Геморрагические состояния и тромбоцитоз
Геморрагическими состояниями (синдромом), ранее известными как геморрагические диатезы, называют склонность к кровоточивости. Она может возникать как спонтанные кровотечения или длительное время протекать латентно, выявляясь при травмах или оперативных вмешательствах.
Причина схематически заключается в нарушениях тромбоцитарного, плазменного или сосудистого механизмов свертывания крови. Но часто имеет значение комбинация нескольких факторов. Соответственно трем основным механизмам нарушения свертывания выделяют коагулопатии (нарушения плазматических факторов свертывания), тромбоцитопении и тромбоцитопатии и сосудисто обусловленные нарушения свертывания (вазопатии). Эти состояния могут быть врожденными и приобретенными.
Схематически группы ДД можно представить следующим образом:
- Врожденные нарушения:
- сосудистые: телеангиоэктазия, генетически детерминированные дисплазии соединительной ткани (синдром Марфана, Элерса — Данлоса и др. Для них же имеет значение тромбоцитарный и, в меньшей степени, плазменный механизм);
- изменение количества и качества тромбоцитов;
- нарушение формирования тромба (гемофилии А, В, С), болезнь Виллебранда — Юргенса, дефицит других факторов свертывания, дисфибриногенемия, нарушения ингибиторов фибринолиза.
- Приобретенные изменения:
- сосудистые (цинга);
- иммунные (болезнь Шенляйн — Геноха, СКВ и др.);
- кортикостероиды;
- инфекции;
- сенильная пурпура;
- изменения количества и качества тромбоцитов, в том числе уремия, болезни печени и селезенки, В12-, фолат- и железодефицитные состояния;
- нарушения формирования тромба при антикоагулянтной и фибринолитической терапии, болезнях печени, волчаночно-подобных состояниях;
- ДВС-синдром;
- острая гипокальциемия (прием цитрата);
- эффект разведения;
- трудотерапия;
- укусы змей и некоторых насекомых;
- лечение препаратами чеснока.
В физиологических условиях существует система нормального гемостаза. Свертывание крови запускается двумя системами. Быстрый путь начинается с освобождения тканевой тромбокиназы. Более медленный начинается с активации внутрисосудистых циркулирующих факторов, например, с адгезии и агрегации тромбоцитов на стенке поврежденного сосуда. Освобождаются АТФ, III фактор тромбоцитов, из плазматических факторов активируется XII, а также запускается каскад свертывания. Конечным продуктом является нерастворимый фибрин-полимер. В организме имеется фибринолитическая система, предохраняющая от неконтролируемого внутрисосудистого свертывания крови. В нее входят ингибиторы свертывания: антитромбин, протеин С и S, фибринолизин, плазмин (табл. 4.1).
Таблица 4.1
Номенклатура и синонимы факторов коагуляции
| № | Наиболее распространенное название | Синонимы |
| I | Фибриноген | — |
| II | Протромбин | — |
| III | Тканевой фактор | Тромбопластин |
| IV | Кальций ионизированный | — |
| V | Проакцеллерин | Лабильный фактор; глобулин-акцеллерин; тромбоген |
| VI | Г ипотетический | — |
| VII | Проконвертин | Стабильный фактор; акцеллятор превращения сывороточного протромбина |
| VIII | Антигемофильный фактор | Антигемофильный глобулин; антигемофильный фактор А; тромбоцитарный профактор 1; тромбопластиноген |
| IX | Плазменный компонент тромбопластина | Фактор Кристмаса; антигемофильный фактор В; тромбоцитарный профактор 2 |
| X | Фактор Стюарта | Фактор Прауера; тромбокиназа |
| XI | Предшественник плазменного тромбопластина | Антигемофильный фактор С |
| XII | Фактор Хагемана | Фактор Гласса; контактный фактор |
| XIII | Фибрин стабилизирующий фактор | Фактор Лаки — Лоранда; плазменная трансглютаминаза; фибринолигаза |
| — | Прекалликреин | Фактор Флетчера |
| Высокомолекулярный кининоген | Фактор контактной активации; фактор Фитцджеральда; фактор Вильямса; фактор Фладжека; фактор Рейда; фактор Вашингтона |
Внимательный расспрос больного, сбор полноценного анамнеза — неотъемлемое условие установления нозологического диагноза геморрагического синдрома. Важно выяснить наследственную предрасположенность. Если ответ на этот вопрос положительный, то с большой долей уверенности можно говорить о коагулопатиях. Но ряд геморрагических состояний наследуется а/p, поэтому не в каждом поколении выявляют больного. Возможны и новые мутации.
Одним из первых этапов диагностики является выяснение самостоятельности или вторичности геморрагического синдрома: принимал ли пациент какие-либо препараты, оказывающие влияние на гемостаз и нет ли у него заболеваний, проявлениями которых может быть синдром кровоточивости, как, например, при СКВ, инфекционном эндокардите, сепсисе, злокачественных опухолях (особенно кроветворной системы). Выяснение возраста, в котором у пациента начались кровотечения, разрешающих факторов (при отсутствии травм или при их наличии, малые оперативные вмешательства) или локализация кровотечений (метроррагии, носовые кровотечения, внутрисуставные кровоизлияния) ведут к правильному диагнозу. Точно так же, как и определение интервала между временем травмы и началом кровотечения. Если существует светлый латентный промежуток, то можно думать о коагулопатии. Немедленное начало кровотечения после травмы свидетельствует в пользу нарушения тромбоцитарного механизма, обычно при этом длительное сдавление кровоточащего участка приводит к тромбированию.
Осмотр больного позволяет выявить особенности геморрагического синдрома: кожные и органные проявления (например, изменения суставов), распространенность и время рецидивирования. Экхимозы и гематомы могут быть как при коагулопатиях, так и при тромбоцитопениях. При последних могут быть и умеренно выраженные петехии, свойственные прежде всего сосудисто-обусловленным геморрагическим синдромам. При тромбоцитопениях они объясняются тем, что эндотелий, постоянно омываемый кровью, сам непосредственно из крови не питается. Его жизнедеятельность поддерживается фагоцитозом тромбоцитов. В случае же их недостатка развивается дистрофия эндотелия, эритроциты под действием внутрисосудистого давления буквально проламывают его и выталкиваются за пределы сосуда.
Для дальнейшей характеристики геморрагического синдрома необходимо искать кровоизлияния на слизистой оболочке, конъюнктивах, внутрисуставно. К геморрагическим проявлениям следует относить кровотечения из десен спонтанные, длительные кровотечения из лунки удаленного зуба, прокрашивание слюны при чистке зубов нежесткой щеткой. Необходимо помнить о существовании возрастных особенностях геморрагического синдрома у детей при коагулопатиях (гемофилия). При этом очередность симптоматики такова: кефалгематома в родах, кровотечение из пупочного остатка, длительное наличие кровяных корочек, кровотечения при прорезывании зубов, экстравазаты (синяки) и гематомы при повышенной активности ребенка (старше года). Геморрагическим проявлением следует считать маточное кровотечение и гематурию при отсутствии заболеваний, симптомами которых они являются (например, ювенильные маточные кровотечения, нефритическая форма гломерулонефрита и т. д.).
Сравнительно в малом количестве исследований можно с большой степенью достоверности выявить наличие, тип и степень нарушения гемостаза.
Длительность кровотечения характеризует тромбоцитарное звено свертывания, хотя она может быть изменена и при сосудистых расстройствах. Для характеристики тромбоцитарного звена свертывания крови проводят пробу Ринера и рекомендуют следующую методику. На плечо накладывается манжетка аппарата Рива — Роччи и создается давление в 40 мм. рт. ст. На пальмарной поверхности предплечья наносят два надреза длиной 9 мм и глубиной 1 мм. Каждые 30 с полоской фильтровальной бумаги определяют, продолжается ли кровотечение. В норме оно прекращается к 4,5-й минуте с пределами колебания в 1,5 мин. Пробу регистрируют как положительную при нарушениях числа и функции тромбоцитов, в том числе при приеме аспирина.
Проба Кончаловского — Румпеля заключается в наложении манжетки на предплечье и поддержании в ней в течение 5 мин артериального давления (АД), среднего между систолическим и диастолическим. Появление петехий свидетельствует о сосудистых или тромбоцитарных расстройствах.
Время свертывания крови (по Фонио, Мас-Магро, Бюркеру и др.) характеризует коагуляционное звено гемостаза. Техника проведения пробы: после прокола пальца скарификатором первую каплю крови удаляют стерильной ватой. Вторую — наносят на часовое стекло, которое помещается в термостат (повсеместно используют предметные стекла, а вместо термостата —- ладонь лаборанта). Сразу после попадания капли на стекло начинается перемешивание капли с помощью палочки с прикрепленной к ее концу тонкой леской или конским волосом (применение инъекционных игл не рекомендуется). С помощью секундомера регистрируют первое появление нитей фибрина (начало свертывания) и образование сгустка (окончание свертывания крови).
Определение числа тромбоцитов лучше проводить в ближайшие 30 мин после забора крови. Обычно их число колеблется в пределах 150⋅109–400⋅109/л.
Использование указанных трех лабораторных тестов (длительность кровотечения, время свертывания крови и определение числа тромбоцитов) может помочь врачу в ДД различных геморрагических заболеваний и позволяет отнести конкретный случай к группе вазопатий, коагулопатий и тромбопений или тромбоцитопатий (табл. 4.2)
Таблица 4.2
Основные практически значимые ДД-признаки различных геморрагических состояний
| Вид геморрагического состояния | Возможная нозологическая форма | Количество тромбоцитов | Длительность кровотечения | Время свертывания крови |
| Коагулопатия | Гемофилия | Норма | Не изменено | Увеличено* |
| Тромбоцитопения | «Идиопатическая тромбоцитопеническая пурпура» | Значительно снижено* | Увеличена* | Не изменено |
| Тромбоцитопатия | Некоторые формы тромбоцитопатий | Норма или незначительно снижено | Может быть увеличено | Может быть увеличено |
| Вазопатия | Геморрагический васкулит | Норма или повышено | Не изменено | Не изменено |
*Маркирующие признаки.
При тромбопении во многих случаях показана пункция костного мозга для определения состояния мегакариопоэза.
Определение функции тромбоцитов, что важно для уточнения формы тромбоцитопатии, достигается изучением их агрегации (при добавлении коллагена, эпинефрила, АТФ), адгезии (на стеклянной пластинке) или способности выделять факторы, например III, фосфолипид мембраны. В ряде случаев необходимо определить наличие антитромбоцитарных антител.
Многие лабораторные пробы основаны на измерении времени между внесением в изучаемый объект активированного фактора и появлением признаков свертывания (фибрина). Частичное тромбопластиновое время определяется рекальцификацией плазмы крови с добавлением фосфолипида, заменяющего тромбоцитарный фактор, и каолина и определением времени начала свертывания. Оно увеличивается при дефиците I, II, V, VI–VIII факторов, а также при применении гепарина. На показатели влияют также прекалликреин, калликреин, кининогены, продукты распада фибрина. Не влияет состояние VII, XIII факторов, III тромбоцитов, их число и функция. Протромбиновое время определяют добавлением тканевой тромбокиназы в рекальцинированную плазму крови пациента, что дает возможность изучить состояние быстрого пути свертывания. Увеличивается при дефиците I, И, V, VII, IX факторов, а также при применении гепарина. Метод может быть использован для контроля применения витамина К, поскольку он влияет на II, V, IX, X факторы. При гемофилии протромбиновое время не изменяется. Тромбиновое время увеличивается при наличии циркулирующего антитромбина или гипо- и дисфибриногенемии (табл. 4.3).
Таблица 4.3
Наиболее вероятные причины геморрагических состояний по результатам проведения первичных скрининговых исследований
| Числотромбоцитов | Время кровотечения | Частичное тромбопластиновое время | Протромбиновое время | Вероятный диагноз | Наиболее частые причины | |
| наследственные | приобретенные | |||||
| Уменьшено | Увеличено | Норма | Норма | Тромбоцитопения | Синдром Олдриха и др. | Идиопатическая тромбоцитопеническая пурпура; медикаментозная, др. вторичные формы |
| Норма | Увеличено | Увеличено | Норма | Болезнь Виллебрандта | — | СКВ; ингибиторы VIII фактора |
| Норма или повышено | Увеличено | Норма | Норма | Нарушение функции тромбоцитов | Тромбастения; дефицит релизинг-факторов | Лекарственные средства; уремия; диспротеинемия; тромбоцитемия |
| Норма | Норма | Увеличено | Норма | Нарушения свертывания по интринзинг-системе | Гемофилия А или В; дефицит прекалликреина, высокомолекулярного кининогена; XI и XII факторов | Ингибиция VІІІс фактора и СКВ |
| Норма* | Норма* | Увеличено | Увеличено | Нарушение свертывания по общему или нескольким патогенетическим путям | Дефицит V и X факторов, протромбина или фибриногена; дисфибриногенемия | Болезни печени; дефицит витамина К; внутрисосудистое свертывание; фибриногенолиз; гепарин |
| Норма | Норма | Норма | Увеличено | Нарушение свертывания по экстринзинг системе | Дефицит VII фактора | |
| Норма | Норма | Норма | Норма | Дефицит VIII фактора; наследственная телеангиоэктазия | Аллергическая пурпура; цинга; лекарственные средства; аутоэритроцитарная сенсибилизация | |
*Может быть изменен при приобретенных состояниях, обусловленных дефицитом нескольких факторов свертывания.
В специализированных лабораториях возможно определение отдельных факторов свертывания, например, уровень фибриногена в норме составляет 2–4 г/л.
Коагулопатии обусловлены нарушениями плазменных факторов свертывания. Для них типичны массивные кожные кровоизлияния, внутрисуставные, внутримышечные, висцеральные кровотечения, развившиеся от минимальнейшей травмы, часто с латентным промежутком. Известны наследственные и приобретенные формы коагулопатий. Приобретенные коагулопатии обычно характеризуются изменением нескольких факторов свертывания, причем на фоне какого-то основного заболевания. При наследственных формах, как правило, качественно или количественно изменяется только один фактор. Теоретически это может касаться любого плазменного фактора свертывания, но 80% всех коагулопатий приходятся на болезнь Виллебранда и гемофилии А и В.
Гемофилия А как дефицит структурно неизмененного VIII фактора свертывания (АГГ — антигемофильного глобулина) — классическая болезнь свертывания, на Х-сцепленный рецессивный характер наследования которой любят указывать как генетики, так и терапевты и, поблескивая интеллектом, ссылаются на пример царствовавшей семьи. Особенно охотно о гемофилии А рассуждают гематологи: они ощущают себя на коне после того, как получили в свои руки АГГ — единственный реальный препарат, позволяющий не только сохранить жизнь больного (это достигалось и криопреципитатами), но сохранить и качество жизни, предотвратить инвалидность.
В настоящее время доказано, что возможен не только дефицит фактора, но и его структурные нарушения. Как классическое Х-сцепленное рецессивное заболевание гемофилия поражает половину мальчиков от матерей-носительниц гена, а половина рожденных в таких семьях дочек становится носительницами гена, 1/3 всех случаев гемофилии рассматривается как новая мутация. Частота гемофилии в популяции по расчетам оказывается близкой к 1:10 000 населения. Но такую массу больных врач в своей практике не видит, так как пенетрантность гена может варьировать, дефицит в какой-то степени компенсируется другими факторами, да и активность фактора должна снизиться до 5–25% нормы, чтобы заявить о себе в легкой форме, проявляясь большей, чем обычно, кровоточивостью после ранений или удаления зубов. Но в практической работе основная масса врачей (и, может быть, оправданно) не любит лезть в лечебно-диагностические дебри, если патологические симптомы «проходят сами». Заболевание средней степени тяжести (уже наверняка диагностируемое) развивается при уровне VIII фактора в 1–4%. Тяжелые формы отмечают при снижении АГГ до 0,8–0,9% нормы. В клинической картине наряду с большими гематомами и характерными для этого заболевания кровоизлияниями в суставы, обусловливающими анемию, билирубинемию, резорбционную лихорадку, известны гематурия, гематомезис, редко — кровохарканье. Тромбопластиновое время увеличивается при снижении уровня VIII фактора ниже 30%. Число тромбоцитов, протромбиновое время и время кровотечения не изменяются.
Гемофилия В проявляется схожей, но значительно более мягкой симптоматикой. Обусловлена дефицитом фактора Хагемана — Кристмаса (IX), выявляют с частотой 1:100 000 населения.
Суммарно гемофилии А+В выявляют даже чаще, чем болезнь Виллебранда. Это связано с тем, что гемофилии — значительно более выраженные состояния, клинически значимые случаи.
Говорят о существовании гемофилии С, отличием которой от гемофилии А является а/p тип наследования. Клиническая картина полностью аналогична, распространенность низкая: не более 1–2% всех типов гемофилий.
Изменения фибриногена (I фактора) могут быть качественными (дисфибриногенемия), обусловленными мутациями гена, что приводит к нарушению агрегации фибрин-мономеров в фибрин-полимер, и количественными (гипо- и афибриногенемия). Последний вариант обусловлен снижением синтеза I фактора. При значительном снижении уровня фибриногена снижается содержание и других факторов. Клиническая картина при поражении I фактора свертывания крови соответствует тяжелому варианту гемофилии.
Болезнь Виллебранда — Юргенса обусловлена дефектом одноименного фактора свертывания и является, видимо, самой частой наследственной коагулопатией. По а/д типу наследуется качественный дефект, а по а/p типу передается количественный дефект этого фактора. Фактор влияет как на свертывание крови, образуя комплекс с VIII фактором, так и на адгезию тромбоцитов, что позволяет некоторым авторам рассматриваете болезнь Виллебранда в группе тромбоцитопатий. Заболевание проявляется соответствующим длительным анамнезом, кровоизлияниями в кожу и слизистую оболочку, кровотечениями после минимальнейших травм, экстракции зуба, а также метроррагиями, носовыми кровотечениями. Применение свежезамороженной плазмы крови надолго восстанавливает нормальное свертывание. В современных условиях возможно введение синтетического производного вазопрессина, выводящего VIII фактор и фактор Виллебранда из депо в организме.
Приобретенная коагулопатия в практике чаще всего развивается при заболеваниях печени. В печени синтезируются и подвергаются разрушению практически все плазменные факторы свертывания. При тяжелых гепатоцеллюлярных процессах снижается синтез факторов свертывания и активируется протеолитическая система, что создает дополнительные условия для кровотечений. Риск развития последних еще более возрастает при спленомегалии, сопровождающейся усиленным разрушением тромбоцитов. Уровень снижения плазменных факторов свертывания при заболеваниях печени происходит параллельно развитию гипоальбуминемии, что можно использовать как опорный клинический признак. Уменьшение объема поступающей в кишечник желчи, дефицит питания, нарушение кишечной флоры в совокупности с поражением паренхимы печени ведут к нарушению обмена витамина К, определяющего синтез II, VII, IX и X факторов свертывания. Восстановление протромбинового времени и увеличение выраженности вышеуказанных факторов в плазме через 12–24 ч после инъекции менадиона натрия бисульфита свидетельствует в пользу холестаза при сохранной паренхиме печени.
Содержание в крови антикоагулянтов повышается при гипернефроме и полипозе кишечника. В ряде случаев эти состояния могут манифестировать с геморрагического синдрома: мелена, гематурия, меноррагия. Но чаще указанные признаки, а также кровоизлияния в места инъекций, в тяжелых случаях и субдуральные гематомы развиваются при неудовлетворительном контроле за антикоагулянтной терапией. В практике это гепарин и кумарины (антагонисты витамина К). Гепарин действует содружественно с антитромбином III как ингибитор тромбина. Одновременно блокируются ІХа и Ха факторы. Контроль состояния свертывания при гепаринотерапии можно провести, определяя частичное тромбопластинове время или тромбиновое время. В специализированных условиях возможно определение уровня гепарина в плазме крови. Антагонисты витамина К блокируют синтез II, VII, IX и X факторов, нарушая реакцию карбоксилирования. Последняя активирует указанные факторы. Ряд препаратов могут не только сами блокировать некоторые факторы свертывания, но и усиливать действие антикоагулянтов. Это декстран, хинидин, индометацин, барбитураты, рифампицин и др.
Синдром диссеминированного внутрисосудистого свертывания (ДВС-синдром) может быть острым и хроническим. В норме существует равновесие между свертывающей и противосвертывающей системой, в частности, фибринолизом. При массивном потреблении факторов свертывания наступает истощение этой системы, развивается тромбоцито- и фибриногенопения, в крови циркулирует большое количество продуктов распада фибрина, которые сами способны блокировать свертывание. Массивный геморрагический синдром при этом сосуществует с тромбозами, эмболиями, нарушением микроциркуляции.
Первичный фибринолиз способны запустить рак простаты, лейкемии, травмирующие обширные операции. В терапевтических целях фибринолиз достигается применением стрепто- и урокиназы.
Приобретенные коагулопатии могут быть обусловлены наличием циркулирующих антител к соответствующим факторам свертывания. В частности, их выявляют у 10–12% больных гемофилией А, что существенно сказывается на успехе применения АГГ. Из других состояний антитела нередко выявляют при СКВ.
Нарушения тромбоцитарного звена могут касаться как числа тромбоцитов (тромбоцитопении и тромбоцитоз), так и их функции (тромбоцитопатии), быть первичными и приобретенными.
Тромбопенией считается уменьшение количества тромбоцитов в периферической крови ниже 150⋅109/л. Но кровотечений при этом числе тромбоцитов еще не возникает. Первые слабовыраженные геморрагические симптомы появляются при уменьшении числа тромбоцитов до 30⋅109/л, при уровне 20⋅109/л они часты, а при значениях 10⋅109/л — обязательны и выражены. Чем быстрее происходит уменьшение числа тромбоцитов, тем раньше, даже при относительно благополучных показателях их содержания в крови, развиваются кровотечения. Возраст пациентов и превалирование старых тромбоцитов в их общем пуле также повышают вероятность возникновения кровотечений. Для тромбоцитопений типичны петехии и экстравазаты (пурпура). В отличие от коагулопатий кровотечение развивается непосредственно после травмы и может быть уменьшено или приостановлено поджатием кровоточащего участка. Возможны кровоизлияния во все органы, но особенно опасны кровоизлияния в мозг.
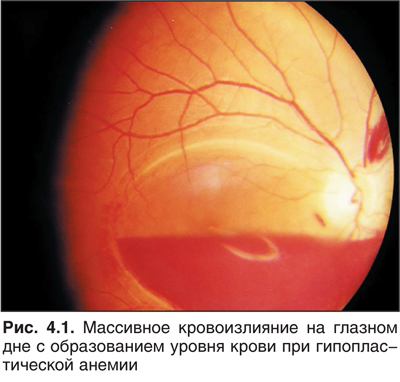
Нарушения пролиферации протекают с уменьшением числа мегакариоцитов в костном мозге и могут возникать при врожденных синдромах Фанкони, Вискотта — Олдрича и аномалии Мей — Хеглина. В двух последних случаях возникают и тромбопатии. Приобретенные нарушения созревания тромбоцитов часто сочетаются с апластическими анемиями (рис. 4.1), инфильтрацией костного мозга опухолевыми клетками. Причем в последнем случае тромбоцитопенический эффект обусловлен несколькими слагающими: вытеснение мегакариоцитов опухолевыми клетками, аутоантителами к циркулирующим кровяным пластинкам, депонированием их и разрушением в увеличенной селезенке.
Разрушение тромбоцитов происходит также на протезах (клапаны сердца, имплантанты сосудов), при включении экстракорпоральных путей кровообращения (гемодиализ, гемосорбция; имеет значение и механизм гемодилюции), при потреблении красных кровяных пластинок в гигантских кавернозных гемангиомах (синдром Казабаха — Меррита), а также в декомпрессированных органах, подвергнутых ранее сжатию.
Тромбоциты усиленно разрушаются при операциях с гипотермией.
Массивные обменные переливания крови могут вести к вторичной тромбоцитопении за счет эффекта разбавления (на определенном этапе может включаться иммунный механизм).
Тромбоцитопению вызывает ионизирующее излучение и целый ряд препаратов.
Непосредственно костный мозг угнетают антиметаболиты, антимитотические средства, противоопухолевые антибиотики, бензены, азидотимидин. Аплазию костного мозга могут обусловить также хлорамфеникол, соли золота, мезантоин, триметадион, фенилбутазон, циметидин.
Селективно угнетают мегакариоциты этанол, эстрогены, толбутамид, хлоротиазиды.
Непосредственно разрушает тромбоциты и запускает иммунный механизм гепарин.
Иммунный механизм формирования тромбоцитопении вызывают дезипрамин, диазепам, дигитоксин, ацетазоламид, гидрохлорохин, карбамазепин, лидокаин, метилдопа, новобиоцин, органические соединения мышьяка, пара-аминосалициловая кислота (ПАСК), соли золота, сульфаметазин, хлорвинил, хлорпропамид, ранитидин, циметидин, хинин, диклофенак.
Вероятный иммунный механизм формирования тромбоцитопении (антитела выявляют не всегда) известен для дисульфирама, кислоты ацетилсалициловой, барбитуратов, спиронолактона, солей висмута, диазоксида, препаратов наперстянки, в том числе дигоксина, инсектицидов, кодеина, мепробамата, нитроглицерина, органических красителей для волос, преднизолона, пропилтиоурацила, а также для резерпина, ртутных диуретиков, стрептомицина, алкалоидов спорыньи, сульфаниламидов, тетрациклинов, тетраэтиламмония, тиомочевины, фенацетина, фенилбутазона, цефалотина, хлорохина, эритромицина.
Ускоренное разрушение тромбоцитов (в норме тромбоцит живет 10 дней), чаще всего приобретенное состояние, возникает при васкулитах, инфекциях, прежде всего при сепсисе, вызванном грамот- рицательной микрофлорой. Любое воспаление при недостатке ингибиторной системы вызывает появление в крови продуктов арахидоновой кислоты — кининов и тромбоксана А2. Тромбоцит существует за счет циклического аденозинмонофосфата. Тромбоксан же блокирует аденилциклазу, переводящую АТФ в циклический аденозинмонофосфат (цАМФ). Поэтому при хроническом воспалении развивается нарушение функции тромбоцитов, они не способны удерживаться в токе крови, агрегируются и формируются микротромбозы. А это обусловливает вторичным тромбоцитопениям потребления.
Механизм влияния различных препаратов на функцию тромбоцитов суммирован в табл. 4.4.
Таблица 4.4
Влияние некоторых препаратов на функцию тромбоцитов
| I. Ингибиторы обмена арахиндоновой кислоты
A. Ингибиторы циклоксигеназы
Б. Ингибиторы синтеза тромбоксана B. Блокаторы высвобождения арахидоновой кислоты (кортикостероиды) II. Активаторы аденилатциклазы
III. Ингибиторы фосфодиестеразы
IV. Антимикробные препараты
V. Антикоагулянты
VI. Кровозаменители
VII. Лекарственные средства, применяемые в кардиологии
VIII. Психотропные препараты и анестетики
IX. Цитостатики
X. Другие
|
Тробоцитопеническая пурпура новорожденных вызывается аллоантителами, вырабатывающимися на материнские антигены. Антитела относятся к классу IgG, проходят через плаценту и разрушают тромбоциты плода. Заболевание развивается, если мать и ребенок имеют разный набор антигенов на поверхности тромбоцитов. Чаще у матери отсутствует антиген PLA-1 (3% американской популяции белых).
Болезнь Верльгофа (идиопатическая тромбоцитопеническая пурпура) может протекать в нескольких формах. Острую форму выявляют преимущественно у детей. Не исключен а/д механизм передачи структурной неполноценности тромбоцитов, облегчающей при наличии дополнительных повреждающих факторов выработку антител. Выявляемые антитела относятся к классу IgG. Имеются сведения, что антитела повреждают не тромбоциты, а тормозят «отшнуровку» кровяных пластинок от мегакариоцитов. У новорожденных от матерей с идиопатической тромбоцитопенической пурпурой (ИТП) описаны преходящие тромбопенические состояния, связанные с трансиммунным переносом антител.
Такой разброс сведений об этиологии заболевания не исключает, что в перспективе будут выделены несколько самостоятельных форм аналогично тому, что произошло с некогда единым понятием гемофилии.
В анамнезе за 2–3 нед до начала заболевания часто отмечается вирусная инфекция. Быстрое уменьшение количества тромбоцитов до 20⋅109/л и ниже сопровождается спонтанными петехиальными высыпаниями, экхимозами, расположение их беспорядочное, цвет варьирует в зависимости от давности кровоизлияния. Геморрагии в склеру, кровотечения из слизистой оболочки, мено- и метроррагии, реже — почечные и кишечные кровотечения дополняют симптоматику. Заболевание может длиться от нескольких дней до месяцев. Через 6 мес выздоравливают до 70% детей. У остальных формируется хроническая форма заболевания.
Болезнь Верльгофа у взрослых пациентов прогностически протекает менее благоприятно. Чаще начинается исподволь с непостоянной петехиальной сыпи на фоне общего хорошего состояния и нормальных размеров селезенки. Женщины поражаются в 3 раза чаще мужчин. Спонтанные ремиссии отмечают не более чем у 10% больных. Количество тромбоцитов уменьшается до 10–70⋅109/л, нередко выявляют гигантские и полуразрушенные пластинки. В пунктате костного мозга — раздражение и омоложение мегакариоцитов.
Тромбоцитопеническая тромботическая пурпура Мошковица (тромботическая микроангиопатия) по ряду признаков может быть отнесена и в группу васкулитов. Фульминантное заболевание с почти обязательным летальным исходом проявляется обвальным развитием симптоматики. Лихорадка, множественные кровоизлияния в кожу (пурпура), кровотечения из слизистой оболочки, гениталий. Нарушения микроциркуляции ведет к спутанности сознания, неврологическим знакам, почечной недостаточности. В анализах крови выраженная тромбоцитопения, анемия, ретикуло- и фрагментоцитоз, лейкоцитоз со сдвигом влево. Длительность жизни тромбоцитов уменьшена в 2,5 раза и едва достигает 4 дней. В пунктате костного мозга количество мегакариоцитов нормальное или даже увеличенное.
ДД необходимо проводить с ДВС-синдромом, при котором, в отличии от тромбоцитопенической тромботической пурпуры, нет выраженного гемолиза.
Тромбоцитопатия — нарушение функциональной способности тромбоцитов при сохранении их количества. При этом возможны как нарушения структуры самих пластинок с недостатком фосфолипидов оболочки и нарушением адгезивных свойств, «гладкостью» клеток при электронной микроскопии и отсутствием псевдоподий, так и отсутствием плазменных факторов. Тромбастении, протекая в целом благоприятнее, чем другие формы геморрагических синдромов (диатезов), возникают, видимо, чаще, чем диагностируются. Есть сведения, что около 20% всех ювенильных маточных кровотечений обусловлены тромбастенией.
Наиболее известным вариантом наследственных тромбастений является тромбастения Гланцмана — Пика, а/p заболевание. Клиническая картина вариабельна и напоминает болезнь Виллебранда. Отмечают нормальное число тромбоцитов в периферической крови, нарушение агрегации тромбоцитов при добавлении коллагена, тромбина, АТФ.
Синдром Бернарда — Сулье (синдром гигантских тромбоцитов) наследуется а/p. Кровяные пластинки резко увеличены в размерах, содержат много гранул, иногда отмечают легкую тромбоцитопению. Адгезионная способность тромбоцитов снижена, но может быть и повышена. Возможны тяжелые висцеральные кровоизлияния. Количество мегакариоцитов не изменено.
Синдром Мей — Хеглина наследуется а/д. Объясняют нарушенной фрагментацией мегакариоцитов. Выявляют редко, протекает с умеренной кровоточивостью. В крови выявляют гигантские тромбоциты и базофильную мелкую зернистость гранулоцитов. Если одновременно уменьшается и количество пластинок, то прогноз значительно ухудшается.
Синдром Вискота — Олдрича наследуется Х-сцепленно рецессивно. Тромбопения ассоциируется с ультраструктурной неполноценностью кровяных пластинок. Геморрагический синдром сочетается с экземами, дерматитами, склонностью к инфекционным заболеваниям, что и определяет плохой прогноз.
Приобретенные нарушения функции тромбоцитов возникают при многих заболеваниях (миелопролиферативные, диспротеинемии, уремия, гепатопатии) и применении большого количества лекарственных препаратов (декстран, индометацин, ристомицин, трициклические антидепрессанты). Патогенетические механизмы разнообразны и выяснены не до конца.
Если тромбоцитопения понятна врачу и расшифровка этого лабораторного признака проводится по достаточно известным алгоритмам, то ТРОМБОЦИТОЗ либо остается незаметным, либо вызывает «диагностическую фрустрацию».
Тромбоцитоз — повышение количества тромбоцитов в периферической крови выше 450⋅109/л. Клинических проявлений при тромбоцитозе обычно не бывает или они мало выражены до уровня тромбоцитов 800⋅109/л.
Тромбоцитоз в зависимости от причин может относиться к одной из трех основных больших групп:
1) автономный или первичный;
2) транзиторный или физиологический;
3) реактивный или вторичный.
Основные причины тромбоцитоза и его наиболее существенные формы:
А. Автономный тромбоцитоз:
- эссенциальный тромбоцитоз (первичная тромбоцитемия). Относится к миелопролиферативным заболеваниям. В 95% случаев протекает с лейкоцитозом. Эритропоэз не изменяется или умеренно активируется. При этом выявляют микросфероцитарную гипохромную анемию. Анемия усиливается при интермиттирующих гастроинтестинальных кровотечениях. Экхимозы и более обширные кровоизлияния в кожу и мышцы легко развиваются после травм. Нарушения микроциркуляции приводят к интенсивной боли в конечностях, поскольку спонтанная агрегация тромбоцитов приводит к микротромбозам. Идиопатический тромбоцитоз бывает очень сложно отдифференцировать от истинной полицитемии и остеомиелофиброза. Микротромбозы при тромбоцитемии со спонтанной агрегацией пластинок предотвращаются применением ацетилсалициловой кислоты в невысоких дозах. Она блокирует циклоксигеназу тромбоцитов, это прерывает синтез тромбоксана А2, который и вызывает агрегацию клеток. При первичном или автономном тромбоцитозе количество тромбоцитов не зависит от регуляторных процессов. Количество и размеры мегакариоцитов значительно увеличиваются. Продукция тромбоцитов может возрастать в 15 раз (тромбоцитемия). Автономная пролиферация может затрагивать только тромбоциты или касаться других клеточных линий, как при миелопролиферативных заболеваниях;
- полицитемия истинная (хронический миелолейкоз). Одновременно увеличивается количество лейкоцитов и эритроцитов;
- миелофиброз генуинный и миелоидная метаплазия;
- хроническая миеломная лейкемия;
- рефрактерная анемия с избытком бластов;
- миелопролиферативные состояния при болезни Дауна.
Б. Реактивный тромбоцитоз:
- При острых состояниях:
- инфекция;
- острое воспаление;
- некроз тканей (в том числе инфаркт миокарда);
- постоперационный.
- При хронических заболеваниях:
- хронические инфекции (остеомиелит, туберкулез);
- желудочно-кишечные заболевания (язвенный колит, регионарный илеит, целиакия);
- хронические воспалительные болезни (РА, узелковый периартериит, острая ревматическая лихорадка, гранулематоз Вегенера);
- гиперостозинфантильный кортикальный (синдром/болезнь Роске-де Тони — Сильвермана. Передается а/д. Спонтанно разрешающееся заболевание периоста у детей грудного возраста. Заболевание манифестирует на 2–4-м месяце жизни, но описаны случаи рентгенологически документированного кортикального гиперостоза уже внутриутробно. У младенца появляются беспокойство, потеря аппетита, лихорадка, болезненность и опухание в области пораженных костей с возможными псевдопарезами. Позднее появляются увеличение и деформации пораженных костей. Лабораторно — гиперостоз, тромбоцитоз, ускорение СОЭ, повышение С-реактивного белка (СРБ) и активности шелочной фосфатазы сыворотки крови. Рентгенологически выявляют кортикальный гиперостоз многих костей. Вероятность его появления и выраженность (по убывающей): нижняя челюсть, большеберцовая кость, лучевая кость, локтевая кость, плечевая, ключица, реже — другие трубчатые кости, еще реже — ребра, лопатка и верхняя челюсть. При гистологических исследованиях на ранних этапах периост замещается тканью, богатой сосудами с выраженной инфильтрацией сегментоядерными лейкоцитами. Затем из этой ткани образуется остеоид с его последующей кальцификацией. Через некоторое время кость резорбируется вплоть до Restituio ad integrum. Весь процесс длится от нескольких недель до нескольких месяцев, но в 25% случаев возможны рецидивы, что приводит к задержке моторного развития ребенка. ДД ювенильного кортикального гиперостоза проводят, прежде всего, с остеомиелитом;
- саркоидоз;
- новообразования (лимфома Ходжкина, рак молочной железы, рак легкого, реже — любая карцинома). При опухолях чаще отмечают реактивный тромбоцитоз (чаще хронический), нередко коррелирующий с активностью основного заболевания. Количество тромбоцитов обычно не превышает 1 млн/мкл. Реактивный тромбоцитоз может возникать в период, когда новообразование клинически ничем не выделяется.
- При ускоренном гемопоэзе:
- острая потеря крови;
- гемолиз;
- по типу синдрома «отдачи» — ребаунд-тромбоцитоз (восстановление после мегалобластной анемии, после супресии костного мозга).
- При замедленном разрушении (аспленические состояния):
- после удаления селезенки;
- после тромбирования селезенки.
В норме селезенка содержит 1/3 общего количества тромбоцитов, которые способны переходить в периферическую кровь. Некоторое время после после удаления селезенки количество тромбоцитов может превышать 500–600⋅109/л, достигая 1000⋅109/л. Повышение числа тромбоцитов обычно отмечают в 1–10-й дни после спленэктомии, достигая максимума в 1–3-ю недели. Постспленэктомический тромбоцитоз спонтанно разрешается в сроки от нескольких недель до нескольких месяцев. Если имеется миелопролиферативное заболевание или гемолитическая анемия, тромбоцитоз может персистировать и дольше. Тем не менее, увеличение количества тромбоцитов — серьезное основание для назначения антитромботической терапии, хотя тромбозы выявляют редко, не чаще чем у 4% гематологически нормальных пациентов.
- Иммунные состояния:
- нефротический синдром;
- реакция «трансплантат» против хозяина.
- Дефицит витамина Е.
- Алиментарный тромбоцитоз (дефицит железа). Количество тромбоцитов при дефиците железа может колебаться от нормального до значительно повышенного, но возвращается к норме через 1–2 нед применения препаратов железа.
- Реакция на лекарственные средства:
- алкалоиды;
- цитроворум-фактор;
- кортикостероиды.
В. Физиологический тромбоцитоз (транзиторный):
- физические упражнения (перегрузки);
- стрессы (гиперадреналемия);
- беременность и роды.
При транзиторном (физиологическом) тромбоцитозе происходит мобилизация тромбоцитов из экстраваскулярных депо. Доступные депо включают селезенку и легкие. Адреналин освобождает 20-50% тромбоцитов. Адреналининдуцированный тромбоцитоз не возникает у больных с удаленной селезенкой, что еще раз подтверждает то, что высвобожденные тромбоциты поступают в кровь из селезенки. Реактивный тромбоцитоз часто реализуется за счет двух механизмов: освобождение тромбоцитов и повышение их образования (табл. 4.5). Реактивный тромбоцитоз подразделяется на острый и хронический. При остром реактивном тромбоцитозе тромбоциты возникают как острофазовые компоненты. Это характерно при острой кровопотере, острых воспалительных заболеваниях, при хирургических вмешательствах. Обычно количество тромбоцитов достигает 500–600⋅109/л, хотя описаны случаи реактивного тромбоцитоза с количеством тромбоцитов в 1000⋅109/л.
Таблица 4.5
ДД эссенциальной тромбоцитемии и реактивного тромбоцитоза
| Показатель | Тромбоцитемия | Реактивный (вторичный) тромбоцитоз |
| Общая масса мегакариоцитов | Значительно увеличена | Незначительно повышена |
| Количество мегакариоцитов | Увеличено | Увеличено |
| Объем мегакариоцита | Увеличен | Уменьшен |
| Степень продукции тромбоцитов | Повышена значительно | Повышена умеренно |
| Общая масса тромбоцитов | Увеличена значительно | Увеличена умеренно |
| Тромбэмболии и/или геморрагии | Типичны | Нехарактерны |
| Продолжительность тромбоцитоза | Обычно персистирует | Часто транзиторный |
| Спленомегалия | У 80% больных | Отсутствует |
| Количество тромбоцитов | >1000⋅109/л | <1000⋅109/л |
| Длительность кровотечения | Часто увеличена | Не изменена |
| Морфология и функция тромбоцитов | Часто изменена | Не изменена |
| Количество лейкоцитов | Увеличено в 95% случаев | Обычно в норме |
Сосудисто обусловленные геморрагические синдромы (вазопатии) — большая и разнородная группа заболеваний, объединенных по основному симптому: наличию геморрагий. Поражения сосудов могут быть наследственными (синдром Гиппель — Линдау, болезнь Ослера), иммунными (геморрагический васкулит), дистрофическими (цинга), инфекционными. Все это обусловливает нарушение проницаемости с выходом эритроцитов и других форменных элементов крови, появление экссудативно-геморрагических знаков на коже с гиперергическими элементами. Степень кровоточивости редко достигает угрожающих значений.
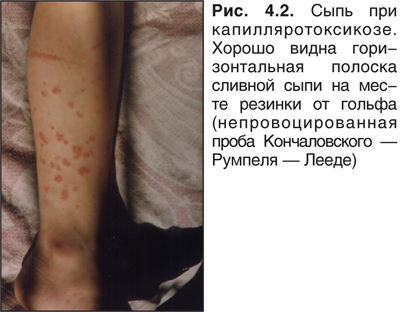
Пурпура Шенляйн — Геноха (капилляротоксикоз, анафилактоидная или иммунокомплексная пурпура) — классический пример иммунокомплексной болезни с выраженным компонентом внутрисосудистого свертывания. Провоцирующими факторами является инфекция, у стариков чаще дисбактериоз с активацией калликреин-кининовой системы. У детей в анамнезе нередко выявляют инфекцию верхних дыхательных путей и ротоглотки. Иногда провоцирующими факторами являются антибиотики, сульфаниламиды, жаропонижающие препараты. На коже появляются экссудативно-геморрагические элементы, часто сливные. Преимущественная локализация — разгибательная поверхность ног и рук, ягодицы, периартикулярные области, реже — на лице и туловище. Проба Кончаловского — Румпеля — Лееде может быть положительной (рис. 4.2). Очень типична схваткообразная боль в животе за счет так называемой гистаминовой интоксикации и поражения сосудов брыжейки и стенки кишечника. Возможны кровавый понос, некроз стенки кишки. Выражены общие симптомы: лихорадка, недомогание, артралгия и опухание голеностопных, коленных, реже — тазобедренных, достаточно редко — локтевых и плечевых и лучезапястных суставов. Экссудата в полости сустава при этом нет, деформаций не остается. Прогностически неблагоприятными являются гематурия и отеки, свидетельствующие о сосудистом процессе в почках с формированием капилляротоксического нефрита.
В анализах крови — лейкоцитоз, ускорение СОЭ.
Как синдром геморрагический васкулит может предшествовать СКВ, криоглобулинемии и другим заболеваниям.
Болезнь Ослера (наследственные геморрагические телеангиоэктазии) наследуется а/д. Гистологически определяют локальные расширения венул и капилляров с истончением стенки. Именно в этих местах и происходит разрыв с последующим кровотечением. Несмотря на то что заболевание является наследственным, телеангиоэктазии обычно формируются к возрасту 20–30 лет и старше. Они выглядят как вишнево-синюшные округлые непульсирующие образования. Излюбленная локализация — конъюнктива, слизистая оболочка носа, губ, десен, неба, желудочно-кишечного и респираторного трактов, мочевого пузыря, печень и селезенка, плечевой пояс, подушечки пальцев, подногтевое ложе. Надавливание на телеангиоэктазии вызывает их побледнение. ДД проводят с «сосудистыми паучками» при заболеваниях печени и с сосудистыми невусами (для них типична центральная пульсация). В паренхиматозных органах сосудистые эктазии представлены в виде артерио-венозных фистул. Отсюда становится понятной клиника заболевания: желудочно-кишечные кровотечения, гематурия, кровавая рвота, кровохарканье.
Цинга, обусловленная дефицитом витамина С, патогенетически объяснима нарушением синтеза гидроксипролина с образованием неполноценного коллагена, что и обусловливает высокую проницаемость сосудистой стенки. Первый симптом — кровоточивость десен при чистке зубов. У людей пожилого возраста ускоряется выпадение зубов. Обычно в современных условиях все-таки не выявляют кожных (патогномонична их связь с волосяными фолликулами) и поднадкостничных кровоизлияний. Можно определить концентрацию аскорбиновой кислоты в гранулоцитах. Следует помнить, что у 70–80% нашего населения отмечают дефицит аскорбиновой кислоты.
Сенильная пурпура (простая пурпура) — прогностически благоприятная форма пурпуры за счет атрофии кожи и дистрофии сосудистой стенки у лиц старческого возраста. При незначительных травмах или спонтанно на шее, лице, разгибательной поверхности предплечий, ног появляются петехии или небольшие экхимозы. После исчезновения экхимозов на коже могут оставаться четко очерченные с неправильными контурами ярко-красные пятна. Проявления пурпуры более отчетливы при АГ и венозных стазах. Паравазальные отложения гемосидерина со временем оставляют на коже коричневатые пятна. Простая пурпура может возникать и у людей молодого возраста, что объясняют наследственными механизмами, и особенно часто — у женщин. У них особенно отчетливо пурпура проявляется перед менструациями. Установление диагноза возможно после исключения васкулитов, тромбоцитопений и -патий, злоупотребления аспирином.
Криоглобулинемия чаще всего развивается как следствие аутоиммунных или хронических инфекционных заболеваний. Развитие процесса связано с комплексом, состоящего из IgG как антигена и IgM или IgG как антитело (смешанная криоглобулинемия). Моноклональная криоглобулинемия развивается при лимфопролиферативных заболеваниях. Клиническая картина характеризуется сосудистой пурпурой, распространяющейся с ног на ягодицы и переднюю брюшную стенку. Длительная пурпура обусловливает коричневую пигментацию. Обострение заболевания сопровождается артралгией, синдромом Рейно. У половины больных развивается нефрит. Степень выраженности проявлений коррелирует с концентрацией криоглобулинов. В противоположность пурпуре Шенляйн — Геноха, кишечные колики возникают очень редко.
В целом же криоглобулинемии подразделяются на следующие типы:
I. Моноклональная с единственным компонентом
Множественная миелома; макроглобулинемия Вальденстрема; хронический лимфолейкоз; неходжкинская лимфома; болезнь тяжелых цепей; эссенциальная (идиопатическая) моноклональная криоглобулинемия.
II. Смешанная моноклональная
Лимфопролиферативные заболевания; аутоиммунные заболевания; гепатит; эссенциальная (идиопатическая) смешанная криоглобулинемия (тип II); прочие состояния.
III. Смешанная поликлональная
- Инфекции
- Вирусные (мононуклеоз; цитомегаловирусный посттрансфузионный синдром; острый гепатит В; лаймовский артрит)
- Бактериальные (подострый бактериальный эндокардит; лепра; острый постстрептококковый нефрит; лимфогранулема венерическая; сифилис; Q-лихорадка)
- Грибковые (кокцидиомикоз)
- Паразитарные (кала-азар; токсоплазмоз; тропическая спленомегалия; эхинококкоз; малярия; шистозомиаз)
2. Аутоиммунные заболевания (СКВ; РА; узелковый периартериит; синдром Шегрена; склеродермия; тиреоидит; полимиозит; фиброэластоз эндокарда; вульгарная пузырчатка; синдром Бехчета; саркоидоз; пурпура Шенлейна — Геноха)
3. Болезни печени (постнекротический лаэннековский цирроз; билиарный цирроз; хронический гепатит)
4. Болезни почек (пролиферативный гломерулонефрит)
5. Семейная
6. Эссенциальная смешанная криоглобулинемия
Близко к криоглобулинемиям стоит пурпура при диспротеинемиях. Клинические признаки пропорциональны вязкости крови. Объясняется это тем, что гиперпротеинемия повреждает эндотелий, обусловливает агрегацию тромбоцитов и выпадение тромбина.
Инфекционные заболевания, особенно тяжелые, сопровождаются геморрагическим синдромом. Это обусловлено инфекционным повреждением сосудистой стенки, тромбоцитопенией, диссеминированным внутрисосудистым свертыванием. В тяжелых случаях развивается фульминантная пурпура как выражение аллергической реакции по типу Санарелли — Шварцмана. В стенках мелких сосудов выявляют фибриноидные некрозы, просвет обтурирован тромбами. Такое течение характерно прежде всего у детей. Но значительно чаще в практике отмечают кожные петехии (тяжелые формы гриппа, лептоспироз, тиф, риккетсиозы, подострый инфекционный эндокардит).