Порушення кровообігу
Содержание
ВИДИ І ПРИЧИНИ ПОРУШЕНЬ КРОВООБІГУ
Розлади кровообігу пов’язані зі змінами функції серця, артерій, мікросудин, вен, порушенням реологічних властивостей крові, проникності гістогематичного бар’єра або нейрогуморальної регуляції. Окремі ланки серцево-судинної системи функціонують у тісному взаємозв’язку, тому зміни, що виникають в одній із них, впливають на функцію всіх інших. При цьому можливі зміни тканинного метаболізму аж до ураження і загибелі клітин і стромальних структур. Порушення кровообігу можуть мати загальний характер, коли змінюється функція всієї кровоносної системи, або ж відноситися до окремих ділянок судинного русла. У морфологічній картині уражень, зумовлених розладами крово- і лімфообігу, поєднуються ознаки як загальні, так і зумовлені структурно-функціональними особливостями цієї тканини.
Загальні і місцеві порушення кровообігу, які відзначають при багатьох захворюваннях, ускладнюють їх перебіг. На сьогодні розрізняють наступні види порушень: артеріальне і венозне повнокров’я (гіперемія), ішемія, стаз крові, тромбоз, ДВЗ-синдром, емболія, інфаркт, кровотеча і крововилив.
Артеріальна гіперемія
Артеріальна гіперемія — підвищене кровонаповнення тканини, зумовлене надмірним припливом артеріальної крові, може виникати в нормальних і патологічних умовах, мати загальний або місцевий характер, розвиватися в здоровому організмі або в умовах патології.
Загальна артеріальна гіперемія спостерігається при еритремії (збільшення кількості еритроцитів у крові) або при збільшенні всього ОЦК (плетора). Шкірні покриви таких хворих набувають вираженого червонуватого відтінку, а рівень АТ підвищується. Проте значно частіше артеріальна гіперемія обмежується яким-небудь конкретним регіоном. У фізіологічних умовах вона пов’язана з функцією судинозвужувальних і судинорозширювальних нервів або з дією фізичних факторів. Інтенсивність кровотоку в мікросудинах зростає у результаті збільшення просвіту периферичних артерій, що сприяє підтримці метаболічних процесів у тканині, що ними живиться. Гіперемія, пов’язана з посиленням функції органів, визначається як функціональна, або робоча.
Механізми розвитку артеріальної гіперемії в умовах патології більш різноманітні. У відповідності з причиною розрізняють ангіоневротичну (нейропаралітичну), колатеральну, постанемічну, вакатну, що виникає внаслідок артеріовенозної нориці, і запальну артеріальну гіперемію.
Ангіоневротична гіперемія пов’язана з порушенням співвідношення процесів збудження і гальмування у відділах вегетативної нервової системи, що регулюють тонус відповідних артерій. Домінування парасимпатичних впливів над симпатичними веде до дилатації артерій і переважання об’єму притоку крові над її відтоком по венозним судинам. Цей вид вазомоторної гіперемії може виникати рефлекторно, наприклад при психомоторних афектах, ушкодженні нервових провідників або сплетень, деяких інфекційних захворюваннях (грип, висипний тиф), для перебігу яких характерне ураження вузлів симпатичної нервової системи. Органи, шкірні покриви, слизова оболонка при артеріальній гіперемії почервонілі, іноді припухлі, температура їх підвищена. Відчуття пульсації гіперемованого поля свідчить про поширення пульсової хвилі на периферичну сітку мікросудин. Зазвичай цей вид артеріальної гіперемії має швидкоминучий, транзиторний характер і не залишає слідів.
Колатеральна гіперемія розвивається при обмеженні прохідності магістрального артеріального стовбура, наприклад при його тромбозі, здавлюванні ззовні, ваді розвитку судини. У результаті притікаюча кров спрямовується в гілки, що відходять ще до перешкоди, переповнює і розширює їх. Формування обхідних шляхів-колатералей визначається темпами обтурації судини, кількістю попередньо існуючих анастомозів і ступенем розвитку чутливої іннервації по ходу судин, що забезпечує більш-менш повне і швидке розкриття колатеральних шляхів. Утворення колатералей стимулюється метаболітами, що надходять з ішемізованої тканини і чинять вазодилататорний ефект.
Судинна мережа, що виявилась у зоні колатеральної гіперемії, поступово пристосовується і перебудовується. Відзначається посилення еластичного та аргірофільного каркаса, гіпертрофія і гіперплазія гладком’язових клітин стінки судин, що потрапили до неї. Колатеральна гіперемія є важливим компенсаторним механізмом, завдяки якому підтримується кровопостачання тканин при закритті основного артеріального стовбура.
Вакатна (від лат. vacuus — порожній) гіперемія є наслідком градієнта барометричного тиску між будь-якою ділянкою тіла або всім організмом і навколишнім середовищем. Прикладом місцевого артеріального повнокров’я такої природи є гіперемія шкіри, викликана за допомогою медичної банки. Загальну вакатну гіперемію відзначають при різкому зниженні тиску в навколишньому середовищі внаслідок швидкого підйому водолазів або кесонних робіт, розгерметизації висотних літальних апаратів. Різке розширення судин у цих умовах може призвести до повнокров’я слизових оболонок, кровотечі з носа й вух, крововиливів із тяжким ураженням внутрішніх органів.
Постанемічна гіперемія розвивається у тих випадках, коли відновлюється прохідність артерій, раніше частково або повністю виключених з кровотоку. Така ситуація буває при знятті хірургічної лігатури, видаленні пухлини або рідини, що здавлювали судину, реканалізації тромбованої або стенозованої артерії. Це може призводити до ішемізації інших органів, зокрема головного мозку, з розвитком непритомного стану (колапсу).
Гіперемія на ґрунті артеріовенозної нориці найчастіше є наслідком вогнепального поранення з ураженням артерії і вени та утворенням сполучення між ними. Артеріальна кров, що спрямовується по новому шляху у вену, підвищує температуру тканини, змінює процеси обміну, сприяє прискореному росту волосся на шкірі.
Запальна гіперемія — обов’язковий компонент запального процесу. В її основі лежить повнокров’я периферичних судин, пов’язане з порушенням процесів обміну і парезом судин. Посилений приплив крові до збудженої ділянки підвищує локальну температуру, а при ураженні шкіри або слизових оболонок фарбує їх у червоний колір. Гіперемія більш виражена у гострій фазі процесу і минає разом з його закінченням.
Венозне повнокров’я
Венозна (застійна, пасивна) гіперемія — патологічна зміна кровообігу, зумовлена утрудненим відтоком венозної крові при збереженні притоку її в тканини по відповідним артеріям. Венозне повнокров’я може бути загальним і місцевим, гострим і хронічним.
Гостра загальна венозна гіперемія виникає при СН, яка швидко розвивається, що може бути наслідком ІМ, токсично-інфекційних уражень серцевого м’яза, внаслідок чого втрачається здатність ефективно підтримувати загальну гемодинаміку або при ураженні мітрального або аортального клапана. Це призводить до переповнення кров’ю ємнісних судин. Швидкість кровотоку у венах і капілярах знижується, а тиск в них значно підвищується. Просвіти всіх мікросудин розширюються, а ті, що раніше не функціонували, розкриваються. У цих умовах максимально дилатуються дрібні вени, венули і венулярні відділи капілярів внаслідок відносно більшого підвищення тиску в дистальних відділах судинної мережі. Так як редукований гемоглобін венозної крові збіднений киснем, тканини відчувають гіпоксію. Проникність стінок мікросудин і розтяжність опорної сполучної тканини підвищуються, а об’єм тканинної рідини істотно збільшується.
Ознаками венозної гіперемії є зниження температури, синюшність (ціаноз) шкіри і слизових оболонок, що пов’язано із заповненням поверхнево розташованих судин кров’ю, що містить відновлений гемоглобін. Ціанотичне забарвлення більш помітне на периферичних частинах тіла — на губах, носі (акроціаноз), кінчиках пальців. У виражених випадках ціаноз поєднується з набряком жирової клітковини і водянкою порожнин. У тканинах відзначають плазматичне просочування (плазморагія), набряк строми, сповільнення і зупинку кровотоку в мікросудинах, численні крововиливи діапедезного характеру. Внаслідок того, що при гострій венозній гіперемії порушується більшість основних функцій крові (транспортна, дихальна, трофічна, екскреторна, підтримка водного балансу тканини), у паренхіматозних органах виникають дистрофічні зміни і розсіяні дрібновогнищеві некрози. Швидко наростаючий застій крові в басейні магістральної вени веде до застійного геморагічного інфаркту — некрозу, що формується на фоні вираженого ціанозу, набряку і просочування тканини кров’ю. Такі інфаркти відзначають при закритті селезінкової, ниркової, брижових вен, тромбозі синусів мозкової оболонки або коронарного синуса.
Венозне повнокров’я проявляється неоднаково в різних органах залежно від архітектоніки судин. У легенях виявляють дилатацію і переповнення кров’ю капілярів, просочування фільтратом крові міжальвеолярних перегородок і заповнення ним значної частини альвеол (інтерстиціальний і альвеолярний набряк), діапедезний вихід еритроцитів. Гостра венозна гіперемія печінки характеризується збільшенням її об’єму. На розрізі тканина органа набуває темно-червоного кольору, стає вологою зі стертим малюнком.
Регургітація венозної крові із серця, що гостро виникає при його дисфункції, супроводжується не тільки повнокров’ям тканин, але й крововиливами в центральних зонах печінкових часточок. Нирки при гострому венозному повнокров’ї збільшуються в об’ємі, маса їх зростає від 250–300 до 400–500 г. Найбільше повнокров’я відзначається в мозковій речовині пірамідок, що пов’язано з внутрішньоорганним перерозподілом кровотоку. Дистрофічні зміни, що розвиваються на фоні набряку тканини нирок, максимально виражені у найбільш чутливих до гіпоксії проксимальних відділах канальцевої системи нефрону. Об’єм селезінки в умовах гострого венозного застою збільшується в 2,5 раза, що супроводжується різким напруженням її капсули, а маса досягає ≥300 г. На розрізі з темно-червоної поверхні тканини селезінки рясно стікає кров. У серцевому м’язі відзначаються надмірне кровонаповнення інтрамуральних вен і мікросудин, капіляростази, набряк інтерстиція, дрібновогнищеві ураження м’язових волокон.
Іноді венозна гіперемія має активний характер. Це виявляють у зонах колатерального венозного повнокров’я, наприклад при обструкції великого венозного стовбура, цирозі печінки або шоку. В останньому випадку в печінці, селезінці, підшкірній жировій клітковині, венах ШКТ, тазової порожнини депонується до 40% усієї крові, що призводить до дефіциту наповнення порожнин серця і фібриляції шлуночків. У цілому досить швидко ліквідована гостра венозна гіперемія залишає незначні наслідки.
Хронічну загальну венозну гіперемію відзначають значно частіше, ніж гостру. Вона є наслідком ХСН, ускладнює багато захворювань з тривалим перебігом, які призводять до прогресуючого порушення функції серця: вроджені і набуті вади серця, кардіоміопатії, хронічна алкогольна інтоксикація, міокардити різного генезу, амілоїдоз серця, деякі системні ураження сполучної тканини, перикардит, хронічні запальні процеси у легенях дифузного характеру та ін. Основними причинами порушення насосної функції серця є:
- Слабкість ураженого міокарда, скорочення якого не можуть забезпечити рівень внутрішньошлуночкового тиску, необхідного для переміщення крові, що притікає до серця, в артерії великого і малого кола кровообігу.
- Декомпенсовані вади серця, коли енергія серцевого скорочення виявляється недостатньою для просування необхідної маси крові через звужені серцеві отвори або коли неповне змикання серцевих клапанів призводить до повернення частини крові в ретроградно розташовану камеру серця та венозні судини.
- Обмеження ємності порожнин серця внаслідок масивного субендокардіального тромбозу (ендокардит Леффлера), фіброеластоза ендокарда, різкого потовщення стінки шлуночків і міжшлуночкової перегородки при субаортальному стенозі (ГКМП).
- Неможливість достатнього розширення порожнин серця під час діастоли через скупчення значного об’єму рідини в порожнині перикарда або її облітерації і кальцинації.
- Зменшення ОЦК і, як наслідок, недостатнє повернення крові до серця.
Тривалий венозний застій підтримує стан тканинної гіпоксії, що разом з гемодинамічними факторами порушує проникність гістогематичного бар’єра і призводить до повторних плазмо- і геморагій, стійкого набряку, дистрофії і мікронекрозам, що постійно повторюються. Внаслідок цього хронічна венозна гіперемія веде до атрофії паренхіматозних органів і прогресуючих склеротичних змін. Склерозування тканини зумовлене стимулюючим впливом хронічної гіпоксії на пластичні процеси в сполучній тканині. Новоутворення глікозаміногліканів і колагенових волокон призводить до поступового нарощування маси та огрубінню строми органів, що витісняє паренхіматозні структури, які атрофуються в умовах гіпоксії. У фокусах мікронекрозу відбувається формування зірчастих рубчиків. Унаслідок прогресуючого характеру таких дифузних і дрібновогнищевих склеротичних змін розвивається ціанотична індурація (застійне ущільнення) органів і тканин. Органи стають щільнішими, збільшуються в розмірах, набувають характерного синюшно-темно-червоного кольору.
Венозний застій більш виражений у нижніх частинах тіла, найчастіше в нижніх кінцівках, а при тривалому дотриманні постільного режиму — в нижніх відділах легень. Це явище називають гіпостазом. Гіпостаз у нижніх кінцівках пов’язаний також із розширенням просвіту вен і недостатністю їх клапанів. Венозна гіперемія, що тривалий час утримується, супроводжується гіпертрофією м’язової оболонки вен. Переповнені кров’ю нерівномірно розширені вени часто стають звивистими, на них з’ являються вузлуваті випинання — варикозні вузли. У шкірі виявляють ціаноз, набряк дерми та підшкірної клітковини, що доповнюється розростанням сполучної тканини, атрофічними змінами дерми та епідермісу.
У печінці хронічна венозна гіперемія супроводжується складною структурно-функціональною перебудовою гістоархітектоніки органа. Поряд зі збільшенням розмірів печінки, закругленням країв і напруженням капсули відзначається ущільнення тканини, на її розрізі з’являється малюнок, що нагадує мускатний горіх внаслідок чергування дрібних зон темно-червоного і сірувато-жовтого кольору. Темно-червоні ділянки відповідають серединним зонам печінкових часточок, де на місці центральних вен, відділів синусоїд, що до них прилягають і печінкових клітин, що лежать між ними, утворюються кров’яні озера, розташовуються елементи проліферуючої сполучної тканини.
На периферії печінкових часточок цитоплазма гепатоцитів набуває зернистого вигляду і містить безліч ліпідних крапельок, що надають тканині жовтуватий відтінок. Така нерівномірність фарбування тканини пояснюється особливостями кровопостачання органа і будовою печінкових часточок. У печінку надходить кров з двох джерел: портальної вени й печінкової артерії. Обидві судини, які послідовно діляться, на останок розгалужуються на капіляри, які проникають між балками з паренхіматозних клітин у печінкові часточки. Тут капіляри, що приносять артеріальну і венозну кров, зливаються, утворюючи синусоїдальні капіляри портальної системи, які впадають у центральну вену печінкової часточки. Центральна вена є дренажною системою, що несе кров у нижню порожнисту вену.
Внутрішньодолькові кровоносні капіляри — синусоїди — на всьому протязі покриті сплощеними ендотеліоцитами з незамкнутими міжклітинними щілинами і позбавлені базальної мембрани. Звернена до них поверхня гепатоцитів утворює численні мікроворсинки, що збільшують всмоктувальну поверхню клітин. Особливості будови стінок синусоїдів і зверненої до них плазмолеми гепатоцитів забезпечує вільний обмін між печінковою паренхімою та кров’ю. Гепатоцити і стінку мікросудини розділяє щілина — простір Діссе, що заповнений плазмою, у якій розміщені зірчасті ретикулоендотеліоцити. Підвищення тиску у венозній системі ретроградно поширюється від магістральних судин на печінкові, збірні та центральні вени, а потім і на синусоїди, викликаючи їх розширення. Проте останні дилатуються тільки в центральних і серединних відділах часточки, тому що ближче до її периферії збільшений венозний тиск врівноважується артеріальною кров’ю, що надходить у синусоїди з капілярів печінкової артерії.
Розширення мікросудин в центрі печінкової часточки супроводжується крововиливами, тоді як у зоні злиття синусоїдів і артеріальних капілярів відзначають тільки набряк. У цих умовах відбувається активування адвентиціальних фібробластів центральних і збірних вен, проліферація клітин синусоїдів, що набувають здатності активно продукувати склеропротеїни. Застій крові при хронічній венозній гіперемії порушує циркуляцію в лімфатичній системі, в судинах і капілярах якої накопичується багата білками та метаболітами лімфа. Утруднення лимфовідтоку і, отже, звільнення внутрішньотканинного середовища від надлишку рідини, що містить продукти тканинного обміну, потенціює гіпоксію і сприяє новоутворенню сполучної тканини в печінці та інших органах.
У міру прогресування процесу в печінці розростається сполучна тканина, що веде до склерозу центральних і збірних вен, портальних трактів, утворенню суцільної базальної мембрани в синусоїдах, вузликової гіперплазії збережених печінкових клітин. Така глибока перебудова судинного русла та стромально-паренхіматозних співвідношень, що супроводжується деформацією органа, визначається як мускатний (серцевий) цироз печінки.
У легенях хронічна венозна гіперемія призводить до так званої бурої індурації. Легені збільшуються в розмірах, їх тканина ущільнюється, набуваючи на поверхні розрізу буруватого забарвлення. Посилюється малюнок міждолькових і міжсегментарних прошарків фіброзної строми, яка не містить капілярів, що пов’язано зі склерозом, зумовленим застоєм лімфи. Склерозовані міжсегментарні прошарки визначаються на рентгенограмі (лінії Керлі). У термінальній стадії процесу, коли розвивається інтерстиціальний і альвеолярний набряк, із поверхні розрізу легеневої тканини рясно стікає піниста рідина, змішана з темною венозною кров’ю. Буре забарвлення легень пов’язане з явищами гемосидерозу — накопичення буро-коричневого пігменту гемосидерину, що утворюється в макрофагах при численних діапедезних крововиливах, а ущільнення тканини являє собою наслідок прогресування фіброзу легеневої тканини.
Бура індурація легень відзначається при мітральній ваді серця, особливо при звуженні лівого AV-отвору, а також при поширеному кардіосклерозі. Їй передує ряд компенсаторно-пристосувальних реакцій з боку артеріального і венозного русла легень, спрямованих на попередження переповнення кров’ю капілярів і легеневих альвеол і порушення функції аерогематичного бар’єра. На початкових етапах процесу це досягається скороченням сфінктерів легеневих вен, що попереджає зворотний закид крові з правого передсердя в легеневі вени, а також заповненням депо великого кола кровообігу, що зменшує масу крові, яка притікає до ПШ серця. Потім виникає спазм дрібних легеневих вен. Паралельно через барорецептори розтягнутих кров’ю устів легеневих вен і лівого передсердя передається імпульс скорочення на артеріоли і дрібні гілки ЛА (веноартеріальний рефлекс Китаєва). Адаптивна веноартеріальна реакція веде до гіпертрофії м’язової оболонки легеневих вен і ЛА, які перебудовуються за типом замикаючих артерій.
Згодом гіпертрофія гладком’язових елементів стінки скорочених судин доповнюється явищами ангіосклерозу. Міоеластофіброз знижує реактивність артерій, артеріол і дрібних вен, обмежуючи їх здатність оберігати капіляри легень від різкого переповнення кров’ю. Настає декомпенсація легеневого кровообігу, і капіляри міжальвеолярних перегородок переповнюються кров’ю. Стінки розтягнутих капілярів вибухають в альвеоли, їх проникність підвищується. Плазма і еритроцити виходять у просвіти альвеол і в міжальвеолярний інтерстицій, еритроцити руйнуються і поглинаються макрофагами, що перетворюють гемоглобін у гемосидерин, який після розпаду макрофагів виявляється в легеневій тканині.
Білки плазми крові, продукти клітинного розпаду, гемосидерин надходять у лімфатичне русло легень, порушуючи його дренажну функцію. У результаті до розладів кровообігу поступово приєднується лімфостаз і лімфатичний набряк проміжної тканини легень. Наростаюча гіпоксія тканин активує фіброцити, які у великій кількості присутні в міжальвеолярних перегородках. Фіброцити трансформуються у фібробласти, довгі сплощені відростки яких проникають у заповнені набряковою рідиною щілини між кровоносними капілярами й стінкою альвеоли. Біля тіл відростків фібробластів із склеропротеїнів, що ними секретуються, формуються колагенові волокна.
Склерозування стінок кровоносних судин, капілярів і міжальвеолярних перегородок разом із набряком ще більше погіршує умови газообміну. Залежно від ступеня і тривалості венозного застою явища склерозу і гемосидерозу в легенях набувають генералізованого характеру. Значні скупчення вільних пігментів гемосидерину та ферритину і навантажених ними сидерофагів виявляються не тільки в міжальвеолярних перегородках та альвеолах, але й по ходу лімфатичних судин і у лімфовузлах. Сідерофаги, що з’являються в мокротинні, визначають як «клітини серцевої вади».
Бура індурація легень призводить до прогресуючого погіршення гемодинаміки в малому колі кровообігу, погіршуючи перебіг основного захворювання.
Нирки при хронічному загальному венозному застої не тільки збільшуються в розмірах, але й ущільнюються, стають ціанотичними. Порушення кровообігу у великому колі призводить до спазму ниркових артеріол, що обмежує клубочкову фільтрацію. У результаті здатність нирок екскретувати натрій і воду знижується, а об’єм плазми крові і тканинної рідини в організмі збільшується, що ще більше погіршує кровообіг і тканинний обмін. На фоні застійного повнокров’я і розширення ниркових вен розвивається лімфостаз і набряк строми. Її набряк найбільш виражений у мозковому шарі, де основна речовина проміжної тканини особливо багата глікозаміногліканами. Клубочки також трохи збільшені і повнокровні. Позаклітинні простори в області тубулярних елементів нефрону розширені внаслідок набряку і вторинного порушення реабсорбції рідини. В умовах наростаючої гіпоксії виникає дистрофія епітелію канальців. На цій стадії процесу можливе огрубіння базальної мембрани і розвиток склерозу, що призводить до альбумінурії.
Індурація селезінки при венозній гіперемії пов’язана із застоєм крові, набряком і склерозуванням строми, а також з відносною непідатливістю фіброзної капсули органа. При хронічній венозній гіперемії капсула селезінки напружена, потовщена, пульпа її ущільнена, поверхня зрізу тканини темно-коричнево-червоного кольору. Мікроскопічно виявляють розширення щілин пульпи та синусів, зумовлене повнокров’ям, збільшенням маси і огрубінням волокнистих структур строми.
В інших органах (ЦНС, ШКТ, ендокринні залози) венозне повнокров’я індукує описаний вище стереотипний комплекс змін: переповнення вен, ціаноз, порушення лімфовідтоку і набряк тканини, плазмо- і геморагії, дистрофічні зміни і ураження клітин паренхіми, прогресуюче склерозування строми.
Місцеве венозне повнокров’я виникає при утрудненні відтоку крові від будь-якої частини тіла або органа. До найбільш частих причин місцевого повнокров’я відносяться звуження і обтурація просвіту вени при тромбозі, хронічному запальному процесі в стінці судини, здавлюванні вени ззовні пухлинною, що розростається або рубцевою тканиною, а також накладення пов’язки, гіпсу, лігатури. Як і при загальному венозному застої, вени, що знаходяться дистальніше перешкоди, розширені, застійні ділянки тіла ціанотичні, температура їх знижена. В основі патологічних змін, що розвиваються безпосередньо в області регіонального венозного застою, лежать принципово ті ж закономірності і механізми, що описані вище. Так, мускатна печінка і її цироз можуть бути наслідком облітеруючого тромбофлебіту печінкових вен (синдром Бадда — Кіарі), а ціанотична індурація нирок або селезінки — тромбозу основної магістральної вени відповідного органа. Іноді місцеве венозне повнокров’я виникає внаслідок формування венозних колатералей, що вимагає значного часу і можливо тільки при поступовому обмеженні відтоку крові по вені. У тих ситуаціях, коли блокада венозного стовбура розвивається швидко, можливе використання тільки раніше існуючих шляхів відтоку крові.
Основними наслідками венозної гіперемії є розвиток циркуляторної гіпоксії, накопичення в інтерстиції білків і плазми, продуктів порушеного обміну та деструкції тканинних структур, які активують процеси фібрилогенезу і призводять до розвитку органосклерозу. Вторинні ускладнення, пов’язані із загальною і місцевою венозною гіперемією, — зниження функціональних можливостей органів, схильність до хронічних запальних процесів. При компенсаторному формуванні колатералей переповнені кров’ю вени різко розширюються, стінка їх стоншується. В умовах хронічного перебігу процесу поєднання атрофії і гіпертрофії гладком’язових елементів венозної стінки в сукупності з перебудовою її сполучнотканинного каркаса призводить до недостатності венозних клапанів, появи варикозних вузлів, а іноді і до небезпечних кровотеч. У нижніх кінцівках, у венах таза подібні зміни сприяють тромбоутворенню (флеботромбоз) і запаленню (флебіт), які часто поєднуються (тромбофлебіт). Прикладом компенсаторного венозного повнокров’я є колатералі, що утворюються при застої крові у воротній вені внаслідок цирозу печінки.
Ішемія
Ішемією називають патологічний стан, зумовлений обмеженням припливу артеріальної крові. Незважаючи на різноманіття безпосередніх причин, що викликають ішемію, прийнято розрізняти наступні її види: ангіоспастичну, компресійну, пов’язану з перерозподілом крові.
Ангіоспастична (рефлекторна) ішемія зумовлюється порушенням вазомоторних центрів і судинозвужувальних нервів, а також дією гуморальних факторів, що циркулюють у крові або утворюються у судинній стінці. Велике значення в розвитку ангіоспастичної ішемії має стан судинного ендотелію, що продукує фактори як констрикції, так і розслаблення судинної стінки, а ураження ендотеліального моношару порушує судинну реакцію на гуморальні впливи. Неадекватне кровопостачання тканини може бути також пов’язане з порушенням розслаблення гладких м’язів артерій, що веде до їх тривалої констрикції.
Обтураційна ішемія — наслідок звуження або закриття просвіту артерії при запальному процесі (облітеруючий ендартеріїт), емболізації, утворенні масивної атеросклеротичної бляшки, тромбу. У механізмах обтураційної ішемії моментом, що сприяє процесу, є збільшення в’язкості крові внаслідок посиленої агрегації її формених елементів або підвищення її зсілості.
Компресійна ішемія розвивається при рубцюванні тканини, стисканні артерії джгутом, лігатурою, скупченням рідини, пухлиною, що розростається.
Ішемія в результаті перерозподілу крові виникає під впливом факторів, які викликають гіперемію в інших ділянках тіла. Наприклад, така причина сонливості після прийому їжі або непритомного стану внаслідок швидкого видалення значного об’єму рідини або масивної пухлини з черевної порожнини. У цих випадках до раніше стиснених тканин і органів спрямовується велика маса крові з одночасним зменшенням кровотоку в судинах головного мозку, шкіри та інших ділянок тіла.
Морфологічні зміни в органах при ішемії виникають внаслідок порушення мікроциркуляції і розвитку гіпоксії. Макроскопічно органи зменшені в розмірах, гіпотермічні, в’ялі, зі зморщеною капсулою. Тканина на розрізі в зоні ішемії тьмяна, більш світла, ніж ділянки, що отримують артеріальну кров.
Залежно від інтенсивності ішемізації тканини, її чутливості до гіпоксії, тривалості останньої зміни можуть розвиватися або на рівні мікроструктур, або у вигляді грубих уражень аж до формування інфаркту. Макроскопічно виявляють компенсаторну дилатацію мікросудин, порушення кровотоку в них, підвищення судинної проникності, ознаки порушення обмінних процесів у паренхіматозних клітинах, деструктивні зміни різних елементів тканини. У стромі органів відзначають набряк, накопичення і перерозподіл глікозаміногліканів, гомогенізацію та руйнування аргірофільних і колагенових волокон.
Ішемія внаслідок спазму артерій зазвичай нетривала і ліквідується без істотних наслідків. У тих випадках, коли ангіоспазм стає затяжним, а тканинний метаболізм характеризується високою потребою в кисні (як наприклад у серцевому м’язі або головному мозку), розвивається інфаркт.
Найбільш часто до тяжких наслідків призводить обтураційна ішемія, що гостро виникає. Її наслідки залежать і від регіонарних особливостей тканини. Якщо відповідна ділянка багато васкуляризована і її кровопостачання здійснюється не однією магістральною судиною, а декількома артеріальними гілками, то передумови для розвитку ішемії зменшуються. Крім того, чим швидше розвивається ішемія, тим менше можливостей для реалізації компенсаторних механізмів, зокрема відкриття вже наявних колатералей і формування нових. При поступовому зменшенні прохідності судини можливості для відновлення кровопостачання тканини за колатералями істотно зростають і наслідки такої ішемії можуть бути незначні. Помірна ішемія, що зберігається тривалий час і не призводить до необоротних некротичних змін тканини, поступово призводить до атрофії паренхіми і склерозу.
Інфаркт
Інфаркт — вогнище некрозу, що розвинулося внаслідок порушення кровообігу. Інфаркт називають також циркуляторним, або ангіогенним некрозом. Термін «інфаркт» (від лат. нафарширувати) був запропонований Вірховим для форми некрозу, при якій змертвіла ділянка тканини просочується кров’ю. Розміри та морфологічні особливості інфаркту визначаються калібром обтурованої судини, наявністю інших порушень кровообігу, на фоні яких він розвивається. При магістральному типі розгалуження артерії інфаркт за своїми контурами нагадує конус, вузька частина якого (вершина) звернена до воріт органа, а основа орієнтована на периферію, до зони термінального розгалуження внутрішньоорганних артерій. Інфаркти такої форми зазвичай виявляють у селезінці, нирці, легенях. В органах з переважанням розсіяного типу розгалуження артерії, наприклад у головному мозку, кишечнику, серці, територія, що постачається кров’ю цієї артерії, не утворює конусоподібних контурів, при цьому інфаркти не мають певної форми.
Види інфаркту
Зона інфаркту може займати весь орган або більшу його частину (тотальний і субтотальный інфаркт) або виявлятися тільки під мікроскопом (мікроінфаркт). За макроскопічними ознаками розрізняють 3 види інфаркту: білий, білий з геморагічним вінчиком і червоний.
Білий (ішемічний) інфаркт формується при непрохідності магістрального артеріального стовбура і запустінні всього судинного русла в його басейні внаслідок недостатнього розвитку судинних анастомозів і колатералей. Найчастіше виявляють у селезінці, іноді в головному мозку, печінці. Зону некрозу добре видно при макроскопічному обстеженні приблизно через 24 год після порушення кровопостачання. Під мікроскопом тканина ущільнена, блідо-жовтого забарвлення, структура тканини не визначається, а утворюючі її елементи зливаються в гомогенну масу. По периферії зона інфаркту відмежовується запальним демаркаційним валом.
Білий інфаркт із геморагічним вінчиком виглядає як ділянка білувато-жовтого кольору, оточена темно-червоною зоною крововиливів. Такий інфаркт розвивається у випадках, коли компенсаторному включенню колатералей і реактивної артеріальної гіперемії судин периферичної зони передує ангіоспазм, що змінюється паралітичним розширенням. У результаті різке повнокров’я судин супроводжується явищами стазу крові і діапедезними крововиливами в некротизовану тканину. Білий інфаркт із геморагічним вінчиком розвивається в серці, селезінці, іноді в нирках.
Червоний (геморагічний) інфаркт зазвичай виявляють у легенях, що пов’язано з особливостями їх кровопостачання. Іноді геморагічний інфаркт виникає на фоні вираженої гіперемії в інших органах: кишечнику, головному мозку, нирках. При червоному інфаркті зона ішемії просочується кров’ю, набуває темно-червоного кольору і чітких меж. Цей ефект виникає, якщо слідом за перекриттям просвіту артерії периферичні судини змертвілої тканини переповнюються кров’ю, що надійшла по колатералям. При венозному застої ретроградне надходження крові з вен у зону ішемії також веде до просочування некротизованої тканини кров’ю.
Геморагічний інфаркт може розвиватися також у результаті вираженого венозного застою при швидкому припиненні відтоку крові по великих венозних стовбурах або одночасному вимиканні з кровотоку великої кількості дрібних вен. Венозні застійні інфаркти виявляють у селезінці при тромбозі вени, що відводить від неї кров, у головному мозку — при порушенні прохідності синусів твердої мозкової оболонки або яремних вен, у серці — при обтурації коронарного синуса тромботичними масами, у тканинах нижніх кінцівок — при перев’язці стегнової вени. Мікроскопічно у вогнищі геморагічного інфаркту відзначають маси гемолізованих еритроцитів, що інфільтрують некротизовану тканину.
Загальні закономірності формування і загоєння інфаркту
Стадія ішемії і некрозу. Розвитку інфаркту передує ішемія. Перші зміни, що зумовлені порушенням кровопостачання, визначаються порушенням тканинного дихання, компенсаторною активацією анаеробного гліколізу, швидким накопиченням метаболітів у клітинах у токсичних концентраціях. Недостатнє відтворення енергії і гістотоксичний ефект ішемії порушують електролітний гомеостаз клітин і пригнічують пластичні процеси, що призводить до прогресуючої дисоціації цитомембран, закислення внутрішньоклітинного середовища, денатурації білків, загибелі та руйнування клітин.
Мікроскопічно при ішемізації виявляють внутрішньоклітинний набряк або, навпаки, дегідратацію цитоплазматичного матриксу. Органели клітин набухають, їх мембрани гомогенізуються і фрагментуються, гранули лабільного глікогену зникають, визначається накопичення ліпідів у вигляді крапель внаслідок їх вивільнення з дисоціюючих фосфоліпідів цитомембран і порушення ліпідного обміну. В лізосомах накопичуються продукти внутрішньоклітинного розпаду. Відбуваються перерозподіл, конденсація або вимивання ядерного хроматину і руйнування ядерець, розплавлення цитоплазматичних рибосом та органел немембранної структури.
Гістохімічно і біохімічно в ішемізованій тканині визначається зниження рівня макроергічних фосфатів, активності окисно-відновних ферментів, нагромадження недоокислених метаболітів, порушення обміну електролітів, зменшення вмісту глікогену, РНК і ДНК, а згодом — накопичення продуктів розпаду стромальних структур. На некротичній стадії інфаркту при мікроскопічному дослідженні ядра клітин не фарбуються, всі структурні елементи тканини зливаються в однорідну масу.
Стадія репаративних змін настає після формування некрозу. По периферії інфаркту завжди існує зона дистрофічних змін і реактивного запалення — так званий демаркаційний вал. Мікроскопічно запальна реакція відзначається вже через декілька годин, а максимум її розвитку доводиться на 3-тю–5-ту добу. Запалення в зоні демаркаційного валу супроводжується виходом формених елементів крові з капілярів. Поступово некротичні маси частково розплавляються під дією протеолітичних ферментів, що вивільнюються з нейтрофільних лейкоцитів, частково піддаються фагоцитозу або резорбуються лімфатичною системою і виводяться її судинами.
Організація зони некрозу — заміщення некротичних мас сполучною тканиною, що вростає з боку демаркаційного валу й до 7–10-ї доби трансформується в грануляційну (юну) сполучну тканину, а згодом дозріває в рубцеву.
Особливості розвитку інфаркту в різних органах
Морфологія інфаркту багато в чому залежить від органної архітектоніки судинної системи. У клінічній практиці найчастіше визначають інфаркт серця (міокарда), головного мозку, кишечнику, легень, нирок і селезінки. Час, необхідний для розвитку інфаркту в різних органах, неоднаковий і залежить від функціональних енерговитрат та філогенетично сформованого метаболізму, що визначає потреби тканини в забезпеченні киснем.
Для розвитку ІМ досить повного припинення його кровопостачання на 20–25 хв, проте ішемія тривалістю 5 хв уже призводить до загибелі окремих м’язових клітин. У реальному житті формування інфаркту серцевого м’яза потребує дещо більшого проміжку часу, тому що в зоні ішемії завжди частково зберігається кровотік по судинним анастомозам і колатералям. Він недостатній для того, щоб повністю запобігти некрозу, але дещо збільшує період його розвитку і обмежує розміри.
Інфаркт зазвичай локалізується в ЛШ, найчастіше в передній стінці. За типом це білий інфаркт із геморагічним вінчиком, що має неправильну форму. Залежно від обсягу та локалізації ураженої тканини міокарда розрізняють дрібно- і великовогнищевий, субепікардіальний, інтрамуральний, субендокардіальний і трансмуральний ІМ, що охоплює всі шари серцевої стінки. В зоні переходу інфаркту на епікард або ендокард розвивається реактивне запалення, яке в першому випадку призводить до фіброзного перикардиту (випіт у порожнину перикарда плазми крові, збагаченої фібрином, і утворення фібринозних нашарувань на епікарді), у другому — до тромбоендокардиту (пристіночний тромбоз відповідно до зони інфаркту).
Формування ІМ починається з ішемічної стадії. Поряд із прогресуючим порушенням метаболізму й дезінтеграцією клітинних мембран відзначають фрагментацію, розтягнення і дезінтеграцію міофібрил кардіоміоцитів. У результаті знижується активність внутрішньоклітинних ензимів, змінюється характер фарбування клітин при використанні основних або кислотних гістологічних барвників, порушується здатність клітин до променезаломлювання в поляризованому світлі і люмінесцентно-мікроскопічні властивості. Ці явища використовують для ранньої діагностики метаболічних та ішемічних ушкоджень серця.
Гістологічні ознаки загибелі клітин — зморщування, набряк і руйнування клітинного ядра, зникнення поздовжньої і поперечної смугастості; гомогенізацію саркоплазми виявляють через 12 год (рис. 2.1).
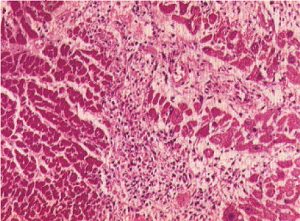 Рис. 2.1. Гострий ІМ
Рис. 2.1. Гострий ІМ
Паралельно з деструктивними змінами робочих клітин міокарда відбувається судинно-тканинна реакція, що характеризується спазмуванням і паретичною дилатацією інтрамуральних артерій та артеріол, плазматичним просочуванням і підвищенням проникності їх стінок, а також порушенням мікроциркуляції з внутрішньосудинною агрегацією еритроцитів, набряком інтерстицію.
При розвитку некрозу кровотік у некротичній зоні припиняється, а в периінфарктній збільшується. Поряд з діапедезними крововиливами в ній відбувається екстравазація лейкоцитів і формується лейкоцитарний вал. У товщі некротичної зони навколо збережених судин іноді виявляють острівці життєздатної тканини, по периферії яких визначають такі ж явища, як і в зоні навколо інфаркту.
Протягом перших 18–24 год від початку патологічного процесу міокард у басейні ураженої артерії відрізняється блідістю на фоні підкреслено нерівномірного кровонаповнення навколишньої тканини. У кінці 1-ї доби ділянка некрозу стає помітна макроскопічно. У зв’язку з безперервною діяльністю серця, високою активністю ферментів, що виділяються з лейкоцитів, на 3-тю–5-ту добу починається розм’якшення (міомаляція) загиблої тканини. Поступове розсмоктування (резорбція) некротизованої маси здійснюється при активній участі мікрофагальних клітин, які з’являються на 4-й день ззовні від лейкоцитарного валу.
Фібропластична реакція інтерстицію також виникає на 4–5-ту добу, а перші волокнисті елементи новоствореної сполучної тканини в зоні інфаркту з’являються ще через 3 доби. Протягом наступного тижня зона некрозу являє собою м’язові волокна, що розпадаються, просочені набряковою рідиною та інфільтровані лейкоцитами, що розпадаються. По її периферії і навколо периваскулярних острівців збереженого міокарда відбувається новоутворення сполучної тканини.
Процес організації триває 2–2,5 міс. У подальшому сполучна тканина, що утворилася на місці некротичних мас, ущільнюється, її судини спустошуються і облітеруються, на місці некрозу утворюється рубець (рис. 2.2).
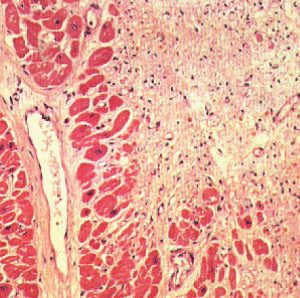
Рис. 2.2. Постінфарктний кардіосклероз
Провідна система серця більш стійка до гіпоксії порівняно з робочим міокардом і здатна довше зберігатися в зонах ішемії, що важливо для відновлення ритмічної роботи серця після екстреної інвазивної антиішемічної терапії.
У нирках зазвичай розвивається білий інфаркт із геморагічним вінчиком. Внаслідок хорошого розвитку судинних анастомозів і колатералей інфаркт виникає тільки при порушеннях прохідності судин більшого калібру, ніж долькова артерія. Характерне розміщення інфаркту — передня латеральна поверхня органа, тому що в цій зоні ниркові артерії розгалужуються не за магістральним, а за розсіяним типом, при якому міжсудинні колатералі виражені значно слабше. Зазвичай інфаркт нирки нагадує за формою конус, повернений основою до капсули, верхівкою до ниркової балії. Однак іноді процес обмежується тільки корою, не зачіпаючи пірамідки, і ураження наближається за формою до квадрата.
Інфаркт нирки часто супроводжується гематурією внаслідок потрапляння крові в сечові канальці при розриві дрібних судин. Ішемічна стадія інфаркту нирки розвивається за загальними закономірностями. Некроз усіх структур ниркової паренхіми настає через 24 год, проте ураження епітелію ниркових канальців виникає значно раніше. Так, уже через 6 год відзначається загибель епітелію звивистих, а через 12 год — прямих канальців нефрону. До цього ж часу по периферії інфаркту розвивається реактивне запалення, що досягає свого максимуму приблизно до 3-ї доби процесу.
Формування демаркаційної зони супроводжується порушеннями кровотоку в мікросудинах, явищами набряку, плазморагіями та діапедезними крововиливами, активною міграцією лейкоцитів. Це призводить через добу до утворення периферичної геморагічної зони інфаркту та лейкоцитарного валу. Приблизно з цього ж часу з’являються макрофаги і починається процес резорбції некротичних мас. На 7-му добу деструктивно-резорбтивні процеси поєднуються з чітко вираженими явищами організації, що через кілька тижнів завершується утворенням щільного сполучнотканинного рубця, рідше — кісти.
У селезінці звичайний морфологічний тип інфаркту — білий (ішемічний). В умовах вираженого венозного застою інфаркт селезінки може бути геморагічним, який набуває протягом декількох діб сірого або білого забарвлення. Ішемічний інфаркт селезінки конічної форми, блідо-жовтого забарвлення. На поверхні капсули органа в області широкої частини цього конуса, а також на межі зони інфаркту розвиваються реактивне запалення, процеси лізису, резорбції та організації некротичних мас. Безпосередньо в зоні некрозу спочатку руйнується червона пульпа, потім фолікули і трабекули. Організація інфаркту здійснюється за загальними закономірностях. Дозрівання постінфарктного рубця супроводжується деформуванням селезінки.
Інфаркт головного мозку в 85–90% випадків є білим, в інших — червоним або змішаного характеру. Білий інфаркт може вражати будь-які відділи мозку. Спочатку це нечітко відмежована ділянка в’ялої або крихкої консистенції сіро-червоного кольору, зі стертим природним малюнком базальних вузлів або кори головного мозку. Геморагічні інфаркти у вигляді невеликих вогнищ червоного кольору локалізуються переважно в межах скупчень сірої речовини, найчастіше в корі. Змішані інфаркти складаються з білих і червоних ділянок, причому останні розташовуються в сірій речовині.
Топографія різних морфологічних типів інфарктів мозку визначена особливостями кровопостачання його різних ділянок. Найчастіше вони виникають у басейні середньої мозкової артерії, рідше — скелетних і базилярних артерій. Геморагічні інфаркти формуються в добре васкуляризованих зонах — скупченнях сірої речовини або в корі головного мозку.
Розвиток інфаркту головного мозку включає ішемічну та некротичну стадії. Ішемічна стадія характеризується дистрофічними змінами нервової тканини, крововиливами і деструкцією клітинних мембран з необоротною дезорганізацією обмінних процесів і електролітного гомеостазу нервових клітин. При мікроскопічному дослідженні визначають лізис глибок базофільної речовини, просвітління цитоплазми, гіперхроматоз і деформацію ядра. У результаті нервові клітини та їх ядра набувають кутастої форми, а цитоплазма гомогенізується, втрачає базофільні включення і просвітлюється. Порушення циркуляції крові в мікросудинах поєднується з перицелюлярним набряком — появою світлого проміжку між капілярною стінкою або тілом нейрона і навколишньою тканиною. Навколо капілярів відзначають набряк відростків гліальних клітин, що їх оточують.
Некротична стадія інфаркту — стадія наростаючого аутоліза ішемізованої тканини мозку. Загибелі нейронів передує їх різке просвітління або ущільнення і перетворення в пікноморфні (ущільнені дегідратовані) клітини, а потім і в гомогенну безструктурну масу. Разом з нейроцитами в деструктивні зміни залучаються і клітини глії. З дрібних судин відбуваються діапедезні крововиливи, невеликі і поодинокі у вогнищах білого інфаркту, численні і такі, що зливаються між собою при геморагічному інфаркті.
До початку 2-ї доби починається резорбція некротизованої нервової тканини. На межі з вогнищем ішемічного ураження накопичуються лейкоцити. Разом з ними в зону некрозу проникають численні активовані астроцити і з’являються зернисті кулі з ліпідними включеннями. Частина астроцитів втрачає цитоплазматичні відростки, в їх цитоплазмі виявляються численні фібрили, що набувають здатності до утворення волокнистих структур. Навколо вогнища некрозу починається новоутворення судин, капілярів і судинних петель.
В організації некротичних мас беруть участь як гліальні, так і сполучнотканинні клітини — фібробласти. Проте в кінцевій стадії процесу при невеликих розмірах інфаркту продукти мезодермальної проліферації повністю витісняються гліоволокнистими структурами, що утворюють рубець. У великих вогнищах серединна зона інфаркту, що організувався, залишається сполучнотканинною, а в центрі рубця, що сформувався, утворюється одна або кілька порожнин, ззовні оточених розростаннями глії.
Інфаркт легень, як правило, має геморагічний характер, причиною чого є подвійне кровопостачання легень і венозний застій. Кров попадає в легені як по бронхіальним артеріям, що входять в систему великого кола кровообігу, так і по артеріям малого кола кровообігу. Між ЛА і бронхіальними артеріями існують численні анастомози, які мають будову артерій замикаючого типу і у звичайних умовах не функціонують. При обтурації досить великої гілки ЛА в її басейн під великим тиском спрямовується кров з бронхіальних артерій по анастомозам, що відкрилися рефлекторно.
Легеневі капіляри, що переповняються кров’ю, різко дилатуються, їх стінки розриваються, кров виливається в інтерстицій альвеолярних перегородок і порожнини альвеол, що імбібує відповідну ділянку тканини. Завдяки автономному артеріальному кровопостачанню бронхи в зоні інфаркту зберігають життєздатність. Нерідко геморагічний інфаркт у легені розвивається на фоні хронічної венозної гіперемії, так як підвищення тиску у великих венах сприяє ретроградному надходженню крові в зону інфаркту.
Інфаркт найчастіше розвивається в периферичних зонах середніх і нижніх відділів легень. При цьому макроскопічно виявляють вогнища більш щільної консистенції, ніж навколишня тканина, конусоподібної форми, основою повернені до плеври, на якій з’являється фібринозний наліт і гіперемія внаслідок реактивного запалення. На розрізі некротизована тканина темно-червоного кольору, злегка зерниста, вибухає над поверхнею.
У 1-шу добу в зоні інфаркту мікроскопічно визначаються набряк і крововиливи у вигляді скупчень частково гемолізованих еритроцитів в інтерстиціальній тканині, в просвітах альвеол і дрібних бронхів, що супроводжується кровохарканням. Потім приєднуються ознаки некрозу стінок альвеол, накопичуються сідерофаги. На 3-тю–4-ту добу інфаркт являє собою гомогенізовану масу із зруйнованих еритроцитів, на фоні яких видно сліди некротизованих альвеолярних перегородок. Розплавлення некротизованої тканини і крові, що вилилася, їх резорбція і організація починаються з периферії і від збережених периваскулярних і перибронхіальних зон. Через 2–8 міс на місці інфаркту залишається рубець або кіста.
Білий інфаркт у легені виявляють рідко. Виникає при порушенні кровотоку в бронхіальних артеріях на фоні утруднення капілярного кровотоку, наприклад внаслідок стискання внутрішньоальвеолярним ексудатом або при ущільненні (гепатизації) легеневої тканини, зумовленого пневмонією.
У кишечнику інфаркт розвивається за типом геморагічного. Найбільш характерна локалізація — басейн верхньої брижової артерії, що у зв’язку з великою довжиною частіше піддається обструкції. Макроскопічно інфаркт кишки має вигляд темно-червоної ділянки, що досить чітко відмежований від неураженого кишечнику. Серозна оболонка в області інфаркту кишки стає тьмяною, на ній з’являються фібринозні нашарування. Стінка кишки потовщена, слизова оболонка синюшна.
Некротичні і реактивні зміни в ішемізованому сегменті кишечнику розвиваються швидко. Через 15–20 хв після припинення кровопостачання в його стінці виявляють виражені мікроциркуляторні порушення: тотальний набряк тканини, уповільнення і припинення руху крові в різко розширених повнокровних капілярах і венулах, численні крововиливи. Через 30 хв у набряклій стромі слизової оболонки кишки з’являються лейкоцити, лімфоцити, розвивається макрофагальна реакція. Протягом 1–1,5 год у стінці кишки відбувається некроз, що починається з виразки її слизової оболонки.
У сітківці ока інфаркт має характер білого, який в умовах венозного застою трансформується в геморагічний. Ділянка ураженої тканини у вигляді конуса повернена верхівкою до зорового диска, зазвичай локалізується в скроневому сегменті. Мікроскопічно виявляють деструкцію внутрішніх шарів сітківки, гангліозних клітин і нервових волокон на фоні порушення мікроциркуляції, набряку та крововиливів.
Дуже рідко відзначають інфаркти в печінці, м’язах, кістках.
Наслідки інфаркту надзвичайно значимі для організму. Так, ураження при ІМ >30% тканини ЛШ супроводжується розвитком гострої СН із зупинкою серця. Ураження провідної системи серця при формуванні некрозу зумовлює тяжкі порушення ритму. При великому трансмуральному інфаркті іноді відбувається вибухання некротизованої ділянки серцевої стінки та її стоншення — розвивається гостра аневризма серця. У деяких випадках десинхронізація процесів міомаляції, резорбції некротичних мас і організації зони інфаркту призводить до розриву аневризми, заповнення порожнини перикарда кров’ю і летального результату. У результаті ІМ можуть виникати розриви міжшлуночкової перегородки, відрив папілярних м’язів, що також призводить до серйозних наслідків. У більш віддалені строки велика рубцева зона, змінюючи геометрію скорочення серця та внутрішньосерцеву гемодинаміку, сприяє розвитку ХСН і загальної венозної гіперемії.
Інфаркт головного мозку супроводжується його набряком, розладом мікроциркуляції та метаболічними порушеннями як у безпосередній близькості від вогнища ураження, так і у віддалених ділянках. Результат інфаркту визначається його розмірами, локалізацією і темпами розвитку патологічного процесу. Смерть таких хворих може бути зумовлена як самим вогнищем ураження в головному мозку, так і причинами, безпосередньо з ним не пов’язаними. Нерідко при повільному формуванні інфаркту хворі помирають не від деструктивних змін, що стосуються життєво важливих центрів головного мозку, а внаслідок СН, пневмонії та іншої патології, що приєднується і ускладнює перебіг інфаркту. Серйозним ускладненням інфаркту головного мозку є крововилив у розм’якшену тканину. Як і набряк мозку, так і збільшення його обсягу внаслідок відновлення кровотоку по судинам у зоні ішемії можуть викликати дислокацію та защемлення стовбура мозку. При успішному результаті на місці інфаркту формується рубець або кіста з більш-менш значними порушеннями функції ЦНС.
Інфаркт кишечнику вимагає обов’язкового хірургічного втручання, тому що кінцевою фазою його розвитку є гангрена із проривом кишкової стінки. Потрапляння вмісту кишечнику в черевну порожнину спричиняє розвиток перитоніту. Причиною перитоніту може стати також інфаркт селезінки, що зазвичай закінчується формуванням грубого рубця, який деформує орган.
Інфаркт легені зазвичай не несе безпосередньої загрози життю хворого. Проте його перебіг може ускладнитися постінфарктною пневмонією, нагноєнням і поширенням запального процесу на плевру з розвитком пневмотораксу та гангрени легені. Однією з найбільш характерних причин нагноєння інфаркту є попадання в судину гнійного ембола. Це викликає гнійне розплавлення тканини легені та утворення абсцесу на місці інфаркту.
При інфаркті нирок, що зазвичай загоюється рубцюванням відповідної ділянки, загрожуючі життю ускладнення виникають при нагноєнні або при великих ураженнях, особливо при симетричних некрозах коркового шару, наслідком яких може бути ГНН.
Стаз крові
Етіологія і патогенез. Стаз (від лат. зупинка) — місцеве припинення течії крові в мікроциркуляторному руслі, головним чином у капілярах, посткапілярах і венулах. Повній зупинці кровотоку в мікросудинах передує його різке уповільнення, що визначається як передстаз. Механізмами стазу крові є зменшення різниці тиску між проксимальними і дистальними відділами мікросудин і підвищення опору в мікросудинах. Залежно від причини розрізняють ішемічний, застійний і справжній капілярний стаз крові.
Зменшення артеріолярно-венулярного градієнта тиску крові призводить до уповільнення кровотоку в мікроциркуляторному руслі і розвитку ішемічного стазу. В основі зменшення артеріолярно-венулярного градієнта тиску можуть лежати також венозна гіперемія та підвищення тиску у венулярному сегменті мікрогемоциркуляторного русла. Справжній капілярний стаз пов’язаний з первинним підвищенням опору кровотоку в мікросудинах внаслідок порушення реологічних властивостей крові та появи перешкод у їх просвітах.
Реологічні властивості крові — в’язкість і плинність у мікросудинах визначаються головним чином агрегаційною здатністю її формених елементів, насамперед еритроцитів, здатних злипатися з утворенням «монетних стовпчиків» і поліморфних конгломератів (рис. 2.3).
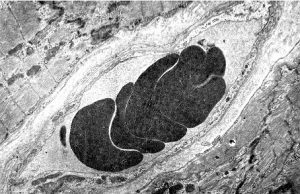
Рис. 2.3. Агрегація еритроцитів у кровоносній мікросудині міокарда
Агрегації еритроцитів сприяють фактори, що підвищують проникність судинної стінки. Вихід у тканину рідини та дрібнодисперсних альбумінів підвищує концентрацію глобулінів і фібриногену в плазмі крові, а також полегшує надходження у венулярні відділи продуктів порушеного метаболізму і біологічно активних речовин із тканин. Агрегація еритроцитів посилюється при зниженні поверхневого потенціалу їх цитолеми і при уповільненні швидкості руху крові в капілярах.
Морфологічна картина. Гістологічно стаз проявляється наявністю розширених мікросудин, заповнених однорідною масою з формених елементів крові, що злиплися. Проте на електронограмах еритроцити ще якийсь час зберігають чіткі контури (див. рис. 2.3), а згодом границі між ними стираються внаслідок гемолізу і процесів згортання крові. Це пояснює оборотність змін при нетривалій зупинці крові в мікросудинах, тоді як стійкий стаз призводить до утворення гіалінових тромбів.
Результати. Стаз крові — явище неспецифічне і виникає під впливом причин загального та місцевого характеру — при патологічних процесах, що супроводжуються інтоксикацією і порушенням кровообігу, при фізичних (термічних, променевих, вібраційних), хімічних (кислоти, луги), біологічних (переливання несумісної групи крові) впливах. Минущі стази, особливо в тканинах, малочутливих до гіпоксії, можуть завершуватися без яких-небудь істотних наслідків. При відстроченому завершенні стазу, коли приплив артеріальної крові відновлюється, але ще зберігається гіпоксія тканин і судин, виникають численні крововиливи шляхом діапедезу і розриву мікросудин. Результатом стазу крові, що тривалий час зберігається в тканинах з високим споживанням кисню, є некробіоз і некроз.
Тромбоз
Тромбозом (від грецького trombosis — згортання) називають прижиттєве порушення природного стану крові в просвітах судин або в порожнинах серця з утворенням згустку, який називається тромбом. В основі тромбозу лежить фізіологічна здатність крові до згортання (гемостазу) при ушкодженні судинної стінки, що є найважливішою захисною реакцією організму, яка зупиняє кровотечу.
При внутрішньосудинному згортанні лімфи також формуються тромби, проте закономірності лімфотромбозу істотно відрізняються. Збереження рідкого стану крові забезпечується антигемостатичними властивостями інтактного ендотелію судин, а також функціональною збалансованістю систем, одна з яких здійснює згортання крові, друга перешкоджає цьому, третя сприяє розчиненню тромбу, що утворився. Завдяки взаємодії цих систем, яка постійно координується нервовою та ендокринною системами, умови для утворення тромбу в нормі відсутні.
Судинна стінка і гемостаз. Інтактний ендотеліальний моношар виконує роль атромбогенного бар’єра між стінкою судини і кров’ю, що циркулює, перешкоджає згортанню крові та тромбоутворенню. Він синтезує і катаболізує метаболіти, що регулюють взаємодію формених елементів крові і факторів гемостазу, що містяться в плазмі та судинній стінці. Атромботичні властивості ендотелію забезпечуються насамперед його глікокаліксом — пристінковим шаром глікопротеїдів, насичених глікозаміногліканами і сіаловими кислотами. Разом з полярними фосфоліпідами плазмолеми ендотеліоцитів вони надають внутрішній поверхні судинної стінки негативний потенціал, такий самий, як і у формених елементів крові. Атромбогенність ендотелію посилюється здатністю кумулювати на поверхні комплекс біологічно активних речовин, що надходять із тканини й елімінуються з крові.
Тромборезистентність ендотелію визначається цілою низкою факторів. Одним із них є зв’язування та активація антитромбіну III, що інгібує тромбін та інші фактори згортання, до яких належать гепаринсульфати, наявні в глікокаліксі ендотеліоцитів, і білок тромбомодулін, що інгібує тромбін та інші фактори коагуляційного каскаду. До факторів тромборезистентності ендотелію належить активація комплексом тромбін — тромбомодулін системи С-протеїну, потужного антикоагулянтного комплексу, що інгібує циркулюючі в крові фактори згортання V–VIII. При цьому протеїн С блокує інгібітор тканинного активатора плазміногена, що посилює фібриноліз. Ендотеліоцити здійснюють також секрецію активаторів плазміногена тканинного і сироваткового (урокіназного) типів, синтез і виділення простацикліну і оксиду азоту (NO) — високоефективних антиагрегантів тромбоцитів і вазодилататорів.
Прокоагулянтні властивості клітин ендотелію пов’язані з вивільненням фактора Віллебранда — макромолекулярного білка, що синтезується і накопичується в специфічних органелах (тільця Вейбела — Палладе). Фактор Віллебранда зв’язує і переносить регуляторний білок — плазмовий фактор VII, а також є рецептором для глікопротеїнів поверхні тромбоцитів. Крім того, ендотеліоцити виділяють тканинний тромбопластичний фактор (фактор ІІІ), стимулятори агрегації тромбоцитів і вивільнення ними біологічно активних речовин.
При ураженні та відторгненні ендотеліоцитів оголюється субендотелій судинної стінки, що активно зв’язує білки плазми і тромбоцити, провокуючи тромбоутворення. У структуру субендотелію входять різні типи колагену, еластин, глікопротеїни і глікозаміноглікани, фібронектин, ламинін, тромбоспондин, що асоціюються з фібриногеном і сприяють адгезії тромбоцитів.
Найбільш потужним стимулятором тромбоцитів є фібрилярний колаген, що здійснює також контактну активацію факторів так званих внутрішніх шляхів згортання крові. Тромбоспондин здатний асоціюватися з волокнами фібрину і полімеризуватися подібно фібриногену. Посилює клітинну взаємодію, перетворюючи оборотну агрегацію тромбоцитів у необоротну, специфічно зв’язується з моноцитами і є молекулярним містком між ними і активованими тромбоцитами в ділянках ушкодження судинної стінки. Фіброкінетин, основний компонент сполучнотканинного матриксу, утворює ковалентні зв’язки з фібрином і здійснює рецептор-опосередковане осадження активованих тромбоцитів.
Тромбоцитарна ланка є найважливішою в системі гемостазу. Участь тромбоцитів у гемостазі зумовлена їх здатністю до адгезії та агрегації, вмістом власних та адсорбованих факторів згортання крові, фізіологічно активних речовин. Поверхня тромбоцитів, як і клітин ендотелію, покрита глікокаліксом. Реактивність тромбоцитів залежить від величини негативного заряду, зумовленого поліаніонними властивостями глікокаліксу і фосфатними групами плазмолеми. Плазмолема тромбоцитів має звичайну для клітинної мембрани будову, утворює численні інвагінації (поверхнево-пов’язану систему каналів), що у багато разів збільшують її площу. На тромбоцитах адсорбуються фактори згортання, імуноглобуліни. Крім того, тромбоцити є джерелом факторів агрегації і дезагрегації формених елементів крові, зокрема фосфоліпідів, тромбоксана А2 — стимулятора агрегації і вазоконстрикції, ряда простагландинів. З ними асоційовані рецепторні і регуляторні білки, у тому числі аденілатциклаза і фосфофоліпаза А2, аденіннуклеотиди, комплекс ферментів, що каталізують утворення і трансформацію арахідонової кислоти в ендопероксиди і кінцеві продукти їх метаболізму.
Будь-які агенти, що змінюють фізико-хімічний стан глікокаліксу та проникність плазмолеми, активують тромбоцити, підвищуючи їх агрегаційну здатність і провокуючи реакцію вивільнення — секрецію в навколишнє середовище вмісту тромбоцитарних гранул, які є депо біологічно активних речовин та адгезивних білків. Тромбоцити містять 2 основних їх типи — α-гранули і щільні тільця. α-Гранули депонують фібриноген, фібронектин, фактор Віллебранда, тромбоспондин, а також фактор росту, що стимулює міграцію та проліферацію гладком’язових клітин судинної стінки, тромбоцитарний фактор ІV (антигепарин), тромбоцитоспецифичні глобуліни. Щільні тільця багаті АДФ та іонізованим кальцієм, містять гістамін, епінефрин, серотонін.
Реакції тромбоцитів на дію активуючих агентів опосредковуються підвищенням концентрації в цитоплазмі іонів кальцію, які депоновані в плазмолемі та тромбоцитарних гранулах, у щільній тубулярній системі, розміщеній у субмембранній зоні поруч з елементами цитолеми. Кальцій надходить у тромбоцити також із навколишнього середовища у вигляді трансмембранної течії. Обов’язковою умовою агрегації тромбоцитів є присутність фібриногену. Фосфоліпіди плазмолеми тромбоцитів є каталізатором для тканинних і плазмових тромбопластинів, попередників тромбіну.
Тому участь тромбоцитів у гемостазі визначається їх здатністю адсорбувати на своїй поверхні плазмові фактори коагуляції, здійснювати секрецію комплексу біологічно активних речовин та адгезивних білків, поставляти в навколишнє середовище комплекси, що активують прокоагулянти, а також міцно асоціюватися з судинною стінкою та один з одним. Роль у гемостазі інших формених елементів, еритроцитів і лейкоцитів зумовлена вмістом у них більшості факторів згортання крові, які залучаються в процес утворення фібрину при ушкодженні судинної стінки.
Загальні закономірності гемостазу. Фактори згортання крові в нормі перебувають у неактивному стані, у формі попередників. Плазмові фактори згортання крові і їх функцій наведені в табл. 2.1.

Активування факторів згортання крові відбувається послідовно, причому фермент, що є продуктом відповідної реакції, діє на свій специфічний субстрат, викликаючи появу іншого ферменту, що починає наступний етап у ланцюзі цього каскадного процесу, що завершується перетворенням розчинного фібриногену в нерозчинний фібрин. Кожний такий етап представляє комплекс реакцій, у яких беруть участь активований коагуляційний фактор — фермент, субстрат — проензимна форма сполученого коагуляційного фактора і ко-фактор — прискорювач реакції. Усі компоненти цих реакцій збираються на фосфоліпідах і утримуються разом іонами кальцію. Такою білково-ліпідною матрицею, на якій збираються та активуються ферментні та інші фактори згортання, є поверхня тромбоцитів.
У механізмі згортання крові можна умовно виділити зовнішній і внутрішній шляхи, тісно пов’язані між собою. Зовнішній шлях запускається при ушкодженні судинної стінки і тканин і вивільненні в кров тканинного фактора згортання (фактор III, тромбопластин). Тромбопластин являє собою ліпопротеїдний комплекс, білкова частина якого працює як ко-фактор фактора VII згортання крові, а фосфоліпідна — служить матрицею для активної форми останнього і його субстрату — фактора X.
Внутрішній шлях згортання формується факторами, що утримуються в крові, активуються при контакті плазми з субендотелієм, зміненими клітинними мембранами, із зарядженою поверхнею або під впливом біогенних амінів і протеаз. Сполучений з калікреїн-кініновою системою, системою комплементу та інших ферментних систем крові. Калікреїн бере участь у взаємодії факторів XII і XI, зв’язуючи внутрішній і зовнішній шляхи згортання крові. Вихідним пунктом внутрішнього шляху є активація фактора Хагемана, за яким послідовно активуються фактори VII, IX, XI. Разом із кальцієм вони утворюють на поверхні активованих тромбоцитів або ушкодженої судинної стінки комплекс, що активує фактор X, на рівні якого поєднуються зовнішній і внутрішній шляхи гемостазу.
Між механізмами обох шляхів згортання крові існують складні взаємини. Невелика кількість тромбіну, що утворюється при активації зовнішнього шляху, стимулює агрегацію тромбоцитів і реакцію вивільнення тромбоцитарних факторів, але його недостатньо для утворення фібрину. При цьому активується фактор V, що є рецептором фактора X, який активується при фіксації на поверхні тромбоцитів. Основна маса фактора X трансформується в активний стан за допомогою більш складного і ефективного внутрішнього шляху гемостазу.
Схема подальшого етапу, загального для обох шляхів згортання крові після активації фактора X, включає стадії утворення тромбіну з протромбіну і згортання фібриногену. Кожна з них здійснюється при участі відповідних активованих комплексів, що складаються з високомолекулярного неферментного білка, активної протеїнази та кальцію. Вони фіксовані на фосфоліпідній або іншій негативно зарядженій підкладці, утвореній поверхнею клітин крові або стінкою судин. Жорсткий зв’язок таких комплексів з фосфоліпідами забезпечує їх оптимальний захист від інгібіторів, вихід у навколишнє середовище тільки кінцевого ферменту в ланцюзі перетворень тромбіну та локалізацію процесу згортання в ушкодженій ділянці. При цьому ферментні фактори запускають аутокаталітичний процес гемостазу, а неферментні компоненти реакції прискорюють їх і забезпечують специфічність дії на субстрати.
Загальний шлях зовнішнього та внутрішнього шляхів згортання крові починається активацією фактора X і завершується поляризацією фібриногену. Субстратом фактора X служить протромбін, синтезований у печінці, від якого послідовно від’єднуються 2 фрагменти і утворюється тромбін — серинова протеїназа. Основні функції тромбіну: обмежений протеоліз фібриногену з наступною полімеризацією утворених фібрин-мономерів у фібрин; стимуляція тромбоцитів та ендотелію; стимуляція синтезу простагландинів; вивільнення адгезивних білків; активування регуляторних білків — факторів згортання крові, а також фібринстабілізуючого фактора XIII. Між новоутвореними полімерами фібрину встановлюються додаткові перехресні зв’язки, що підвищує їх еластичність і резистентність до дії фібринолітичних агентів.
При активуванні гемостазу в 1 мл крові може утворитися приблизно 150 од. тромбіну — кількість, достатня для згортання декількох її літрів. Проте в організмі рідкий стан крові зберігається навіть при масивних травмах. Це забезпечується складною системою, що запобігає ланцюговій реакції, яка могла б призвести до згортання всієї маси крові в серці та судинах. Тромбоутворенню перешкоджає антикоагулянтна система, що включає як фактори, які утворюються безпосередньо при активації гемостазу, так і існуючі незалежно від нього. Вона функціонально сполучена з системою фібринолізу, що розчиняє утворені тромби.
Антигемостатична система крові включає наступні механізми:
- Зниження локальної концентрації факторів згортання за допомогою вимивання і розведення в кровотоці.
- Виснаження частини факторів згортання, що залишаються у фокусі ушкодження, за рахунок їх утилізації.
- Звільнення крові від активованих факторів згортання внаслідок їх елімінації і катаболізму гепатоцитами та мононуклеарною системою. Цей механізм може бути ефективний тільки при збереженні циркуляції в зоні ушкодження.
- Інгібування активних факторів і ко-факторів крові фізіологічною протизсідальною системою, що регулює рівень тромбіну. У крові циркулює складний набір протеаз та інших біохімічних інгібіторів, що взаємодіють з одним або декількома факторами коагуляції. До їх числа відноситься основний плазмовий інгібітор ферментів — антитромбін III, що у присутності гепарину інактивує тромбін, фактори згортання XII, XI, X, IX і кініноген. Протеїн С, що набуває під дією тромбіну здатності до протеолізу, інактивує фактори згортання V, VIII, XI, XII. Швидкість інактивації зростає при зв’язуванні факторів з тромбомодуліном на поверхні ендотеліоцитів у присутності іонів кальцію і фосфоліпідів. Крім того, протеїн С блокує активацію комплементу, нейтралізує тканинний інгібітор плазміногена, що прискорює його перетворення в плазмін, який у свою чергу здійснює лізис згустків фібрину, і т.д. Таким чином, система біохімічної регуляції гемостазу функціонально поєднує механізми, спрямовані як на активацію факторів згортання крові, так і на блокування їх активних форм.
- Лізис фібрину протизсідальною системою, що здійснює ферментативний і неферментативний фібриноліз. Ця система активується при надмірному нагромадженні тромбіну, її ефекторною ланкою є викид у кров гепарину та активаторів фібринолізу з тканинних джерел і клітин крові. Фібриноліз має внутрішній і зовнішній механізми активації; перший забезпечується лейкоцитарними протеазами і плазміногеном, що перетворюється в плазмін за участю фактора XII і калікреїну. Внутрішній ферментативний механізм фібринолізу запускається тканинними кінінами, які синтезуються головним чином ендотелієм та активуються при утворенні комплексів з фібрином.
Неферментативний фібриноліз ініціюється за допомогою викиду в кровотік гепарину, що зв’язується з тромбіном, фібриногеном та іншими тромбогенними протеїнами, з катехоламінами. Комплекси, що утворюються, мають протизгортальну активність, розщеплюють нестабілізований фібрин, блокують полімеризацію його мономерів, а також є антагоністами фактора ХIII, що стабілізує свіжепреципітований фібрин. Продукти ферментативного та неферментативного лізису фібрину набувають властивості дезагрегантів і антикоагулянтів.
Залежно від масштабів ушкодження і ступеня участі окремих компонентів системи згортання крові розрізняють судинно-тромбоцитарний і коагуляційний механізми, тісна взаємодія яких забезпечує надійність гемостазу. Судинно-тромбоцитарний механізм гемостазу зупиняє кровотечу з периферичних судин невеликого калібру при обмеженій участі другого механізму. При цьому відзначають швидкоминучий спазм травмованих судин внаслідок рефлекторного викиду в кровотік катехоламінів і підвищення тонусу вегетативної нервової системи. Слідом за цим відбувається накопичення тромбоцитів у зоні ушкодження, їх адгезія до ранової поверхні з послідовним розвитком усіх фаз активування — формування псевдоподій, розпластуванння і реакцією вивільнення.
Накопичення необоротно агрегованих тромбоцитів, які протягом 1–3 с адгезуються до ушкоджених ендотеліальних клітин або до субендотелію, що оголився, забезпечує формування гемостатичного тромбу. Це поєднується з вторинним спазмом ушкоджених судин, зумовленим виділенням із тромбоцитів цілого ряду біологічно активних речовин, запуском процесів преципітації фібриногену та формування волокон фібрину, активуванням антикоагулянтних і фібринолітичних механізмів, що координують процес гемостазу.
Коагуляційний механізм гемостазу, що реалізується при ушкодженні великих судин, загалом аналогічний описаному вище. Також починається рефлекторною реакцією судинної стінки, опосередкованою нейрогуморальною системою регуляції, і адгезією тромбоцитів у зоні ушкодження. Виділення судинно-тканинного і коагуляційного механізмів гемостазу досить умовно, тому що вони функціонально поєднані і сполучною ланкою є тромбоцити, що являють собою центр формування тромбу.
Морфологія і види тромбів. За морфологічними особливостями розрізняють тромби білі (аглютинаційні), червоні, змішані (шаруваті) і гіалінові.
Білий тромб виникає у відділах судинної системи зі швидкою течією крові, наприклад у порожнинах серця і на стулках його клапанів, в аорті і коронарних артеріях. Утворюється при зниженні атромбогенних властивостей ендотелію і накопиченні в крові факторів, що стимулюють тромбоцити, являє собою сухувату світло-сіру масу з тьмяною гофрованою поверхнею щільної консистенції, спаяний зі стінкою судини, легко кришиться при спробі відділення. Основу білого тромбу становлять тромбоцити, що склеїлися з судинною стінкою і між собою. Тромбоцитарні конгломерати формують коралоподібні фігури, орієнтовані перпендикулярно течії крові, простори між якими заповнені мережею фібрил фібрину зі скупченнями нейтрофільних лейкоцитів.
Шарувате відкладення тромбоцитів зумовлено чергуванням фаз тромбоутворення з переважанням адгезії та аглютинації тромбоцитів і полімеризації мономерів фібрину на їх поверхні, що відіграє роль матриці. Під час реакції вивільнення, що супроводжує активування та аглютинацію тромбоцитів, разом з адгезивними протеїнами і біологічно активними речовинами з них виділяється фермент ретрактозим. Фермент викликає скорочення гладком’язових клітин судинної стінки і ущільнює тривимірну сітку, утворену волокнами фібрину, чим забезпечує консолідацію всіх його елементів. Тромб втрачає частину рідини, місцями відокремлюючись від судинної стінки, щілини, що в ньому виникли, полегшують тромболізис і процес організації.
Червоний тромб утворюється внаслідок підвищення потенціалу гемокоагуляційних механізмів при відносно невисокій активності тромбоцитів і зниженні антиагрегаційних властивостей судинної стінки. Найбільш часта локалізація червоних тромбів — ємнісні судини з відносно низькою швидкістю кровотоку. Внаслідок високих темпів утворення і меншого вмісту тромбоцитів червоний тромб легше відокремлюється від судинної стінки. Він пухкий з гладкою вологою, лише місцями гофрованою поверхнею, що надає йому подібність з посмертним згустком крові. Новоутворені тромби цього типу темно-червоного забарвлення, згодом набувають бурого відтінку; їх поверхня втрачає блиск. Структурну основу червоного тромбу становить тривимірна сітка волокон фібрину різної товщини, петлі якої заповнені аглютинованими і різного ступеня олуженими еритроцитами з незначною домішкою лейкоцитів і невеликих скупчень тромбоцитів. Проте кораловидні фігури, утворені ними в білих тромбах, відсутні.
Змішаний тромб включає ділянки, що за своєю структурою відповідають білому або червоному тромбу. Чим повільніше тромбоутворення, тим краще виражена скелетна частина тромбу, утворена коралоподібно розгалуженими агрегаціями тромбоцитів, що характерна для білого тромбу, і тим менші зони коагуляції крові, що представлені сіткою полімеризованого фібрину, осередки якого заповнені зсілими еритроцитами з вкрапленням інших формених елементів. Присутність у змішаних тромбах світлих і темних ділянок надає їм строкатий шаруватий вигляд як на поверхні, так і на розрізі. Такі тромби найчастіше виявляють в артеріях різного калібру, великих венах, аневризмах серця і артерій. Так само, як і червоні тромби, вони мають у судинах подовжену форму.
Макроскопічно в них розрізняють голівку, зазвичай конічної або сплощеної форми, щільно з’єднану зі стінкою судини, що відповідає за своєю будовою білому тромбу. Голівка тромбу переходить у тіло (власне змішаний тромб), що продовжується в пухкий, нещільно зв’язаний з ним вільно розміщений у просвіті судини хвіст, який являє собою червоний тромб.
Зв’язок змішаного тромбу із судинною стінкою і описані вище особливості будови відрізняють його від посмертного згустку крові. Найбільших розмірів змішані тромби досягають у великих венах, де, як правило, розміщуються за течією крові. Такий тромб може починатися в стегновій вені, де його голівка щільно прикріплена до судинної стінки, тіло (змішаний тромб) продовжується в зовнішню клубову вену, переходячи в пухкий темно-червоний хвіст, що іноді досягає нижньої порожнистої вени.
Гіаліновий тромб являє собою однорідну гіаліноподібну масу, що утворюється при аглютинації і деструкції еритроцитів, лейкоцитів і преципітованих білків плазми крові в дрібних периферичних судинах. Вміст фібрину в гіалінових тромбах порівняно невеликий, а присутність його мінлива. Утворенню гіалінового тромбу часто передує стаз крові в мікросудинах.
Тромби класифікують також залежно від їх локалізації, відношення до просвіту судини, у якій вони сформувалися, і етіологічних факторів, що сприяли тромбоутворенню. Тромби, що тільки частково обмежують просвіт судини, називають пристінковими, що повністю закривають його — обтуруючими. Для останніх характерний розвиток як у дистальному, так і в проксимальному напрямку за течією крові. У тих випадках, коли такий тромб має будову шаруватого або змішаного, визначення місця, де почалося його утворення і відповідно розташована голівка, викликає значні труднощі.
Пристінкові тромби зазвичай виявляють у просвітах великих судин, у камерах серця і на клапанах при атеросклерозі та запальних процесах (тромбартеріїт, тромбоендокардит, тромбофлебіт), при венозній гіперемії, що супроводжується сповільненням кровотоку (марантичні тромби). Патологічна дилатація артерій або камер серця (аневризми), варикозне розширення вен також сприяють тромбоутворенню (дилатаційні тромби). Обтуруючі тромби найбільш характерні для дрібних судин. Часто при наростанні пристінкового тромбу шляхом нашарування тромботичних мас, що знову утворюються, можлива закупорка магістральних судин — коронарних артерій серця або кишечнику, великих артерій головного мозку, печінкових, стегнових та інших вен. Такий тромбоз називають прогресуючим.
Проміжне положення між пристінковими та обтуруючими тромбами за впливом на кровотік займають так звані аксіальні тромби, які, прикріплюючись вільною частиною до судинної стінки тільки в області голівки і частково тіла, істотно обмежують прохідність судини. У передсерді великий ростучий тромб, відірвавшись від стінки, може залишатися в його порожнині у зваженому стані, набуваючи під дією кровотоку кулястої форми (кулясті тромби). Фактором, провокуючим тромбоз, може стати розростання пухлини, що проникає в просвіт вени і утворює поверхню, на якій ініціюється тромбоутворення (пухлинні тромби).
Фактори розвитку тромбозу. Ініціювання тромбозу визначається загальними і місцевими передумовами, при поєднанні яких порушується рівновага процесів про-, антикоагуляції та фібринолізу. Найбільш істотними факторами загального характеру, що зумовлюють схильність до тромбоутворення, є порушення гемодинаміки при СН, зміни складу крові при захворюваннях системи крові, інфекційно-алергічних процесах, патологічних нейрогуморальних реакціях (хронічний стрес) і порушеннях кровообігу із схильністю до ангіоспастичних явищ.
Із місцевих факторів, що сприяють тромбозу, слід назвати насамперед зміни судинної стінки та локальні порушення гемодинаміки. Зміни судинної стінки, що здійснюють тромбогенний ефект, мають різну природу, проте у всіх випадках відбувається ушкодження судинного ендотелію, що призводить до втрати його антигемостатичних властивостей. Безпосередніми причинами цього може стати механічне ушкодження або запалення, що запускає судинно-тромбоцитарний механізм гемостазу, до якого приєднуються гемокоагуляційні процеси. Такі ж є наслідки розпаду атеросклеротичної бляшки, ангіоспазму, різкого підвищення рівня АТ і судинної проникності з наступним відшаруванням і десквамацією ендотеліоцитів, що оголює субендотелій. Тромбозу сприяє також поява завихрень в потоці крові, що травмують ендотеліальний моношар і тромбоцити.
Уповільнення швидкості кровотоку створює сприятливі умови для агрегації тромбоцитів до судинної стінки і обмежує вимивання факторів, що ними виділяються. Про важливе значення цих змін для розвитку тромбозу свідчать у 5 разів частіша локалізація тромбів у місцях розгалужень судин або атеросклеротичних бляшок, що деформують їх стінку, частіше тромбування вен, ніж артерій з типовою локалізацією у нижніх кінцівках, синусах венозних клапанів, варикозних розширеннях і аневризмах судин і серця. Проте більшість із названих передумов не має абсолютного значення для тромбозу, і тільки їх поєднання з гострим або хронічним порушенням згортальної і протизгортальної систем стає достатньою умовою для його розвитку.
Результати тромбозу, як і його безпосередні причини або будова тромбів, неоднакові. При неускладненому розвитку тромбу в ньому відзначають асептичне розплавлення (аутоліз), що наступає як під впливом літичних ферментів (катепсинів, гідролаз, пептидаз), що вивільняються з поліморфно-ядерних лейкоцитів і тромбоцитів, так і внаслідок фібринолізу, зумовленого дією плазміну та пептидаз плазми крові.
Розплавлення тромбів починається із серединної зони, де накопичується найбільша кількість ензимів. Кашкоподібний детрит, що утворюється, і напіврідкі маси в білому тромбі жовтуватого відтінку в червоному — набувають червоно-коричневого забарвлення в результаті великої кількості еритроцитів. Іноді продукти аутолізу потрапляють у кровотік і переносяться течією крові. Дрібні тромби можуть аутолізуватися повністю.
Паралельно з аутолізом до кінця 1-ї доби починається організація тромбу, в якій бере участь судинна стінка. У тих ділянках тромбу, які пізніше інших втягуються в асептичний аутоліз, у перші 4 доби відбуваються розпад і гомогенізація формених елементів крові і ниток фібрину зі злиттям детриту в гіаліноподібну масу.
На 2-гу добу відзначають проліферацію ендотеліоцитів судинної стінки, які ніби наповзають на поверхню тромбу, поступово вкриваючи її. Поряд з цим відзначають розмноження клітин інтими, накопичення активованих макрофагів, некротичні зміни ще збережених лейкоцитів і проникнення фібропластичних елементів у тромб. У наступні дні явища лізису детриту і виражена макрофагальна реакція поєднуються із вростанням у тромб тяжів проліферуючих ендотеліоцитів, з яких потім утворюються кровоносні капіляри. В організації тромбу разом з фібробластами та макрофагами беруть активну участь недиференційовані гладком’язові клітини судинної стінки, що продукують глікопротеїни і колаген.
Організація тромбу починається з його голівки, поширюючись потім на тіло. Новоутворені судини з’єднуються з vasa vasorum або з просвітом тромбованої судини. По мірі дозрівання сполучної тканини в тромбі з’являються щілини і канали, покриті ендотелієм (каналізація тромбів), а з 5-го тижня виявляють диференційовані судини (васкуляризація тромбу), з яких іноді формуються судинні порожнини (кавернозна трансформація тромбу). Каналізація і васкуляризація тромбу частково відновлюють прохідність судини. Еволюція тромбу завершується дозріванням новоутвореної сполучної тканини в рубцеву і наступним формуванням фіброзно-м’язової бляшки, що стенозує просвіт судини. При порушенні процесу організації у гіалінізовані ділянки тромбу випадають солі кальцію, що призводить до петрифікації тромботичних мас. У венах цей процес іноді завершується петрифікацією — утворенням каменів (флеболітів).
Значення тромбозу для організму неоднозначне. Тромби, що утворюються при ушкодженнях судин, захищають організм від фатальної крововтрати, організація тромботичних мас в аневризмах серця і судинах попереджає розриви їх стінки. Проте у більшості випадків, коли тромбоз розвивається як патологічний процес, існує погроза виникнення його більш-менш небезпечних ускладнень. Це визначається локалізацією та швидкістю утворення тромбу, ступенем обмеження просвіту судини, наявністю або відсутністю колатералей, а також наступною еволюцією тромбу, що утворився.
Найнебезпечніші ускладнення тромбозу зумовлені:
- Локальними порушеннями кровотоку внаслідок обмеження прохідності просвіту тромбованої судини.
- Здатністю тромбу або його частини відокремлюватися від стінки судини і переноситися потоком крові на значні відстані (тромбоемболія) при млявому розвитку процесів організації або внаслідок аутолізу.
- Інфікуванням тромбу та переходом асептичного аутолізу в септичний.
Обтурація тромбом магістральної судини при недостатньому розвитку колатералей викликає ішемію або венозну гіперемію з можливими несприятливими наслідками. У той же час поступове, розтягнуте за часом формування пристінкового тромбу навіть у великих артеріальних стовбурах не обов’язково призводить до тяжких наслідків, наприклад до розвитку інфаркту, тому що в цих випадках кровотік частково встигає відновитися за рахунок колатералей. Небезпека ускладнень при тромбозі різко зростає при його прогресуючому розвитку, що свідчить про істотні загальні порушення регуляції гемостазу та кровообігу. Наслідками цього можуть бути ріст і перетворення тромбів з пристінкового або аксіального в обтуруючий або швидке збільшення хвоста, що нещільно зв’язаний з тілом, виникнення в різних судинах численних тромбів, слабко фіксованих до судинної стінки. Відрив від стінки всього або частини такого тромбу перетворює його в тромбоембол, що вільно мігрує з течією крові. Розвиток тромбоемболії можливий при будь-якій локалізації тромбів, проте найчастіше це відзначають при флеботромбозі, тромбофлебіті або тромбозі порожнин і особливо вушок серця.
Аутоліз тромбу буває не тільки асептичним. Потрапляння в нього гноєтворних бактерій зумовлює септичне розплавлення тромботичних мас із наступним поширенням утворених інфікованих продуктів розпаду по організму, що викликає тромбобактеріальну емболію судин та утворення вогнищ гнійного запалення в різних органах і тканинах.
У патологоанатомічній практиці нерідко виникає необхідність диференціювати тромби від посмертних згустків крові, які також бувають білі або змішані та іноді мають досить значну подібність з тромбами. Така схожість визначається подібністю механізмів, що зумовлюють посмертне згортання крові. Вважається, що до остаточної зупинки метаболічних процесів, що протікають у судинній стінці, в ній відбувається нагромадження і дифузія в просвіт судини АДФ з наступною активацією тромбоцитів і запуском внутрішнього шляху згортання крові. Разом з тим відмінність умов, у яких це відбувається, від процесу тромбоутворення в живому організмі знаходить висвітлення в морфології посмертних згустків і тромбів.
ДВЗ-синдром
ДВЗ-синдром — синдром дисемінованого внутрішньосудинного згортання крові — патологічна ситуація, безпосередньо пов’язана з дисфункцією системи гемостазу організму. Основна відмінність локального тромбозу та ДВЗ-синдрома полягає у відносно обмеженому характері цих змін у першому випадку та їх генералізації з переважною локалізацією в мікроциркуляторному руслі — в другому. Тромбоз великих судин може розвиватися як компонент або фінал цього синдрому. Відповідно до етіопатогенетичних особливостей синдрому переважають порушення прокоагулянтної або судинно-тромбоцитарної ланки гемостазу або обидві вони активуються в однаковому ступені.
ДВЗ-синдром неспецифічний і розвивається при численних захворюваннях і патологічних станах: серцево-судинних (великовогнищевий ІМ, вроджені «сині» вади серця, СН та ін.), усіх видах шоку, включаючи кардіогенний. ДВЗ-синдром входить до числа ускладнень трансфузії несумісної крові, травматологічної, інфекційно-септичної, онкологічної, акушерської, ятрогенної, аутоімунної та імунокомплексної патології, алергічних реакцій, отруєнь гемокоагулянтами та гемолітичними отрутами, великих оперативних втручаннях, у тому числі із застосуванням АШК.
Патогенез ДВЗ-синдрому визначається активацією факторів згортання крові з наступним їх виснаженням, надмірною стимуляцією фібринолізу, що супроводжуються масивними кровотечами, що вкрай важко купіруються, та крововиливами. Безпосередні причини розвитку ДВЗ-синдрома неоднозначні. Найчастіше його факторами стають продукти гемолізу, амніотична рідина, ендотоксини, прогресуючий ацидоз, протеолітичні ензими, елагова кислота, надмір АДФ, деякі ліпідні фракції плазми і циркулюючі імунні комплекси, гіперадреналінемія, загальні гемодинамічні порушення та ін. В основі активації цими факторами системи коагуляції і зниження ефективності процесів, що гальмують згортання крові, лежать два основних механізми: вивільнення в кровотік тканинних факторів та альтерація ендотеліального моношару.
Багаточисленні фактори, що ініціюють ДВЗ-синдром, часто тісно взаємозв’язані. Навіть обмежене ушкодження ендотелію стимулює прокоагулянтну активність за допомогою експресії тканинного фактора. Серед причин, здатних провокувати ушкодження ендотелію, особливе значення мають гіпоксія, ацидоз і шок як найбільш характерні для кардіологічної патології. Продукція тканинного тромбопластину різко зростає при розширенні зони альтерації ендотелію з оголенням субендотеліальних структур, активуванням тромбоцитів і внутрішнього шляху згортання крові через контактний XII фактор Хагемана, що діє на калікреїн-кінінову систему, фібриноліз, систему комплементу. Це супроводжується блокадою фагоцитарної функції мононуклеарів, які у фізіологічних умовах підтримують рівновагу процесів гемостазу, елімінуючи з крові розчинні комплекси фібрину.
Ініціація ДВЗ-синдрому безпосередньо пов’язана з дією як тромбопластину і тромбіну, так і ендотеліотропних медіаторів. У результаті цього коагуляція і тромбогенез виявляються первинними процесами з наступною активацією і агрегацією тромбоцитів, які вивільняють біологічно активні речовини в поєднанні з інтенсивним використанням факторів згортання крові. При цьому компенсаторна активація тромбіном протизгортальної системи, що в нормі забезпечує адекватне підвищення антикоагулянтної та фібринолітичної дії, виявляється недостатньою. Розповсюджений мікротромбогенез супроводжується зростанням активності системи фібринолізу з появою в крові плазміну, що гідролізує фібрин, інактивує фактори V, VIII, IX, XI і знижує їх концентрацію в крові.
Протеїнази тромбін і плазмін зумовлюють преципітацію фібрину. У той же час вони розщепляють його і фібриноген з утворенням ранніх і пізніх продуктів деградації, які перешкоджають полімеризації фібрину-мономера і викликають дисфункцію тромбоцитів. Разом з тим деяка частина фібрин-мономерів полімеризується в мікросудинах і, захоплюючи формені елементи крові, провокує реакцію фібрин — еритроцити та мікроангіопатогенну гемолітичну анемію з вивільненням у кров фосфоліпідів і АТФ, які є індукторами ДВЗ. Частина тромбоцитів, попередньо активованих різними індукторами, включаючи тромбін і колаген, зв’язується в цих мікротромботичних комплексах, вивільняючи тромбоспондин, фібропластин та інші адгезивні білки, що також сприяє виснаженню захисних механізмів протизгортальної системи. Надмірне споживання факторів згортання, високий рівень розчинних комплексів фібрину, тромбоцитопенія, дисфункція протизгортальних механізмів призводять до реалізації вторинних процесів гіперкоагуляції, недостатності гемостазу, кровотечам і крововиливам.
У розвитку ДВЗ-синдрому розрізняють 4 стадії, що деякими дослідниками розглядаються скоріше як форми цього патологічного процесу. Перша стадія морфологічно характеризується масованим мікротромбозом з блокуванням мікросудин, зумовленим гіперкоагуляцією і внутрішньосудинною агрегацією формених елементів крові на фоні активації плазмових систем гемостазу.
Морфологічними еквівалентами цієї стадії ДВЗ-синдрому є фібринові, гіалінові, глобулярні, тромбоцитарні, лейкоцитарні, еритроцитарні (червоні) мікротромби, склад і будова яких не відповідає структурі тромбів у макросудинах. Фібринові мікротромби, яким при морфологічній діагностиці ДВЗ-синдрому приділяється вирішальна роль, як і гіалінові, складаються переважно з фібрину з більш-менш значною домішкою фібриногену (рис. 2.4).
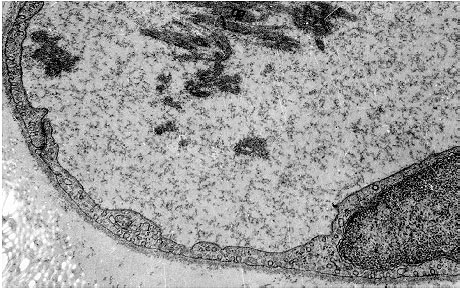
Рис. 2.4. Преципітати фібрину в просвіті кровоносної мікросудини
Каркасом глобулярних мікротромбів служать агрегіровані еритроцити з явищами гемолізу, на вилужених оболонках яких відкладаються фібринові маси. Тромбоцитарні або пластинчасті мікротромби, поряд з компактно розміщеними кров’яними пластинками, включають поодинокі еритроцити, лейкоцити і нитки фібрину. Фактором, що сприяє утворенню таких мікротромбів є альтеративні зміни ендотеліоцитів.
Білі або лейкоцитарні тромби частіше утворюються при ДВЗ-синдромі інфекційної етіології, розміщуючись переважно в дистальних відділах мікрогемоциркуляторного русла. Червоні тромби, головним компонентом яких є вилужені сладжовані еритроцити та преципітати фібрину, виявляють у всіх ділянках мікрогемоциркуляторного русла.
Друга стадія ДВЗ-синдрому, коагулопатія споживання, визначається тромбоцитопенією, внутрішньоваскулярною преципітацією фібрину зі зниженням у крові вмісту фібриногену та інших плазмових факторів згортання. Проявляється як гіпер-, так і гіпокоагуляцією у вигляді кровотеч та ознак геморагічного діатезу. Коагулопатія споживання є наслідком розповсюдженого мікротромбозу з підвищеним використанням факторів згортання крові і видалення коагулятів фібрину з кровотоку фагоцитами, а також клітинами печінки і селезінки.
Активація фібринолізу в третій стадії ДВЗ-синдрому забезпечує відновлення гемоперфузії в мікрогемоциркуляторному руслі, звільняючи просвіти мікросудин від тромботичних мас. Разом з тим поява в крові плазміну — високоактивної протеази, що розщеплює фібриноген і фібрин, сприяє вторинному формуванню так званих гіалінових мікротромбів.
Четверта, відновлювальна, стадія зводиться до залишкових проявів попередньої блокади мікросудин у вигляді дистрофічних і некротичних змін тканин, що найбільш постраждали, завершується одужанням або при несприятливому перебігу процесу — розвитком ГНН, недостатності надниркових залоз, гострої печінкової, легеневої або іншої органної недостатності.
Клініко-морфологічна картина ДВЗ-синдрому визначається характером етіологічних факторів, їх інтенсивністю та тривалістю дії, адекватністю та ефективністю лікувальних заходів. Незважаючи на генералізований характер патологічного процесу, в загальній картині часто домінують регіональні порушення з переважним ураженням легень (68%), нирок (66%), селезінки та печінки (відповідно 52 і 50%).
Часте ушкодження легень пояснюється їх функцією своєрідного судинного фільтра. У кровоносних капілярах легень затримуються продукти альтерації та сторонні частки, що є тригером для запуску ДВЗ-синдрому, провокуючи внутрішньосудинну коагуляцію, агрегацію, сладж і аглютинацію формених елементів крові з утворенням усіх варіантів мікротромбів. Численні поліморфні за складом мікротромби викликають явища дистрофії в паренхіматозних органах, а при затяжному перебігу процесу — некробіотичні і некротичні зміни. Мікрогемодинамічні порушення при значній поширеності і тривалості здатні призводити до органної недостатності, що різко погіршує клінічну картину. Інтенсивне розповсюджене мікротромбоутворення іноді ускладнюється тромботичною оклюзією схильних до цього артерій м’язового і м’язово-еластичного типу, наприклад при атеросклерозі або системних ураженнях сполучної тканини.
Облігатним компонентом клініко-морфологічної картини крові при ДВЗ є тромбогеморагічний синдром, що майже в 40% випадків має перебіг з вираженою крововтратою, численними точкоподібними або розповсюдженими крововиливами в різних органах. Найчастіше це легеневі альвеоли, товща надниркової залози, паренхіма печінки, селезінки, нирок, субендо- і субепікардіальні зони в серці. Відзначають також появу дрібно- і великоплямистого геморагічного висипу на шкірі, численних крововиливів у місцях ін’єкцій та операційних розрізів.
У ЦНС мікронекрози, що найчастіше викликані фібриновими тромбами, поєднуються з внутрішньотканинними і інтраоболонковими крововиливами. У ШКТ розповсюджений мікротромбоз і крововиливи в слизову оболонку призводять до гострих виразок шлунка, ерозивного гастриту, ентероколіту. Моно- або поліорганна недостатність, гіпофібриногенемія і тромбогеморагічний синдром часто доповнюються постгеморагічною анемією і вираженою гіпотензією, порушеннями серцевого ритму і мозковою симптоматикою.
Залежно від темпів розвитку та особливостей перебігу розрізняють гостру, підостру та хронічну форми ДВЗ-синдрому. Генералізований характер гострої форми внаслідок швидкого масованого надходження тромбопластинового компонента в кровотік в період від декількох годин до доби призводить до шокового стану: затемнення свідомості, гіпотензія, гостра поліорганна недостатність, часто з явищами вогнищевого панкреонекрозу та ерозивно-виразкового ентероколіту.
Підгостра форма розвивається протягом декількох діб, а іноді й 1 тиж. Мозаїчність супутньої симптоматики свідчить про поліорганний процес, проте у клінічній картині найчастіше домінують ознаки переважаючого ураження якого-небудь одного органа або системи. У цілому помірно виражені ознаки тромбогеморагічного синдрому можуть різко посилюватися, набуваючи вираженого генералізованого характеру, у випадку приєднання навіть невеликого екзо- або ендогенного стимулу, наприклад при онкологічній, ятрогенній патології, затяжних коронарних кризах.
Хронічний ДВЗ-синдром, що супроводжує запальні та аутоімунні процеси (гепатит, панкреатит, пневмонію, системні ураження сполучної тканини, онкопатологію, ІХС), може тривати місяцями. Послаблюючись на період ремісії та підсилюючись при загостренні, ДВЗ-синдром може впливати на клінічну картину основного захворювання.
Результат ДВЗ-синдрому визначається причиною, ступенем вираженості та характером розвитку. При найбільш тяжкій, гострій формі, що майже в 50% випадків загрожує летальним кінцем, вирішальне значення має своєчасність діагностики та збалансованість лікувальних заходів, орієнтованих на оптимізацію процесів гемокоагуляції. При менш тяжких затяжних формах ДВЗ-синдрому акцент переміщується на адекватну терапію провокуючих його захворювань і процесів.
Емболія
Емболією (від греч. — вторгнення, вставка) називають патологічний процес переміщення з течією крові субстратів (емболів), які відсутні в нормальних умовах і здатні обтурувати судини, викликаючи гострі регіонарні порушення кровообігу. Для класифікації емболій використовується ряд ознак: характер і походження емболів, їх обсяги, шляхи міграції у судинній системі, а також частота повторення емболії у конкретного хворого.
Залежно від місця виникнення емболи переміщаються:
- З порожнини лівого передсердя, ЛШ або магістральних судин у периферичні відділи великого кола кровообігу. Такі ж шляхи міграції емболів із легеневих вен, що попадають у ліві відділи серця (ортоградна емболія).
- Із судин різного калібру венозної системи великого кола в праве передсердя, ПШ і далі по течії крові в артерії малого кола кровообігу.
- Із гілок портальної системи у ворітну вену печінки.
- Проти течії крові у венозних судинах значного калібру (ретроградна емболія). Це відзначають у випадках, коли питома вага тромбу дозволяє йому перебороти рушійну силу потоку крові, у якому він перебуває. Через нижню порожнисту вену такий ембол може опускатися в ниркову, клубову і навіть стегнові вени, викликаючи їх обтурацію.
- З вен великого кола в його артерії, минаючи легені, що стає можливим при наявності вроджених або набутих дефектів у міжпередсердній або міжшлуночковій перегородці, а також при невеликих розмірах емболів, здатних проходити через артеріовенозні анастомози (парадоксальна емболія).
Джерелами емболії можуть бути тромби та продукти їх руйнування; вміст розпаду пухлини або кісткового мозку; жир, що вивільняється при ушкодженні жирової клітковини або кісток; частки тканин, колонії мікроорганізмів, вміст навколоплідних вод, сторонні предмети, пухирці газу та ін. У відповідності з природою емболів розрізняють тромбоемболію жирову, тканинну, бактеріальну, повітряну, газову емболію та емболію сторонніми предметами.
Тромбоемболія — найпоширеніший вид емболії. Тромб, що фіксований не міцно, або його частина можуть відокремитися від місця прикріплення і перетворюватися в ембол. Цьому сприяють раптове підвищення АТ, зміна ритму серцевих скорочень, фізичне навантаження, що різко зросло, коливання внутрішньочеревного або внутрішньогрудного тиску (при кашлі, дефекації). Іноді причиною мобілізації тромбу стає його розпад при аутолізі. Ембол, що вільно рухається, заноситься течією крові в судину, просвіт якої менше розмірів ембола, і фіксується в ньому внаслідок ангіоспазму.
Найчастіше тромбоемболію відзначають у ємнісних судинах великого кола кровообігу, в першу чергу у венах нижніх кінцівок і малого таза (венозна тромбоемболія). Тромбоемболи, що тут утворилися, як правило, заносяться течією крові в систему ЛА. Артеріальну емболію у великому колі кровообігу виявляють у 8 раз рідше. Її основними джерелами є тромби, локалізовані у вушку лівого передсердя, між трабекулами ЛШ, на стулках клапанів, що утворилися в зоні інфаркту, аневризми серця, аорти або її великих гілок, що сформувалися на атеросклеротичній бляшці.
Стан, при якому відзначається підвищена схильність до внутрішньосудинного тромбоутворення і повторних тромбоемболій, визначається як тромбоемболічний синдром. Цей синдром розвивається при поєднаному порушенні механізмів, що контролюють процеси гемостазу і підтримують плинність крові, з іншими загальними та місцевими факторами, що сприяють тромбоутворенню. Цей стан відзначають при тяжких оперативних втручаннях, онкологічній патології, захворюваннях серцево-судинної системи.
Жирова емболія виникає внаслідок потрапляння в кров крапельок власного або стороннього нейтрального жиру. Причинами цього є травма скелету (закриті переломи або вогнепальні поранення довгих трубчастих кісток, численні переломи ребер, кісток таза), великі ушкодження м’яких тканин з розтрощенням підшкірної жирової клітковини, тяжкі опіки, інтоксикації або електротравми, жирова дистрофія печінки, закритий масаж серця, деякі види наркозу. Жирова емболія може виникати також при введенні хворому лікувальних або діагностичних препаратів на масляній основі.
Жирові краплі зазвичай потрапляють у легені і затримуються в дрібних судинах і капілярах. Частина жирових крапельок проникає через артеріовенозні анастомози у велике коло кровообігу й розноситься кров’ю у головний мозок, нирки та інші органи, блокуючи їх капіляри. При цьому макроскопічні зміни в органах відсутні. Проте цілеспрямоване дослідження гістологічних препаратів із використанням барвників, що виявляють жир, дозволяє діагностувати жирову емболію у більшості подібних ситуацій.
Тканинну (клітинну) емболію відзначають при попаданні в кровоток часток тканин, продуктів їх розпаду або окремих клітин, які стають емболами. Тканинна емболія виникає при травмах, проростанні злоякісних пухлин у просвіт судини, виразковому ендокардиті. Емболами можуть стати комплекси кістковомозкових клітин і мегакаріоцити, обривки дерми, м’язової тканини, частки печінки, мозку, продукти деструкції стулок клапанів серця або комплекси пухлинних клітин. Можлива також емболія навколоплідними водами, що містять рогові лусочки і попадають у капіляри легень; при неповному відшаруванні плаценти, коли емболами стають ворсинки хоріона, що опинилися у венах матки. Загроза тканинної емболії існує також у випадках, коли порушується методика проведення пункційної біопсії внутрішніх органів, неправильно виконується катетеризація великих вен.
Бактеріальна (мікробна) емболія ускладнює запальні процеси, викликані піогенною або грибковою мікрофлорою, відзначається при зараженні найпростішими або тваринними паразитами. Колонії мікроорганізмів, друзи грибів, патогенні амеби, що потрапили в кровотік, затримуються у легенях або обтурують периферичні судини великого кола кровообігу, що живлять тканини нирок, печінки, серця, головного мозку та інших органів. На новому місці можливий розвиток патологічного процесу, подібний до того, що виявився джерелом емболії.
Повітряна емболія виникає при потраплянні в кров пухирців повітря, які мігрують у судинному руслі, затримуються в місцях розгалуження дрібних судин і капілярів і обтурують просвіт судини. У виражених випадках можлива блокада більших судинних гілок і навіть скупчення піни, утвореної повітрям і кров’ю у порожнині правого серця. У зв’язку з цим при підозрі на повітряну емболію розкриття порожнини серця роблять не виймаючи його з грудної клітки, під водою, заповнивши нею розкриту порожнину перикарда.
Причиною повітряної емболії є ушкодження вен, у які повітря засмоктується внаслідок негативного тиску крові. Найчастіше це відзначають при пораненні яремних або підключичних вен, відкритій травмі синусів твердої мозкової оболонки, баротравмі легень. Повітря може надходити в зіяючі після пологів вени внутрішньої поверхні матки. Загроза повітряної емболії існує при операціях на серці з використанням АШК, під час розкриття грудної клітки або накладанні діагностичного або лікувального пневмоперитонеума, а також при необережному внутрішньовенному введенні лікарських засобів.
Газова емболія при певній подібності з повітряною має дещо інші механізми розвитку. В її основі лежать зміни розчинності газів у рідині пропорційно тиску в середовищі. Так, при швидкому підйомі водолазів, що перебували на значній глибині, при швидкісному підйомі в розгерметизованому висотному літальному апараті гази повітря або спеціальної дихальної суміші, розчинені в крові, вивільняються (ефект «газованої води») і, вільно циркулюючи в ній, стають джерелом емболії. У зв’язку з більш значним об’ємом крові, що перебуває у великому колі, ніж у малому, зміни в його басейні виражені сильніше.
Емболія сторонніми предметами можлива внаслідок проникнення їх у судинне русло при вогнепальних пораненнях (осколки, дріб, кулі), іноді при потраплянні в судини уламків катетерів. Значно частіше джерелом цього виду емболії виявляються вапно, кристали ХС, що містяться в атероматозній масі, яка потрапляє в кровотік при руйнуванні атеросклеротичних бляшок або їх виразковому ураженні. Емболія сторонніми предметами, що мають велику питому вагу, може бути ретроградною. Такі емболи здатні переміщатися при зміні положення тіла.
Значення для організму і наслідки емболії визначаються розмірами і кількістю емболів, маршрутами їх міграції в судинній системі та характером матеріалу, що їх утворює. Залежно від величини емболів розрізняють емболію великих судин і мікроциркуляторного русла (ДВЗ-синдром). Усі емболії, за винятком повітряної і газової, є ускладненням інших захворювань, перебіг яких вони обтяжують аж до летального кінця. Найчастіше виявляють венозну тромбоемболію, при якій емболи, залежно від свого розміру, затримуються в периферичних гілках ЛА, викликаючи геморагічні інфаркти легень, або закривають їх просвіт уже в початкових відділах, що призводить до раптової смерті. Проте відзначаються випадки, коли тромбоемболи при порівняно невеликому діаметрі, але значній довжині під дією течії крові обтурують судини значно більшого калібру, ніж їх власний, або затримуються в місці розгалуження загального легеневого стовбура.
Основним патогенетичним фактором, що визначає клініку ТЕЛА і її великих гілок, є різке підвищення опору кровотоку в малому колі кровообігу. Раптове відключення судин супроводжується місцевою рефлекторною вазоконстрикцією, що іноді поширюється на всю артеріальну систему легені. Ця реакція поглиблюється масивним викидом катехоламінів і підвищенням в’язкості крові внаслідок стресу. Підйом тиску в ЛА може стати причиною різкого перевантаження правого серця з дилатацією його порожнин і розвитком гострого легеневого серця. Спазм артерій при їх раптовій механічній обтурації відзначають не тільки в легенях. Так, причиною зупинки серця при закупорці ЛА є і гостре легеневе серце, і рефлекторний спазм коронарних артерій серця. При заносі емболів у невеликі гілки ЛА, внаслідок чого не було летального кінця, на ЕКГ відзначають зміни, характерні для гострої коронарної недостатності.
Виражений ангіоспазм ушкоджує судинний ендотелій, посилює адгезію та агрегацію тромбоцитів, включає каскадний процес гемостазу і зумовлює трансформацію ембола в зростаючий тромб (емболотромбоз). Наслідком тромбоемболії артерій великого кола кровообігу є ішемізація відповідних органів і тканин з наступним розвитком інфарктів. Інфікування тромбів істотно ускладнює наслідки тромбоемболії, джерелами якої вони стають, тому що в місцях фіксації таких емболів до змін, пов’язаних із порушенням кровообігу, приєднується гнійне запалення (тромбобактеріальна емболія).
Наслідки жирової емболії визначаються обсягом жиру, що потрапив у кров, і залежать від вихідних фізико-хімічних властивостей крові, стану ліпідного обміну та системи гемостазу. При травмах значна частина жирових крапель формується з ліпідів крові, що супроводжується різким підвищенням її коагуляційної активності. У зв’язку з цим жирову емболію часто розглядають як варіант травматичної коагулопатії. Летальний кінець при жировій емболії може бути також наслідком закупорки капілярів і циркуляторної гіпоксії головного мозку. При невеликому обсязі обтурованих мікросудин жирова емболія має перебіг без істотної клінічної симптоматики. Жир, що потрапив до легень, частково розщеплюється або омилюється макрофагами і виводиться через дихальні шляхи. У більш тяжких випадках можливе приєднання пневмонії, а при вимиканні ⅔ легеневих капілярів розвивається гостра легенева недостатність із загрозою зупинки серця.
Тканинну (клітинну) емболію відзначають у судинах великого кола кровообігу частіше, ніж у малому. Найбільше практичне значення має емболія клітинами злоякісної пухлини, що лежить в основі гематогенної дисемінації пухлинного процесу. У результаті клітини пухлини можуть заноситися течією крові практично у будь-який регіон і давати початок новому вогнищу розростання пухлини. Це явище отримало назву метастазування, а вогнища пухлинного росту, що виникли в результаті, називають метастатичними. При поширенні подібних клітин із течією лімфи говорять про лімфогенне метастазування. Аналогічний механізм лежить в основі гематогенного поширення патогенної мікрофлори з появою фокусів інфекції у будь-якому місті великого або малого кола кровообігу, куди заносяться бактеріальні емболи, — у легенях, нирках, селезінці, головному мозку, серцевому м’язі. Бактеріальні емболії часто супроводжуються клінічними і біохімічними ознаками ДВЗ-синдрому.
Наслідки газової емболії можуть варіювати від стертих форм кесонної хвороби до тяжких і навіть з летальним кінцем порушень у системі кровообігу та внутрішніх органах, головним чином у головному мозку. Кесонна хвороба розвивається при пасивному виділенні газу, розчиненого в крові, у результаті різкого зниження тиску навколишнього середовища. Пухирці газу, що утворюються, — емболи потрапляють у мікросудини ЦНС і внутрішніх органів, скелетних м’язів, шкіри, слизових оболонок, порушуючи їх кровопостачання і викликаючи кесонну хворобу. У подальшому можуть з’явитися численні дрібні крововиливи та дрібні некрози в різних органах, зокрема в головному мозку і серці, наслідком чого є паралічі та порушення серцевої діяльності.
Скупчення значного об’єму газу, що виділився з крові, в камерах серця може стати причиною блокади кровотоку і летального кінця, тому що повітряний міхур, що стискається та розширюється, унеможливлює перекачування серцем крові. Газова емболія може ускладнювати газову гангрену, що виникає при інфікуванні рани анаеробною інфекцією, коли гази, що накопичуються в ураженій тканині, прориваються у кровотік.
Кровотеча
Кровотеча — вихід крові з просвітів судин або порожнин серця при порушенні цілісності їх стінки або підвищенні її проникності. Варіанти цього процесу різноманітні, їх диференціюють залежно від причини, механізмів і місця виникнення, від того, куди виливається кров із судинного русла, від характеристики процесу залежно від часу виникнення. Так, якщо кров виливається назовні, говорять про зовнішню кровотечу, якщо в порожнину організму — про внутрішню.
До зовнішніх кровотеч належать також кровохаркання, кровотеча з носа, блювання кров’ю, виділення крові з калом, з порожнини матки. Прикладами внутрішньої кровотечі є надходження крові в черевну порожнину (гемоперитонеум), у порожнину перикарда (гемоперикардіум) або плеври (гемоторакс).
Накопичення крові в результаті внутрішньої кровотечі безпосередньо в тканинах називають крововиливом. При цьому залежно від місця кровотечі, її об’єму та швидкості можливе просочування тканини кров’ю, що розсуває її клітинні і стромальні елементи, заповнює периваскулярні зони (геморагічна інфільтрація), або утворення порожнини заповненої кров’ю, що супроводжується деструкцією тканинних структур (гематома). Площинні крововиливи в шкірі або слизових оболонках називають синцями, а дрібні точкові — петехіями або екхімозами.
Гематоми утворюються в результаті ушкодження вен або артерій. Чим вищий тиск крові в ушкодженій судині і менше опір навколишньої тканини, тим більша ймовірність утворення гематоми, а не геморагічної інфільтрації. У м’яких тканинах стегна, в ділянці сідниць гематома може уміщати 100–200 мл і більше, в заочеревинній клітковині 1–2 л крові. Іноді між ушкодженою артерією і порожниною, що виникла в тканинах, зберігається прямий зв’язок, пульсова хвиля передається на кров, що утримується в ній. Гематома в такому випадку називається пульсуючою.
В основі кровотеч лежать наступні механізми: розрив, роз’їдання або різке підвищення проникності судинної стінки при відсутності її видимих ушкоджень.
Кровотеча шляхом розриву стінки судини або серця відбувається в результаті травми або патологічного процесу, що знижує її міцність. Це можуть бути захворювання запального або аутоалергічного характеру, при яких уражуються і некротизуються гладком’язові і волокнисті структури судинної стінки (сифіліс, туберкульоз, медіонекроз аорти, ревматизм, атеросклероз, інфаркт та ін.). Розриву сприяє раптове підвищення тиску, що часто виявляється пусковим фактором при наявності інших сприятливих змін. Такий механізм спонтанних надклапанних розривів аорти при медіонекрозі, розривів вроджених і набутих аневризм різних судин, коли стінки їх змінені і стоншені внаслідок деструкції еластичного каркаса і рубцювання. Розриви подібного характеру найбільш часті при ІМ, в артеріях еластичного типу, менш адаптованих до різких перепадів тиску, ніж артерії з добре розвиненим м’язовим шаром, у венах, які розміщені під слизовими оболонками або на поверхні, що випинається в просвіт порожнистих органів, наприклад стравоходу або прямої кишки.
Іноді тяжка внутрішня кровотеча виникає при розриві капсули і тканини органа внаслідок підвищення внутрішньоорганного тиску або патологічної перебудови його структури. Такі наслідки розривів печінки і її капсули при травмі, метастазах раку, стінки й судин матки при ускладнених пологах, селезінки і її капсули при портальній гіпертензії, кров’яних інфекціях (малярія, поворотний тиф).
Кровотеча в результаті роз’їдання стінки судин (арозійна кровотеча) виникає при вторинному її залученні в патологічний процес. Це відзначають у випадку інфільтративного росту злоякісної пухлини, що руйнує судинну стінку, під дією протеолітичних ферментів у вогнищі нагноєння, на дні виразки шлунка під впливом шлункового соку, у процесі розпаду некротизованої тканини пухлинного вузла або при специфічному запаленні (казеозний некроз і розпад стінки туберкульозної каверни), внаслідок руйнування стінки та судин фаллопієвої труби ворсинками хоріона при позаматковій вагітності, у кишечнику, як ускладнення черевного тифу або дизентерії.
Кровотеча шляхом діапедезу являє собою просочування крові через судинну стінку при підвищенні її проникності. Еритроцити і плазма крові виходять із судин через розширені міжендотеліальні стики і тимчасові трансендотеліальні канальці, утворені при злитті мікропіноцитозних пухирців. Після проходження еритроцита, ніби шляхом «переливання» у периваскулярну зону, міжклітинна щілина відновлюється до вихідного стану. Діапедезні геморагії виникають, як правило, з артеріол, венул, капілярів і проявляються у вигляді дрібних крововиливів з геморагічною інфільтрацією, іноді вони набувають системного характеру (геморагічний синдром).
Причини діапедезних крововиливів численні та різноманітні. Основними факторами їх виникнення є гіпоксія, інтоксикація та аутоалергічні процеси, порушення нейросудинної регуляції, реологічних властивостей крові та системи гемостазу, підвищення внутрішньосудинного тиску, авітамінози, радіаційні впливи. У зв’язку з цим діапедезні крововиливи спостерігаються при різних захворюваннях серцево-судинної системи, ураженнях головного мозку, гіпертонічній хворобі і симптоматичних гіпертензіях. Також відзначаються при колагенових і ряді тяжких інфекційних захворювань, при запальних процесах, викликаних високотоксичними речовинами або мікроорганізмами, при хворобах системи кровотворення, коагулопатіях, гострій формі променевої хвороби.
Діапедезні крововиливи лежать в основі розвитку петехій та екхімозів. При атеросклерозі можливі діапедезні крововиливи в атеросклеротичні бляшки. Розповсюджені діапедезні крововиливи спонтанного характеру, що відзначаються при цілому ряді захворювань, є одним з їх провідних проявів і визначаються як геморагічний діатез.
Підвищена кровоточивість дрібних судин пов’язана з комплексом змін, що включають підвищення проникності судинної стінки, порушення складу та властивостей крові, що знижують її згортання. Так, при гострій променевій хворобі відбувається включення всіх цих патогенетичних ланок. Геморагічні діатези при спадковій патології (недостатність або відсутність тих або інших факторів згортання крові) мають первинний характер, тоді як при загальних порушеннях в організмі, наприклад при хворобах системи кровотворення, С-авітамінозі, деяких інфекціях, отруєннях арсеном, фосфором, грибами, укусах змій, введенні в організм надмірних доз антикоагулянтів, променевій хворобі і т.д., вони є ускладненнями основного захворювання.
Залежно від джерела розрізняють серцеві, артеріальні, венозні, артеріально-венозні і капілярні кровотечі. За строками вони можуть бути первинними і вторинними, ранніми, пізніми, рецидивуючими або повторними.
Первинні кровотечі мають місце при будь-якому проникаючому ушкодженні судинної стінки і за механізмом свого виникнення відносяться до геморагій. Вторинні кровотечі походять із судин, травмованих у момент ушкодження тканини або якщо відбулася деструкція судини при подальшому перебігу патологічного процесу, наприклад внаслідок роз’їдання при нагноєнні. Небезпечна вторинна кровотеча відзначається також при септичному розплавленні тромбу, що закрив дефект, утворений у судинній стінці.
Результат і значення кровотечі для організму визначаються видом, особливостями джерела, тривалістю, інтенсивністю та об’ємом крововтрати. Іноді кровотеча має фізіологічний (менструальна геморагія) або компенсаторний характер (вікарні носові кровотечі при порушеннях менструального циклу). Проте у більшості випадків воно є патологічним процесом, шкідливі наслідки якого для організму пропорційні об’єму крововтрати.
Найнебезпечніші кровотечі з порожнин серця або великих судин, які, як правило, призводять до раптової смерті. Небезпека швидкої масивної крововтрати полягає не стільки у втраті еритроцитарної маси, скільки в різкому зменшенні ОЦК. Це призводить до глибоких гемодинамічних порушень, коли захисні механізми організму виявляються недостатніми для попередження геморагічного шоку. У результаті смерть від гострої кровотечі настає протягом декількох хвилин без ознак загальної анемії, перш ніж встигає мобілізуватися кров з депо та периферичних судин.
Ушкодження судин порівняно невеликого калібру при зниженні гемостатичних властивостей крові або відсутності кваліфікованої допомоги призводить до тривалої кровотечі, що триває протягом декількох діб, з втратою значного сумарного об’єму крові. Смерть від гострого недокрів’я, що настає, зумовлена тканинною гіпоксією. Так, знекровлювання головного мозку спочатку призводить до колапсу, а зниження САТ до 70 мм рт. ст. і втрата 40% гемоглобіну викликають гостру анемію головного мозку з загрозою смерті. Тканини при такій кровотечі стають блідими, тьмяними, селезінка зморщується, її пульпа стає в’ялою. У нирках різке повнокров’я мозкової речовини поєднується із знекровлюванням коркового шару внаслідок рефлекторного спазму міждолькових артерій і скидання крові по розташованим на межі артеріовенозним анастомозам.
Незначна за разовим об’ємом, але довготривала (протягом місяців і років) рецидивуюча кровотеча, наприклад при пептичній виразці шлунка, порушенні менструального циклу, неспецифічному виразковому коліті, геморої, істотно не порушує загальну гемодинаміку, але призводить до розвитку хронічної постгеморагічної анемії.
При крововиливах, поряд із об’ємом крововтрати, великого значення набуває його локалізація. Так, навіть порівняно невеликий крововилив у ЦНС при АГ, розриві аневризми мозкових артерій призводить, як правило, до летального кінця. Разом з тим масивні крововиливи в клітковину або м’язи часто не являють прямої небезпеки для організму.
Кров, що інфільтрує тканину, резорбується макрофагами і видаляється лімфатичними шляхами; кров, що утворює гематому, — згортається. Гемоглобін вивільняється з еритроцитів і трансформується спочатку в макрофагах внутрішньоклітинно в гемосидерин, а через 7–8 діб — у розміщений поза клітинами гематоїдин, що надає вмісту гематоми жовто-коричневі кольори. У гематомі гемосидерин розміщений по периферії, а гематоїдин — у центрі. При великих крововиливах відзначають так звану гемолітичну жовтяницю, що виникає у результаті перетворення гематоїдину в білірубін.
Зсіла кров піддається організації, що починається на 2–4-ту добу з периферії утвореної порожнини. При великих її розмірах формується фіброзна сумка, що містить детрит, який поступово розріджується та резорбується (кістозна гематома). Кров, що вилилася в порожнині організму або утворила гематому, втрачає свої бактерицидні властивості і стає живильним середовищем для мікрофлори. У результаті виникає серйозна загроза нагноєння, що може різко погіршити стан хворого. Розсмоктування крові, що потрапила в черевну, плевральну або перикардіальну порожнину, супроводжується утворенням великої кількості спайок.
Численні крововиливи в тканини при геморагічних діатезах призводять до вторинних змін. На поверхні шкірних покривів і слизових оболонок, що безпосередньо стикаються з повітрям, іноді виникають ерозії і виразки, у головному мозку — численні петехії або геморагічні інсульти, у легенях можливе утворення ателектазів.
ЛІТЕРАТУРА
- Благодаров В.Н., Гавриш О.С., Конончук М.А. та ін. (2000) Патологічна анатомія в рисунках та схемах. ВАТ «Атлант UMC», Київ, 302 с.
- Зербино Д.Д., Лукасевич Л.Л. (1989) Диссеминированное внутрисосудистое свертывание крови: факты и концепции. Медицина, Москва, 256 с.
- Общая патология человека: Руководство для врачей (1990) Под ред. А.И. Струкова, В.В. Серова, Д.С. Саркисова. В 2 т. Т. 1. 2-е изд., перераб. и доп. АМН СССР. Медицина, Москва, 448 с.
- Пальцев М.А., Аничков Н.М. (2001) Патологическая анатомия. Учебник. В 2 т. Т. 2, ч. 1. Медицина, Москва, 736 с.
- Патологическая анатомия. Курс лекций. Учебное пособие (1998) Под ред. В.В. Серова, М.А. Пальцева. Медицина, Москва, 640 с.
- Струков А.І., Сєров В.В. (2004) Патологічна анатомія. Підручник (Пер. з рос.). 4-те вид., стереотипне. Факт, Харків, 864 с.
- Шлопов В.Г. (2004) Патологічна анатомія. Нова книга, Вінниця, 758 с.