Дилатаційна кардіоміопатія
Содержание
Визначення
Дилатаційна кардіоміопатія (ДКМП) — захворювання серцевого м’яза, що характеризується дилатацією і систолічною дисфункцією ЛШ при відсутності порушень наповнення (гіпертензія, клапанні вади) або ІХС, здатних викликати глобальне погіршення систолічної функції. Може також відзначатися дилатація і дисфункція ПШ, проте це не є обов’язковим для встановлення діагнозу.
Епідеміологія
Частота виявлення ДКМП у різних регіонах неоднакова, тому визначити її справжню поширеність важко. Ідіопатичну ДКМП відзначають у 0,4 випадку на 1 тис. населення, щорічно виявляють 0,08 нового випадку на 1 тис. населення, що становить приблизно 25% всіх випадків кардіоміопатії і є причиною щорічної смерті 10 тис. хворих. Чоловіки хворіють у середньому в 3 рази частіше, ніж жінки.
Етіологія
Етіологія ДКМП остаточно не встановлена. Багато дослідників дотримуються поліетіологічної гіпотези походження захворювання — достатньо описано випадків розвитку ДКМП, яка є кінцевим результатом різних патологічних процесів. Виділяють ідіопатичну, сімейну (або генетичну), вірусну (і/або імунну) і асоційовану з відомими серцево-судинними захворюваннями ДКМП.
Ідіопатична ДКМП, що є первинним захворюванням міокарда невідомої етіології, розвивається у 40% хворих. Сімейна ДКМП головним чином асоціюється з мутаціями в генах цитоскелета і екстрацелюлярного матриксу.
Останнім часом досягнутий значний прогрес в області молекулярної генетики ДКМП. У 20% хворих захворювання успадковується або виявляється в сімейному анамнезі. При спадкових формах виявлено аутосомно-домінантний, аутосомно-рецесивний, а також пов’язаний з Х-хромосомою гена або мітохондріальною трансмісією тип успадкування. Мутації виявлені в генах кардіоміоцитів, які кодують контрактильні білки або їх регулюючі елементи, включаючи компоненти саркомера, цитоскелета, а також різні механізми, що забезпечують узгодження процесів збудження — скорочення, бета-адренергічні шляхи проведення і процеси, що призводять до дефіциту енергетичних механізмів, у тому числі мутації мітохондрій, метаболізм глікогену, обміну кальцію, а також транскрипторну регуляцію.
Виникнення ДКМП пов’язують з варіантами мутацій гена актину, важливе значення також приділяється патології гена білка дистрофіна, що входить до складу комплексу, який зв’язує м’язовий цитоскелет кардіоміоцита з екстрацелюлярним матриксом.
Результати дослідження CARDIGENE (1999) свідчать, що при ДКМП мають значення генетичні порушення шляхів ендотеліна і поліморфізм гена ендотелінових рецепторів типу А — перший ідентифікований генетичний чинник ризику розвитку захворювання.
Існує вірусно-імунологічна теорія виникнення ДКМП. При ДКМП виявлений ряд порушень імунної регуляції, включаючи гуморальну і клітинну аутоімунну реактивність по відношенню до міоцитів, зниження вмісту і клітинної активності природних кілерів і відхилення у діяльності супресорних клітин. Наявність порушень імунної регуляції і великої кількості антиміокардіальних антитіл при ДКМП узгоджується з цією гіпотезою: при імунологічному дослідженні підвищення титрів антитіл до вірусу Коксакі В3 виявляли у 40% хворих з ДКМП і тільки у 2% — в групі здорових осіб, при цьому в ендоміокардіальних біоптатах не визначали ознак міокардиту. У 50% хворих ДКМП виявляють антитіла до міокарда, у 49% — антиінтерфібрилярні антитіла, у 30% — сенсибілізацію до серцевого антигену. Активація аутоімунних процесів призводить до утворення антитіл до міозину і β1-адренорецепторів.
Причиною пригнічення активності природних кілерів може бути первинне порушення їх дозрівання, детерміноване антигенами системи HLA, у хворих з ДКМП найчастіше виявляють гаплотипи HLA B27, HLA A2, HLA DQ4 і HLA DR4, що вказує на спадкову схильність до захворювання та свідчить про можливу його імунну основу. Незважаючи на виявлення порушень гуморального імунітету, в цілому вірусно-інфекційно-аутоімунна гіпотеза на сьогодні залишається не доведеною.
Патогенез
Патогенез ДКМП треба насамперед розглядати на молекулярному рівні, оскільки порушується експресія генів, що призводить до зміни фенотипу. Виділяють кілька механізмів, які лежать в основі розвитку захворювання: одиничний генний дефект; поліморфні зміни генів-модифікаторів (у гені АПФ та інших компонентах РААС і β2-адренергічних рецепторах); порушення експресії нормальних генів, що кодують білки, які регулюють скорочувальну функцію серця або формують структуру його порожнин.
Мутації генів, що кодують білки позаклітинного матриксу, можуть бути причиною послаблення механічного взаємозв’язку між матриксом і кардіоміоцитами, можливим наслідком чого є прогресуюча дилатація серця. В кардіоміоцитах, що залишилися, відбувається ушкодження внутрішньоклітинних органел, у тому числі і відповідальних за енергетику клітини. Внаслідок цього відбувається швидке розщеплення АТФ, що веде не тільки до порушення процесу скорочення, але й до контрактури міокарда в результаті нестачі енергії і кальцієвого перевантаження.
Певна роль у розвитку фіброзу приділяється деградації нормального колагенового матриксу металопротеїназами, активація яких відбувається внаслідок активації прозапальних цитокінів і експозиції вільних кисневих радикалів (оксидантного стресу).
При ушкодженні міокарда і дилатації серця відбувається активація різних компенсаторних механізмів, спрямованих на нормалізацію серцевої діяльності (схема 9.1).

Схема 9.1. Компенсаторні механізми при розвитку СН при ДКМП
Довгострокові компенсаторні механізми включають розвиток гіпертрофії життєздатних кардіоміоцитів, що залишились, і зміну геометрії камер серця, що становить суть ремоделювання ЛШ.
Гіпертрофія міокарда не досягає адекватного ступеня і не відповідає потребам дилатованого серця, оскільки скорочувальна активність гіпертрофованого міокарда на одиницю маси нижче, ніж у здоровому серці. Поступово відбувається зміна геометрії шлуночка від нормальної еліпсоїдної до сферичної форми (ремоделювання) і поступове переважання дилатації над гіпертрофією його стінки. Серцевий викид підтримується тахікардією і більшим об’ємом діастолічного наповнення, що в свою чергу підвищує внутрішньоміокардіальне напруження як у систолу, так і в діастолу та збільшує потребу міокарда в кисні. Ці процеси є пусковим механізмом активації РААС, стимулюють виділення норадреналіну симпатичними терміналями, а також секрецію НУП.
Тривала надмірна активація РААС спричиняє ушкоджуючу дію, наслідком якої є потенціювання активності інших нейрогормональних систем, коронарна та системна вазоконстрикція, безпосередня токсична, ушкоджуюча дія ангіотензину II на кардіоміоцити, що призводить до їх дисфункції та загибелі.
Тривала активація САС чинить ряд негативних ефектів на серцево-судинну систему, включаючи підвищення потреби в кисні, зменшення сили скорочення (внаслідок підвищення ЧСС), підвищення концентрації катехоламінів у плазмі крові, зниження чутливості β-адренорецепторів до катехоламінів, пригнічення функції мітохондрій, порушення окислювально-відновної рівноваги, що реалізується шляхом дії через β-рецептори серця і цАМФ.
У результаті порушень з боку β-адренергічних шляхів проведення, значною мірою модулюючих функцію серця на рецепторному та клітинному рівнях, відбувається значне зменшення кількості та щільності β1-рецепторів в ураженому серці, тоді як щільність β2-рецепторів залишається практично без змін. У сукупності з підвищенням концентрації блокуючих G-протеїнів і посиленням процесів β-рецепторного фосфорилування ці зміни посилюють порушення скорочувальної функції ураженого міокарда.
Активація нейрогормонів, цитокінів, перебудова біомеханіки міокарда зумовлюють зміну генної експресії, загибель кардіоміоцитів, клітинне ремоделювання, що в свою чергу є причиною дисфункції міокарда та його ремоделювання. Можливо, певний генотип первинно призводить до активації нейрогормонів і цитокінів. Велику роль відіграють ендотелін-1 і ФНП-α.
Порушення насосної функції серця при ДКМП може бути зумовлено зменшенням кількості самих кардіоміоцитів. Активація процесів, пов’язаних з механічним перерозтягненням стінок ураженого серця, з ефектами системи нейрогуморальної регуляції і цитокінами відображається у прогресуючій втраті контрактильних елементів шляхом апоптозу та некрозу.
Патологічна анатомія
Захворювання характеризується різкою дилатацією всіх порожнин серця, маса серця може досягати 1000 г і більше.
У порожнинах передсердь і шлуночків часто (за даними розтину >50%) виявляють пристінкові тромби. AV-отвір значно розтягнутий, стулки клапана злегка білуваті, подовжені, як і сухожильні нитки. Сосочкові м’язи гіпертрофовані, часто різко склерозовані.
Специфічних змін, що виявляються мікроскопічно, немає, визначають нерівномірну гіпертрофію кардіоміоцитів з великими неправильної форми ядрами, також помітна вогнищева жирова дистрофія м’язових волокон, дезорганізація міофібрил, втрата міофіламентів, мікроцитоліз, колапс строми, інтерстиціальний фіброз. Новоутворені колагенові структури характеризуються спотвореним співвідношенням між типами колагену і порушенням архітектоніки взаємного розташування волокон. При ДКМП переважає колаген I+III типу, помірний вміст колагену VI типу та знижений вміст колагену IV типу.
Відзначають виражені дегенеративні і некробіотичні зміни кардіоміоцитів (зникнення поперечної смугастості, вакуолізація ядер, некроз, помірний вміст глікогену). Нерідко визначають запальні інфільтрати, при гістологічному дослідженні біоптатів міокарда в інтерстиціальній тканині міокарда нараховують від 5 до 10 лімфоцитів при збільшенні в 400 або 200 разів відповідно.
Клінічна картина
Домінуючим клінічним симптомом є наростаюча СН. Як правило, ранні стадії захворювання виявляють випадково при профілактичному рентгенологічному або ехоКГ-дослідженні. Період від появи перших симптомів до виникнення розгорнутої клінічної картини становить зазвичай 2 роки.
Клінічні симптоми зумовлені дисфункцією ЛШ, ПШ або обох шлуночків. У ⅓ хворих можливий біль за грудиною, нерідко — різні порушення ритму, характерними є епізоди тромбоемболії судин великого і малого кола кровообігу.
При прогресуванні захворювання та появі декомпенсації симптоми загалом характерні для СН.
Діагностика
На ЕКГ специфічних ознак немає, можуть виявлятися порушення внутрішньошлуночкової провідності, блокада лівої ніжки пучка Гіса, зниження вольтажу зубця R, можливі порушення ритму серця, в тому числі екстрасистолічна аритмія, пароксизмальна тахікардія, фібриляція передсердь. За даними добового моніторування ЕКГ часто виявляють тяжкі шлуночкові порушення ритму, які не виявляються при звичайній реєстрації ЕКГ.
При рентгенологічному дослідженні виявляють кардіомегалію, застійні зміни в малому колі кровообігу (рис. 9.1).

Рис. 9.1. Рентгенограма хворого з ДКМП
ЕхоКГ є основним методом діагностики, за допомогою якого визначають розміри ЛШ, ступінь підвищення кінцевого діастолічного тиску, порушення його систолічної і діастолічної функції. У хворих з ДКМП відзначають виражену дилатацію ЛШ (рис. 9.2) і лівого передсердя (рис. 9.3), товщина їх стінок нормальна або зменшена. Важливим є також визначення наявності дилатації ПШ, вимір систолічного тиску в ПШ за ступенем трикуспідальної регургітації за допомогою допплєрівського дослідження. Часто виявляють внутрішньопорожнинні тромби.

Рис. 9.2. ДКМП. М-режим
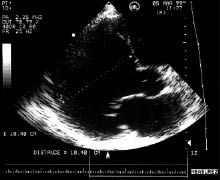
Рис. 9.3. ДКМП. В-режим, парастернальна позиція, довга вісь
Ремоделювання міокарда асоціюється з порушенням гемодинаміки і зменшенням ФВ, ударного об’єму, збільшенням об’ємів та підвищенням тиску в камерах серця. Часто виявляють дилатацію кільця мітрального і тристулкового клапанів та ознаки клапанної регургітації.
Дані імунологічних досліджень свідчать про достовірне зниження активності природних кілерів, підвищення рівня ФНП-α порівняно зі здоровими особами, а також про наявність специфічних циркулюючих антитіл, які є важливими маркерами ДКМП: антиміозинові антитіла (до важких ланцюгів α- і β-міозину), антимітохондріальні антитіла (до М7, до аденіннуклеотидного транслокатора — ферменту внутрішньої мембрани мітохондрій серця, що здійснює перенос АТФ і АДФ між цитоплазмою кардіоміоцитів і матриксом цих органел), антитіла до β-адренорецепторів.
При проведенні прижиттєвої біопсії міокарда виявляють неспецифічні дегенеративні зміни різного ступеня, міоцитоліз, вогнища некрозу.
Характерні морфологічні ознаки ДКМП при гістологічному дослідженні відсутні. Як правило, виявляють множинні вогнища фіброзного заміщення міокарда, іноді запальні інфільтрати.
При радіонуклідній вентрикулографії визначають розширення порожнин серця, порушення локальної скоротності на фоні дифузного зниження скоротності міокарда, значне зниження ФВ ЛШ і ПШ.
При сцинтиграфії міокарда з 201Tl виявляють дифузні та вогнищеві дефекти накопичення ізотопу. При сцинтиграфії міокарда з 67Gl ізотоп накопичується у вогнищах запалення при міокардиті і не накопичується при ДКМП.
Катетеризацію порожнин серця проводять, якщо діагноз залишається під сумнівом після неінвазивного дослідження. Серцевий викид може бути нормальним або зниженим, ФВ знижена, на ангіограмі визначається дифузний гіпокінез. Підвищення кінцево-діастолічного тиску в ЛШ відзначають в пізній стадії захворювання. Під час катетеризації можна провести біопсію міокарда кожного шлуночка.
До діагностичних критеріїв ДКМП належать підтверджувальні ознаки і ті, що її виключають. До підтверджувальних ознак належать прогресуюча СН, резистентна до лікування; кардіомегалія з наявністю відносної недостатності мітрального і тристулкового клапанів; тромбоемболічний синдром; порушення ритму і провідності; відносно молодий вік; відсутність ознак запального процесу; відсутність зв’язку захворювання з інфекційним або будь-яким іншим етіологічним чинником.
До ознак, що виключають ДКМП, належать нормальні розміри серця, наявність симптомів ІХС (обструкція >50% просвіту основних коронарних артерій), документована АГ (>160/100 мм рт. ст.), вроджені вади серця і набуті зміни клапанного апарату серця, вказівки в анамнезі на хронічне вживання алкоголю (>40 г в день для жінок і >80 г в день для чоловіків протягом понад 5 років) з ремісією алкогольної кардіоміопатії після 6 міс абстиненції, системні захворювання, ураження перикарда.
Лікування
Повинне бути спрямоване на рішення наступних основних завдань:
- запобігання прогресування СН, подальшого ремоделювання серцево-судинної системи (зменшення нейроендокринної, клітинної активації);
- зменшення вираженості симптоматики (покращання систолічної функції серця, периферичного кровообігу);
- покращання якості життя (покращання переносимості фізичних навантажень);
- збільшення тривалості життя (поліпшення прогнозу);
- зменшення кількості випадків госпіталізації;
- зменшення кількості ускладнень.
Лікування включає немедикаментозні підходи (дієта, обмеження прийому рідини та кухонної солі, фізичні навантаження), застосування лікарських препаратів (діуретиків, інгібіторів АПФ, блокаторів β-адренорецепторів, серцевих глікозидів, блокаторів рецепторів ангіотензину II, антагоністів альдостерону, інотропних агентів, антикоагулянтів, антиаритмічних засобів), застосування допоміжних пристроїв для підтримки функції ЛШ, а також сучасні хірургічні втручання (кардіоміопластика, відновлення мітрального і тристулкового клапана, часткова вентрикулектомія ЛШ і трансплантація серця).
Важливе значення в лікуванні хворих з СН має зниження постнавантаження шляхом нормалізації рівня АТ, а також корекція анемії для підвищення транспорту кисню тканинам.
За останні роки кардинально змінилися погляди на значення фізичної активності: встановлено, що тривала малорухливість може призводити до атрофії скелетних м’язів, подальшого погіршення переносимості фізичних навантажень, венозного тромбозу, легеневої емболії і прогресування симптомів СН.
Робоча група по кардіологічній реабілітації разом з Робочою групою по СН Європейського товариства кардіологів у 2001 р. опублікували рекомендації по проведенню фізичних тренувань у хворих з ХСН.
До відносних протипоказань щодо використання фізичного навантаження у хворих із стабільною СН належать:
- Збільшення маси тіла на 1,8 кг і більше за попередні 1–3 дні;
- Супутня постійна або періодична терапія добутаміном;
- Зниження САТ при навантаженнях;
- IV ФК по NYHA;
- Складні порушення ритму в стані спокою або якщо такі з’являються при навантаженні;
- Вихідна ЧСС ≥100 уд./хв.
До абсолютних протипоказань належать:
- Прогресивне погіршення переносимості фізичного навантаження або задишка в стані спокою або при навантаженні протягом 3–5 попередніх днів;
- Значна ішемія при невеликих навантаженнях (50 Вт);
- Неконтрольований цукровий діабет;
- Гостре системне захворювання або лихоманка;
- Наявність емболії в анамнезі;
- Тромбофлебіт;
- Активний перикардит або міокардит;
- Регургітація на клапанах, що вимагає хірургічного втручання;
- Фібриляція передсердь, що виникла знову.
Відносні критерії, при яких рекомендується починати навантажувальні тренування:
- компенсована СН протягом принаймні 3 тиж;
- здатність розмовляти без задишки (з частотою дихання <30 за 1 хв);
- ЧСС у спокої не більше 100 уд./хв;
- незначна втома;
- серцевий індекс не менше 2,1 хв/м2 (для хворих, яким проводиться інвазивне моніторування);
- рівень ЦВТ <12 мм рт. ст. (для хворих, яким проводиться інвазивне моніторування).
Відносні критерії, при яких необхідно змінити або припинити тренування:
- виражена задишка або втома;
- частота дихання >40 за 1 хв під час вправ;
- поява III тону або хрипів у легенях;
- посилення хрипів у легенях;
- посилення другого компонента II тону;
- недостатній пульсовий тиск (<10 мм рт. ст. між САТ і ДАТ);
- зниження АТ (>10 мм рт. ст.) під час збільшення навантажень;
- збільшення кількості суправентрикулярних або шлуночкових екстрасистол під час навантажень;
- профузне потовиділення, блідість, сплутаність свідомості.
Медикаментозне лікування ДКМП по суті є лікуванням СН.
Діуретики залишаються засобами першого ряду, оскільки це єдина група препаратів, які можуть адекватно контролювати затримку рідини при СН. При помірній СН лікування зазвичай починають із застосування тіазидних діуретиків, при більш вираженій — застосовують петльові діуретики (торасемід, фуросемід або етакринову кислоту). Комбінація діуретиків, що діють на різні ділянки нефрону, дозволяє попередити розвиток резистентності, а також виникнення побічних ефектів.
Інгібітори АПФ відіграють ключову роль у довгостроковій терапії хворих з ДКМП і систолічною дисфункцією незалежно від ступеня її вираженості.
Інгібітори АПФ слід застосовувати з обережністю у хворих з дуже низьким системним АТ (САТ <80 мм рт. ст.), значно підвищеним рівнем креатиніну в сироватці крові (>3 мг/дл), підвищеним рівнем калію в сироватці крові (>5,5 ммоль/л). Починати лікування інгібіторами АПФ треба із застосування низької дози під контролем рівня АТ, подвоюючи дозу кожні 3–7 днів при хорошій переносимості і титруючи її до цільової.
Найбільш частими побічними ефектами інгібіторів АПФ, які потенційно обмежують їх застосування, є артеріальна гіпотензія, погіршення функції нирок, сухий кашель, ангіоневротичний набряк. Можлива заміна інгібіторів АПФ антагоністами рецепторів ангіотензину II.
Усім пацієнтам з СН внаслідок систолічної дисфункції ЛШ необхідно призначати блокатори β-адренорецепторів (метопролол, бізопролол, карведилол, небіволол), крім випадків, коли вони протипоказані або погано переносяться.
Лікування блокаторами β-адренорецепторів починають з низьких доз — з ⅛ середньої терапевтичної дози, подвоюючи дозу кожні 2–3 тиж до оптимальної. Терапію у хворих з ДКМП слід починати в стаціонарі під наглядом лікаря. Початок лікування блокаторами β-адренорецепторів може супроводжуватися клінічним погіршенням у 10–20% хворих, що проявляється зниженням переносимості фізичного навантаження, збільшенням явищ застою в легенях, посиленням втомлюваності, збільшенням периферичних набряків і, як наслідок, погіршенням якості життя. Проте при подальшому лікуванні ознаки короткочасного гемодинамічного погіршення зникають.
У хворих з вираженою СН використовують антагоністи альдостерону (спіронолактон) як засіб додаткового впливу на нейрогуморальні механізми розвитку СН, що поліпшують прогноз.
Серцеві глікозиди (дигоксин) показані хворим з ДКМП, у яких виражені явища декомпенсації, для зменшення симптомів СН. У пацієнтів з тахісистолічною формою фібриляції передсердь застосування серцевих глікозидів дозволяє знизити частоту скорочень шлуночків. Відміна серцевих глікозидів у хворих з ДКМП як з синусовим ритмом, так і фібриляцією передсердь і низькою ФВ призводить до наростання симптомів декомпенсації серцевої діяльності.
Непрямі антикоагулянти (варфарин) показані пацієнтам з постійною формою фібриляції передсердь, тромбоемболічними ускладненнями в анамнезі, при наявності тромбів у порожнинах серця.
Імплантація кардіовертера-дефібрилятора при наявності рецидивуючої фібриляції шлуночків або стійкої шлуночкової тахікардії та резистентності до антиаритмічних препаратів, а також імплантація трикамерного електрокардіостимулятора в режимі DDDR у хворих зі значними порушеннями внутрішньошлуночкової провідності та десинхронізацією скорочення шлуночків при тяжкій СН, що рефрактерна до медикаментозної терапії, входить у перелік додаткового асортименту медичних послуг хворим з ДКМП (наказ МОЗ України № 436 від 03.07.2006 р.).
Патогенетична терапія не розроблена. Застосування імунодепресантів у цьому випадку не виправдане.
Оскільки прогноз при ДКМП несприятливий і будь-яка терапія не запобігає летальному кінцю, ці хворі являють собою найбільшу групу реципієнтів для трансплантації серця, що дозволяє збільшити тривалість життя. На успішний результат оперативного втручання можна розраховувати у хворих без супутніх системних захворювань, порушень психіки, високого необоротного опору легеневих судин, у пацієнтів віком молодше 60 років.
ЛІТЕРАТУРА
- Амосова Е.Н. (1999) Кардиомиопатии. Книга плюс, Киев, 422 с.
- Капустян Л.Н., Рябенко Д.В., Вигонтина О.Г. и соавт. (2008) Hsp90 как аутоантиген при дилатационной кардиомиопатии. Укр. кардіол. журн., 6: 75–78.
- Коваленко В.Н., Несукай Е.Г. (2001) Некоронарогенные болезни сердца. Практическое руководство. Морион, Киев, 480 с.
- Коваленко В.Н. (ред.) (2008) Руководство по кардиологии. Морион, Киев, 1424 с.
- Мравян С.Р., Канвар С., Голухова Е.З. (1997) Клинико-инструментальные показатели в оценке прогноза миокардита и дилатационной кардиомиопатии. Кардиология, 7: 67–72.
- Наказ № 436 Міністерства охорони здоров’я України від 03.07.2006 р. «Про затвердження протоколів надання медичної допомоги за спеціальністю «Кардіологія» (2006) Укр. кардіол. журн., 6; 89–115.
- Оганов Р.Г., Фомина И.Г. (ред.) (2006) Болезни сердца: руководство для врачей. Литтерра, Москва, 1328 с.
- Ardehali H., Kasper E.K., Baughman K.L. (2005) Diagnostic approach to the patient with cardiomyopathy: whom to biopsy. Amer. Heart J., 149: 7–12.
- Ardehali H., Qasim A., Cappola T. et al. (2004) Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Amer. Heart J., 147: 919–923.
- Aukrust P., Ueland T., Lien E. et al. (1999) Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Amer. J. Cardiol., 83: 376–382.
- Burkett E.L., Hershberger R.E. (2005) Clinical and genetic issues in familial dilated cardiomyopathy. J. Amer. Coll. Cardiol., 45: 969–981.
- Charron Ph., Tesson F., Poirier O. et al. (1999) Identification of a genetic risk factor for idiopathic dilated cardiomyopathy. Europ. Heart J., 20: 1587–1591.
- Chojnowska L., Ruzyllo W. (2000) Rodzinna kardiomiopatia przerostowa. Kardiol. Pol., 53(III): 88–95.
- Elliot P. (2000) Cardiomyopathy. Diagnosis and management of dilated cardiomyopathy. Heart, 84: 106–112.
- Elliot P., Anderson B., Arlustini E. et al. (2008) Classification of the cardiomyopathy; a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J., 29: 270–276.
- Galderisi M., Mondillo S. (2007) Echocardiography in clinical practice. One Way S.r.l., 120 p.
- Giordano F.J. (2005) Oxygen, oxidative stress, hypoxia and heart failure. J. Clin. Invest., 115: 500–508.
- Graham R.M., Owens W.A. (1999) Pathogenesis of inherited forms of dilated cardiomyopathy. N. Engl. J. Med., 341: 1759–1762.
- Gustafson A.B., Gottlieb R.A. (2004) Mechanisms of apoptosis in the heart. J. Clin. Immunol., 23: 447–459.
- Hannuksela J., Leppilampi M., Peuhkurinen K. et al. (2005) Hereditary hemochromatosis gene (HFE) mutations C282Y, H63D and S65C in patients with idiopathic dilated cardiomyopathy. Eur. J. Heart Fail., 7: 103–108.
- Kamisago M., Sharma S.D., DePalma S.R. et al. (2000) Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med., 34: 1688–1696.
- Keller D.I., Carrier I., Schwartz K. (2002) Genetics of familial cardiomyopathies and arrhythmias. Swiss. Med. Weekly, 132: 401–407.
- Mahon N.G., Murphy R.T., MacRae C.A. et al. (2005) Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann. Intern. Med., 143: 108–115.
- Manolio T.A., Baughman K.L., Rodeheffer R. et al. (1992) Prevalence and etiology of idiopathic dilated cardiomyopathy. Amer. J. Cardiol., 69: 1458–1466.
- Maron B.J., Towbin J.A., Thiene G. et al. (2006) Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation, 113: 1807–1816.
- Mogensen J., Murphy R.T., Shaw T. et al. (2004) Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Amer. Coll. Cardiol., 44: 2033–2040.
- Morita H., Seidman J., Seidman C.E. (2005) Genetic causes of human heart failure. J. Clin. Invest., 115: 518–526.
- Nelson G.S., Berger R.D., Febics B.J. et al. (2000) Left ventricular or biventricular pacing improves cardiac function at diminished energy cost in patients with dilated cardiomyopathy and left bundle-branch block. Circulation, 102: 3053–3059.
- Report of the WHO/ISFC task force on the definition and classification of cardiomyopaties (1996) Circulation, 93: 841–842.
- Tesson F., Charron P., Peuchmaurd M. et al. (1999) Characterization of a unique genetic variant in the beta-1 adrenoceptor gene and evaluation of its role in idiopathic dilated cardiomyopathy. J. Mol. Cell Cardiol., 31: 1025–1032.
- Topol E.J. (Ed.) (2007) Textbook of cardiovascular medicine. 3th ed. Williams&Wilkins, Lippincott, 1628 p.