаза) та катепсин G також беруть участь у деградації суглобового хряща, саме тому активно вивчаються сполуки, що потенційно здатні здійснювати інгібуючий вплив на ці ферменти. (Steinmeyer J. et al., 1996). Так, ПМН еластаза є відповідальною за деградацію не лише еластину, а й інших компонентів позаклітинного матриксу, зокрема фібронектину, ламініну, протеогліканів та колагену IV типу. Значення катепсину G полягає як у прямому, так і непрямому деградуючому впливі на позаклітинний матрикс фіброзного кільця, таким чином зазначений фермент бере участь у дегенеративно-дистрофічних процесах у хребті (Konttinen Y.T. et al., 1999). Значення цієї сполуки у дегенеративних процесах у суглобах остаточно не вивчене.
Аспартат протеїнази. За даними A.L. Sapolsky та співавторів (1973) катепсин D виявляється у 2 або 3 рази більшій кількості у суглобовому хрящі хворих на первинний ОА порівняно з групою контролю (хрящова тканина здорових осіб). Катепсин D було виявлено у хондрокластах, що знаходяться поряд з матриксом хряща при ендохондральній осифікації, а також гіпертрофованих хондроцитах суміжних з остеохондральними з’єднаннями (Nakase et al., 2000).
У тканинах суглоба людини виявлені фізіологічні інгібітори серинових і цистеїнових протеаз. Активність інгібітору АП-1 (іАП-1), а також цистеїнових протеаз знижена у хворих на ОА (Martel-Pelletier J. et al., 1990; 1991). Аналогічно ММП/ТІМП — саме дисбалансом між сериновими та цистеїновими протеазами та їх інгібіторами можна пояснити підвищену активність ММП у суглобовому хрящі хворих на ОА. Крім того, ММП здатні активувати один одного. Наприклад, стромелізин-1 активує колагеназу-1, -3 і желатиназу 92 кД; колагеназа-3 активує желатиназу 92 кД; ММП-МТ активує колагеназу-3, а желатиназа-72 кД потенціює цю активацію; ММП-МТ також активує желатиназу 72 кД (Murphy G. et al., 1987; Ogata Y. et al., 1992; Atkinson S.J. et al., 1995; Knauper V. et al., 1996; 1997; d’Оrtho M.P. et al., 1997).
На сьогодні активно вивчається роль субродини ММП — АDAMTS — протеаз, відповідальних за деградацію агрекану. Наразі виявлено АDAMTS-1; -4; -5; -9 та -15, серед яких найбільш активними є ADAMTS-4 та -5, що ізольовані від хряща та більш відомі як агреканаза-1 та агреканаза-2 відповідно (Nagase H., Kashiwagi M., 2003; Porter et al., 2005). Встановлено, що стимулюючий вплив на деякі ADAMTS гени здатні здійснюювати фактори росту, прозапальні цитокіни та гормони. Так, ТФР-β може стимулювати виділення фібробластоподібними синовіоцитами ADAMTS-4 і не впливає на експресію ADAMTS-5 (Yamanishi Y. et al., 2002). Стимуляція хондроцитів або експлантатів хряща ІЛ-1 модулює здатність ADAMTS-4 руйнувати агрекан (Patwari P. et al., 2005). Регуляторний вплив гормонів на ADAMTS може відображати зданість трийодтироніну (Т3) підвищувати експресію мРНК ADAMTS-5, не впливаючи на ADAMTS-4, з подальшим деградуючим впливом на агрекан (Makihita S. et al., 2003).
Хоча ТІМП є переважно інгібіторами ММП широкого спектра, їх вплив на ADAMTS протеази є більш селективним. Так, ADAMTS-4 та -5 пригнічуються впливом ТІМП-3, але нечутливі до ТІМП-1; -2; -4.(Arner A.C. et al., 1999; Kashiwagi M. et al., 2001; Hashimoto T. et al., 2003). На ADAMTS-1 інгібуючий вплив здатен здійснювати ТІМП-3, а також ТІМП-2, але водночас ця агреканаза є нечутливою до дії ТІМП-1 або -4. (Rodriguez-Manzaneque J.C. et al., 2002). Враховуючи такий пригнічуючий профіль, ТІМП-3 можна сміливо вважати фізіологічним інгібітором агреканаз хряща (Gendron C. et al., 2003).
Цитокіни можна розділити на три групи — деструктивні (прозапальні), регуляторні (у тому числі протизапальні) й анаболічні (фактори росту).
Типи цитокінів (за: van den Berg W.B. et al., 1999)
| Деструктивні |
|
| Регуляторні |
|
| Анаболічні |
|
Деструктивні цитокіни, зокрема ІЛ-1, індукують збільшення вивільнення протеаз і пригнічують синтез протеогліканів і колагенів хондроцитами. Регуляторні цитокіни, зокрема ІЛ-4 і -10, пригнічують продукцію ІЛ-1, збільшують продукцію антагоніста рецептора ІЛ-1 (ІЛ-1РА) та знижують рівень і NО-синтази у хондроцитах. Таким чином, ІЛ-4 протидіє ІЛ-1 за трьома напрямками: 1) знижує продукцію, перешкоджає його ефектам; 2) збільшує продукцію основного «скавенджера» ІЛ-1РА та 3) знижує продукцію основного вторинного «месенджера» NO. Крім того, ІЛ-4 знижує ферментативну деградацію тканини. В умовах in vivo оптимальний терапевтичний ефект досягається при комбінації ІЛ-4 та ІЛ-10. Анаболічні цитокіни, такі як ТФР-β і ІФР-1, реально не перешкоджають продукції чи дії ІЛ-1, а проявляють протилежну активність, наприклад стимулюють синтез протеогліканів і колагену, пригнічують активність протеаз, а ТФР-β ще й пригнічує вивільнення ферментів і стимулює їх інгібітори.
Прозапальні цитокіни відповідають за підвищений синтез і експресію ММП у суглобових тканинах. Вони синтезуються в синовіальній оболонці, а потім дифундують у суглобовий хрящ через синовіальну рідину. Прозапальні цитокіни активують хондроцити, які у свою чергу також здатні виробляти прозапальні цитокіни. В уражених ОА суглобах роль ефектора запалення відіграють головним чином клітини синовіальної мембрани. Саме синовіцити макрофагального типу секретують протеази і медіатори запалення. Серед них у патогенезі ОА найбільше задіяні ІЛ-1β, ФНП-α, ІЛ-6, ЛІФ та ІЛ-17.
Біологічно активні речовини, що стимулюють деградацію суглобового хряща при ОА (Carrabba M. et al., 1996)
|
Дані літератури свідчать, що ІЛ-1β і, можливо ФНП-α, — головні медіатори деструкції суглобових тканин при ОА (Pelletier J.P. et al., 1997). Однак досі не відомо, чи діють вони незалежно один від одного, чи між ними існує функціональна ієрархія. На моделях ОА у тварин показано, що блокада ІЛ-1 ефективно запобігає деструкції суглобового хряща (van de Loo F.A. et al., 1995; Caron J.P. et al., 1996), тоді як блокада ФНП-α призводить лише до послаблення запалення в тканинах суглоба (van de Loo F.A. et al., 1995; Plows D. et al., 1995). У синовіальній мембрані, синовіальній рідині та хрящі хворих виявлено підвищені концентрації обох цитокінів (Wood D.D. et al., 1983; Pelletier J.P. et al., 1989; 1993; 1997; Chikanza I.C. et al., 1993; Farahat M.N. et al., 1993). У хондроцитах вони здатні збільшувати синтез не тільки протеаз (головним чином ММП і АП), але й мінорних колагенів, наприклад І і ІІІ типу, а також зменшувати синтез колагенів ІІ й ІХ типу і протеогліканів (Martel-Pelletier J. et al., 1999). Ці цитокіни також стимулюють активні форми кисню і такі медіатори запалення, як ПГE2. Результатом таких макромолекулярних змін у суглобовому хрящі при ОА є неефективність репаративних процесів, що призводить до подальшої деградації хряща.
Вищезазначені прозапальні цитокіни модулюють процеси пригнічення/активації ММП при ОА. Так, дисбаланс між рівнями ТІМП-1 і ММП у хрящі при ОА може опосередковувати ІЛ-1β, тому що дослідження in vitro продемонструвало, що підвищення концентрації ІЛ-1β призводить до зниження концентрації ТІМП-1 і збільшення синтезу ММП хондроцитами (Martel-Pelletier J. et al., 1994). Синтез АП також модулюється ІЛ-1β. Стимуляція in vitro хондроцитів суглобового хряща із використанням ІЛ-1 викликає дозозалежне збільшення синтезу АП і різке зниження синтезу іАП-1 (Martel-Рelletier J. et al., 1991). Здатність ІЛ-1 зменшувати синтез іАП-1 і стимулювати синтез АП є потужним механізмом генерації плазміну та активації ММП. Крім того, плазмін є не лише ферментом, що активізує інші ферменти, він також бере участь у процесі деградації хряща шляхом прямого протеолізу. Крім того, ІЛ-1β здатен разом із ФНП-α стимулювати експресію індукованої NO-синтази, що викликає підвищену секрецію оксиду азоту.
M.E. Goldring та співавтори (2004) продемонстрували супресивний вплив ІЛ-1β на цілу низку генів, відповідальних за пригнічення диференціації хондроцитів, включаючи COL2A1.
ІЛ-1β синтезується у вигляді неактивного попередника з масою 31 кД (пре-ІЛ-1β), а потім, після відщеплення сигнального пептиду, перетворюється в активний цитокін з масою 17,5 кД (Mosley B. et al., 1987; Siders W.M. et al., 1993). Cвій вплив ІЛ-1β здійснює через низку факторів транскрипції, таких як нуклеарний фактор-каппа В (NF-κB), C/EBP, AP-1 та родину ETS (Masuko-Hongo K., 2004). У тканинах суглобів, включаючи синовіальну мембрану, синовіальну рідину і суглобовий хрящ, ІЛ-1β виявляють в активній формі, а в дослідженнях in vivo продемонстрована здатність синовіальної мембрани при ОА секретувати цей цитокін (Pelletier J.P. et al., 1995). Деякі серинові протеази здатні перетворювати пре-ІЛ-1β у його біоактивну форму (Black R.A. et al., 1988). У ссавців такі властивості виявлені лише у однієї протеази, що належить до родини цистеїнових аспартатспецифічних ферментів і називається ІЛ-1β-конвертуючий фермент (ІКФ, або каспаза 1). Цей фермент здатний специфічно перетворювати пре-ІЛ-1β у біологічно активний зрілий ІЛ-1β з масою 17,5 кД (Black R.A. et al., 1988; Kronheim S.R. et al., 1992). ІКФ — це профермент із молекулярною масою 45 кД (р45) (Black R.A. et al., 1988; Kronheim S.R. et al., 1992), який локалізується у клітинній мембрані. Після протеолітичного розщеплення проензиму р45 утворюються дві субодиниці, відомі як р10 і р20, яким властива ферментативна активність (Wilson K.P. et al., 1994).
ФНП-α також синтезується у вигляді мембранозв’язаного попередника з масою 26 кД; шляхом протеолітичного відщеплення він вивільняється з клітини у вигляді активної розчинної форми з масою 17 кД (Aggrawal B.B. et al., 1985; Gearing A.J. et al., 1994). Протеолітичне відщеплення здійснюється ФНП-α-конвертуючим ферментом (ФНП-КФ), який належить до родини адамалізинів (Black R.A. et al., 1997). A.R. Amin та співавтори (1997) виявили підвищену експресію мРНК ФНП-КФ у суглобовому хрящі хворих на ОА.
Біологічна активація хондроцитів і синовіцитів ІЛ-1 і ФНП-α опосередковується зв’язуванням зі специфічними рецепторами на поверхні клітин — ІЛ-Р і ФНП-Р. Для кожного цитокіну ідентифіковано два типи рецепторів — ІЛ-1Р І та ІІ типу (Slack J. et al., 1993) і ФНП-Р І (р55) та ІІ (р75) типів (Tartaglia L.A., Goeddel D.V., 1992). За передачу сигналів у клітинах тканин суглобів відповідають ІЛ-1РІ та р55 (Martel-Pelletier J. et al., 1992; Arend W.P., 1993; Westacott C.I. et al., 1994; Sadouck M. et al., 1995; Alaaeddine N. et al., 1997). ІЛ-1Р І типу має дещо більшу афінність до ІЛ-1β, ніж до ІЛ-1α; ІЛ-1Р ІІ типу — навпаки, має більшу спорідненість до ІЛ-1α, ніж до ІЛ-1β. Досі залишається нез’ясованим, чи може ІЛ-1Р ІІ типу опосередковувати сигнали ІЛ-1, чи він служить лише для конкурентного інгібування зв’язку ІЛ-1 з ІЛ-1Р І типу (Martel-Pelletier J. et al., 1999). У хондроцитах і синовіальних фібробластах хворих на ОА виявляють велику кількість ІЛ-1РІ та р55 (Martel-Pelletier J. et al., 1992; Westacott C.I. et al., 1994; Sadouck M. et al., 1995; Alaaeddine N. et al., 1997), що у свою чергу пояснює високу чутливість цих клітин до стимуляції відповідними цитокінами (Martel-Pelletier J. et al., 1992). Цей процес призводить як до підвищення секреції протеолітичних ферментів, так і до деструкції суглобового хряща.
Не виключається участь ІЛ-6 у патологічному процесі при ОА. В основі цього припущення є такі спостереження:
- ІЛ-6 збільшує кількість клітин запалення в синовіальній мембрані (Guerne P.A. et al., 1989);
- ІЛ-6 стимулює проліферацію хондроцитів;
- ІЛ-6 посилює ефекти ІЛ-1 відносно підвищення синтезу ММП і пригнічення синтезу протеогліканів (Carroll G.J., Bell M.C., 1993).
Проте ІЛ-6 здатний індукувати продукцію ТІМП (Lotz M., Guerne P.A., 1991), але не впливає на продукцію ММП, тому вважають, що саме цей цитокін бере участь у процесі стримування протеолітичної деградації суглобового хряща, який здійснюється за механізмом зворотного зв’язку.
Ще одним представником родини ІЛ-6 є ЛІФ — цитокін, який виробляється хондроцитами, отриманими від хворих на ОА, у відповідь на стимуляцію прозапальними цитокінами ІЛ-1β і ФНП-α (Lotz M. et al., 1992; Campbell I.K. et al., 1993; Hamilton J.A. et al., 1993). ЛІФ стимулює резорбцію протеогліканів хряща, а також синтез ММП і продукцію NO (Carroll G.J., Bell M.C., 1993). Роль цього цитокіну при ОА остаточно не з’ясована.
ІЛ-17 — гомодимер масою 20–30 кД (Yao Z. et al., 1995) що має ІЛ-подібну дію, проте значно менш виражену. ІЛ-17 стимулює синтез і виділення ряду прозапальних цитокінів, у тому числі ІЛ-1β, ФНП-α, ІЛ-6, а також ММП у клітинах-мішенях, наприклад у макрофагах людини (Jovanovic D. et al., 1998). Крім того, ІЛ-17 стимулює продукцію NO хондроцитами (Attur m.G. et al., 1997). Як і ЛІФ, роль ІЛ-17 у патогенезі ОА недостатньо вивчена.
Неорганічний вільний радикал NO відіграє важливу роль у деградації суглобового хряща при ОА. Хондроцити, отримані від хворих на ОА, виробляють велику кількість NO як спонтанно, так і після стимуляції прозапальними цитокінами порівняно з нормальними клітинами (Pelletier J.P. et al., 1996). Високий вміст NO виявлено в синовіальній рідині й сироватці крові хворих на ОА (Farrell A.J. et al., 1992; Mcinnes I.B. et al., 1996) — це результат збільшення експресії та синтезу індукованої NО-синтази (іNOС) — ферменту, відповідального за продукцію NO (Mcinnes I.B. et al., 1996 ; Grabowski P.S. et al., 1997). Нещодавно була клонована дезоксирибонуклеїнова кислота (ДНК) хондроцитспецифічної іNOС, визначена амінокислотна послідовність ферменту (Charles I.G. et al., 1993). Амінокислотна послідовність вказує на 50% ідентичність і 70% подібність з іNOС, специфічної для ендотелію та нервової тканини.
NO пригнічує синтез макромолекул ПКМ суглобового хряща та стимулює синтез ММП. Більше того, збільшення продукції NO супроводжується зниженням синтезу антагоніста ІЛ-1Р (ІЛ-1РА) хондроцитами (Pelletier J.P. et al., 1996). Таким чином, підвищення рівня ІЛ-1 і зниження — ІЛ-1РА призводить до гіперстимуляції NO хондроцитів, що в свою чергу веде до посилення деградації хрящового матриксу. Є повідомлення про терапевтичний ефект in vivo селективного інгібітору іNOС відносно прогресування експериментального ОА (Stefanovic-Racic M. et al., 1994; Connor J.R. et al., 1995; Pelletier J.P. et al., 1998).
При ОА внаслідок деградації матриксу значно збільшується кількість фрагметів фібронектину, які в свою чергу здатні активувати хондроцити через інтегринові рецептори (переважно α5β1-інтегрин), стимулюючи синтез ММП. Крім того фрагменти фібронектину можуть стимулювати або активувати інші фактори (NO, ПГЕ2) і катаболічні та прозапальні цитокіни (ІЛ-6, ІЛ-8), які посилюють ураження хряща (Homandberg G.A., 1997).
За результатами останніх досліджень описана роль адипоцитокінів (або адипокінів) як медіаторів запалення. Адипокіни (резистин, лептин, адипонектин тощо) є розчинними медіаторами, що секретуються переважно жировою тканиною, але не тільки. Так, A. Schaffler та співавтори (2003) виявили адипокіни у синовіальній рідині та детально описали функцію кожного (табл. 5.4).
Таблиця 5.4
Адипоцитокіни та остеоартроз
| Медіатор | Функція |
| Лептин | Експресія лептину хрящем позитивно корелює зі ступенем тяжкості ОА
|
| Адипонектин |
|
| Вістафін | Продукується хондроцитами людини
Підвищується експресія при стимуляції ІЛ-1
|
| Резистин | Індукує розвиток ОА при введенні у колінний суглоб мишам |
Найбільшою у синовіальній рідині є концентрація адипонектину, водночас найбільш вивченим є лептин. Так, лептин виявлено у остеофітах та хрящі пацієнтів хворих на ОА, а його рівні у синовіальній рідині корелюють з ІМТ. Лептин виражено стимулює анаболічні функції хондроцитів через індукцію синтезу ІФР-1 та ТФР-β (Dumond H. et al., 2003), а також синтез протеогліканів та колагену. Лептин посилює здатність прозапальних цитокінів до виділення NO (Otero M. et al., 2005). За даними останніх досліджень вістафін розглядається як продегенеративний медіатор хряща (Gosset M. et al., 2008).
Природні інгібітори цитокінів здатні безпосередньо перешкоджати зв’язуванню цитокінів із рецепторами клітинних мембран, знижуючи їх прозапальну активність. За способом природні інгібітори цитокінів можна розділити на три класи.
До першого класу інгібіторів відносять антагоністи рецепторів, які перешкоджають зв’язуванню ліганда з його рецептором шляхом конкуренції за зв’язувальний центр. Досі такий інгібітор знайдений тільки для ІЛ-1 — це вищезазначений конкурентний інгібітор системи ІЛ-1/ІЛ-1Р ІЛ-1РА (Arend W.P., 1993; Dinarello C.A., 1996). ІЛ-1РА блокує багато ефектів, які спостерігаються у тканинах суглобів при ОА, включаючи синтез ПГ синовіальними клітинами, продукцію колагенази хондроцитами і деградацію ПКМ суглобового хряща.
ІЛ-1РА виявляють у різних формах — одній розчинній (рІЛ-1РА) і двох міжклітинних (мкІЛ-1РАІ і мкІЛ-1РАІІ) (Arend W.P., 1993). Афінність розчинної форми ІЛ-1РА у 5 разів перевищує таку в міжклітинних форм (Martel-Pelletier J. et al., 1999). Незважаючи на інтенсивний науковий пошук, функція останніх залишається невідомою. Досліди in vitro показали, що для пригнічення активності ІЛ-1β необхідна концентрація ІЛ-1РА, що в 10–100 разів перевищує норму, в умовах in vivo потрібне підвищення концентрації ІЛ-1РА у тисячі разів (Pelletier J.P. et al., 1995; Campion G.V. et al., 1996; Joosten l.A. et al., 1996; Bakker A.C. et al., 1997). Цей факт може частково пояснити відносний дефіцит ІЛ-1РА і надлишок ІЛ-1 у синовії хворих на ОА.
Другий клас природних інгібіторів цитокінів представлений розчинними рецепторами цитокінів. Прикладом таких інгібіторів у людини, що мають відношення до патогенезу ОА, є рІЛ-1Р і рр55 (Giri J.G. et al., 1990; Lantz M. et al., 1990). Розчинні рецептори цитокінів становлять вкорочені форми нормальних рецепторів, зв’язуючись із цитокінами, вони перешкоджають їх зв’язуванню з мембраноасоційованими рецепторами клітин-мішеней, діючи за механізмом конкурентного антагонізму.
Основним попередником розчинних рецепторів є мембранозв’язані ІЛ-1РІІ. Афінність рІЛ-1Р відносно ІЛ-1 та ІЛ-1РА різна. Так, рІЛ-1РІІ має більшу спорідненість до ІЛ-1β, ніж до ІЛ-1РА, а рІЛ-1РІ — проявляє більшу афінність до ІЛ-1РА, ніж до ІЛ-1β (Arend W.P., 1993; Svenson M. et al., 1993; Dinarello C.A., 1996).
Для ФНП також існують два типи розчинних рецепторів — рр55 і рр75, як і розчинні рецептори ІЛ-1, вони утворюються шляхом «шедингу» (скидання). В умовах in vivo обидва рецептори виявляють у тканинах уражених суглобів (Cope A.P. et al., 1992; Chikanza I.C. et al., 1993; Roux-Lombard P. et al., 1993). Роль розчинних рецепторів ФНП у патогенезі ОА дискутується. Припускають, що в низьких концентраціях вони стабілізують тривимірну структуру ФНП і підвищують період напіввиведення біоактивного цитокіну (Arderka D. et al., 1992), в той час як високі концентрації рр55 і рр75 можуть знижувати активність ФНП шляхом конкурентного антагонізму (Higuchi M., Aggrawal B.B., 1992). Імовірно, рр75 може виступати як переносник ФНП, полегшуючи його зв’язування з мембраноасоційованим рецептором (Tartaglia L.A. et al., 1993).
Третій клас природних інгібіторів цитокінів представлений групою протизапальних цитокінів, до яких відносять ТФР-β, ІЛ-4, ІЛ-10 і ІЛ-13. Протизапальні цитокіни знижують продукцію прозапальних, а також деяких протеаз, стимуюють продукцію ІЛ-1РА і ТІМП (Hart P.H. et al., 1989; Lacraz S. et al., 1992; de Waal Malefyt R. et al., 1993; Jenkins J.K. et al., 1994; Jovanovic D. et al., 1998).
РЕПАРАЦІЯ СУГЛОБОВОГО ХРЯЩА І ФАКТОРИ РОСТУ В ПАТОГЕНЕЗІ ОА
Завдяки прогресу біотехнології, зокрема технології клонування, останнім часом інтенсивно поповнюється перелік факторів росту, які, будучи анаболічними факторами, відіграють важливу, але не до кінця вивчену роль у патогенезі ОА (табл. 5.5).
Першою групою факторів росту, про яких йдеться нижче, є ІФР. Вони у великій кількості містяться в сироватці крові, мають ряд загальних властивостей з інсуліном. ІФР-2 — більш характерний для ембріональної стадії розвитку, тоді як ІФР-1 — домінуючий представник групи у дорослої людини. Обидва представника цієї групи діють шляхом з’єднання з рецепторами ІФР І типу. Якщо функція ІФР-2 залишається не відомою, значення ІФР-1 вже визначено — він здатний стимулювати синтез протеогліканів хондроцитами і значно пригнічувати катаболічні процеси в суглобовому хрящі. ІФР-1 — головний анаболічний стимул для синтезу протеогліканів хондроцитами, наявний у сироватці крові та синовіальній рідині (Schalkwijk J. et al., 1989). ІФР-1 — важливий фактор для культивування хондроцитів в експериментальних моделях ОА in vitro (Schalkwijk J. et al., 1989; Tyler J.A., 1989; Tesch G.H. et al., 1992; van Beuningen H.M. et al., 1993). Припускають, що ІФР-1 потрапляє в синовіальну рідину із плазми крові. Крім того, нормальні хондроцити продукують обидва фактори — експресія ІФР-1 і ІФР-2 виявлена в синовіальній оболонці та хрящі хворих на ОА (Middlton J.F., Tyler J.A., 1992; Keyszer G.M. et al., 1995). У нормальному хрящі ІФР-1 не має мітогенних властивостей, однак здатний стимулювати проліферацію клітин в ушкодженому матриксі, що свідчить про участь у репаративних процесах (van den Berg W.B. et al., 1999).
Таблиця 5.5
Деякі фактори росту та їх вплив на суглобовий хрящ за даними літератури (за: van den Berg W.B. et al., 1999)
| Фактор росту | Стимуляція синтезу протеогліканів | Вплив на фенотип чи диференціювання хондроциту | Наявність у тканинах суглоба при ОА |
| ІФР | Tyler J.A., 1989
Schalkwijk J. et al., 1989 Tesch G.H. et al., 1992 van Beuningen H.M. et al., 1993 |
Bohme K. et al., 1995
Wroblewski J. et al., 1995 Ballock R.T. et al., 1993 |
Middelton J.F. et al., 19921
Keyszer G.M. et al., 19952 Denko C.W. et al., 19963 |
| ТФР-α | van den Kraan P.M. et al., 1992
Morales T.I., 1994 van Beuningen H.M. et al., 1994 |
Bohme K. et al., 1995
Ballock R.T. et al., 1993 Wu L.N.Y. et al., 1992 |
Fava R. et al., 19893
Schlaak J.F. et al., 19963 Chambers M.G. et al., 19971 |
| КМП і ХМП* | Luyten F.P. et al., 1992
Lietman S.A. et al., 1997 Erlacher L. et al., 1998 van Beuningen H.M. et al., 1998 |
Chen P. et al., 1995
Hiraki Y. et al., 1991 Enomoto-Iwamoto M. et al., 1998 |
Erlacher L. et al., 19981 |
*ХМП — хрящовий морфогенетичний протеїн; 1у хрящі; 2у синовіальній тканині; 3у синовіальній рідині.
Дії ІФР-1 і ІФР-2 контролюються різними ІФР-зв’язувальними білками (ІФР-ЗБ), які також виробляються хондроцитами (Martel-Pelletier J. et al., 1998). ІФР-ЗБ можуть виконувати функцію переносника, а також мають блокувальну ІФР-активність. Ізольовані із суглобового хряща хворих на ОА клітини виробляють надлишкову кількість ІФР-ЗБ, що свідчить про блокування ними ефектів ІФР (Dore S. et al., 1994). J. Martel-Pelletier та співавтори (1998) показали, що хоча синтез ІФР-1 у хрящі при ОА зростає, хондроцити слабко реагують на стиляцію ІФР-1. Виявилося, що цей феномен пов’язаний (принаймні частково) з підвищенням рівня ІФР-ЗБ. ІФР-ЗБ має високу спорідненість з ІФР і є важливим біомодулятором його активності. На сьогодні вивчено сім типів ІФР-ЗБ, порушення регуляції ІФР-ЗБ-3 і ІФР-ЗБ-4 відіграє важливу роль при ОА (Marlet-Pelletier J. et al., 1998).
Біологічно активні речовини, що стимулюють репарацію і пригнічують деградацію суглобового хряща (за: Carrabba M. et al., 1996, зі змінами)
|
Ще одна категорія факторів росту, що проявляють різну дію щодо хондроцитів, включає фактор росту, отдержаний із тромбоцитів (ФРОТ), фактор росту фібробластів (ФРФ) і ТФР-β. Ці фактори виробляються не тільки хондроцитами, але й активованими синовіцитами (Remmers E.F. et al., 1991; Nalashima M. et al., 1994). ФРФ має як анаболічні, так і катаболічні властивості залежно від концентрації та стану суглобового хряща (Sah R.L. et al., 1994). ФРОТ бере участь у підтримці гомеостазу ПКМ суглобового хряща, хоча не має явних мітогенних властивостей. Цей фактор росту, як відомо, має здатність посилювати синтез протеогліканів і зменшувати їх деградацію (Schafer S.J. et al., 1993).
ТФР-β становить особливий інтерес щодо вивчення його ролі в патогенезі ОА. Він є членом великої ТФР-родини, має загальні фунціональні та сигнальні властивості з нещодавно відкритими факторами росту кісткових морфогенетичних білків (КМБ).
ТФР-β — плейотропний фактор: з одного боку, він має імуносупресивні властивості, з іншого — це хемотаксичний фактор і потужний стимулятор проліферації фібробластів (van den Berg W.B. et al., 1999). Унікальними властивостями ТФР-β є здатність пригнічувати вивільнення ферментів із різних клітин і значно збільшувати продукцію інгібіторів ферментів (наприклад ТІМП) (Wright J.K. et al., 1991; Gunter M. et al., 1994). ТФР-β вважають важливим регулятором ушкодження тканини внаслідок запалення. Так, у тканині суглобового хряща ТФР-β значно стимулює продукцію матриксу хондроцитами (van der Kraan P.M. et al., 1992; Morales T.I., 1994; van Beuningen H.M. et al., 1994), особливо після преекспозиції цим фактором. Нормальний хрящ нечутливий до ТФР-β. У хворих на ОА ТФР-β стимулює продукцію агрекана і малих протеогліканів у суглобовому хрящі (Roughley P.J. et al., 1994; CS-Szabo G. et al., 1995).
ТФР-β виробляється багатьма клітинами, зокрема хондроцитами. Він вивільняється в латентній формі, зв’язаній із спеціальним білком, який називається «білок, асоційований із латентністю» (БАЛ). Дисоціація з цим білком здійснюється протеазами, які у великій кількості виробляються у запалених тканинах. Не враховуючи ТФР-β, який продукується активованими клітинами, запаси латентної форми цього фактора є важливим елементом реактивності ТФР-β у тканині після локального ушкодження. ТФР-β у значній кількості міститься в синовіальній рідині, синовіальній мембрані та хрящі суглоба, ураженого ОА (Fava R. et al., 1989; Schlaak J.F. et al., 1996; Chambers M.G. et al., 1997). У ділянках ушкодженої тканини, де є запальні інфільтрати, виявляють коекспресію ФНП та ІЛ-1, тоді як у ділянках з явищами фіброзу виявляють винятково експресію ТФР-β (Chu C.Q. et al., 1991).
Інкубація культури хондроцитів, одержаних від хворих на ОА, із ТФР-β викликає значне підвищення синтезу протеогліканів цими клітинами. Стимуляція ТФР-β нормальних хондроцитів викликає підвищення синтезу протеогліканів лише після багатьох днів інкубації. Можливо, цей час необхідний для зміни фенотипу клітин під впливом ТФР-β (наприклад для зміни так званої компартменталізації протеогліканів: знову створені протеоглікани локалізуються тільки навколо хондроцитів).
Відомо, що активація синтезу факторів росту, зокрема ТФР-β — важлива ланка в патогенезі фіброзу нирок і печінки, формуванні рубця при загоєнні рани (Border W.A., Ruoslahti E., 1992). Збільшення навантаження на хондроцити in vitro призводить до гіперпродукції ТФР-β, тоді як зменшення синтезу протеогліканів після імобілізації кінцівки може бути нівельоване ТФР-β (Zerath E. et al., 1997). ТФР-β індукує формування остеофітів у крайовій зоні суглобів як механізм пристосування до зміни навантаження. ІЛ-1, викликаючи помірний запальний процес у синовії у відповідь на ушкодження суглоба, сприяє утворенню хондроцитів зі зміненим фенотипом, які продукуються у надмірній кількості (van den Berg W.B. et al., 1999).
Повторні локальні ін’єкції рекомбінантного ТФР-β у значних концентраціях призвели до розвитку ОА у мишей лінії C57Bl — до утворення остеофітів, що характерно для ОА людини, і до значної втрати протеогліканів у зоні «хвилястої границі» (van den Berg W.B. et al., 1999).
Для розуміння того, як надлишок ТФР-β викликає вищеописані зміни хряща, необхідно відмітити, що експозиція ТФР-β індукує характерний фенотип хондроцита зі зміною підкласу синтезованих протеогліканів і порушенням нормальної інтеграції елементів ПКМ. І ІФР-1, і ТФР-β стимулюють синтез протеогліканів хондроцитами, що культивуються в алгінаті, проте останній ще й індукує так звану компартменталізацію протеогліканів (van Оsch G.J.V.M. et al., 1998). Крім того, виявлено, що ТФР-β підвищує рівень колагенази-3 (ММП-13) в активованих хондроцитах, що відрізняється від загальних уявлень про ТФР-β як фактор, який, навпаки, зменшує вивільнення деструктивних протеаз (Moldovan F. et al., 1997). Хоча не відомо, чи бере участь ТФР-β-індукований синтез ММП-13 у патогенезі ОА. ТФР-β не тільки стимулює синтез протеогліканів, але й сприяє їх відкладанню у зв’язках і сухожиллях, збільшуючи скутість і зменшуючи обсяг рухів у суглобах (Kawaguchi H. et al., 1992; Hoshi K. et al., 1997).
КМП — члени суперродини ТФР-β. Деякі з них (КМП-2; -7 і -9) мають властивість стимулювати синтез протеогліканів хондроцитами (Luyten F.P. et al., 1992; Lietman S.A. et al., 1997; van Beuningen H.M. et al., 1998). Ефекти КМП здійснюються шляхом зв’язування зі специфічними рецепторами на поверхні клітин; сигнальні шляхи ТФР-β і КМП дещо відрізняються (Yamashita H. et al., 1996; Attisano L., Wrana J.L., 1998). Подібно до ТФР-β, передача сигналу від КМП здійснюється через комплекс рецепторів серинової/треонінової кінази І і ІІ типу. В цьому комплексі рецептор ІІ типу трансфосфорилюється і активує рецептор І типу, який передає сигнал сигнальним молекулам, названим Smad (Attisano L., Wrana J.L., 1998). Після одержання сигналу Smad швидко фосфорилюється. На сьогодні відомо, що в сигнальному шляху КМП фосфорилються Smad-1, -5 і -8, а в сигнальному шляху ТФР-β — Smad-2 і -3. Потім названі Smad асоціюються з Smad-4, який є загальним для сигнальних шляхів всіх представників суперродини ТФР-β. Цей факт пояснює наявність перехресних функцій у членів суперродини ТФР-β, а також явище взаємного пригнічення сигнальних шляхів ТФР-β і КМП шляхом конкуренції за загальні компоненти. Нещодавно ідентифікований ще один клас Smad-білків, який представлений Smad-6 і -7. Ці молекули виступають як регулятори сигнальних шляхів ТФР-β і КМП (Hayashi H. et al., 1997).
Незважаючи на те що стимулюючий вплив КМП на синтез протеогліканів відомий давно, їх роль у регуляції функції суглобового хряща залишається суперечливою. Це пов’язано з відомою здатністю КМП викликати дедиференціювання клітин, стимулювати кальцифікацію та утворення кісткової тканини (van den Berg W.B. et al., 1999). M. Enomoto-Iwamoto та співавтори (1998) показали, що взаємодія КМП з КМП-рецептором ІІ типу необхідна для підтримки диференційованого фенотипу хондроцитів, а також контролю їх проліферації та гіпертрофії (Enomoto-Iwamoto M. et al., 1998). За даними L.Z. Sailor та співавторів (1996) КМП-2 підтримує фенотип хондроцитів у культурі протягом 4 тиж, не викликаючи їх гіпертрофії. КМП-7 (ідентичний остеогенному протеїну-1) тривалий час підтримує фенотип зрілих хондроцитів суглобового хряща, що культивуються в алгінаті (Flechtenmacher J. et al., 1996).
Введення КМП-2 і -9 у колінні суглоби мишей на 300% збільшувало синтез протеогліканів, причому значно більше, ніж ТФР-β (van Вeuningen H.M. et al., 1998). Однак стимулювальний ефект виявився тимчасовим, і через кілька днів рівень синтезу повернувся до вихідного. ТФР-β викликав більш тривалу стимуляцію синтезу протеогліканів (рис. 5.6), що, ймовірно, пов’язано з аутоіндукцією ТФР-β і сенсибілізацією хондроцитів до цього фактора.
ТФР-β відповідає за утворення хондрофітів, що можна вважати небажаним ефектом його дії, КМП-2 також сприяє утворенню хондрофітів, але в іншій ділянці суглобового краю (головним чином в ділянці ростової пластинки) (van Beuningen H.M. et al., 1998).

Рис. 5.6. Різна кінетика впливу ТФР-β і КМП-2 на синтез протеогліканів хондроцитами (за: van den Berg W.B. et al., 1998)
Хрящові морфогенетичні протеїни (ХМП-1 і -2) — ще одні із представників суперродини ТФР-β, необхідні для утворення хрящової тканини в період розвитку кінцівок (Luyten F.P., 1997). Мутація гена ХМП-1 викликає хондродисплазію (Thomas J.T. et al., 1996). Можливо профіль дії у ХМП більш селективний, спрямований на хрящ. Незважаючи на те що ТФР-β і ХМП здатні стимулювати хондроцити, вони можуть проявляти дію і на багато інших клітин, тому їх використання для репарації хряща може супроводжуватися побічними ефектами. ХМП обох типів виявляють у хрящі здорових і уражених ОА суглобів, вони сприяють репарації ПКМ суглобового хряща після ферментативної деградації, підтримуючи нормальний фенотип (Erlacher L. et al., 1998).
Синергізм факторів росту. Один фактор росту здатний індукувати сам себе, а також інші фактори росту, ця взаємодія має тонку регуляцію. Так, ФРФ разом з іншими факторами росту забезпечує більш ефективну репарацію суглобового хряща після травматичного дефекту (Frenz D.A. et al., 1994; Otsuka Y. et al., 1997). ІФР-1 разом із ТФР-β у значно більшому ступені індукують нормальний фенотип хондроцитів при культивуванні їх in vitro (Tsukazaki T. et al., 1994; Yaeger P.C. et al., 1997). Продемонстровано, що ТФР-β перешкоджає продукції ІФР-1 і ІФР-ЗБ, а також дефосфорилює рецептор ІФР-1, стимулює зв’язування ІФР-1 (van den Berg W.B. et al., 1999). В інтактному хрящі мишей виявлене явище синергізму ІФР-1 з багатьма факторами росту (Verschure P.J. et al., 1994). Однак слабку реакцію хондроцитів на ІФР-1 не можна нівелювати використанням у комбінації з іншими факторами росту (Verschure P.J. et al., 1994).
Взаємодія анаболічних і деструктивних цитокінів. Фактори росту демонструють складну взаємодію з ІЛ-1. Так, преекспозиція хондроцитів ФРФ збільшує вивільнення протеаз після експозиції ІЛ-1; можливо, це пов’язане зі збільшенням експресії рецепторів ІЛ-1 (Chandrasekhar S., Harvey A.K., 1989). Фактор росту, отриманий із тромбоцитів, також стимулює ІЛ-1-залежне вивільнення протеаз, однак він знижує ІЛ-1-опосередковане пригнічення синтезу протеогліканів (Smith R.J. et al., 1991). Ймовірно, що деякі фактори росту можуть одночасно стимулювати процес репарації хряща й сприяти його руйнуванню. Інші фактори росту, наприклад ІФР-1 і ТФР-β, стимулюють синтез суглобового матриксу і пригнічують ІЛ-1-опосередковане руйнування суглобового хряща, тобто їх активність пов’язана лише з репарацією тканини. Така взаємодія не залежить від преекспозиції хондроцитів ІЛ-1. Відзначимо, що кінетика ефектів ІЛ-1 і ТФР-β може бути різною: здатність ТФР-β пригнічувати деградацію суглобового хряща послабляється його повільною дією на мРНК ТІМП (Lum Z.P. et al., 1996). З іншого боку, відзначається підвищення рівня іNOС і NO при відсутності ТФР-β (Vodovotz Y. et al., 1996). Враховуючи NO-залежність супресорного ефекту ІЛ-1 відносно синтезу протеогліканів хондроцитами (van de Loo F.A.J. et al., 1998), можна пояснити, чому ми спостерігаємо значно більш сильну протидію ТФР-β-залежному пригніченню синтезу протеогліканів порівняно з руйнуванням протеогліканів in vivo.
У дослідженні на мишах, яким внутрішньосуглобово вводили ІЛ-1 та фактори росту, продемонстровано, що ТФР-β значно протидіє ІЛ-1-опосередкованому пригніченню синтезу протеогліканів суглобового хряща, тоді як КМП-2 не здатний на таку протидію: його стимуляторний потенціал повністю пригнічувався ІЛ-1 навіть за умови високої концентрації КМП-2 (van Beuningen H.M. et al., 1994; Glansbeek H.L. et al., 1997). Слід відмітити, що при відсутності ІЛ-1 КМП-2 значно інтенсивніше стимулював синтез протеогліканів, ніж ТФР-β (рис. 5.7).
Рис. 5.7. Синтез протеогліканів суглобового хряща на наступний день після 1-ї та 3-ї внутрішньосуглобових ін’єкцій ІЛ-1:1 і 3 — при відсутності ІЛ-1, 2 і 4 — при наявності ІЛ-1 (за: van Beuningen H.M. et al., 1994)
Крім впливу на синтез протеогліканів, ТФР-β також значно впливає на ІЛ-1-індуковане зниження вмісту протеогліканів у хрящі. Можливо, залежно від відносної концентрації ІЛ-1 і ТФР-β вміст протеогліканів знижується або підвищується. Відзначимо, що описану вище протидію ІЛ-1 і ТФР-β спостерігали в товщі хряща, однак поблизу хондрофітів по краях суглобових поверхонь такого явища не відзначали. Утворення хондрофітів індукується ТФР-β, який впливає на хондрогенні клітини, що знаходяться в періості, викликаючи розвиток хондробластів і відкладання протеогліканів (van Вeuningen H.M. et al., 1994; 1998). Вірогідно, ці хондробласти не чутливі до ІЛ-1.
H.L. Glansbeek та співавтори (1998) вивчали здатність ТФР-β і КМП-2 протидіяти пригніченню синтезу протеогліканів у суглобах мишей із зимозаніндукованим артритом (тобто в моделі «чистого» ІЛ-1-індукованого запалення). Внутрішньосуглобове введення ТФР-β значно протидіяло пригніченню синтезу протеогліканів, викликаного запаленням, у той час як КМП-2 був практично не здатний протидіяти цьому ІЛ-1-залежному процесу. Повторні ін’єкції ТФР-β у колінний суглоб досліджуваних тварин значно стимулювали синтез протеогліканів хондроцитами, сприяли збереженню існуючих протеогліканів виснаженого запаленням хряща, але не пригнічували запальний процес.
При дослідженні протеоглікансинтезуючої функції хондроцитів із використанням експериментальних моделей ОА у тварин завжди відзначали підвищення вмісту і стимуляцію синтезу протеогліканів (Adams M.E., Brandt K.D., 1991; van der Kraan P.M. et al., 1992; Gaffen J.D. et al., 1995) на ранніх стадіях ОА, на відміну від запальних моделей, у яких спостерігається значне пригнічення синтезу (ІЛ-1-залежний процес) (van de Loo A.A.J. et al., 1995; 1998). Підвищення активності анаболічних факторів, зокрема факторів росту, що спостерігається при ОА, нівелює дія таких супресорних цитокінів, як ІЛ-1. Серед факторів росту найбільше значення має ТФР-β; КМП-2 навряд чи відіграє значну роль у цьому процесі. Хоча ІФР-1 здатний стимулювати синтез протеогліканів in vitro, в умовах in vivo таку властивість не спостерігають при локальному застосуванні ІФР-1. Можливо, це пов’язано з тим, що ендогенний рівень цього фактора росту є оптимальним. На більш пізніх стадіях ОА з’являються ознаки пригнічення синтезу протеогліканів, що, ймовірно, пов’язано з домінуючою дією ІЛ-1, а також неможливістю факторів росту протидіяти йому внаслідок зниження їх активності.
Аналіз експресії факторів росту у мишей лінії STR/ORT зі спонтанним ОА продемонстрував підвищення рівня мРНК ТФР-β та ІЛ-1 в ушкодженому хрящі (Chambers M.G. et al., 1997). Необхідно відмітити, що активація ТФР-β з латентної форми є важливим елементом репарації тканини. Розуміння ролі ТФР-β ускладнює результати дослідження експресії рецепторів ТФР-β II типу у кроликів лінії ACL (Boumediene K. et al., 1998). Відразу після індукції ОА виявлено знижений рівень цих рецепторів, що свідчить про недостатню сигнальну функцію ТФР-β. Відзначимо, що у дефіцитного за рецептором ТФР-β II типу мишей виявлені ознаки спонтанного ОА, що також свідчить про важливу роль сигнальної функції ТФР-β у погіршенні репарації хряща і розвитку ОА (Serra R. et al., 1997).
Абсолютний вміст факторів росту в суглобах хворих на РА або ОА може свідчити про їх вірогідну роль у патогенезі цих захворювань. Проте незважаючи на те що в суглобах при ОА і РА виявляють високі концентрації факторів росту, характер процесів деградації та репарації при обох захворюваннях зовсім різний. Імовірно, існують інші, ще не ідентифіковані фактори, що відіграють основну роль у патогенезі цих хвороб, або інші аспекти досліджуваних явищ визначають перебіг процесів деградації та репарації в тканинах суглобів (наприклад експресія певних рецепторів на поверхні хондроцитів, розчинні рецептори, що зв’язують білки, або дисбаланс анаболічних і деструктивних факторів).
РОЛЬ ВІДКЛАДАННЯ КРИСТАЛІВ У ПАТОГЕНЕЗІ ОА
У 30–60% хворих на ОА виявляють кристали основного фосфату кальцію (ОФК) у суглобовій рідині (Dieppe P.A. et al., 1972; Gibilisco P.A. et al., 1985). На думку A. Swan та співавторів (1994), кальцієвмісні кристали знаходяться в суглобовій рідині у значно більшої кількості хворих на ОА, проте занадто дрібні розміри або мала кількість кристалів не дозволяють ідентифікувати їх за допомогою загальноприйнятих методик. Наявність кристалів ОФК у суглобовій рідині корелює з рентгенологічними ознаками дегенерації суглобового хряща (Halverson P.B., McCarty D.J., 1986) і асоціюється з більшим об’ємом випоту порівняно з випотом у колінних суглобах без кристалів (Carroll G.J. et al., 1991). Дослідження факторів, що впливають на рентгенологічне прогресування гонартрозу, показало, що відкладання кристалів пірофосфату кальцію дигідрату (ПФКД) є предиктором несприятливого клінічного й рентгенологічного результату (Ledingham J. et al., 1995). При дослідженні пацієнтів похилого віку виявлено, що ОА асоціюється із хондрокальцинозом, особливо в латеральному тибіофеморальному відділі колінного суглоба і у перших трьох п’ястно-фалангових суглобах (Sanmarti R. et al., 1996). Часто у хворих на ОА виявляють обидва типи кристалів — ОФК і ПФКД (Dieppe P. et al., 1978).
Клінічно дегенерація суглобового хряща, зумовлена відкладанням кальцієвмісних кристалів, відрізняється від такої при первинному ОА (Ryan L., McCarty G.M., 1997). Якби кристали були простим епіфеноменом дегенерації хряща, їх виявляли б у суглобах, які найчастіше уражуються первинним ОА, тобто в колінних, кульшових, дрібних суглобах кистей. Навпаки, хвороби відкладання кристалів частіше вражають нетипові для первинного ОА суглоби — плечові, променезап’ясткові, ліктьові. Наявність кристалів у суглобовій (випотній) рідини асоціюється з більш тяжкою дегенерацією суглобового хряща. Обговорюється питання про те, що є причиною, а що наслідком — відкладання кристалів або дегенерація хряща (Bjelle A., 1981). Проміжну позицію займає таке припущення: первинна аномалія метаболізму хряща веде до його дегенерації, а вторинне відкладання кристалів прискорює його деградацію (так звана теорія ампліфікаційної петлі) (Doherty M. et al., 1982).
Точний механізм ушкодження суглобового хряща кальцієвмісними кристалами не відомий, його окремі елементи наведені нижче. Теоретично кальцієвмісні кристали можуть безпосередньо ушкоджувати хондроцити. Проте при гістологічному дослідженні кристали рідко локалізуються поблизу хондроцитів, ще рідше поглинаються ними (Schumacher H., 1988). Найбільш імовірним є фагоцитоз кристалів клітинами синовіального покриття з подальшим виділенням ними протеолітичних ферментів або секрецією цитокінів, що стимулюють виділення ферментів хондроцитами. Ця концепція підтверджується дослідженням ролі ПФКД-індукованого синовіту в розвитку швидко прогресуючого ОА при пірофосфатній артропатії (Fam A.G. et al., 1995). У ході цього дослідження кроликам з ОА, індукованим частковою латеральною меніскектомією, у правий колінний суглоб вводили кристали ПФКД (1 або 10 мг) 1 раз на тиждень. Виявилося, що після 8 ін’єкцій у правому колінному суглобі були значно серйозніші зміни порівняно з лівим. Інтенсивність синовіального запалення корелювала із внутрішньосуглобовими ін’єкціями кристалів ПФКД та їх дозою. Незважаючи на те що використані в цьому дослідженні дози кристалів ПФКД перевищують такі in vivo, результати свідчать про роль ПФКД-індукованого запалення в прогресуванні ОА при пірофосфатній артропатії.
Потенційні механізми індукування ушкодження суглобового хряща кальцієвмісними кристалами пов’язані з їх мітогенними властивостями, здатністю індукувати ММП і стимулювати синтез ПГ.
Мітогенний ефект кальцієвмісних кристалів. При кристаласоційованих артропатіях часто виявляють проліферацію клітин синовіального покриття, причому самі кристали лише частково відповідальні за цей процес (Garancis J.C. et al., 1981). Збільшення кількості синовіальних клітин супроводжується посиленням секреції цитокінів, які сприяють хондролізу і викликають секрецію протеолітичних ферментів. Концентрації кристалів ОФК, що виявляються при патології суглобів у людини, дозозалежно стимулюють мітогенез культури фібробластів шкіри, що знаходяться у фазі спокою, синовіальних фібробластів собак і мишей (Barnes D., Colowick S., 1977; Cheung H.S. et al., 1984). Кристали ПФКД, урату, сульфату, карбонату і дифосфату кальцію стимулюють ріст клітин (Barnes D., Colowick S., 1977; Cheung h., McCarty G.M., 1985). Початок і пік включення (3Н)-тимідину, що індукуються цими кристалами, зміщені на 3 год порівняно зі стимуляцією клітин сироваткою крові. Ймовірно, цей період необхідний для фагоцитозу і розчинення кристалів. Додавання контрольних кристалів того ж розміру (наприклад брильянтового пилу або часточок латексу) не стимулювало мітогенез. Кристали урату натрію моногідрату мали слабкі мітогенні властивості і значно поступалися таким урату кальцію, що свідчить про важливе значення вмісту кальцію в кристалах у мітогенезі. Синтетичні кристали ОФК мали такі ж мітогенні властивості, як і кристали, одержані від пацієнтів із хондрокальцинозом (Cheung H. et al., 1984). Мітогенний ефект кальцієвмісних кристалів не був результатом підвищення вмісту кальцію в навколоклітинному живильному середовищі in vitro, оскільки розчинення кристалів ОФК у живильному середовищі не стимулювало включення (3Н)-тимідину фібробластами (Cheung H., McCarty G.M., 1985).
Один із вірогідних механізмів ОФК-індукованого мітогенезу такий: аномальна проліферація синовіальних клітин може бути пов’язана (принаймні частково) з ендоцитозом і внутрішньоклітинним розчиненням кристалів, що призводить до підвищення концентрації Са2+ у цитоплазмі клітин і активації кальцієзалежного шляху, що веде до мітогенезу. На підтримку цієї концепції виступає необхідність прямого контакту клітина — кристал для стимуляції мітогенезу, оскільки експозиція культури клітин кристалам викликала ріст клітин, а експозиція клітин, позбавлених можливості такого контакту, не викликала їх росту (Bowen-Pope D., Rubin H., 1983). Для вивчення необхідності фагоцитозу кристалів, що відбувається при взаємодії клітина — кристал, клітини культивували з 45Са-ОФК і (3Н)-тимідином. Виявилося, що клітини із вмістом 45Са-ОФК включали набагато більшу кількість (3Н)-тимідину, ніж клітини без мічених ОФК (Borkowf A. et al., 1987). У культурі макрофагів пригнічення цитохалазином ендоцитозу кристалів викликало інгібіцію розчинення кристалів, що також підкреслює необхідність фагоцитозу (Owens J. et al., 1986).
Кальцієвмісні кристали розчинні в кислоті. Після фагоцитозу кристали розчиняються в кислому середовищі фаголізисом макрофагів. Хлорохін, амонію хлорид, бафіломіцин А1 і всі лізосомотрофні агенти, що підвищують лізосомальну рН, дозозалежно пригнічують внутрішньоклітинне розчинення кристалів і поглинання (3Н)-тимідину у фібробластах, культивованих із кристалами ОФК (McCarty G.M. et al., 1998).
Додавання ОФК-кристалів до багатошарової культури фібробластів викликало негайне десятиразове підвищення вмісту внутрішньоклітинного кальцію, яке поверталося до вихідного рівня через 8 хв. Джерелом кальцію переважно був позаклітинний іон, оскільки кристали ОФК були додані в безкальцієве живильне середовище. Подальше підвищення концентрації внутрішньоклітинного кальцію спостерігалося через 60 хв і тривало не менше 3 год. Тут джерелом кальцію були фагоцитовані кристали, розчинені у фаголізосомах (Halverson P. et al., 1998).
Визначено, що мітогенний ефект ОФК-кристалів схожий на такий у факторі росту, отриманому із тромбоцитів, у якості фактора росту; подібно до останнього, ОФК-кристали проявляють синергізм відносно ІФР-1 і плазми крові. Блокада ІФР-1 знижує мітогенез клітин у відповідь на ОФК (Cheung H. et al., 1986). P.G. Mitchell та співавтори (1989) показали, що індукція ОФК кристалами мітогенезу фібробластів Balb/c-3T3 вимагає наявності серинової/треонінової протеїнкінази С (ПКС) — одного з головних медіаторів сигналів, генерованих при зовнішній стимуляції клітин гормонами, нейротрансмітерами і факторами росту. Зниження активності ПКС у клітинах Balb/c-3T3 пригнічує ОФК-опосередковану індукцію протоонкогенів c-fos і c-myc, але не впливає на стимуляцію цих онкогенів, опосередковану фактором росту, отриманим із тромбоцитів (Mitchell P.G. et al., 1989).
Підвищення вмісту внутрішньоклітинного кальцію після розчинення фагоцитованих кристалів — не єдиний сигнальний шлях для мітогенезу. Коли фактори росту, такі, як фактор росту, отриманий із тромбоцитів, зв’язуються зі своїм мембранним рецептором, стимулюється фосфоліпаза С (фосфодіестераза), яка гідролізує фосфатидилінозитол 4,5-бісфосфат з утворенням внутрішньоклітинних месенджерів — інозитол-3-фосфату і діацилгліцеролу (Berridge M., 1987). Перший вивільняє кальцій з ендоплазматичної сітки, модулюючи активність кальцієзалежних і кальцій/кальмодулінзалежних ферментів, таких як протеїнкінази і протеази (Rasmussen H., 1986).
R. Rothenberg і H. Cheung (1988) повідомили про підвищену деградацію фосфоліпазою С фосфатидилінозитолу 4,5-біфосфату в синовіальних клітинах кроликів у відповідь на стимуляцію ОФК-кристалами. Останні значно підвищують вміст інозитол-1-фосфату в клітинах із міченим (3Н)-інозитолом; пік був досягнутий протягом 1 хв і зберігався близько 1 год.
Діацил-гліцерол — потенційний активатор ПКС (Kikkawa U., Nishizuka Y., 1986). У зв’язку з тим, що ОФК-кристали підвищують активність фосфоліпази С, що веде до акумуляції діацилгліцеролу, можна чекати підвищення активації ПКС. P.G. Mitchell та співавтори (1989) порівнювали вплив ОФК-кристалів і фактора росту, отриманого із тромбоцитів, на синтез ДНК фібробластами Balb/c-3T3. У культурі клітин ПКС була інактивована інкубацією клітин із туморпідтримувальним форболовим диефіром (ТФД), аналогом діацилгліцеролу. Тривала стимуляція низькими дозами ТФД знижує активність ПКС, тоді як одноразова стимуляція високою дозою — активує. Стимуляція ОФК-кристалами синтезу ДНК була пригнічена після інактивації ПКС, що свідчить про важливість цього ферменту в ОФК-індукованому мітогенезі. Раніше G.M. McCarty та співавтори (1987) продемонстрували зв’язок мітогенної відповіді фібробластів людини на ОФК-кристали з активацією ПКС. Однак ОФК-кристали не активують фосфатидилінозитол-3-кіназу або тирозинкіназу, підтверджуючи той факт, що механізм активації клітин кристалами ОФК є селективним (McCarty G.M. et al., 1987).
Проліферацію клітин контролює група генів, названих протоонкогенами. Білки foc і myc, продукти протоонкогенів c-fos і c-myc локалізовані у клітинному ядрі й пов’язані зі специфічними послідовностями ДНК (Kelly K. et al., 1983; Cochran B. et al., 1984). Стимуляція 3Т3-фібробластів ОФК-кристалами призводить до експресії c-fos протягом декількох хвилин, яка досягає максимуму через 30 хв після стимуляції. Індукція транскрипції c-myc ОФК-кристалами або фактором росту, отриманим із тромбоцитів, відбувається протягом 1 год і досягає максимуму через 3 год після стимуляції. Не менше 5 год клітини підтримують підвищений рівень транскрипції c-fos і c-myc (Cheung H.S. et al., 1989). У клітинах з інактивованою ПКС стимуляція c-fos і c-myc кристалами ОФК або ТФД значно пригнічена, в той час як індукція цих генів фактором росту, отриманим із тромбоцитів, не змінюється.
Представники родини мітогенактивованих протеїнкіназ (МАПК) — ключові регулятори різних внутрішньоклітинних сигнальних каскадів. Один із підкласів цієї родини — р42/р44 — регулює проліферацію клітин за допомогою механізму, що включає активацію протоонкогенів с-fos і c-jun. ОФК- і ПФКД-кристали активують протеїнкіназний шлях передачі сигналу, в якому беруть участь обидва р42 і р44, що свідчить про роль цього шляху в мітогенезі, індукованому кальцієвмісними кристалами (Nair D. et al., 1997).
Нарешті, в ОФК-індукованому мітогенезі бере участь транскрипційний ядерний фактор κВ (NF-κВ), який вперше був описаний як ген легкого ланцюга імуноглобуліну κ (Igκ). Це індукований транскрипційний фактор, важливий для багатьох сигнальних шляхів, оскільки він регулює експресію різних генів. Індукція NF-κВ зазвичай сполучена з вивільненням із цитоплазми інгібіторних білків, названих IκВ. Слідом за індукцією NF-κВ відбувається транслокація активного транскрипційного фактора в ядро. Кристали ОФК індукують NF-κВ у Balb/c-3T3 фібробластах і фібробластах шкіри людини (McCarty G.M. et al., 1987).
Кілька шляхів можуть бути залучені в передачу сигналу після активації NF-κВ, однак усі вони включають протеїнкінази, які фосфорилюють (і в такий спосіб деградують) ІκВ (Barnes P., Karin M., 1997). Ґрунтуючись на результатах досліджень in vitro, раніше припускали, що ІκВ є субстратом для кіназ (наприклад ПКС і протеїнкіназа А) (Baeuerle P.A., Baltimore D., 1988; Shirakawa F., Mizel S.B., 1989). Проте нещодавно був ідентифікований ІκВ-кіназний комплекс великої молекулярної маси (Mercurio F. et al., 1997). Ці кінази специфічно фосфорилюють серинові залишки ІκВ. Активація NF-κВ ФНП-α і ІЛ-1 вимагає ефективної дії Nf-κВ-індукуючої кінази (НІК) і IκВ-кінази (Woronicz J.D. et al., 1997). Молекулярний механізм активації НІК на сьогодні не відомий. Незважаючи на те що кристали ОФК активують і ПКС і NF-κВ, не відомо, у якій мірі ці два процеси можуть бути зв’язані. Оскільки модифікація IκВ-кінази здійснюється шляхом фосфорилювання, не виключається роль ПКС в індукції кристалами ОФК NF-κВ шляхом фосфорилювання та активації IκВ-кінази (Didonato J.A. et al., 1997). У підтримку цієї концепції може служити пригнічення інгібітором ПКС стауроспорином ОФК-кристаліндукованого мітогенезу й експресії NF-κВ (McCarty G.M. et al., 1987). Аналогічно стауроспорин може пригнічувати IκВ-кіназу, а отже, пригнічувати протеїнкіназу А та інші протеїнкінази (Tamaoki T., 1991).
Таким чином, механізм ОФК-кристаліндукованого мітогенезу у фібробластах включає не менше двох різних процесів (рис. 5.8):
1) швидка мембранозв’язана подія, яка призводить до активації ПКС і МАПК, індукції NF-κВ і протоонкогенів;
2) більш повільне внутрішньоклітинне розчинення кристалів, що призводить до підвищення внутрішньоклітинного вмісту Са2+, а в подальшому — до активації ряду кальцієзалежних процесів, що стимулюють мітогенез.
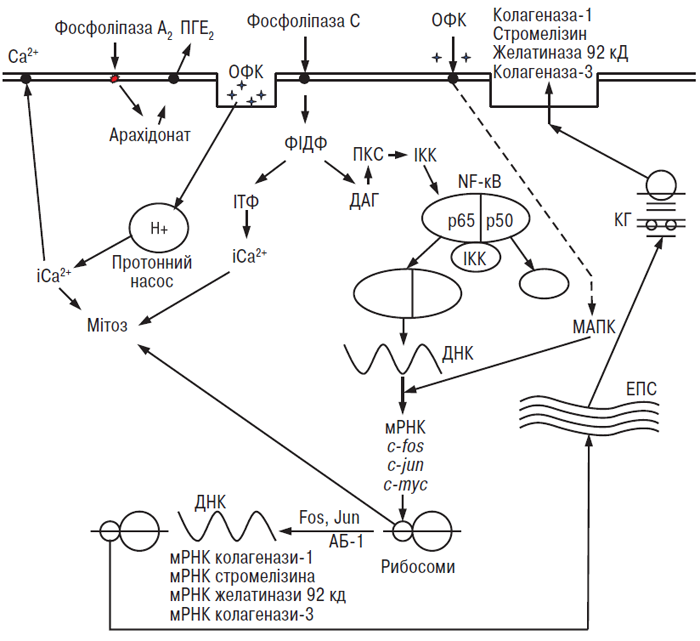
Рис. 5.8. Деякі біологічні ефекти кальцієвмісних кристалів (за: McCarty G.M. et al., 1987–1998). ОФК — основний фосфат кальцію; ЕПС — ендоплазматична мережа; ПКС — протеїнкіназа С; ДАГ — діацилгліцерол; ФІДФ — фосфатидил-інозитолдифосфат; ІТФ — інозитолтрифосфат; NF-κВ — ядерний фактор κВ; МАПК — мітоген-активована протеїнкіназа; IКК — IκВ-кіназа; КГ — комплекс Гольджи
Індукція ММП кальцієвмісними кристалами. Медіаторами ушкодження тканини кальцієвмісними кристалами є ММП — колагеназа-1, стромелізин, желатиназа 92 кД і колагеназа-3.
З урахуванням зв’язку між вмістом кристалів ОФК і деструкції тканин суглобів було висловлено гіпотезу, згідно з якою кристали ОФК і, можливо, деякі колагени фагоцитуються синовіальними клітинами. Стимульовані синовіцити проліферують і секретують протеази. Ця гіпотеза була перевірена in vitro шляхом додавання природних або синтетичних кристалів ОФК, ПФКД та інших у культуру синовіцитів людини або собаки. Рівень активності нейтральних протеаз і колагеназ дозозалежно зріс і був приблизно у 5–8 разів вищим порівняно з контрольною культурою клітин, які культивували без кристалів (Cheung H. et al., 1981).
У клітинах, культивованих у середовищі, що містить кристали, виявлено коіндукцію мРНК колагенази-1, стромелізину і желатинази-92 кД з подальшою секрецією ферментів у середовище (McCarty G.M. et al., 1992; 1998). Кристали ОФК також викликали акумуляцію мРНК колагенази-1 і колагенази-2 у зрілих хондроцитах свині з подальшою секрецією ферментів у середовище (Mitchell P.G. et al., 1992; McCarty G.M. et al., 1997).
G.M. McCarty та співавтори (1998) вивчали роль внутрішньоклітинного розчинення кристалів у кристаліндукованій продукції ММП. Підвищення лізосомального рН за допомогою бафіломіцину А1 пригнічувало внутрішньоклітинне розчинення кристалів, а також послабляло проліферативну відповідь фібробластів людини на кристали ОФК, але не пригнічувало синтез і секрецію ММП (McCarty G.M. et al., 1998).
Ні кристали ОФК, ні ПФКД не індукували продукцію ІЛ-1 in vitro, на відміну від кристалів урату натрію (Malawista S. et al., 1985). Сучасні дані чітко свідчать про пряму стимуляцію продукції ММП фібробластами і хондроцитами при контакті з кальцієвмісними кристалами (McCarty G.M. et al., 1999).
Клінічна картина ОА свідчить про значну роль ММП у прогресуванні хвороби. Наявність кальцієвмісних кристалів посилює дегенерацію тканин уражених суглобів.
Стимуляція синтезу ПГ. Поряд зі стимуляцією росту клітин, секрецією ферментів кальцієвмісні кристали викликають вивільнення ПГ із культур клітин ссавців, особливо ПГЕ2 (Cheung H. et al., 1981; McCarty G.M., Cheung H., 1985; Dayer j.M. et al., 1987). Вивільнення ПГЕ2 у всіх випадках відбувається протягом першої години після експозиції клітин кристалам. R. Rothenberg (1987) визначив, що основними джерелами арахідонової кислоти для синтезу ПГЕ2 є фосфатидилхолін і фосфатидилетаноламін, а також підтвердив, що фосфоліпаза А2 і ЦОГ — домінуючі шляхи продукції ПГЕ2.
У відповідь на дію кристалів ОФК також може виділятися ПГЕ1 (Rashad S. et al., 1989). G.M. McCarty та співавтори (1993; 1994) вивчали вплив ПГЕ2, ПГЕ1 та його аналога мізопростолу на мітогенну відповідь фібробластів людини на кристали ОФК. Усі три агенти пригнічували мітогенну відповідь дозозалежним шляхом, причому ПГЕ1 і мізопростол проявляли більш виражену інгібуючу активність. ПГЕ1 і мізопростол, але не ПГЕ2 пригнічували акумуляцію мРНК колагенази у відповідь на дію кристалів ОФК (McCarty G.M. et al., 1993; 1994).
M.G. McCarty і H. Cheung (1994) досліджували механізм ОФК-опосередкованої активації клітин ПГЕ. Автори показали, що ПГЕ1 (сильніший індуктор внутрішньоклітинного цАМФ, ніж ПГЕ2) пригнічує ОФК-індукований мітогенез і продукцію ММП через цАМФ -залежний шлях передачі сигналу. Вірогідно, підвищення продукції ПГЕ, індуковане кристалами ОФК, послаблює їх інші біологічні ефекти (мітогенез і продукцію ММП) за механізмом зворотного зв’язку.
Запалення, індуковане кристалами. Кальцієвмісні кристали часто виявляють у синовіальній рідині у хворих на ОА (McCarty G.M. et al., 1999), однак епізоди гострого запалення з лейкоцитозом відзначають рідко як при ОА, так і при кристаласоційованих артропатіях (наприклад синдром «плечовий суглоб Мілуоки») (McCarty G.M. et al., 1999). Флогістичний потенціал кристалів може бути модифікований за участю ряду інгібіторних факторів. R. Terkeltaub та співавтори (1988) продемонстрували здатність сироватки й плазми крові значно пригнічувати відповідь нейтрофільних гранулоцитів на кристали ОФК. Факторами, які викликають таке пригнічення, є кристали, що зв’язують білки. Дослідження одного з таких білків — α2-HS-глікопротеїну (АHSГ) — показало, що АHSГ є найбільш потужним і специфічним інгібітором відповіді нейтрофільних гранулоцитів на кристали ОФК. АHSГ — сироватковий протеїн печінкового походження; відомо, що порівняно з іншими білками сироватки крові він у відносно високих концентраціях утримується в кістковій тканині й тканині, що мінералізується (Triffit J., 1978). Крім того, АHSГ наявний у синовіальній рідині без ознак запалення, а також виявлений на кристалах ОФК у нативній синовіальній рідині (Terkeltaub R. et al., 1988). Таким чином, не виключається ймовірність модулювання АHSГ флогогенного потенціалу кристалів ОФК в умовах in vivo.
Враховуючи вищесказане, наводимо дві схеми патогенезу ОА, запропоновані W.B. van den Berg та співавторами (1999) (рис. 5.9) і M. Carrabba та співавторами (1996) (рис. 5.10), які поєднують механічні, генетичні та біохімічні фактори.
Рис. 5.9. Схема участі ТФР-β у патогенезі ОА (за: van den Berg W.B. et al., 1999)
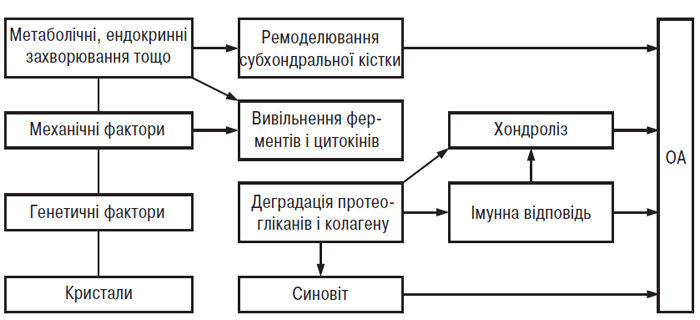
Рис. 5.10. Схема патогенезу ОА (за: Carrabba M. еt al., 1996)