Анемии
Анемия — одно из наиболее распространенных патологических состояний человека. Но в практике любого врача-негематолога анемия чаще всего является синдромом, признаком другого заболевания.
Анемия — снижение уровня гемоглобина и/или эритроцитов в объеме крови. По уровню снижения содержания гемоглобина выделяют легкую степень анемии (110–90 г/л), среднюю (90–70 г/л) и тяжелую (ниже 70 г/л).
Среднее количество эритроцитов в периферической крови у мужчины составляет 4,5–5 млн/мм3 (=⋅1012/л) и 4–5 млн/мм3 (=⋅1012/л) у женщин. Уровень гемоглобина для мужчин в норме составляет 160±20 г/л, для женщин — 140±20 г/л. У детей число эритроцитов и концентрация гемоглобина практически не зависят от пола, но определяются возрастом. В первые дни жизни показатели гемоглобина и гематокрита выше, чем у взрослых. При условии нормально протекавшей беременности и хорошего обеспечения матери железом, определяющее значение имеет время перевязки пуповины. После рождения уровень гемоглобина и количество эритроцитов достаточно быстро снижаются. Наименьшие величины отмечают в конце 2-го месяца жизни (табл. 3.1).
Таблица 3.1
Изменение показателей гематокрита и некоторых других характеристик красной крови в зависимости от возраста (Natan D., Orkin S., 2003)
| Возраст | Гематокрит, % | Ретикулоциты, ‰ | Средний диаметр эритроцитов | МСV, мкм3 | МСН, пкг | МСНС, % |
| 1 день | 56 (54–59) | 42 (15–65) | 8,0±0,4 | 106±7 | 35,5±1,5–2,5 | 33,5±1,1–11,7 |
| 3 дня | 58 (56–62) | 41 (13–60) | — | 106±6 | — | 33,4±2,0 |
| 5 дней | 60 (58–62) | 30 (10–50) | — | — | — | — |
| 7 дней | 58 (56–61) | 10 (5–15) | 8,1±0,2 | 103±7 | 35,5±1,6–2,5 | 34,5±1,1–1,7 |
| 2 нед | 55 (53–58) | 8 (3–13) | — | 100±5 | — | 34,2±2,0 |
| 4 нед | 44 (41–48) | 8 (3–13) | 7,9±0,2 | 100±6 | 33,5±2,0 | 34,2±1,5 |
| 2 мес | 37 (34–39) | 8 (3–15) | 7,4±0,2 | 96±8 | 30±2,0 | 34,0±1,7 |
| 4 мес | 35 (31–38) | 10 (5–25) | 7,3±0,3 | 88±6 | 29±2,0 | — |
| 6 мес | 37 (34–39) | 8 (3–13) | 7,3±0,2 | 77±7 | 26±2,5 | 33,5±2,0 |
| 9 мес | 36 (34–39) | 8 (3–13) | — | 76±7 | — | — |
| 1 год | 37 (33–40) | 8 (3–13) | 7,1±0,2 | 73±8 | 23,5±3,7 | 32,5±2,4 |
| 2–6 лет | 38 (34–41) | 5 (1–13) | — | 76±8 | 26,0±3,0 | 32,7±2,0 |
| 7–12 лет | 41 (37–43) | 5 (1–13) | — | 79±8 | 27,0±3,0 | 33,7±1,8 |
| 13–17 лет (муж.) | 44 (39–47) | 5 (1–13) | — | 78±8 | 28,0±3,0 | 34,0±3,0 |
| 13–17 лет (жен.) | 41 (36–44) | 5 (1–15) | — | 79±8 | 29,0±3,0 | 34,0±3,0 |
| Взрослые (муж.) | 46 (40–49) | 3 (1–14) | 7,2±0,3 | 87 ( 82–92) | 32,0 (27–35) | 35,0±4,0 |
| Взрослые (жен.) | 41 (36–44) | 6 (1–14) | 7,4±0,3 | 85 (81–90) | 33 (27–34) | 34,1±4,0 |
Если количество эритроцитов, концентрация гемоглобина или гематокрит уменьшаются, возникает анемия. Скрининговыми показателями, обязательными для всех больных с анемией или при ее исключении, являются определение количества эритроцитов, уровня гемоглобина, среднего объема эритроцита, среднего содержания гемоглобина в эритроците, цветного показателя, определение количества ретикулоцитов, морфология эритроцитов на мазке крови. Изменения этих показателей могут быть достаточно специфичным признаком (табл. 3.2).
Таблица 3.2
Наиболее распространенные морфологические изменения эритроцитов и их клиническое значение
| Тип изменений | Характеристика | Физиологическое значение | Возможные клинические причины |
| Макроцит | Диаметр >8,5 мкм. Содержание гемоглобина достаточное | • Молодые эритроциты («акселератная» генерация? Ранняя потеря ядра)
• Нарушение созревания мегалобластов при измененном синтезе ДНК |
Ускоренный эритропоэз, дефицит витамина В12 или фолиевой кислоты |
| «Тонкий» макроцит | Диаметр увеличен, но средний обмен эритроцита (МСV) — в норме, часто — гипохромия (см. мишеневидные клетки) | Повышение концентрации лецитина и холестерина в мембране | Патология печени постслленэктомический синдром |
| Микроцит | Диаметр <7,0 мкм | Выделить:
а) нормальное насыщение гемоглобином б) нормальной формы |
См. ниже |
| Гипохромные клетки | Обычно микроциты | Нарушение синтеза гемоглобина:
• дефицит железа • нарушение синтеза глобина • нарушение синтеза порфирина |
Железодефицитные анемии при хронических заболеваниях (?). Талассемия. Гемоглобинопатии С и Е. Сидеробластная анемия |
| Нормоцит | Содержит ядро | Предстадия эритроцита | Напряженный эритропоэз (особенно при острых гемолитических анемиях); состояния после спленэктомии |
| Тельца Жолли | Включения, чаще по периферии, темные, пыльцеобразные | Остатки ядра | Тяжелые мегалобластные анемии; на фоне спленомегалии; состояния после спленэктомии |
| Тельца Гейнца | Темно-красные неправильно расположенные включения (при суправитальной окраске) | Денатурированный гемоглобин | глюкозо-6-фосфатдегидрогеназа-недостаточность; нестабильный гемоглобин; талассемия |
| Базофильная зернистость | Темные включения | Рибосомы | β-талассемия; свинцовая и другие интоксикации |
| Сидероциты | Темные грубые 3–4 включения в эритроцитах | Гранулы железа | Состояния после спленэктомии |
| Мишеневидные клетки | Соотношение поверхности к объему повышено. Тонкие клетки с циркулярной окраской в центре и по периферии, разделенной бледной зоной | • Спленэктомия нарушает потерю липидов ретикулоцитами
• Накопление холестерина и фосфолипидов на эритроцитах • Врожденное состояние |
То же, что и при гипохромии; постспленэктомия; заболевания печени (особенно — обструктивная желтуха); дефицит лецитин-холестерин- ацилтрансферазы |
| Лептоциты | Тонкие гипохромные клетки. Диаметр — в норме. МСV снижен | Талассемия | |
| Сфероциты | Круглые, чаще мелкие эритроциты без просветления в центре | • Нарушения мембраны эритроцитов
• Эритроцитарные фрагменты после столкновения с нитями фибрина, стенками патологически измененных сосудов и искусственными предметами в русле циркуляции |
Наследственный сфероцитоз (Минковского — Шофара); приобретенная аутоиммунная гемолитическая анемия |
| Эллиптоциты | Эллипсоидные клетки, гипохромии нет | • Наследственные нарушения
• Приобретенные нарушения |
Наследственный эллиптоцитоз; при различных анемиях, особенно мегалобластных |
| Серповидные клетки | Появляются при недостатке кислорода | Молекулярная агрегация HbS | Отмечается при гемоглобине S, J, C-Harlem, C-Capetown |
| Шистоциты | Каскообразные или треугольные клетки. Размер ниже нормы. Фрагменты эритроцитов | Гемолитическая микроангиопатическая анемия; гемолитическая анемия в ответ на действие физических агентов; уремия; злокачественная гипертензия | Появляются после разрушения эритроцитов о нити фибрина, измененные стенки сосудов, искусственные предметы в русле кровообращения |
| Каплевидные эритроциты | Напоминают грушу или каплю. Обычно микроциты, часто гипохромны | Поврежденные или фрагментированные эритроциты | Миелопролиферативные заболевания; большая талассемия; тяжелый дефицит железа |
| Акантоциты («шпорообразные») | Фестончатые; до 5–10 «шпор»-выростов на поверхности клетки (толщина, длина и расположение вариабельны) | При заболеваниях печени соотношение холестерина/лецитина в мембране эритроцитов повышено. В неионных детергентах могут принимать нормальную форму | Абеталипопротеинемия; болезни печени с гемолитической анемией; дефицит пируваткиназы; постспленэктомический синдром (редко) |
| Эхиноциты (шипообразные клетки) | Имеют 10–30 «шпор»-выростов, равномерно расположенных по поверхности эритроцита | Может быть результатом интра- и/или экстрацеллюллярных изменений. Итог накопления жирных кислот и/или лизолецитина на поверхности эритроцитов | Новорожденные; дефицит пируваткиназы; дефицит фосфоглицераткиназы; уремия |
| Стоматоциты | Униконкавные клетки (в противоположность биконкавным — двояковогнутым — нормальным). Щелевидное просветвление (стома) в центре | • Наследственно обусловленные. Первичный дефект структуры или функции мембраны с нарушением катионной проницаемости, содержания или потока
• Приобретенные нарушения катионного потока или состава |
• Наследственный сфероцитоз
• Реже: алкогольный цирроз; алкоголизм; обструктивные заболевания печени; новообразования; артефакт |
| Ксероциты | Плотные, дегидратированные, неравномерно сокращенные клетки. Возможно выпадение гемоглобина по периферии | Результат потери воды | Семейный ксероцитоз |
Дальнейшая дифференциация анемий проводится на основании определения диаметра эритроцита, среднего содержания (МСН — mean corpuscular haemoglobin) и средней концентрации гемоглобина (МСНС). Содержание гемоглобина в эритроцитах характеризуется «цветным индексом». Но лучше определять содержание гемоглобина в эритроците в абсолютных величинах:
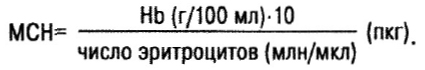
В норме МСН=27–34 пкг (нормохромные показатели).
Средняя концентрация гемоглобина в отдельном эритроците (МСНС — mean corpuscular haemoglobin concentration):
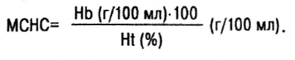
В норме МСНС=31–35 г/100 мл, или 310–350 г/л.
То есть учитывается соотношение гемоглобина и объема эритроцитарной массы и определяется среднее насыщение эритроцита гемоглобином. Но при гипохромных анемиях со сниженным содержанием гемоглобина в эритроцитах в комбинации с уменьшением размеров клеток МСНС может не измениться.
На основании определения диаметра (объема) эритроцита выделяют микро-, нормо- и макроцитарные анемии. Средний объем эритроцита (МСѴ — mean corpuscular volume):
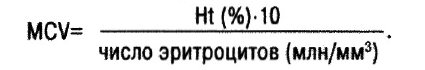
В норме МСV=85–95 fl.
Трактовку результатов анализа крови следует проводить с учетом методики подсчета форменных элементов. В последние годы все чаще клинический анализ крови проводят на автоматических счетчиках, что значительно повышает точность подсчета, но не отменяет значения данных, получаемых с помощью световой микроскопии. В основу работы автоматических счетчиков крови положен принцип прерывания постоянного электрического или светового потока отдельными форменными элементами. Это осуществляют с помощью двух методов.
- Электрический импеданс используют в приборах Culter, ТОА, Hoffman La Roche, Cell-Dyn, Instrumentation Laboratory Collect, Sequoia-Turner, Backer, Clay Adams Ultra-Floe 100. Клетки крови, будучи плохими проводниками электричества, выходя из точечного отверстия — апертуры, — кратковременно изменяют электрическое поле, создаваемое двумя электродами, находящимися по обе стороны апертуры. Амплитуда возникающего сигнала пропорциональна объему замещенного электролита. Это позволяет подсчитать количество и объем каждой клетки, проходящей через апертуру.
- Принцип оптической детекции используют в автоматических анализаторах Technicon и Ortho diagnostic systems. Оптический поток создается лазерами. При прохождении клетки через световой поток происходит его отражение, рассеивание и поглощение. Изменение светового луча фиксируют датчиками, переводящими световой импульс в электрический. Амплитуда импульсов зависит от размеров объема, свойств поверхности и структуры клетки.
Но большинство автоматических счетчиков не определяют молодые формы лейкоцитов, нормобласты и ретикулоциты. Эти данные можно получить только при микроскопии. Судить об увеличении или уменьшении количества форменных элементов крови можно только по абсолютному значению их показателей. RBC (число эритроцитов) при автоматическом подсчете определяют без лизиса лейкоцитов. Увеличение количества лейкоцитов более 50 000/мкл может привести к ошибочному завышению показателей, гемоглобин (г/л) определяют спектрометрически гемоглобинацидным методом. Мутность раствора (лейкоцитоз, гиперлипидемия) может привести к искусственному завышению показателя. МСѴ прямым измерением определяют только в приборах серии Technicon. В остальных счетчиках эта величина расчетная. Кривая распределения эритроцитов по объему должна быть унимодальной. Бимодальное распределение свидетельствует об аномальных эритроцитах. Искусственное завышение показателя может возникать при холодовой агглютинации, гипергликемии.
Для практических целей необходимо определение нескольких параметров, поскольку только их содружественная динамика позволяет избежать ложных заключений. Например, для железодефицитной анемии типичны снижение гемоглобина, гематокрит — на нижней границе нормы, число эритроцитов чаще не изменяется. При талассемии гемоглобин снижается незначительно, гематокрит в пределах нижней границы нормы, число эритроцитов повышено. При истинной полицитемии гемоглобин на верхней границе нормы, показатели гематокрита и эритроцитов — значительно повышены.
По среднему содержанию гемоглобина в эритроците выделяют нормо-, гипо- и гиперхромные состояния. Так, для анемии Минковского — Шофара характерно уменьшение диаметра эритроцитов и небольшое снижение содержания гемоглобина в эритроцитах. Концентрация гемоглобина снижается только при тяжелых состояниях, например, при кризах или дефиците железа.
Для работы врача важно не только определение указанных показателей, но и изучение мазков крови. Например, для оценки состояния костного мозга важно знать концентрацию ретикулоцитов. Обычно в периферической крови их содержится 1–2%. Индекс продуктивности костного мозга определяют как частное от произведения числа ретикулоцитов (в %) на гематокрит (в %), где делитель — 45. В норме показатель равен 1. Его повышение свидетельствует о раздражении костного мозга. Здоровый костный мозг способен за сутки повысить свой индекс продуктивности в 2–3 раза, а при гемолитической анемии этот индекс может достигать 6–9 (табл. 3.3).
Таблица 3.3
ДД-значение изменений показателей красной крови при разных заболеваниях (по К. Rhyner с изменениями, 2000)
| Показатель | Клиническое состояние | ||||||
| Норма | Острая кровопотеря | Почечная анемия | Цирроз печени | Мегалобластная анемия | Компенсированная гемолитическая анемия | Декомпенсированная гемолитическая анемия | |
| Эритроциты, 1012/л | 5 | 2,0 | 1,7 | 3,5 | 2,0 | 4,0 | 2,6 |
| Гематокрит, % | 45 | 25 | 15 | 42 | 30 | 42 | 30 |
| MCV | 90 | 120 | 90 | 120 | 150 | 120 | 120 |
| Полихромазия | Нет | Да | Нет | Нет | Нет | Нет | Да |
| Ретикулоциты % |
1 | 15 | 3 | 4 | 2,5 | 6 | 30 |
| абс⋅109/л | 50 | 300 | 50 | 140 | 50 | 240 | 780 |
| Индекс продуктивности | 1 | 6*(3) | 1 | 3** | 1 *** | 5 | 16*(18) |
*Высокий показатель получается из-за выраженной полихромазии. Объективное его значение в 2 раза ниже; **ретикулоцитоз при макроцитозе свидетельствует против пернициозной анемии; ***небольшое число ретикулоцитов контрастирует с эритропоэтическим гиперактивным неэффективным костномозговым кроветворением (интрамедуллярный гемолиз).
Кроме мазков крови, едва ли не решающее значение имеет изучение пунктата костного мозга. Определение степени клеточности и типов клеток позволяет уточнить патогенез, например, с учетом мегалобластных форм, полинуклеарных эритробластов и т. д. Необходима и окраска на железо с помощью красителя берлинского синего.
Вообще же, при диагностике анемии («олигоцитемии») необходимо учитывать соотношение трех величин; 1) общая масса эритроцитов; 2) объем циркулирующей крови; 3) гематокрит. Изменение объема циркулирующей крови может симулировать олигоцитемию или, наоборот, маскировать ее (табл. 3.4).
Таблица 3.4
Изменение некоторых гематологических и биохимических показателей при нарушениях водно-электролитного баланса
| Состояние | Внеклеточное пространство | Внутриклеточное пространство | |||||
| Количество эритроцитов; Нb | Ht,% | Содержание белка в плазме крови, г/л | Содержание натрия в плазме крови, ммоль/л | MCV, мкм3 | Средняя концентрация Нb эритроцитов*, % мкм2 | ||
| Норма | (жен.) 3,9–4,8⋅1012/л, 12,6–16,2 г% или 7,8–10,2 ммоль/л
(муж.) 4,0–2,3⋅1012/л, 13,5–16,5 г% или 8,6–10,4 ммоль/л |
39–52
40–52 |
65–80 | 137–142 | 84–95 | 31–37 | |
| Дегидратация изотоническая | Дефицит воды | ↑ | ↑ | ↑ | ↑ | В норме | В норме |
| Дефицит плазмы крови | ↑ | ↓ | В норме или ↓ | В норме | В норме | В норме | |
| Кровопотеря | В норме или ↓ | В норме или ↓ | ↑ | В норме | В норме | В норме | |
| Дегидратация гипертоническая | Дефицит воды | ↑ | ↑↑↑ | ↑ | ↑↑↑ | ↑ | ↑ |
| Дегидратация гипотоническая | Дефицит натрия | ↑ | ↑↑ | ↑ | ↓ | ↑ | ↓ |
| Гидратация гипертоническая | Избыток натрия | ↓ | ↓↓ | ↓ | ↑ | ↓ | ↑ |
| Гидратация изотоническая | Избыток жидкостей | ↓ | ↓ | ↓ | В норме | В норме | В норме |
| Гидратация гипотоническая | Избыток воды | ↓ | ↓↓↓ | ↓ | ↓↓↓ | ↑ | ↑ |
Показатель гематокрита диспропорционально низок при гидремии беременных, гипергидратации в случаях застойной сердечной недостаточности или олигурической почечной недостаточности, хронических заболеваниях и гипоальбуминемии, длительном обездвиживании пациента.
Относительное уменьшение объема плазмы крови (гематокрит может быть высоким, нормальным или низким, но относительно высок по отношению к общей массе эритроцитов) происходит при дегидратации с преимущественной потерей солей (протрагированная диарея особенно у детей, холера, обструкция пилорического канала, перитонеальный диализ гипертоническими растворами, диабетический ацидоз, несахарный диабет с ограничением потребления воды).
Уменьшение как объема плазмы крови, так и массы эритроцитов (гематокрит — в норме; масса эритроцитов уменьшена) возникает при острой кровопотере, микседеме, болезни Аддисона, пангино- питуитаризме, иногда — при раке.
Анемии можно подразделить на:
- острые постгеморрагические, развивающиеся после острых наружных или внутренних кровотечений;
- хронические постгеморрагические, развивающиеся при длительной, медленной кровопотере (например, из желудочно-кишечного тракта);
- гемолитические, характеризующиеся уменьшением длительности жизни эритроцита;
- с нарушенным созреванием. Эритропоэз в костном мозгу идет как гиперпластический, с большим количеством ретикулоцитов. Однако большая часть клеток в процессе созревания гибнет (интрамедуллярный гемолиз), так что весь эритропоэз обозначается как неэффективный. Самым ярким примером является В12— и фолиеводефицитные состояния. Сопутствующим анемизирующим фактором является умеренное снижение продолжительности жизни эритроцитов;
- с нарушенной пролиферацией (снижение способности образовывать новые клетки крови). Число эритробластов уменьшается. Важнейшими причинами являются нарушения костного мозга, дефицит железа и эритропоэтина. Костномозговая недостаточность возникает при злокачественных процессах, в том числе метастатических, лучевом или медикаментозном поражении. Уровень эритропоэтина определяют функциональным состоянием почки. Наряду с состоянием почки (например, при анемии брайтиков) имеет значение гипоксия, определяемая по концентрации гемоглобина, насыщению гемоглобина кислородом, сродство гемоглобина к кислороду, почечный кровоток. В дальнейшем эритропоэтин действует на эритропоэтинчувствительные клетки, запуская их дифференциацию.
Все перечисленные патогенетические механизмы, особенно последние три, нередко действуют сочетанно. Например, при гемолитических анемиях восполнение разрушенных эритроцитов возможно только при достаточном поступлении железа, при его недостатке возникает нарушение пролиферации.
Особенностью ДД анемии у новорожденных является выбор наиболее вероятных состояний, исходя из возраста ребенка. В 1-е сутки жизни это постгеморрагическая или аутоиммунная гемолитическая анемия. После 1-х суток жизни необходимо учитывать прежде всего постгеморрагическую анемию, аутоиммунную гемолитическую анемию, врожденные инфекции (цитомегаловирусную, краснушную, токсоплазмоз, сифилис), бактериальный сепсис, врожденные нарушения мембраны эритроцитов или клеточного метаболизма, синдром Даймонда — Блекфана.
Причинами постгеморрагических анемий у новорожденных являются:
- явные кровотечения (разрыв или ранение пупочного канатика; предлежание плаценты; отслойка плаценты; ранение плаценты во время кесарева сечения);
- скрытые кровотечения до рождения (фетоматеринская трансфузия; трансфузия близнец-близнецу);
- скрытые внутренние кровотечения (ретроперитонеальное; внутричерепное; разрыв печени, селезенки).
Постгеморрагическая острая анемия возникает после достаточно массивных кровопотерь. Острая кровопотеря обычно сопровождается бледностью, шумом в ушах, склонностью к обморокам в положении стоя, однако организм достаточно быстро адаптируется, и указанные симптомы выражены не всегда. Внутренние кровотечения, обусловливающие хронические постгеморрагические анемии, представляют значительные сложности в диагностике. Кровотечения при анкилостомидозе, пептической язве двенадцатиперстной кишки могут продолжаться длительное время без рвоты, а дегтеобразный стул появляется спустя 4–7 дней. В крови вначале сохраняется нор- моцитоз при возникающем ретикулоцитозе. Через 3 сут увеличивается объем эритроцитов, снижение концентрации железа в сывоторотке крови приводит к ретикулоцитопении. Как следствие нарушения формирования клеток появляются дегенеративные формы: гипохромные микроциты. В тяжелых случаях при продолжающемся кровотечении в мазках выявляют анизо- и пойкилоциты. Поэтому в терапевтической практике при описанной картине крови необходимо искать хроническое кровотечение прежде всего в желудочно-кишечном тракте.
Хроническая анемия, развивающаяся медленно, не имеет патогномоничных клинических признаков. Одышка при нагрузке, а позже и в покое, учащение приступов стенокардии или их появление у ранее интактного пациента, тахикардия, шум в сердце, перемежающаяся хромота являются признаками гиперкинетического типа кровообращения.
Основные ДД-этапы по главным группам анемий отражены на схеме 3.1.
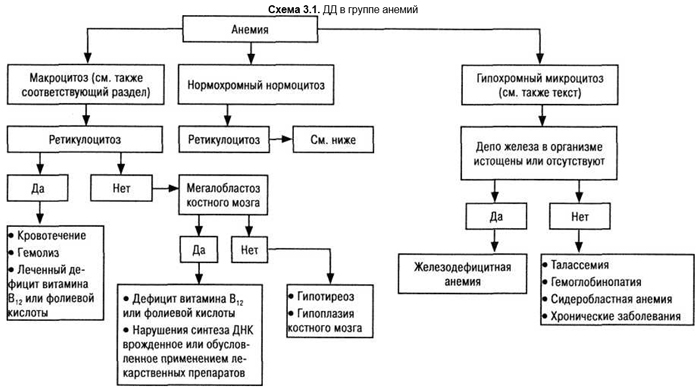
Успешная ДЦ начинается с изучения мазков крови. Анамнез, клинико-лабораторные исследования позволяют во многих случаях отказаться от обременительных этапов (например, исследование костного мозга) (табл. 3.5).
Железодефицитные анемии в наших условиях составляют самую распространенную группу. Железодефицитные анемии — это прежде всего анемии истощения. Все иные причины железодефицитных анемий (кровопотери) могут усугублять состояние, но не являются ведущими в общем спектре причин. Железодефицитные анемии распространены в районах с низким потреблением животного белка, высокой частотой распространения кишечных паразитов. Чем беднее семья, тем вероятнее железодефицитные состояния у ее членов.
Таблица 3.5
ДД-признаки важнейших типов анемии (по J. Rastetter, Н. Begeman, 1999)
| Показатель | Гипохромная (железодефицитная) анемия | Гемолитические анемии | Пернициозная анемия | Апластическая анемия | |
| Врожденная | Приобретенная | ||||
| Возраст | Любой | С детства | Все возрастные группы | Чаще 50–70 лет | Острые формы обычно до 20 лет, хронические — 50–60 лет |
| Лол | Чаще женщины | Муж.=жен. | Тепловые антитела: жен.>муж.
Холодовые антитела: жен.<муж. |
Несколько чаще возникает у мужчин | Несколько чаще возникает у мужчин |
| Кожные покровы и слизистые оболочки | Бледные | Желтушные | Желтушные | Светло-желтые | Бледные |
| Язык | Иногда глоссит | Без особенностей | Без особенностей | Часто атрофия слизистой оболочки,глоссит | Без особенностей |
| Желудочный сок | Часто снижена кислотность | Без особенностей | Без особенностей | Как правило, анацидность | Без особенностей |
| Селезенка | Не увеличена | Часто увеличена | Часто увеличена | Увеличена | — |
| Нервная система | Иногда парестезии | При массивном гемолизе возможен так называемый нейрогемолитический синдром | Как при врожденных гемолитических анемиях | Изменена | Без особенностей |
| Другие клинические проявления | Изменения ногтей, возможен синдром Плуммер — Вильсона | Желчнокаменная болезнь; башенный череп и др. | Желудочно-кишечные расстройства | Склонность к инфекциям и кровотечениям | |
| МСН | Снижен | В норме | В норме | Повышен | В норме |
| Форма и толщина эритроцитов | Анулоцитоз, анизоцитоз | Диагностически значимые изменения (микроциты, эллиптоциты и т. д.) | Дегенеративные формы | Мегалоцитоз, анизо- и пойкилоцитоз | Анизо- и пойкилоцитоз |
| Кривая Прайс — Джонса | Расширение основания | При микросфероцитарной анемии сдвинута влево, в остальных случаях — без особенностей | Обычно смещена влево | Расширена, сдвиг вправо | Расширена |
| Осмотическая резистентность эритроцитов | Не изменена | Снижена, при талассемии — расширена | Часто снижена | В норме или небольшое снижение | В норме |
| Гранулоциты | В норме | Часто число увеличено | В норме или увеличение | Лимфоцитоз; число гранулоцитов снижено; полисегментарные ядра | Снижено значительно; относительный лимфоцитоз |
| Тромбоциты | В норме | В норме | В норме | Снижены | Значительное снижение |
| Костный мозг | Усиленный эритроцитопоэз со сдвигом влево (макробластный костный мозг) | Усиленный эритропоэз, нормобластный костный мозг | Как при врожденных гемолитических анемиях | Мегалобласты; гигантские формы гранулоцито- постических клеток; полисегментарные ядра в мегакариоцитах | Пустой костный мозг; в начале нередко мегалобластные изменения |
| СОЭ | Норма или незначительно
повышена |
Повышена | Значительно повышена | Умеренно или значительно повышена | Значительно повышена |
| Уровень билирубина в сыворотке крови | В норме или снижен | Повышен за счет непрямой фракции | Повышен за счет непрямой фракции | Незначительно повышен | В норме или снижен |
| Уробилин | В норме | Часто — уровень значительно повышен | Уровень значительно повышен | Уровень значительно повышен | В норме |
| Железо в сыворотке крови | Значительно снижено | В норме или значительное повышение; при пароксизмальной ночной гемолитической анемии уровень железа резко снижен | Как при врожденных формах | Уровень чаще повышен или в норме | Уровень чаще резко повышен |
В организме человека содержится около 3–5 г железа. Из этого количества всего несколько миллиграмм приходится на жизненно важное железо в дыхательной цепи. 2/3 всех запасов железа составляет гемоглобин (1/300 молекулярной массы гемоглобина приходится на железо.) Оставшаяся 1/3 находится в виде гемосидерина и ферритина в печени, селезенке и костном мозге в ретикуло-эндотелиальных клетках и в клетках паренхимы печени. Гемосидерин прекрасно выявляется окраской берлинским синим. В миоглобине содержится около 0,2 г железа. В плазме крови железо связано со специфическим транспортным белком — трансферрином. Каждая молекула трансферрина может содержать 2 атома железа. Концентрация трансферрина определяет общую железосвязывающую способность плазмы. Обычно только 1/3 трансферрина насыщена железом, так что в плазме крови циркулирует около 2–4 мг железа. Ежесуточно разрушается приблизительно 25–30 мл из общего объема эритроцитарной массы. Железо, освободившееся из разрушенных красных кровяных клеток, связывается с транспортным белком и переносится в гемоглобинформирующие клетки красного костного мозга. Суточные потери (эпителий кожи, кишечника, моча) составляют у мужчин около 1 мг. Дополнительно к этому количеству женщины теряют в каждый день менструации приблизительно 0,7 мг железа. В оптимально сбалансированном пищевом рационе содержится не более 16 мг железа, а усвоиться может всего 1,6–3,2 мг. Отсюда вполне понятно, почему 80% всех железодефицитных анемий приходится на женщин. При обследовании больших групп населения железодефицитные анемии выявлены у 18% девочек-подростков, 35–55% женщин детородного возраста, у 15–25% женщин в менопаузальный период и только у 2–4% молодых мужчин.
Железо, содержащееся в плазме или в сыворотке крови большей частью связано с трансферрином. Концентрация железа в норме составляет 100 мкг/100 мл. Половые различия начинаются с пубертатного возраста: содержание железа у мужчин составляет 80–150 мкг/100 мл, у женщин 70–130 мкг/100 мл. Уровень железа зависит от общего содержания железа в организме, особенно от насыщенности депо, поступления и расхода железа. При сбалансированном обмене концентрация железа остается в очень узких границах независимо от питания, физической нагрузки и т. д. Тем не менее, утром показатели на 10–30% выше, чем вечером. При распаде гемоглобина (например, гемолитические анемии) уровень железа в сыворотке крови возрастает. И, наоборот, при регенерации эритропоэза, после терапии витамином В6 у пациентов с пиридоксин-зависимой анемией уровень железа в плазме крови снижается. Таким образом, гиперсидеринемия характерна для усиленного распада гемоглобина, сниженного или неэффективного эритропоэза (апластическая или пернициозная анемия), при сидероахрестических анемиях (нарушение усвоения железа), нарушении утилизации железа в ретикуло-эндотелиальной системе, в первые дни после острой кровопотери, при остром гепатите, гемохроматозе. Гипосидеринимия наблюдается при общем дефиците железа, инфекциях и злокачественных опухолях, усиленном эритропоэзе и атрансферринемии. Уровень железа в сыворотке крови и железосвязывающая способность плазмы (сыворотки) крови представлены в табл. 3.6.
Таблица 3.6
Концентрация железа в сыворотке (плазме) крови и ненасыщенная железосвязывающая способность при некоторых состояниях
| Состояние | Железо в сыворотке крови, мкг/100 мл | Железосвязывающая способность, мкг/100 мл |
| Норма | 100 | 200 |
| Последний триместр беременности | 85 | 420 |
| Железодефицитные состояния | 30 | 410 |
| Инфекции,хроническое воспаление,онкологические заболевания | 50 | 150 |
| Нефротический синдром | 40 | 120 |
| Талассемия, порфирия, некроз гепатоцитов | 210 | 30 |
| Гемохроматоз, гемосидероз | 270 | Полное насыщение |
Железо всасывается преимущественно в двенадцатиперстной кишке. Соотношения между необходимым человеку объемом железа, его потерями, особенно при заболеваниях, в период беременности, кормления грудью, росте и небольшим по протяженности отделу желудочно-кишечного тракта, где только при условии интактной стенки кишки возможно всасывание этого железа, делает понятным широкую распространенность железодефицитных состояний. Итак, равновесие в обмене железа нарушается:
- недостаточным поступлением железа;
- нарушенным всасыванием железа;
- повышенными потерями железа;
- повышенными потребностями в железе.
Возможна и чаще наблюдается в практике комбинация нескольких факторов.
В отечественной и зарубежной практике используют следующие термины: железодефицитные состояния, включающие латентный дефицит железа (иногда выделяют и прелатентный дефицит железа), и железодефицитную анемию.
Сохраняющийся некоторое время отрицательный баланс железа вначале обусловливает потребление его запасов в организме, что обеспечивает нормальный гемопоэз. Эта стадия носит название латентного дефицита железа. Клинически латентный дефицит железа проявляется повышенной утомляемостью и чувствительностью к холоду, предрасположенностью к инфекционным заболеваниям, лабильностью настроения, головной болью. Объективно диагностируют атрофию слизистой оболочки рта и языка; хейлит; нарушения глотания из-за атрофии слизистой оболочки или стенозирующего эзофагита (синдром Плуммера — Вильсона); извращенный аппетит (поедание мела); отмечают сухую морщинистую кожу; ломкие ногти; ложкообразные ногти (койлонихия); сухие, ломкие, неопрятно торчащие волосы.
Лабораторно латентный дефицит железа проявляется:
- уровень гемоглобина не ниже 120–140 г/л;
- насыщение трансферритина ниже 16%;
- уровень ферритина в сыворотке крови 10–40 нг/мл;
- недостаточные запасы железа или его отсутствие в костном мозгу;
- свободный протопорфирин эритроцитов 0,7 ммоль/л, но средний объем эритроцита и среднее содержание гемоглобина в эритроците еще не изменяются.
При дальнейшем снижении содержания железа в организме снижается концентрация железа в сыворотке крови ниже 9 мкмоль/л, общая железосвязывающая активность сыворотки крови повышается. В мазках крови регистрируют гипохромные микроциты (схема 3.2), анизоциты. Формируется манифестная железодефицитная анемия, при выраженных формах которой определяются и мишеневидные клетки, что вследствие общности морфологических критериев затрудняет ДД с талассемией. Число ретикулоцитов увеличивается, эритропоэз может быть повышен с большим числом базофильных форм (левый сдвиг). Нередко вследствие общего раздражения костного мозга развивается тромбоцитоз. Сыворотка крови светлая в противоположность желтоватой при пернициозной анемии.
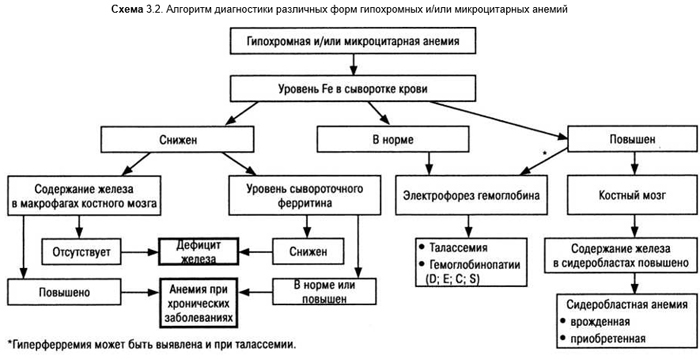
Недостаточное поступление железа редко бывает единственной причиной анемии. Тем более, что в регионах с некалорийной и однообразной пищей неизбежно присутствует инфекционный фактор с повреждением слизистой оболочки кишечника, частые повторные беременности, аборты, глистные инвазии, то есть все то, что обусловливает дефицит железа в организме. Как преимущественный фактор недостаток поступления железа чаще возникает у маленьких детей и одиноких стариков.
Нарушенное всасывание железа свойственно пациентам после гастрэктомии, больным целиакией и спру. Связь между дефицитом железа и ахлоргидрией многообразна.
А. Длительный дефицит железа обусловливает вначале обратимые, а затем и необратимые повреждения слизистой оболочки. В этих случаях выявляют кровопотери, отсутствуют семейные случаи и антитела к париетальным клеткам.
Б. Случаи с первичным поражением желудка и, соответственно, вторичным по отношению к этому дефицитом железа. Чаще отмечают у женщин, нет связи с кровопотерями. Семейная концентрация заболевания и наличие антител свидетельствуют о генетических и иммунных факторах.
Повышенные потери железа. Прежде всего (и чаще всего) — это маточные кровотечения. При отсутствии других симптомов у женщины в период менструации можно остановиться на этом диагнозе. В любом возрасте должны быть срочно исключены кровоточащие опухоли желудочно-кишечного тракта, болезнь Ослера, голубой невус. Прием кортикостероидов и салицилатов также может обусловить желудочные и кишечные кровотечения. Железодефицитная анемия с повышением СОЭ позволяет исключать дивертикулез толстого кишечника и карциному слепой кишки. Нельзя забывать о банальнейшем геморрое. Исследование кала на скрытую кровь, и особенно эндоскопия, во многих случаях могут прояснить ситуацию. По диагностической ценности рентгеноконтрастные исследования целесообразно выполнить после эндоскопических. Значительные потери железа с кровью наблюдаются при глистных инвазиях, например, ancylostoma duodenalis. Реже причиной усиленных потерь железа могут быть целиакия и спру за счет стремительного разрушения клеток эпителия кишечника. Потери железа с мочой увеличиваются при гемат- и гемоглобинуриях.
Повышенная потребность в железе у детей и беременных. Если, как было указано выше, человек в оптимальных условиях может усвоить в сутки 1,6–3,2 мг железа, то растущий плод, плацента и родовое кровоточение требуют поступления 2,1мг железа в сутки в течение всей беременности. При диагностике анемии беременных необходимо принять во внимание, что объем плазмы крови в этот период жизни женщины увеличивается на 40%, вследствие чего концентрация гемоглобина 100–110 г/л может рассматриваться как физиологическая. Но всегда следует помнить, что эти показатели могут быть «последним рубежом», и необходимо определять насыщение трансферрина железом.
В связи с тем, что микроциты выявляют не только при железодефицитных состояниях, ДЦ может быть проведена по следующим пунктам:
- Дефицит витамина В6. Нерациональное применение туберкулостатических препаратов (редко).
- Сидеробластные анемии. Микросфероцитарными являются только редкие наследственные формы.
- Талассемия. Регион проживания пациента и электрофорез гемоглобина позволяют установить диагноз.
- Истинная железодефицитная анемия протекает со снижением содержания железа в организме. Типично отсутствие железа в костном мозгу и повышение трансферрина.
- Снижение потребления железа в кроветворных органах. Опухоли, воспалительные и инфекционные заболевания. Микроцитоз развивается при длительном снижении насыщения трансферрина до 15%.
Для гемолитических анемий вне зависимости от их происхождения общим является резко сниженная продолжительность жизни эритроцита. Максимальная продолжительность жизни эритроцита в норме составляет 120 дней. Эритроцит разрушается быстрее при его собственных дефектах или при наличии экстракорпускулярных повреждающих факторах. Существует несколько классификаций гемолитических анемий. Учитывается тип повреждения, врожденность или приобретенность дефекта, место гемолиза (внутри- или внесосудистый). В России традиционно используют классификацию гемолитических анемий, предложенную Л.И. Идельсоном (1981). В основу построения классификации положен патогенетический принцип. Все гемолитические анемии разделяют на наследственные и приобретенные. Наследственная гемолитическая анемия, в свою очередь, разделяется на три группы:
- Мембранопатии.
- Ферментопатии.
- Гемоглобинопатии.
В любом случае развитие гемолитической анемии начинается с повреждения стенки клетки. Его отмечают при:
- спленомегалии;
- механическом разрушении эритроцитов искусственными клапанами сердца или при гемосорбциях;
- врожденных или приобретенных дефектах мембраны;
- непосредственном влиянии комплементсвязывающих антител;
- нарушенной энергетике клеток при энзимопатиях; разрушении моноцитами эритроцитов, несущих на себе антитела;
- элиминации белковых преципитатов при гемоглобинопатиях или талассемии.
Не каждое ускорение разрушения эритроцитов обусловливает гемолитическую анемию. Анемия начинается тогда, когда костный мозг оказывается не в состоянии восполнять убыль эритроцитов. Поэтому выделяют компенсированную и декомпенсированную гемолитическую анемию. Повреждение мембраны эритроцитов ведет к деформациям клетки и к нарушениям ионных каналов. Поврежденные эритроциты разрушаются в селезенке. Усиленное разрушение дефектных эритроцитов в селезенке обусловливает ее рабочую гипертрофию. Последнее приводит к включению печени в процесс разрушения красных кровяных телец. Эритроциты разрушаются в клетках системы фагоцитирующих мононуклеаров. Очень редко отмечают внутрисосудистый гемолиз, что проявляется гемоглобинемией и гемоглобинурией. Несмотря на то что диагноз «гемолитическая анемия» несложен, около 15% всех больных достаточно долго наблюдаются с первоначально ошибочным диагнозом «хронический гепатит» (на первое место необоснованно выдвигают желтуху).
Признаками гемолитической анемии являются:
- ретикулоцитоз выше 2%. Выявляют при каждой хронической гемолитической анемии. В момент кризов может быть не установлен, необходимы исследования в динамике;
- уменьшение количества эритроцитов;
- нормохромный или гиперхромный характер анемии. Единственное исключение — талассемия, сопровождающаяся гипохромным цветовым показателем;
- изменение морфологии эритроцитов. Например, важно выявление сфероцитов, выявляемых даже при иммунном гемолизе, или фрагментоцитов при механических разрушениях.
- умеренная гипербилирубинемия (не выше 85 мкмоль/л). Уровень билирубина не превышает фильтрационный порог, и при гемолитических анемиях билирубин никогда не определяют в моче;
- гемоглобинемия. Обычно уровень гемоглобина в сыворотке крови не превышает 30 мг/л. При гемолитических анемиях с разрушением эритроцитов в клетках ретикуло-эндотелиальной системы свободный гемоглобин в сыворотке крови повышается несущественно, но значительно повышается при внутрисосудистом гемолизе. При уровне гемоглобина в сыворотке крови в 1000 мг/л его выявляют и в моче. Гемосидеринемия свойственна только хроническим гемолитическим анемиям;
- уровень гаптоглобина снижается только при внутрисосудистом гемолизе. Гаптоглобин, образуя комплексы с гемоглобином, препятствует потерям гемоглобина через почки. Комплекс гаптоглобин — гемоглобин разрушается в макрофагально-макроцитарной системе. Образование же нового гаптоглобина происходит относительно медленно. При ДД-оценке показателя необходимо помнить, что снижение содержания гаптоглобина происходит и при заболеваниях печени:
- повышение уровня железа в сыворотке крови;
- повышенный эритропоэз в костном мозгу;
- необходимо проведение пробы Кумбса для исключения иммунного генеза гемолитической анемии;
- при каждой не определенной анемии желательно проведение пробы на тепловую резистентность эритроцитов. Положительный результат позволяет исключать пароксизмальную ночную гемоглобинурию. Дополнительно необходимо провести окраску мочевого осадка берлинским синим на наличие гемосидерина, выявляемого при хроническом внутрисосудистом гемолизе;
- традиционно применяют пробу на осмотическую резистентность эитроцитов, оказывающуюся сниженной, например, при наследственной сфероцитарной анемии. Однако в современных условиях предпочтительней инкубационный тест, позволяющий отдифференцировать целый ряд гемолитических анемий;
- единственный достоверный современный критерий гемолиза — подтверждение укорочения времени жизни эритроцита по Сr51 или DF-P32. Одновременно определяется не только длительность жизни эритроцитов, но и где происходит их преимущественный гемолиз. Следует принять во внимание, что уменьшение времени жизни эритроцитов происходит практически при всех тяжелых формах анемий, в том числе и при железодефицитной, не говоря уже о пернициозной анемии;
- в современных условиях возможно определение креатина эритроцитов, что достоверно отражает средний возраст эритроцитарной массы. С помощью этого состояния можно установить субклинические формы гемолитических анемий, не выявляющихся с помощью определения уровня ретикулоцитов.
Иногда за гемолитическую анемию ошибочно принимают следующие состояния:
- анемия и ретикулоцитоз, кровотечения; восстановительный период после дефицита железа, витамина В12 или фолиевой кислоты, костномозговой недостаточности (особенно после злоупотреблений алкоголем);
- анемия и желтуха, интрамедуллярный гемолиз; кровоизлияния в полости или в ткани тела;
- алкогольная желтуха без анемии;
- миелофиброз, метастазы в костный мозг;
- миоглобиноурия.
Гемолитические анемии относятся к так называемым нормохромным нормоцитарным анемиям (схема 3.3).
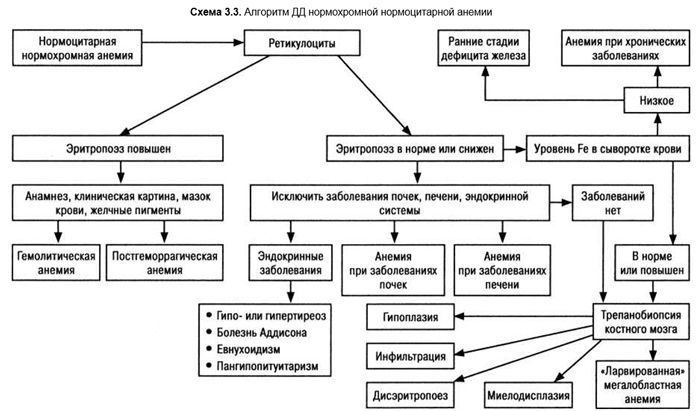
Наследственная сфероцитарная гемолитическая анемия (болезнь Минковского — Шоффара) — а/д передающийся дефект белка эритроцитарной оболочки (ген локализован на 8-й хромосоме), результатом чего является повышение проницаемости мембраны для натрия. Ионы натрия проводят за собой воду, и эритроцит становится похожим на переполненный напряженный шарик. В мазке крови они выглядят как маленькие интенсивно окрашивающиеся сфероциты без типичного для нормальных эритроцитов сниженной интенсивности окраски в центре. Микросфероциты отмечают в небольших концентрациях и при других видах анемий. Дефектные эритроциты легко разрушаются в селезенке. Разрушение может происходить остро, анемия усиливается и развивается желтуха. Присоединение инфекции (прежде всего — парвовирусов) может вызвать угнетение красного костного мозга, и анемия нарастает без желтухи. Возможны мегалобластные кризы за счет вторичного дефицита фолиевой кислоты.
Заболевание возникает во всех странах. В Северной Европе частота сфероцитарной гемолитической анемии составляет 1:5000 новорожденных. Но существует большое число асимптомных носителей гена. Сниженная осмотическая резистентность эритроцитов выявлена у 1% всех доноров.
Анемия развивается только при уменьшении продолжительности жизни эритроцитов на 15–20 дней ниже нормы. Спленомегалия — практически обязательный симптом.
Из-за сгущения желчи часто развиваются камни желчного пузыря. Это служит основанием для первоначальных ошибочных диагнозов «хронический гепатит» или «желчнокаменная болезнь». Скрытые гемолитические анемии как причина холелитиаза занимают особое место у детей и молодых пациентов. Поэтому у каждого ребенка или подростка с желчнокаменной болезнью необходимо исключить скрытый гемолиз.
Уровень ретикулоцитов при сфероцитозе почти постоянно повышен, хотя нормальные показатели ретикулоцитоза не исключают сфероцитарную анемию. В период кризов число лейкоцитов как реакция на разрушенные клетки может быть повышенной, в период ремиссии белая кровь не меняется. СОЭ повышена. При отстаивании крови вследствие большого числа ретикулоцитов между темно-красной колонкой эритроцитов и желтой сывороткой крови определяется розовый слой (шлейф). Аналогичную картину выявляют при любых состояниях, протекающих с высоким ретикулоцитозом. Осмотическая резистентность эритроцитов (минимальная) снижена, хотя у 10% больных может быть нормальной. Средний объем эритроцитов уменьшен, уровень лактатдегидрогеназы (ЛДГ) повышен. Значительно снижена концентрация гаптоглобина. При тяжелых формах микросфероцитарной анемии развиваются вторичные костные деформации как отражение избыточно пролиферировавшего костного мозга.
Спленэктомия, убирая орган максимального гемолиза, практически излечивает больных, не меняя причину.
Эллиптоцитоз наследуется а/д. В США частота эллиптоцитоза составляет 3–5 на 10 тыс. новорожденных, чаще среди представителей негроидной расы. В эндемичных по малярии областях его распространенность достигает 0,6%. Протекает благоприятно. Анемия формируется редко, гемолиз в большинстве случаев компенсированный и провоцируется беременностью, инфекцией, дефицитом витамина В12, отторжением трансплантанта. Диагноз основывается на изучении мазков периферической крови, где выявляют до 80% эллиптоцитов, в то время как в норме концентрация эллипсовидных эритроцитов не превышает 10%. ДД-сложности возникают при других заболеваниях, протекающих со вторичным эллиптоцитозом. Прежде всего, это относится к мегалобластной анемии и железодефицитным состояниям. Но при этих заболеваниях отсутствуют узкоэллиптические клетки, столь характерные для эллиптоцитоза. При этом число эллиптоцитов никогда не достигает величин, свойственных наследственным аномалиям оболочки эритроцита. Далее увеличение количества эллиптоцитов известно при острых постгеморагических анемиях, сифилисе, туберкулезе легких, септическом эндокардите, дефиците пируваткиназы, миелодиспластическом синдроме, хроническом миелолейкозе, опухолевых анемиях. При истинной полицитемии эллиптоциты выявляют за несколько лет до развития миелофиброза. ДД первичного и вторичного эллиптоцитоза основывается на анамнезе и клинической картине. Только в очень редких случаях требуются исследования в специализированных лабораториях.
Стоматоцитоз. Известен еще как меланезийский или южно-африканский овалоцитоз. В соответствующих регионах до 15–30% населения являются носителями этого состояния. Анемия чаще нормохромная. Диагноз этого наследственного состояния основывается, как и в предыдущем случае, на изучении мазков крови. Стоматоциты выглядят как эллипсовидные клетки с центральным щелевидным дефектом окраски, напоминающим приоткрытый рот (stoma). Мембрана стоматоцитов ригидная. Число антигенов группы крови на поверхности резко уменьшено, что может вызвать большие сложности при серологическом маркировании группы крови и озадачить врача.
Клиническая картина напоминает сфероцитарную гемолитическую анемию. Анемия и желтуха проявляются чаще в пубертатный период. Из аномалий конституции характерны изменения черепа, высокое небо, монголоидная внешность, брахифалангия.
Несфероцитарная семейная гемолитическая анемия обусловлена наличием патологического липида в оболочке эритроцита, что обусловливает высокую проницаемость оболочки для катионов с последующим ее разрывом.
Ферментопатии (энзимопатии) как причина гемолитических анемий выделены в отдельную группу сравнительно недавно. Была отмечена группа семейных гемолитических несфероцитарных анемий, при которой спленэктомия не приносила облегчения. Развитие биохимии позволило уточнить причину, выявив дефекты обмена, способные вызвать различной степени тяжести хронические или (после воздействия провоцирующих факторов) острые гемолитические анемии.
Единственный источник энергии для эритроцита — глюкоза. Причем эритроцит, переносящий кислород, сам этот кислород не потребляет. Эритроцит является единственной клеткой нашего организма (если клеткой может быть названа структура без ядра), живущей за счет анаэробного гликолиза путем гликолитического расщепления глюкозы до лактата (цикл Майерхофа — Эмбта) вместо аэробного цикла Кребса. Другой путь — гексозомонофосфатный. Первый поставляет аденозинтрифосфат (АТФ) для катионных каналов и важнейших процессов поддержания мембраны, второй — НАДФН (никотин-аденин-динуклеотид-фосфат — важнейший «энергетический аккумулятор» клетки, макроэргическое соединение). Задача второго пути — поддержание достаточного уровня редуцированного глютатиона, защищающего гемоглобин и мембрану от окислительного разрушения, проще говоря, от токсического действия кислорода. Поэтому и все энзимные дефекты сводятся либо к нарушениям гликолитического цикла, где основным ферментом является пируваткиназа, либо к нарушениям системы гексозомонофосфата.
- Энзимопатии цикла Майерхофа — Эмбта — хронические благоприятно протекающие заболевания, наследующиеся а/p. Достоверные описания известны только для недостаточности пируваткиназы. В зависимости от степени тяжести анемии выявляют различной выраженности анизо/пойкилоцитоз и полихромазию.
- Энзимопатии гексозного цикла (гексозомонофосфатный путь). Наиболее частым состоянием является дефицит глюкозо-6-фосфат-дегидрогеназы (Г-6-ФД). Чаще и ярче симптоматика развивается у мальчиков в возрасте 1,5 года–3 лет. По мере роста и взросления клиническая картина становится мягче. В некоторых группах населения до 40% мужчин являются носителями этого гена (на X хромосоме). Вероятней всего, дефицит Г-6-ФД обеспечивал своим носителям наивысшую резистентность по отношению к малярии по сравнению с основной группой населения. Клиническая выраженность заболевания может быть чрезвычайно различной. Хронический гемолиз, преимущественно компенсированный, возникает очень редко. Гемолитические кризы провоцируются медикаментами, преимущественно ацетилсалициловой кислотой, аскорбиновой кислотой, налидиксовой кислотой, нитрофуранами, фенацетином. В Средиземноморье кризы провоцировались употреблением в пищу конских бобов (ѵісіа fava), отсюда и другое название этой болезни — фавизм. Подробное перечисление препаратов и состояний, способных вызвать гемолиз при Г-6-ФД-недостаточности указан в табл. 3.7.
Таблица 3.7
Некоторые медикаменты и состояния, обусловливающие гемолиз у пациентов с недостаточность Г-6-ФД
| Противомалярийные препараты | Памахин, пентахин, плазмоцид, примахин, хинакрин, хинин, хиноцид |
| Сульфаниламидные препараты | Сульфаниламид, N2-ацетилсульфаниламид, сульфацетамид, сульфаметокосипиридазин, салицилазометоксипиридазин,сульфиоксазол, сульфипиридин |
| Нитрофураны | Нитрофурантоин, фуразолидон, фуральдотон, нитрофуразон |
| Жаропонижающие препараты | Ацетилсалициловая кислота (в высоких дозах), ацетанилид, ацетофинидин, антипирин, аминопирин, n-аминосалициловая кислота, сульфоны |
| Противотуберкулезные препараты | Тизониазид, фтивазид, ПАСК |
| Другие вещества | Бобы, хлорамфеникол, димеркапрол, метиленовый синий, фенилгидразин, пробенецид, ацетилфенилгидразин, хинидин, витамин К (в высоких дозах водорастворимых аналогов), хлорохин, налидиксовая кислота, ориназа |
| Инфекции | Респираторные вирусные инфекции, инфекционный гепатит, инфекционный мононуклеоз, бактериальная пневмония |
| Другие состояния | Диабетический ацидоз |
Гемолиз сопровождается гемоглобинурией, затем отмечается увеличение числа ретикулоцитов и ремиссия. Морфологически в период повреждения клеток выявляют тельца Гейнца и разной степени выраженности метгемоглобинемия.
Из редких форм энзимопатий этого типа известны дефицит 6-фосфоглюконат-дегидрогеназы, глютатион-редуктазы, глютатион-пероксидазы и дефицит восстановленного глютатиона. Клинические проявления не отличаются от дефицита Г-6-ФД. Анемия умеренно выражена, часто вне кризов отсутствует, кризы провоцируются приемом лекарственных препаратов (табл. 3.8).
Таблица 3.8
Энзимопатии, обусловливающие гемолитическую анемию
| Энзим | Реакция | Тип наследования | Примечание | |
| Энзимы цикла Эмбден — Майера | Гексокиназа | Глюкоза -> Г-6-Ф | а/р | Определение технически затруднено.
Активность фермента высока в молодых эритроцитах |
| Глюкозо-фосфат изомераза | Г-6-Ф -> Фр-6-Ф | а/р | ||
| Фосфофруктокиназа | Фр-6-Ф -> Фр-1,6-ДФ | а/р | Редко. Возможно сочетание с мышечной слабостью | |
| Альдолаза | Фр-6-ДФ -> Триозы | ? | Единичные сообщения | |
| Триозо-фосфат изомераза | Дигидроксиацетон-фосфат -> глицеральгид-3-фосфат | а/р | Редко. Сочетается с задержкой умственного развития и эхиноцитозом | |
| Фосфоглицераткиназа | 1,3-дифосфоглицерат -> 3-фосфоглицерат | Х-сцепленно | Редко. Сочетается с умственной неполноценностью | |
| Энолаза | 2-фосфоглицерат -> фосфоэнолпируват | а/р | Единичные сообщения | |
| Пируваткиназа | Фосфоэнолпируват -> пируват | а/р | Большинство случаев врожденной несфероцитарной гемолитической анемии | |
| Энзимы глюкозомонофосфатного пути | Г-6-ФД | Г-6Ф -> 6-фосфоглюконат | Х-сцепленно | Наиболее частая энзимопатия, предрасполагающая к гемолизу |
| Энзимы синтеза глютатиона | γ-Глютамилцистеин синтетаза | Глютамат + цистеин -> γ-глютамилцистеин | а/р | Редко. Сочетается с гиперрефлексией и дискоординацией движений |
| Глютатион-синтетаза | γ-глютамилцистеин + глицин -> глютатион | а/р | Редко. Сочетается с оксипролинурией | |
| Энзимы цикла Раппопорт — Люберинга | 2,3-ДФГ-мутаза | 1,3-ДФГ-> 2,3-ДФГ | ? | Крайне редко. Дискутируется |
| Энзимы катионной помпы | Аденозин-3-фосфатаза | АТФ -> АДФ | ? | Крайне редко. Дискутируется |
| Энзимы пурин/пиримидинового пути | Пиримидин 5’нуклеотидаза | Цитидиловая кислота -> цитозин + фосфат + рибоза | а/р | Нередко. Базофильная зернистость эритроцитов |
| Аденозин диаминаза | Аденозин -> инозин + NH3 | а/д | Повышение активности ведет к дефициту АМФ -> АДФ -> АТФ в клетках | |
| Аденилат киназа | 2АДФ -> АМФ + АТФ | а/р | Редко | |
| Энзимы липидного метаболизма | Лизолецитин ацилтрансфераза | Лизолецитин + R-COO– -> лецитин | а/д | Единичные сообщения |
Условные обозначения: Г-6-Ф — глюкозо-6-фосфат; Фр-6-Ф — фруктоза-6-фосфат; Фр-1,6-ДФ — фруктоза-1,6-дифосфат; Г-6-ФД — глюкозо-6-фосфат дегидрогеназа; 2,3-ДФГ — 2,3-дифосфатглюкоза; АТФ — аденозин-3-фосфат; АДФ — аденозиндифосфат; АМФ — аденозинмонофосфат.
Гемоглобинопатия — итог замены «нормальной» аминокислоты на другую, не свойственную здоровому человеку. Человеческий гемоглобин состоит из двух пар идентичных полипептидных цепей, причем каждая из них несет молекулу гема. Известны 4 различных полипептидных цепи: альфа (141 аминокислота), бета, гамма и дельта — по 146 аминокислот. У взрослого человека в крови в норме содержатся следующие виды гемоглобинов: НbА (а2β2, 97%), HbA2 (a2d2 1–3%), HbF (а2γ2 1–2%). В патологическом типе гемоглобина S на β-цепи в 6-м положении электрически заряженный глутамин заменен на нейтральный валин. Сегодня известны около 160 аналогичных «ошибок». Если наряду с патологическими цепями формируются и нормальные, клинические симптомы отсутствуют. Даже многие гомозиготные формы без провоцирующих факторов ничем клинически не проявляются. Решающее значение для диагноза имеет электрофорез гемоглобина. Во многих случаях это лабораторное исследование позволяет удостовериться в наличии патологического гемоглобина, но не идентифицировать его. Положение спасает химический анализ протеина. О гемоглобинопатии можно думать при следующих состояниях:
- гемолитические анемии, особенно острые гемолитические кризы при приеме лекарственных препаратов. Возникают при так называемых нестабильных гемоглобинах, выпадающих под воздействием препаратов в виде крупных преципитатов. Исключения гемоглобинопатий требуется и при хронических гемолитических анемиях со спленомегалией. Анемии могут быть как компенсированными, так и декомпенсированными. Последнее объясняется различной степенью сродства (аффинитета) кислорода и гемоглобина. Если аффинитет высокий, уровень кислорода в тканях недостаточный, компенсаторно развивается полиглобулия. При низком аффинитете кислородное обеспечение тканей может не страдать.
- метгемоглобинемия. Возникает как при дефиците Г-6-ФД, так и в энзимно неизмененных клетках за счет медикаментозно обусловленных повреждений. Характерны цианоз с эритроцитозом без сердечно-легочных расстройств.
Особая форма гемоглобинопатии, клинически и морфологически не отличимая от малой талассемии, возникает при кроссинговере части бета-цепи с дельта-цепью.
Наиболее частый патологический гемоглобин — HbS. Наследуется а/д и выявляется практически только у лиц негроидной рассы. Развивается тяжелая серповидно-клеточная анемия (дрепаноцитоз), которая у гомозиготных носителей ведет к смерти еще в детском возрасте. Деоксигенированные эритроциты теряют способность менять форму при прохождении через мельчайшие капилляры, возникают тяжелейшие расстройства микроциркуляции, проявляющиеся одышкой, рвотой, кишечными коликами, некрозами кожи и внутренних органов.
Продолжительность жизни гетерозиготных носителей не отличается от общей популяции. Клиническая картина достаточно неспецифична. Начальные проявления болезни в 50% случаев регистрируют к концу первого года жизни. Это — слабость, боль в суставах, головокружение, тошнота, лихорадка. Бледность слизистых оболочек сочетается с серозеленым прокрашиванием склер. Живот болезненный.
Деформация эритроцита объясняется выпадением в осадок (преципитат) патологического гемоглобина при снижении парциального давления кислорода, что практически обязательно в венозной крови. В артериальной крови у гетерозиготов, в отличие от гомозиготов, форма эритроцитов приближается к нормальной. Диагноз основывается на электрофорезе гемоглобина или на инкубации капли крови в обедненной кислородом атмосфере. В последнем случае выявляют типичные серповидные эритроциты.
Талассемия — гемоглобинопатия, при которой происходит не замена аминокислот, а снижение синтеза тех или иных полипептидных цепей. Более известна β-талассемия (анемия кули). Чаще поражаются жители Средиземноморья (Италия, Греция, Турция). На территории бывшего СССР относительно нередко выявляют в Азербайджане, но случаи этого заболевания описаны и в Центральной России. Диагноз легко устанавливается при электрофорезе гемоглобина. Альфа-талассемия — гетерозиготное состояние. Возможны тип 1 (генотип —/аа) и тип 2 (-а/аа). Распространенность гетерозиготных форм значительно шире, но диагностика затруднена, поскольку при электрофорезе патологический гемоглобин движется как нормальный. Крезил-бриллиантовая окраска помогает выявить патологический тип гемоглобина. Большая талассемия — гомозиготное состояние. Общие клинические проявления несущественно отличаются от других видов гемолитических анемий. Также отмечают умеренную желтуху, увеличение селезенки. Ретикулоцитоз выражен, выявляют остеопороз, диагональную исчерченность метафизов, расширение диплоэтического слоя в плоских костях. За счет перестойки костей типичен «щеточный череп»: вертикальная исчерченность плоских костей, напоминающая щетку. Остеопороз с возможностью спонтанных переломов (позвонки). Диагностически ценно выявление в периферической крови мишеневидных эритроцитов. В таких клетках гемоглобин сосредоточен по периферии, а в центре — небольшое ядрышко как «яблочко» в мишени. Осмотическая резистентность, что очень необычно, повышена. Активность ЛДГ повышена, гаптоглобин резко снижен. При тяжелых формах в крови циркулируют эритробласты, фрагментоциты. Неэффективность эритропоэза объясняется интрамедуллярным гемолизом, поэтому достаточно часто, даже без переливаний крови, развивается гемохроматоз и гемосидероз. Он проявляется грязно-серым прокрашиванием кожи, повышением уровня железа и ферритина в крови, практически полным насыщением трансферрина. В далеко зашедших случаях «интоксикации железом» отмечается полигландулярная недостаточность, экзо- и эндокринная недостаточность поджелудочной железы. Наиболее частой причиной смерти является сердечная недостаточность.
При электрофорезе резко снижено содержание гемоглобина А (в норме 97%), повышено содержание HвF, значительно повышается контрентрация НвА2. При гомозиготной альфа-талассемии, клинически неотличимой от β-формы, выявляют гемоглобин Н.
Малая талассемия является гетерозиготной формой. Анемия микроцитарная, гипохромная, мишеневидные эритроциты выявляют редко. В периферической крови нет эритробластов. Гемолиз не выражен. Гиперплазия селезенки несущественна или отсутствует. Необходима ДД с железодефицитными состояниями. В противоположность железодефицитным анемиям уровни трансферрина и железа в сыворотке крови нормальны или даже повышены. Но решающим является повышение уровня НвА2 и HвF. Талассемия может протекать одновременно с другими гемоглобинопатиями, и тогда говорят о двойном гетерозиготном состоянии.
Пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы — Мекелли) названа по основному симптому: гемоглобинурии, развивающейся во сне. Хотя, как оказалось, даже при тяжелом гемолизе с выраженным ретикулоцитозом гемоглобинурия может отсутствовать (25%). Заболевание считается нечастым (1:100 тыс.–1:500 тыс. населения). Но поскольку диагностика затруднена, то истинная частота состояния не известна. На возраст 30–40 лет приходится 54% всех больных. Первые жалобы — слабость и утомляемость. Позднее присоединяется желтуха, которая может длиться годами, пока не появится характерное черно-коричневое прокрашивание утренней порции мочи (у 75% больных). Кроме этого, беспокоят лихорадка, тошнота, рвота, головная и загрудинная боль, боль в пояснице и животе. Боль в животе порой настолько интенсивна, что приводит пациентов на хирургический стол по поводу вероятных холелитиаза, пептической язвы двенадцатиперстной кишки, аппендицита, странгуляционной непроходимости. Боль обусловлена, видимо, микротромбозами. Это же предрасполагает к частым тромбофлебитам, описанным у этих больных. Тромбозы ведут к синдрому Бада — Киари и обструкции вен мозга (50%). Поэтому основная причина смерти больных с пароксизмальной ночной гемоглобинурией — тромботические и тромбогеморрагические осложнения, а не анемия. Вариабельность симптоматики очень велика. Гемолитические кризы, лихорадка и боль в животе провоцируются инфекцией, менструациями, приемом препаратов железа, вакцинацией, введением рентгенконтрастных препаратов, оперативными вмешательствами. С момента первых манифестаций болезни до диагноза могут пройти годы. Степень анемии варьирует, хотя обычно она резко выражена при не увеличенной селезенке. Поэтому при каждой анемии неустановленной причины, даже при отсутствии ретикулоцитоза, надо помнить об анемии Маркиафавы — Меккелли. В основе заболевания лежит дефект мембраны эритро-, грануло- и тромбоцитов, происходящий из стволовых клеток. Дефект ведет к активации комплемента. В крови выявляют макроцитоз. Длительная потеря железа с мочой завершается тяжелым железодефицитным состояниям. Часто развиваются лейко- и тромбопения. Эритропоэз в костном мозгу резко усилен. Железо в моче легко выявляется окраской берлинским синим. При длительном течении возможна аплазия костного мозга с панцитопенией. Проба Кумбса отрицательна. Скрининг-тест — тепловая резистентность. Патогномоничен тест на кислотную устойчивость. ДД, наряду со всеми формами гемолитических анемий, синдромом гемоглобинурии, тромбозами вен, проводится прежде всего с вторичными формами тепловых аутоиммунных гемолитических анемий.
Острая гемолитическая анемия при тяжелой гипофосфатемии развивается при длительном голодании, рвоте и поносах, особенно у алкоголиков. В итоге формируется резкий дефицит АТФ в эритроцитах, последние теряют эластичность и легко разрушаются.
Приобретенные аутоиммунные гемолитические анемии обусловлены разрушением эритроцитов иммуноглобулинами. Тип антител и характер связывания комплемента во многом определяют клиническую картину. Различают следующие типы аутоиммунных анемий:
- классическая форма, обусловленная антителами, максимальная активность которых находится в пределах нормальной температуры человеческого тела (тепловые антитела);
- холодовые аглютинины;
- пароксизмальная холодовая гемоглобинурия.
Этиология этих заболеваний разнообразна. Перекрестно реагирующие антитела (анти-1-антитела) ответственны за развитие анемии после инфекции микоплазмы пневмонии. Для диагноза аутоиммунной гемолитической анемии большое значение имеет положительная проба Кумбса. Она позволяет выявить на мембране эритроцитов неполные антитела, не ведущие к спонтанной аглютинации. Ложноположительные результаты получают очень редко, но специфичность недостаточно высока. Положительную реакцию отмечают при некоторых медикаментозных гемолитических анемиях. Чувствительность определения антиэритроцитарных антител может быть в несколько раз повышена применением агрегат-гемаглютинизационной пробы (АГА-проба).
Тепловая аутоиммунная гемолитическая анемия характеризуется полиморфной клинической картиной. Женщины болеют в 2 раза чаще мужчин. Начало может быть как острым, так и стертым, едва заметным, течение хроническое, волнообразное. Самоизлечение крайне редко. Степень ретикулоцитоза и билирубинемии отражают тяжесть гемолиза. На биохимическом уровне выявляют гемоглобинемию, но гемоглобинурию отмечают только в период обострения. Гибель эритроцитов происходит экстравазально, и при хроническом течении анемии увеличивается селезенка. В периферической крови выявляют макроцитоз (средний объем эритроцитов более 100 мкм3), сфероцитоз, причем их объем больше, чем при врожденном сфероцитозе. В период обострения — лейкоцитоз, количество тромбоцитов может быть увеличено, хотя в большинстве случаев их не выходит за пределы нормы. Сочетание идиопатической аутоиммунной гемолитической анемии с аутоиммунной тромбопенией (иногда с лейкопенией) известно как синдром Фишера — Эванса. Путем исключения основного заболевания, на фоне которого могла бы развиться аутоиммунная гемолитическая анемия, дифференцируют вторичные или симптоматические формы от первичных или идиопатических. Вторичные формы тепловыхаутоиммунных гемолитических анемий обычно предшествует дебюту основного заболевания и чаше развиваются при лимфопролиферативных заболеваниях, особенно при хроническом лимфолейкозе, СКВ, узелковом периартериите, редко — при карциноме, иногда при неспецифическом язвенном колите и болезни Крона.
У детей вторичная тепловая аутоиммунная гемолитическая анемия может развиться в ответ на вирусную, бактериальную, реже — грибковую инфекцию.
Холодовые аглютинины бывают и в норме, но титр их не превышает 1:64. При заболеваниях титр колеблется от 1:8000 до 1:1 000 000. Уровень иммунологически активных иммуноглобулинов может быть настолько завышен, что при электрофорезе регистрируется пик М. Патогенез заключается в том, что иммуноглобулины М анти-І, реже анти-і, (гемолизин) при низких температурах связывают комплемент, что приводит к разрушению эритроцитов. Реакция начинается при температуре <30 °C, оптимум — около 0 °C. Проба Кумбса положительна. Повышение температуры сопровождается элиминацей антител с клеточной мембраны, но фиксированный комплемент на оболочке эритроцитов по-прежнему сохраняется. Чаще выявляют идиопатические формы. Гемолитический криз начинается, как правило, через несколько дней после охлаждения. В благоприятных условиях через 1–2 нед заболевание спонтанно завершается фенотипическим выздоровлением. Картина крови нормализуется через 3–4 нед. Вторичные симптоматические варианты свойственны злокачественным новообразованиям лимфоидной системы.
Доброкачественные транзиторные формы описаны после инфекционного мононуклеоза и микоплазменной пневмонии.
Взаимодействие эритроцитов, комплемента и иммуноглобулинов, естественно, происходит в наиболее зябких местах: кончики пальцев, носа, мочки ушей. В жарком климате все проявления самопроизвольно прекращаются, хотя причина, безусловно, сохраняется. Разнообразные клинические варианты можно свести к следующим проявлениям:
1. Хроническая гемолитическая анемия.
2. Бледность и цианоз кончиков пальцев, носа, ушных раковин, особенно на холоде. Возможны некрозы, что заставляет проводить ДД с васкулитами и болезнью Рейно.
3. После длительного нахождения на морозе — приступы гемоглобинурии. (Дифференцировать с холодовой гемоглобинурией типа Ландштейнера — Доната!) Обычно клиническая картина достаточно мягкая. Самый примечательный симптом — синее ливедо или резкая («трупная») бледность губ, носа, ушных раковин, щек, пальцев. Боль беспокоит мало, скорее — это ощущение резкого озноба, онемения. В тепле вся симптоматика исчезает за несколько минут. Типично анамнестическое указание на резкое потемнение мочи после пребывания на холоде. При идиопатическом течении сохраняется бледность и субиктеричность. Селезенка и печень увеличиваются незначительно. При длительном гемолизе селезенка становится плотной и легко доступной пальпации.
Пароксизмальная холодовая гемоглобинурия обусловлена взаимодействием с комплементом анти-Р-специфичных IgG-антител, так называемый гемолизин Ландштейнера — Доната. Выявляют очень редко. В 1/3 случаев обусловлена сифилисом, хотя персистенция гемолизина может сохраняться и после лечения. Клиническая картина заключается в том, что после длительного пребывания на холоде развивается боль в животе, спине, конечностях, потрясающий озноб, после чего возникает гемоглобинурия. В этот период проба Кумбса положительна. Степень снижения уровня гемоглобина отражает тяжесть заболевания.
Гемолитические анемии при изоантителах развиваются как результат ответа на чужие эритроциты, так что гемолиз рассматривается как посттрансфузионная реакция. В первую очередь — это переливание несовместимой крови по системе АВ0. В относительно «физиологических» условиях этот тип гемолитической анемии выявляют у новорожденных от резус-конфликтной беременности. В этих случаях иммунизация резус-негативной матери осуществляется либо эритроцитами резус-положительного плода, либо предшествующими трансфузиями и абортами. Наиболее реактогенным является резус-фактор Д, затем С, наименее реактогенен Е. Следует помнить, что есть и другие группы крови: К, М. Диагноз не вызывает сомнения, если временной интервал между переливаниями крови и гемолизом ничтожен, иногда же светлая пауза затягивается до нескольких суток.
Иммунные медикаментозные гемолитические анемии развиваются при приеме некоторых лекарственных препаратов.
1. При длительном приеме альфа-метил-допы и L-допы в высоких дозах вырабатываются IgG-антитела аналогичные таковым при аутоиммунных анемиях теплового типа. Проба Кумбса сохраняется положительной и несколько месяцев спустя после отмены препаратов.
2. Гаптеновый тип является необычным осложнением перорального применения пенициллина в высоких дозах. Антибиотик связывается с мембраной эритроцитов, в случае выработки антител (IgG) к пенициллину развивается гемолиз. Проба Кумбса положительна только в период приема антибиотика.
3. Прием изониазида, хлорпромазина, фенацетина, хинина, хинидина и некоторых других лекарственных препаратов может сопровождаться образованием комплекса антиген — (медикамент) — антитело, который совершенно неспецифично и обратимо фиксируется на эритроците. Необратимое присоединение комплемента вызывает лизис оболочки клетки, эритроцит уничтожается неспецифически, по типу «ненужного свидетеля». Проба Кумбса положительна только в период приема лекарственных препаратов. До 20% всех иммунных гемолитических анемий обусловлены медикаметозными воздействиями.
Гемолитические анемии при химических повреждениях эритроцитов наиболее известны при приеме парацетамола и фенацетина. Гемоглобин разрушается за счет перекисного окисления, образуются мет- и сульфогемоглобин, а также типичные внутренние тельца Гейнца. Повреждающий эффект определяется дозой и длительностью приема, хотя на собственной практике мы убедились, что он во многом индивидуален. В казуистических случаях при злостном применении препаратов развивается почечная недостаточность. Диагноз подтверждается выявлением продуктов обмена фенацетина в моче. Эти же препараты способны вызывать гемолиз и при гемоглобинопатии.
Анемии при интоксикациях свинцом обусловлены как снижением продукции эритроцитов, так и их гемолизом. При незначительной интоксикации или в дебюте воздействия гемолиз может играть решающую роль.
Дифференциальная диагностика анемии с тельцами Гейнца (внутренними тельцами). Внутренние тельца представляют собой конечный продукт распада преципитированного гемоглобина, выявляются при окраске на ретикулоциты или метилголубым, могут наблюдаться у 0,5–0,8% здоровых, чаще у новорожденных, что становится понятным в свете постнатальной смены типа гемоглобина и уменьшения его количества. В большом количестве внутренние тельца выявляют после спленэктомии. В патологических условиях внутренние тельца — результат повреждения клетки, часто в сочетании с врожденными дефектами эритроцитов. Возможно развитие анемии любой степени тяжести, массивный внутрисосудистый гемолиз влечет за собой гемоглобинемию и гемоглобинурию. «Идиопатические тельца Гейнца» развиваются при еще не идентифицированных типах энзимо- или гемоглобинопатий. В других случаях внутренние тельца образуются при гемоглобинопатиях, протекающих с острым гемолизом (реже — хроническим) после применения лекарственных препаратов. Большая альфа-талассемия сопровождается феноменом внутренних телец за счет образования β4-тетрамера (НвН), который и образует преципитаты. В формировании телец имеют значение энзимные дефекты, прежде всего гексозмонофосфатного типа после приема различных лекарственных препаратов или бобов (фавизм), а также интоксикации химическими соединениями с образованием связей с амино-, нитро- или гидроксильными группами.
Гемолитические анемии при механических повреждениях эритроцитов типичны для сеансов гемосорбции, гемодиализа, операциях с искусственным кровообращением и особенно у пациентов с имплантированными искусственными клапанами сердца. Решающее значение имеет материал, с которым соприкасаются эритроциты. Показателем гемолиза является уровень ЛДГ в сыворотке крови. Гемолиз, как правило, компенсированный. В периферической крови выявляют фрагменты эритроцитов (фрагментоциты), в моче при окраске берлинским синим — гемосидерин. Декомпенсация развивается только при больших потерях железа с мочой и проявляется уменьшением среднего объема эритроцитов и ретикулоцитопенией или их полным отсутствием. У людей с искусственными клапанами сердца часто формируются билирубиновые камни желчного пузыря. Профилактика: ограничение мясной пищи в рационе, водная нагрузка, прием желчегонных средств перед сном, то есть мероприятия, предотвращающее сгущение желчи.
Микроангиопатические гемолитические анемии характеризуются частой почечной недостаточностью, наличием в крови необычных, причудливых по форме эритроцитов и их фрагментов. Гемоглобинемия и гемоглобинурия дополняют картину. Общее в патогенезе — механическое повреждение эритроцитов фрагментами фибрина или патологически измененными сосудами. Этиология разнообразна.
- Гемолитико-уремический синдром (синдром Гассера) — наиболее частое состояние этой группы. Представляет собой тяжелую микроангиопатическую гемолитическую анемию, возникающую преимущественно у детей (чаще болеют дети в возрасте до 3 лет) с тромбопенической пурпурой и острой почечной недостаточностью в результате тромбирования микроциркуляторного русла почек. Нозологически различают: А) классическую форму гемолитико-уремического синдрома у маленьких детей без связи с определенной инфекцией; Б) не только у маленьких детей развивающиеся варианты заболевания, но также ассоциированные с бактериальной (псевдомонады, пневмококки, сальмонеллы, кишечная палочка O-156, шигеллы) или вирусной инфекцией (Коксаки-, адено-, ЕСНО-вирусы) инфекцией; В) наследственные а/p формы с манифестацией как в детском, так и во взрослом возрасте; Г) гемолитико-уремический синдром взрослых, описываемый после родов, при злокачественной гипертонии, реакциях отторжения трансплантата, приеме гормональных контрацептивов, циклоспорина А, митомицина. Хотя ряд авторов под гемолитико-уремическим синдромом в узком смысле слова понимают только варианты А, Б и В.
Локальное диссеминированное внутрисосудистое свертывание, лежащее в основе этого синдрома, сопровождается разрывами эритроцитов о фибринные наложения на стенках сосудов. Одновременно развивается тромбоцитопения потребления разной степени выраженности. Превалируют острые формы с олигоурией и анурией. Описаны и хронические варианты, диагноз которых затруднен и основывается на исследования мочи относительно продуктов расщепления фибрина.
Выделяют основные и дополнительные диагностические признаки.
Основные признаки:
- Микроангиопатическая гемолитическая анемия с концентрацией гемоглобина <9 г/дл, фрагментированными эритроцитами, гипербилирубинемией за счет непрямой фракции, ретикулоцитозом, повышением уровня ЛДГ.
- Тромбоцитопения чаще <50 000/мкл с петехиями и кровотечениями.
- Острая почечная недостаточность с протеин/цилиндр/гематурией, требующая обязательного проведения гемодиализа.
Дополнительные признаки:
- При детских вариантах — острое начало болезни после кишечной или респираторной инфекции.
- Частые желудочно-кишечные расстройства, боль в животе.
- Церебральная симптоматика (не обязательна) в результате уремических расстройств водно-электролитного равновесия (нарушения сознания, судороги, кома).
- Внутрисосудистое свертывание.
- Артериальная гипертензия (нетипична для детских форм).
- При гистологических исследованиях отек эндотелия, расширение субэндотелиального пространства, тромбы. При классических вариантах гемолитико-уремического синдрома поражены преимущественно гломерулы, при других формах — смешанный гломерулярно-артериальный тип.
- Прогноз при классических вариантах относительно хороший при адекватной терапии острой почечной недостаточности. При других формах — прогноз значительно хуже (АГ, хроническая почечная недостаточность). Наиболее плохой прогноз при митомицин-индуцированных гемолитико-уремических состояниях.
- Тромботическая тромбоцитопеническая пурпура (синдром Мошковица) по клиническим характеристикам примыкает к гемолитико-уремическому синдрому. Весьма вероятно, что этот синдром объединяет гетерогенные состояния. Клинические проявления видны из названия синдрома, практически обязательно развивается почечная недостаточность. В отличие от синдрома Гассера (гемолитико-уремического синдрома) резко выражены неврологические проявления. Возможны фульминантное течение, хронические и рецидивирующие формы. Описаны семейные случаи. Выделяют идиопатическую тромботическую тромбоцитопеническую пурпуру и вторичные ее формы при коллагенозах, инфекциях, опухолях, вакцинациях, на фоне приема медикаментов, при беременности.
- Синдром Шульмана — Упсхава. Врожденный вариант пурпуры Мошковица с а/р (?) наследованием. Причина заключается в наличии генетического дефекта еще не идентифицированного плазменного фактора, который в норме должен определять образование и созревание тромбоцитов. В случае отсутствия этого фактора с детства после инфекционных атак или стрессовых ситуаций появляются острые эпизоды тромбоцитопении. Из-за нарушенного взаимодействия тромбоцитов с эндотелием на внутренней поверхности сосудов образуются тромбы, о которые и разрушаются эритроциты. Переливание свежезамороженной плазмы крови ведет к нормализации числа тромбоцитов, эритроцитов и увеличению времени жизни тромбо- и эритроцитов. В отличие от пурпуры Мошковица не бывает неврологических изменений.
- Злокачественная гипертензия сопровождается повреждением эндотелия, где и начинает откладываться фибрин. Последний и создает ту острую бугристую поверхность, о которую «бьются» эритроциты. До сих пор дискутируется вопрос, является ли микроангиопатическая гемолитическая анемия при эклампсии и преэклапсии результатом ДВС или итогом первичного повышения АД? Видимо, два этих мнения не являются конкурирующими.
- Метастазирование злокачественных опухолей может сопровождаться диссеминированным внутрисосудистым свертыванием, что, как указано выше, приводит к повреждению эритроцитов.
- Микроангиопатические гемолитические анемии наблюдаются также при молниеносном течении геморрагического васкулита, узелковом периартериите, остром гломерулонефрите, отторжении почечного трансплантанта.
Аутоиммуный гемолиз при инфекционных состояниях типичен для септических процессов, особенно при инфекции гемолитическими стрепто- и стафиллокками. Из грамотрицательной флоры большое значение имеет гемолитическая кишечная палочка, выявляемая у детей, ослабленных взрослых и стариков. Инфекции, вызываемые плазмодиями, бартонеллами, микоплазмами, а также мононуклеоз, корь (реже — гепатит) способны сопровождаться аутоимунным гемолизом. Возбудители газовой гангрены могут вызвать гемоглобинурию.
Гемолитические анемии при обменных нарушениях известны прежде всего в педиатрии. У взрослых эти анемии развиваются при заболеваниях почек. Патогенез таких анемий сложен: токсическое повреждение костного мозга, дефицит эритропоэтина и легкий микроангиопатический гемолиз. Чаще развивается при хроническом пиелонефрите. В отличие от нормоцитарной почечной анемии, гемолитическая анемия сопровождается ретикуло- и макроцитозом. Таким образом, при каждой анемии неясной этиологии необходимо определять креатинин и мочевину сыворотки крови. У мужчин пожилого возраста причиной сочетания уремии и анемии может быть рак предстательной железы.
Печеночным анемиям присущ макроцитоз со средним корпускулярным объемом в ПО мкм3. При этом не выявляют дефицита витамина В12 или фолиевой кислоты. Гематокрит составляет 35–40%, ретикулоцитоз — около 3–5%. Выявляют шиповатые эритроциты типа плода каштана или дурмана, напоминающие акантоциты. Нередко определяют и мишеневидные клетки. Легкая компенсированная гемолитическая анемия возникает из-за приобретенного дефекта мембраны эритроцитов, когда вследствие дефицита лецитил-холестерин-ацетил- трансферазы повышается соотношение холестерин/ лецитин. Описанная картина характерна при циррозе печени, алкогольной липодистрофии, хроническом гепатите.
Острая гемолитическая анемия у алкоголиков с жировой дистрофией печени носит название синдрома Циве. Типичны желтуха, ретикулоцитоз, гиперлипид- и гиперхолестеринемии. Влияние алкоголя на эритропоэз многопланово. Имеет значение дефицит белков, витаминов и железа из-за плохого питания, синдрома мальабсорбции и блокады обмена. Отражением этого являются большое количество сидеробластов в костном мозгу. При коррекции питания и прекращении употребления алкоголя анемия обычно исчезает, появляющийся при этом ретикулоцитоз служит основанием для ошибочной диагностики синдрома Циве. Алкоголизм сопровождается также тромбоцитопенией, а гипохромная анемия при циррозе печени должна быть поводом для исключения кровоточащих язв пищевода. Поскольку цирроз сопровождается и спленомегалией, то нельзя исключать и этот фактор развития анемии у алкоголиков.
Акантоцитоз развивается при врожденной α-β-липопротеинемии в сыворотке крови. Из-за дефицита этого липопротеина в оболочке эритроциты принимают форму плодов каштана, выглядят как шиповатые клетки. Длительность жизни таких эритроцитов по сравнению с обычными формами не изменена. Заболевание проявляется стеаторреей, пигментным ретинитом, атаксией и умственной отсталостью.
Гиперспленизм сопровождается усиленным разрушением эритроцитов. Любое увеличение органа воспалительной, неопластичной этиологии, при портальной гипертензии, болезнях накопления сопровождается замедлением кровотока в органе и разрушением клеток. Интенсивность разрушения эритроцитов может быть количественно измерена с помощью меченых эритроцитов Сr51. Имеет значение не только собственно увеличение органа, но и затруднение оттока из селезенки, например, при любых объемных процессах, поддавливающих селезеночную вену. Причинами усиленного разрушения клеток в гиперплазированной селезенке является механическое затруднение движения клеток, дефицит глюкозы, ацидоз и макрофагальная реакция.
ДД феномена красной мочи.
Поскольку некоторые анемии сопровождаются изменением цвета мочи, то требуется ДД этого феномена.
Если центрифугирование красной мочи сопровождается приобретением естественной окраски над осадочным слоем, то можно говорить о гематурии. Если этого не происходит, то необходимо исключать другие причины.
В физиологических условиях моча приобретает красный цвет после обильного употребления свеклы, что наблюдается приблизительно у 10–15% населения. Но тот же самый феномен отмечается у 80% людей с железодефицитной анемией.
Алкаптоурия может придать моче темный цвет и развивается при выделении с мочой гомогентизиновой кислоты, естественного продукта обмена белка. В результате рецессивного генетического дефекта гомогентизиновая кислота не разрушается и попадает в мочу. Отличием от гемоглобинурии является потемнение мочи на свету (постепенно) или при добавлении щелочи (мгновенно).
Миоглобинурия сопровождаетсяотделением темно-коричневой мочи. Миоглобинурия может развиваться у практически здоровых людей после тяжелейших физических нагрузок. Миоглобинурия, рецидивирующая после умеренных физических нагрузок, не сопровождающаяся повышением уровня лактата крови, типична для синдрома Мак — Ардла, мышечной формы гликогеноза с дефицитом мышечной фосфорилазы. Тяжелые ожоги, поражения электрическим током, плохо леченный острый миозит (болезнь Унферрихта) и синдром раздавливания сопровождаются миоглобинурией за счет массивного попадания миоглобина в кровь при разрушениях мышц. Типичное осложнение — почечная недостаточность.
Пароксизмальную миоглобинурию отмечают очень редко. Требуется ДЦ с маршевой гемоглобинурией, поскольку в том и в другом случае изменение окраски мочи развивается после физических нагрузок. Но при пароксизмальной миоглобинурии в период приступа развивается мышечная слабость вплоть до паралича, болезненность при пассивных движениях. Диагноз подтверждает наличие миоглобина в моче. В отличие от гемоглобинурии повышается активность трансаминаз, креатинфосфокиназы и ЛДГ в сыворотке крови.
Гемоглобинурии периодически могут сопровождать каждую гемолитическую анемию, для диагностики этого состояния необходимо провести бензидиновую пробу. Решающим в формировании гемоглобинурии является степень внутрисосудистого гемолиза. Острые атаки фавизма, гемолиза при гемоглобинопатии сопровождаются гемоглобинурией. Чаще гемоглобинурию выявляют при пароксизмальной ночной гемоглобинурии Маркиафавы — Меккелли (см. выше). Маршевая гемоглобинурия развивается после длительной физической нагрузки и не сопровождается другими патологическими проявлениями. Причина — механическое повреждение эритроцитов в сосудах конечностей. Пароксизмальная холодовая гемоглобинурия (см. выше).
Если бензидиновая реакция с темной мочой отрицательна, то необходима ДД с порфирией, миоглобинурией, алкаптоурией и окрашиванием мочи выделяющимися компонентами пищи.
Анемии с нарушенным созреванием эритроцитов в костном мозгу характеризуются несоответствием большого числа эритробластов в костном мозгу и ретикулоцитопенией в периферической крови. Это обусловлено гибелью части зреющих эритробластов.
Иначе это явление обозначается как «неэффективный эритропоэз». Уровень билирубина в крови повышен без признаков периферического гемолиза. Биохимически типичен высокий уровень обмена железа в плазме крови при плохом включении меченного железа в эритроциты. Анизо- и пойкилонитоз дополняют картину. Описываемое состояние отмечают при большой талассемии и мегалобластных анемиях. Поскольку мегалобластные анемии только часть так называемых макроцитарных анемий, то в ДД необходимо учитывать этот генеральный синдром (табл. 3.9, схема 3.4).
Таблица 3.9
ДД-признаки мегалобластных анемий
| Болезнь, синдром | Симптом и состояние | ||||||||||
| Анемия мегалобластная | Лейкоцитопения | Тромбоцитопения | Ахилия | Неврологическая симптоматика | Уровень витамина В12 в сыворотке крови | Уровень фолиевой кислоты в сыворотке крови | Проба Шиллинга | Особенности | Установленная или вероятная причина | Что делать | |
| Истинная пернициозная анемия | ++ | + | + | Очень часто | Очень часто | ↓↓ | Иногда повышена | Патологически изменена | — | IF-нарушение синтеза | Назначить витамин В12 парентерально |
| Пернициозная анемия при ботриоцефаллезе | + | + | + | Не типична | Не характерна | ↓ | Часто снижена | Патологически изменена | Членики лентеца или яйца в испражнениях | Потребление витамина В12 паразитом | Лечение глистной инвазии; витамин В12 внутрь или парентерально |
| Пернициозная анемия беременных | + | +/- | +/- | Часто | Нет | Часто снижен | Снижена | Чаще в норме | Умеренный гемолиз; спленомегалия | Многофакторна | Назначить Fe; В12; фолиевую кислоту |
| Пернициозная анемия после резекции желудка | + | + | + | Да | Редко | Прогрессирует снижение | В норме | Чаще патологически изменена | Дефицит железа | Нарушение всасывания витамина В,2 | Назначить препараты Fe; В12 парентерально |
| Пернициозная анемия при стриктурах или дивертикулах тонкой кишки, межкишечных аностомозах | +/- | +/- | Иногда | Нет | Часто снижена | — | Ренгенологическая картина | Дефицит витамина В12 | Назначить препараты В12 и фолиевой кислоты парентерально; антибиотики | ||
| Мальабсорбция | +/- | +/- | +/- | Иногда | Нет | ↓ | ↓ | Чаще патологически изменена | По основному заболеванию | Нарушение всасывания витамина В12 и фолиевой кислоты | В12, фолиевая кислота и Fe парентерально |
| Миелодиспластический синдром | + | + | + | Нет | Нет | В норме | В норме | В норме | Высока вероятность трансформации в лейкоз | ? | — |
| Мегалобластная анемия при гемобластозах | +/- | +/- | +/- | Не характерна | Нет | Чаще в норме | Часто снижена | Чаще в норме | — | Дефицит фолиевой кислоты или нарушения утилизации | — |
| Пернициозная анемия при циррозе печени | +/- | +/- | Иногда | Редко | Часто снижен | Часто снижен | Чаще патологически изменена | — | Снижена резорбция и накопление витамина В12, резорбции фолиевой кислоты | — | |
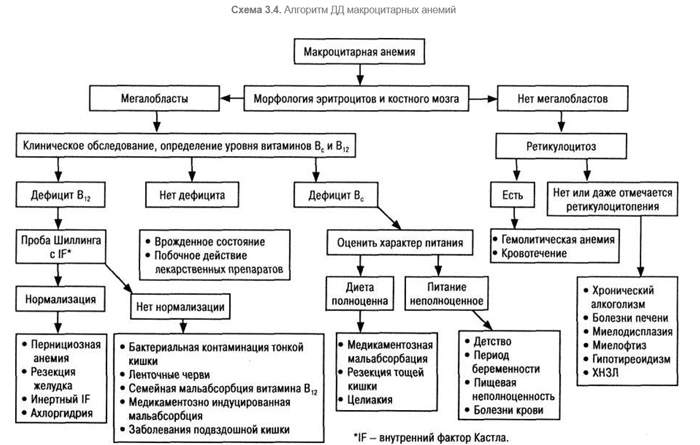
Мегапобластная анемия — это практически всегда дефицит фолиевой кислоты (В6) или витамина В12. Клинико-гематологическая картина при этих двух дефицитарных состояниях следующая:
- интрамедуллярный гемолиз обусловливает легкую желтушность. При дефиците кобаламина рано седеют волосы;
- достаточно часто регистрируют неврологические расстройства. Фуникулярный миелоз протекает с субъективными ощущениями бегания мурашек, щекотания кончиков пальцев, нарушениями походки, положительным симптомом Бабинского, нарушениями глубокой и поверхностной чувствительности, атаксией и парезами. Почти всегда отмечают легкие изменения психики, но возможны и психозы. Причем скрытый дефицит В12 может проявляться только неврологической симптоматикой без изменений в крови;
- типична панцитопения, хотя тромбоцитопения редко достигает критических значений. Поскольку витамин В12 и фолиевая кислота определяют деление клеток, нарушения, кроме кроветворных органов, касаются прежде всего быстрорастущих тканей. Формируется атрофия слизистой оболочки желудочно-кишечного тракта, язык выглядит гладким, с атрофической поверхностью. Реже выявляют болезненный глоссит (гунтеровский);
- вследствие неэффективного эритропоэза повышена активность ЛДГ сыворотки крови и уровень железа. Оба показателя приходят в норму через несколько дней после начала терапии;
- мегалобласты нередко овоидной формы, имеют корпускулярный объем 120 мкм3 и более, сочетаясь с анизо- и пойкилоцитами. В костном мозгу также много мегалобластов;
- наряду с эритромегалобластами в периферической крови и костном мозге выявляют гранулоциты с избыточным количеством сегментов в ядре (6 и более), гигантские палочкоядерные нейтрофильные гранулоциты и гигантские метамиелоциты.
ДД дефицита витамина В12 (кобаламина) и фолиевой кислоты основывается на следующих признаках:
- введение фолиевой кислоты в физиологических дозах (1–2 мг/сут) или витамина В12 (250–500 мг/сут) вызывает терапевтический эффект уже на 4–5-й день лечения;
- определение уровня витамина В12 и фолиевой кислоты в сыворотке крови. Повышение уровня витамина В12 типично для хронического миелобластного лейкоза, истинной полицитемии, остеомиелосклероза. При пернициозной В12-дефицитной анемии уровень фолиевой кислоты, как правило, не изменен (табл. 3.10).
Таблица 3.10
Уровни витамина В12 и фолиевой кислоты у здоровых и у пациентов с их дефицитом
| Показатель | Норма* | Дефицит витамина В12 | Дефицит фолиевой кислоты |
| Витамин В12 в сыворотке крови, нг/л | 450 (160–1000) | 38 (10–110) | 190 (50–500) |
| Фолаты в сыворотке крови, мкг/л | 10 (6–21) | 17 (4,5–37) | 3 |
| Фолаты эритроцитов | 316 (166–640) | 146 (26–395) | 100 |
*Показатели нормы варьируют в зависимости от методик определения и в разных лабораториях.
Возможны проблемы в клинической оценке показателей концентрации витамина В12 в сыворотке крови.
Ложноположительное заключение о высоком уровне В12 (скрытый дефицит):
- погрешности лаборатории;
- предшествующая инъекция витамина В12;
- аномальные протеины сыворотки крови (болезни печени, хронический миелолейкоз).
Ложное заключение о низком уровне витамина В12:
- артефакт радиоактивности сыворотки крови (после сканирования с галлием или технецием);
- дефицит фолиевой кислоты;
- нормальная беременность;
- прием оральных контрацептивов;
- миелома или апластическая анемия;
- дефицит транскобаламина I;
- витамин С в высоких дозах;
- взаимодействие лекарств (антибиотики, хлорпромазин, антифолаты) с реактивами;
- тяжелый дефицит железа (?).
Однако лабораторные показатели не являются абсолютно определяющими для диагноза. При дефиците кобаламина отклонения показателей крови частично или полностью корригируются применением фолиевой кислоты. При дефиците фолатов нарушения будут частично или полностью корригироваться витамином В12. Но поражение спинного мозга при дефиците кобаламина будет прогрессировать, даже если показатели крови будут восстанавливаться фолатами. Таким образом, ответ на терапию является конечным определением типа дефицита независимо от результатов лабораторных исследований.
Патогенез дефицита витамина В12 имеет несколько вариантов. Витамин В12 (EF, внешний фактор Кастла) синтезируется микроорганизмами и присутствует в любых продуктах животного происхождения. Его всасывание в нижних отделах тонкой кишки происходит только в комплексе с гликопротеином желудочного сока (IF, внутренний фактор Кастла). Но взаимоотношения внутреннего фактора Кастла (IF) и цианокобаламина (EF) очень непростые, что и вносит нюансы в патогенез В12-дефицитной анемии. Для полноценного взаимодействия внешнего и внутреннего факторов нужна полноценная поджелудочная железа. Когда кобаламин поступает в желудок, с ним тут же прочно связывается не IF, а кобаламинсвязывающий белок R-типа. Белок R-типа не позволяет всасываться витамину В12. Когда комплекс кобаламинсвязывающего белка с витамином В12 поступает в двенадцатиперстную кишку, трипсин и химотрипсин расщепляют белок R-типа, но не меняют структуру IF. Освобождающийся от белка R-типа витамин В12 переходит к IF и только после этого всасывается. То есть при недостаточной функции поджелудочной железы всасывания витамина В12 не происходит, так как белок R-типа не расщепляется. Итак, дефицит витамина В12 развивается при следующих состояниях:
- Отсутствие внутреннего фактора при пернициозной анемии (болезнь Бирмера), состояние после частичной или тотальной гастрэктомии, врожденный дефект синтеза внутреннего фактора.
- Паразитарное или микробное потребление в кишечнике при глистных инвазиях ленточными цепнями, при синдроме слепой петли, фистулах и дивертикулезах кишечника.
- Мальабсорбция комплекса B12-IF в подвздошной кишке при целиакии, первичном или тропическом спру, болезни Крона, синдроме Золлингера — Эллисона и тяжелой панкреатической недостаточности, болезни Гресбека (врожденное нарушение всасывания комплекса B12-IF в сочетании с протеинурией), терапия неомицином, парааминосалициловой кислотой, дифенилгидантоином, фенобарбиталом, фенилбутазоном, нитрофуранами.
- Длительное и упорное вегетарианство ведет к ограничению поступления в организм витамина В12. До развития дефицита витамина В12 при этом проходит 8–12 лет.
Для ДД причин дефицита витамина В12 (дефицит фактора IF или нарушения всасывания комплекса B12-IF) применяют пробу Шиллинга. Перорально применяют 1 мкг радиоактивного В12 и через 2 ч вводят 100 мкг «холодного» витамина для насыщения рецепторов В12. При нормальной функции почек через 24 ч выводится 8–30% принятой дозы. При пернициозной анемии выделяется менее 2%. Тест повторяют через несколько дней, при этом В12 назначают вместе с IF. Если нормализация не наступает, то говорят о мальабсорбции. Патогенез и клиническая симптоматика мегалобластных анемий суммирована на схеме 3.5.
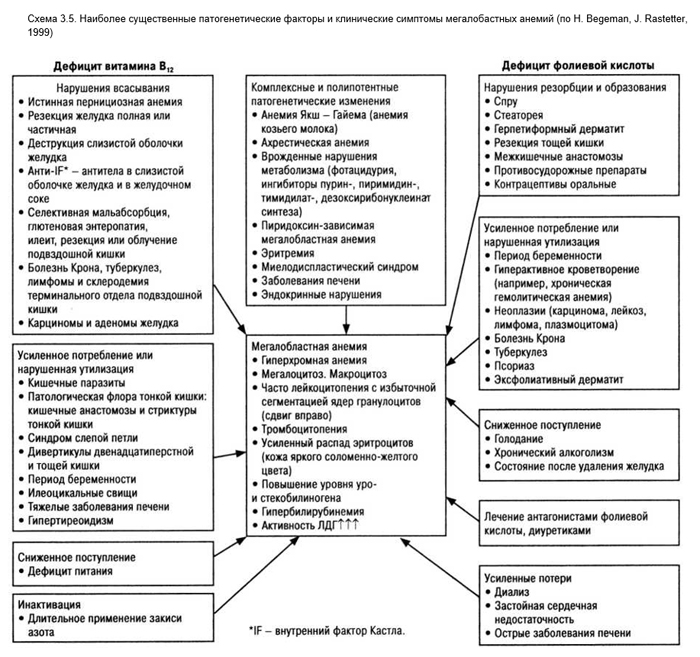
Пернициозная анемия — наиболее частая форма мегалобластных анемий. Среди больных больше женщин (3:2). Достаточно широко распространена в средних широтах, распространенность повышается к северу. Реже выявляют в Африке, у арабов, очень редко — в Японии. Причиной является заболевание желудка, видимо, с наследственно определенным нарушением иммунитета. В пользу этой гипотезы свидетельствуют лимфоплазмоцитарные инфильтраты в атрофической слизистой оболочке желудка, циркулирующие антитела к париетальным клеткам (95%), антитела против IF в сыворотке крови и желудочном соке у 65%, нередкая ассоциация пернициозной анемии с другими аутоиммунными заболеваниями (например, с тиреоидитом Хашимото и тогда заболевание рассматривается как H-B-G-синдром (Hypoparathyroid-Biermer-Gonadedysgenesy). Семейный вариант этого состояния развивается у девочек-подростков и девушек. Является вариантом аутоиммунного полигландулярного синдрома).
Наконец, возможен парадоксальный ответ на кортикостероидную терапию (применимо только в эксперименте) с развитием пернициозной анемии. Антитела к IF патогномоничны для пернициозной анемии. Но антитела к париетальным клеткам находят при многих других заболеваниях: сахарном диабете, заболеваниях щитовидной железы и т. д. Заболевание развивается медленно, манифестирует чаще в возрасте старще 60 лет. У африканцев манифестирует раньше — в возрасте около 40 лет. Казалось бы, можно думать об особенностях питания и массовых глистных инвазиях, но более раннее заболевание пернициозной анемией отмечают и у представителей негроидной расы, проживающих в США. У детей известно в единичных наблюдениях и, видимо, в ряде случаев заболевание описывалось как анемия Якша — Гайема. Желудочный сок даже после стимуляции не содержит соляной кислоты. Часто возникают расстройства стула, боль в животе. Дистрофия не типична, СОЭ значительно повышена. У небольшого числа пациентов с пернициозной анемией развивается рак желудка, о чем свидетельствует микроцитарная анемия и положительная бензидиновая проба при исследовании кала. В практике чаще приходится диагностировать уже леченную пернициозную анемию, когда первоначально ошибочно устанавливали другой диагноз и неприцельно назначали разнообразные противоанемические препараты. Диагноз основывается на исследованиях желудочного сока (ахлоргидрия), гастроскопии с биопсией, поисках антител, пробе Шиллинга.
Состояния, протекающие с дефицитом фолиевой кислоты, изолированно в развитых регионах возникают крайне редко. В практике известны следующие причины:
- врожденные варианты нарушения обмена фолиевой кислоты;
- прием антагонистов фолиевой кислоты: метотрексат (блокирует дигидроредуктазу фолиевой кислоты), дифенилгидантоин, длительный прием барбитуратов, антиовуляторных гормональных контрацептивов и др.;
- повышенный расход витамина в период беременности, гемолитических анемиях, многочисленных хронических заболеваниях;
- мальабсорбция; резекция тощей кишки, поскольку фолиевая кислота всасывается в верхних отделах тонкого кишечника, спру тропическое или идиопатическое, целиакия;
- алкоголизм вследствие комплекса причин: дефицит питания и мальабсорбция.
Дефицит фолиевой кислоты по сравнению с дефицитом витамина В12 развивается очень быстро. При потреблении фолатов менее 5 мкг/сут, их дефицит развивается через несколько недель. Содержание фолатов в сыворотке крови снижается за 7 дней, а клинические изменения (мегалобластная анемия, крайне выраженная депрессия) появились в ближайшие 4 мес. Сведения о лабильности уровня фолиевой кислоты в крови особенно интересны в связи с сообщениями о том, что 50% всех пороков развития нервной трубки связаны с дефицитом фолатов.
Мегалобластная анемия беременных является результатом комбинации дефицита железа, нарушений всасывания витамина В12 и фолиевой кислоты.
Редкие формы мегалобластных анемий развиваются при лечении цитостатическими препаратами (циклофосфамидом и 6-меркаптопурином), при длительном приеме противосудорожных препаратов (прежде всего у детей), при алкоголизме. Последняя причина обусловлена не только дефицитом фолиевой кислоты, но и прямым токсическим действием этанола с одновременным развитием тромбоцитопении, гранулоцитопении и нарушением обмена железа с аккумуляцией гемосидерина в эритробластах. Врожденные мегалобластные анемии более известны в педиатрии.
Это врожденная оротоацидурия, обусловленная дефицитом двух ферментов, участвующих в синтезе уридина и проявляющаяся уже в первые годы жизни тяжелой мегалобластной анемией, нарушением физического и психического развития.
При редком синдроме Леш — Нихана существует дефект пуринового обмена, мегалобластная анемия сочетается в этих случаях с подагрой и умственной отсталостью.
Особенности нарушения обмена витамина В12 в детском и юношеском возрасте суммированы в табл. 3.11.
Таблица 3.11
Детские и юношеские формы дефицита кобаламина
| Характер нарушения | Возраст в дебюте заболевай | Известная частота | Уровень В12 и (В12СС) в сыворотке крови | Тест Шиллинга | Тип наследования | Клиническая картина |
| Врожденный дефицит IF | После 1 -го года жизни | >50 случаев | Низкий (норма) | Как у взрослых с ПА | а/р | Бледность, стоматит, диарея (или запор), спленомегалия, фуникулярный миелоз, нарушения ЦНС, мегалобластная анемия |
| Функционально инертный IF | 10–13 лет | Единичные сообщения | Низкий | Изменен с и без IF | а/р | См. выше |
| Врожденный дефицит ТК II | 1–2-й месяцы жизни | Очень редко | Норма (отсутствует В12СС) | Изменен с и без IF | а/р? | Уменьшение массы тела, диарея, бледность, стоматит, панцитопения, мегалобластная анемия |
| Функционально инертный ТК II (ТК II Cardeza) | 5–12-е годы жизни | Очень редко (описано и у сиблингов) | Повышен | Изменен с и без IF | а/р? | Афтозный стоматит, панцитопения, мегалобластная анемия |
| Селективная мальабсорбция К (синдром Иммерслунд — Гресбека) | В первые 2 года жизни | Относительно нередко* | Низкий | Изменен с и без IF | а/р? | Бледность, желтуха, тошнота, запор, глоссит, спленомегалия, нейропатия, обычно — задержка психического развития, панцитопения, мегалобластная анемия, протеинурия |
| Ювенильная ПА | Вторая декада | <15 | Низкий (высокая) | Как у взрослых с ПА | Не известен | Как у взрослых с ПА |
К — кобаламин; ТКII — транскобаламин II; В12СС — В12-связывающая способность; ПА — пернициозная анемия.
*85–90% всех детских и юношеских форм дефицита кобаламина.
Сидеробластные анемии — неоднородная по этиологии и патогенезу группа. Им присущи такие общие черты:
- Общее содержание железа в организме повышено и даже без трансфузий формируются гемосидероз и гемохроматоз с поражением миокарда, печени, селезенки, поджелудочной железы.
- Сидеробласты составляют до 60–80%, типична их морфология. Гемосидерин откладывается в виде грубых кристаллов митохондриально вокруг ядра, что получило название кольцевых сидеробластов.
Сидеробластные анемии (ранее — сидероахрестические) (табл. 3.12). Общим для них является нарушение утилизации железа при синтезе гема еще на уровне протопорфирина. Не утилизированное железо откладывается в клетках-предшественниках эритроцитов в костном мозгу в нормобластах (преимущественно). Железо откладывается в основном перинуклеарно в свободной форме (увеличивается цитоплазматический ферритин), в лизосомах и митохондриях (сидеросомы). Это и придает клеткам типичный вид, за что их и называют кольцевые сидеробласты. Анемии чаще протекают тяжело. Мегалобластический костный мозг с гиперплазированным эритропоэтическим ростком. Эритропоэз неэффективный, поэтому число ретикулоцитов невелико. Содержание железа и ферритина в сыворотке крови повышено. Показана терапия витамином В6 в высоких дозах (100 мг в/в в течение 10 дней), в итоге развивается ретикулоцитоз с полной или частичной ремиссией.
Таблица 3.12
ДД-признаки сидеробластных анемий и эритремии
| Показатель | Истинная сидеробластная анемия | Эритремия | Анемия при свинцовой интоксикации | Дефицит пиридоксина | Талассемия |
| Эритроцитопоэз | Усилен. Качественно не изменен (очень редко мегалобластные изменения) | Усилен. Мегалобласты. Гигантские формы. Полиплоидия. Два ядра | Вначале повышен. Затем снижен. Базофильная зернистость эритроцитов. Нарушение митоза в эритробластах | Усилен. Морфологически не изменен | Усилен с картиной гемолиза. При большой талассемии — мегалобластные изменения |
| Количество сидеробластов | Значительно повышено, 80% и более — кольцевые формы | Повышено | Значительно повышено | Значительно повышено | Умеренно повышено. Патологические кольцевидные формы выявляют редко |
| Количество сидероцитов | Резко повышено | Повышено | Значительно повышено | Значительно повышено | Незначительно повышено |
| PAS-реакция | Умеренная активность в единичных нормобластах | Высокая активность в нормобластах | Незначительно и редко | Незначительно и редко | Незначительно и редко |
| Ядерные формы в крови | Крайне редко | Типично | Крайне редко | Практически нет | Очень редко |
| Экстрамедуллярное кроветворение | Нет | Да | Нет | Нет | Нет |
| Нарушение обмена порфирина | Наследственная: содержание копропорфирина в эритроцитах повышено; протопорфирина в эритроцитах — в норме. Приобретенная: содержание копропорфирина в эритроцитах незначительно повышено; протопорфирина в эритроцитах резко повышено. | Аналогично приобретенным истинным (идиопатическим) сидеробластным анемиям | Крайне высокая концентрация гамма- аминолевуленовой кислоты в моче | Содержание протопорфирина и других предшественников протопорфирина в эритроцитах повышено | Содержание протопорфирина в эритроцитах повышено; копропорфирина в эритроцитах умеренно повышено |
| Сидероз, гемахроматоз | Может быть | Отсутствует | Нет | Нет | Нет |
| Тест с нагрузкой триптофаном | Норма | — | — | Патологичен |
- Пиридоксинзависимые сидеробластные анемии развиваются исключительно у мужчин. Выявляют только микроцитарные варианты.
- Усиленное потребление пиридоксина отмечают при применении гормональных (эстрогеновых) контрацептивов, пенициламина, противосудорожных препаратов.
- Наследственные сидеробластные анемии, передающиеся Х-сцепленно. Микроцитоз сочетается со спленомегалией. Субъективно пациенты вначале очень мало интересуются своим состоянием, поскольку оно их не беспокоит. Анемия минимальна. Только позднее появляются жалобы, обусловленные как хронической анемией, так и присоединившимся гемохроматозом, увеличением селезенки и печени. У женщин-кондукторов в анализах крови выявляют минимальные изменения. Известны также а/д и а/р передающиеся наследственные сидеробластные анемии; изолированная с неустановленным типом наследования, ассоциированная с митохондриальной цитопатией (синдром Пирсона).
- Приобретенные сидеробластные анемии (рефрактерные анемии при миелодиспластическом синдроме) манифестируют в детском, зрелом и пожилом возрасте. Картина крови характеризуется выраженным анизоцитозом. Возможна трансформация в миелобластный лейкоз.
- Приобретенные химически индуцированные сидеробластные анемии развиваются при применении хлорамфеникола, изониазида, циклосерина, хлорамбуцила, бусульфана, пиразинамида, злоупотребления алкоголем. Препараты вызывают обратимую блокаду эритропоэза (табл. 3.13). В костном мозгу наряду с вакуолизированными эритробластами выявляют сидеробласты с грубыми преципитатами гемосидерина. Обратимая сидеробластная анемия возможна также при дефиците меди (алиментарном или как токсическое действие цинка) и при гипотермии.
Таблица 3.13
Влияние различных химических соединений на определенные этапы синтеза гема с возможным развитием в итоге сидеробластных приобретенных анемий*
| Показатель | Синтез порфобилиногена из аминолевуленовой кислоты | Синтез порфобирина из порфобилиногена |
| Азот | Не влияет | Блокирует |
| Фторид натрия | Блокирует | Не влияет |
| Малоновая кислота | Блокирует | Блокирует |
| Альфа-кетоглутаровая кислота | Блокирует | Блокирует |
| Аденозинтрифосфорная кислота | Блокирует | Активизирует |
| Новарсенбензол | Блокирует | Блокирует |
*Вторичные сидеробластные анемии развиваются при хроническом полиартрите, коллагенозах, микседеме, матастазирующих карциномах, истинной полицитемии, лейкозах и миеломе.
Рефрактерные анемии с гиперпластическим костным мозгом — большая группа гетерогенных приобретенных анемий. Диагностическими признаками являются:
- Анемии с неэффективным, гиперпластическим, нередко мегалобластическим эритропоэзом.
- Отсутствие типичных сидеробластов с кольцевым отложением железа.
- Отсутствие цитологических признаков лейкемии.
- Безуспешность применения фолиевой кислоты, витамина В12, андрогенов, пиридоксина.
Если же анемия сопровождается лейко- и тромбопенией или нарушениями грануло- и тромбопоэза, то есть основания для диагноза «миелодиспластический синдром».
Дисэритропоэтические анемии могут быть врожденными, манифестирующими с раннего детства, и приобретенными. Общий признак — неэффективность гиперпластического эритропоэза с интенсивным депонированием железа в тканях. Более тонкие цитологические и биохимические характеристики позволяют выделить несколько форм этого состояния. Приобретенные дисэритропоэтические анемии рассматриваются как прелейкемические или эритролейкемические синдромы и описываются в разделе миелодиспластического синдрома. Часто выявляют спленомегалию, тромбо- и гранулоцитопению, эритробласты в периферической крови могут отсутствовать. Решающее для диагноза — выявление в костном мозгу патологического эритропоэза.
Анемии вследствие нарушения пролиферации характеризуются снижением клеточности костного мозга. Основные факторы патогенеза — повреждения костного мозга или недостаточное поступление железа в гемоглобинсинтезирующие клетки, неадекватная стимуляция костного мозга эритропоэтином. Часто возникает комбинация нескольких причин и различной степени укорочение средней продолжительности жизни эритроцитов. Возникает несоответствие между количеством эритробластов и степенью анемии. Обязательным признаком является ретикулоцитопения. Уровень билирубина в сыворотке крови не повышен. Уровень железа в плазме крови в норме, но включение радиоактивного железа в эритроциты замедлено или полностью отсутствует (апластическая анемия), нормальное (почечная анемия), или повышено при железодефицитных анемиях.
Апластические анемии (идиопатическая или токсическая панцитопения, панмиелофтиз, приобретенная панмиелопатия) — состояние с нарушением созревания всех трех ростков костного мозга: эритроидного, грануло- и тромбоцитарного. Заболевание поражает все возрастные группы, отсутствуют половые различия. В качестве этиологических факторов называют лекарственные препараты, (прежде всего цитостатики, золото, хлорамфеникол, сульфаниламиды, карбамазепин, ацетилсалициловая кислота, тиреостатики, фенотиазины, сульфанилгуаниды), большие дозы облучения, особенно широких областей тела, химические вещества (ароматические соединения, органические растворители). Все это повреждает стволовые клетки костного мозга. Наряду с этим апластическая (или гипопластическая) анемия может развиться при вирусных гепатитах, инфицировании вирусами Эпштейна — Барр, ВИЧ, лихорадки Денге, микобактериозах, диффузном эссенциальном фасциите, болезни Симмондса и склерозе щитовидной железы в период беременности. У детей известно сочетание панкреатической недостаточности и (гипо)апластической анемии.
Заболевание нередко начинается исподволь, могут пройти годы, прежде чем сформируется полная клиническая картина. Увеличение печени, селезенки и лимфатических узлов (ЛУ) не типично. Анемия макроцитарная, небольшой анизо- и пойкилоцитоз. Уровень железа в сыворотке крови и эритропоэтина повышен. Костный мозг беден клетками, хотя иногда выявляют гиперпластические островки. В современных условиях решающее значение в лечении имеет трансплантация костного мозга.
Из врожденных апластических анемий известны синдромы Фанкони и Даймонда — Блекфана.
Синдром Фанкони (анемия Фанкони). Передается а/p. Развитие обусловлено врожденным генетическим дефектом в одном из генов анемии Фанкони. Локализация генетического дефекта определяет вариант заболевания или комлепементарную группу, обозначаемую как группа А, В, С, D, Е, F, G. Частота заболеваемости анемией Фанкони составляет в среднем 1:360 000 населения. Но в Италии больных с анемией Фанкони значительно больше. Состояние обусловлено, вероятно, сниженной репаративной способностью ДНК. Проявляется низким ростом (отставание в росте — с 1-го года жизни), гиперхромно-макроцитарной анемией со склонностью лейко/тромбоцитопении и кровотечениями. Клинические признаки появляются в 8–9-летнем возрасте (размах времени дебюта от новорожденности до 48 лет) и прогрессируют. Костный мозг гипопластичен, заполнен жировыми клетками. Нередко выявляют агенезию селезенки. С 5–10 лет появляется склонность к частым инфекционным заболеваниям. Гипо/аплазия большого пальца; удвоение, реже — гипоплазия или полное отсутствие лучевой кости; синдактилия. Усиленная генерализованная пигментация кожи. Редко возникают пороки сердца, микрофтальмия, пороки почек. У мальчиков — крипторхизм, микропенис. У 20–30% больных физических дефектов нет. Чем младше ребенок на момент манифестации заболевания, тем выше частота уродств и многообразнее их спектр. В 20–30-летнем возрасте типична смерть от лейкоза или рака кожи. Причем у мужчин с анемией Фанкони вероятность смерти от опухолей в 3–4 раза выше, чем у женщин. При хромосомном анализе — повышенная ломкость хромосом после краткосрочной культивации лейкоцитов в диэпоксибутане. Результаты считаются настолько патогномоничными для анемии Фанкони, что необходимо провести пробу для исключения синдрома Фанкони при любой апластической анемии.
Высокая вероятность перестройки хромосом (еще не заметная у грудных детей, но более выраженная с возрастом). Сниженный ответ лейкоцитов на стимуляцию. Уже с рождения регистрируют нарушения G2-фазы клеточного цикла.
Сочетание анемии Фанкони и врожденного ди- скератоза известно, как синдром Цинзера — Фанкони.
Анемия Фанкони без костных дефектов известна как синдром Эстрена — Дамешека.
Анемия Блекфана — Даймонда — парциальная красноклеточная аплазия — известна как аплазия или гипоплазия только эритроцитарного ростка, грануло- и тромбопоэз не нарушены. Не исключаются а/д или а/p типы передачи, но, как правило, наиболее весомо обсуждается приобретенный вариант заболевания. В качестве причин называют внутриутробное повреждение эритроцитарного ростка, дефект триптофанового обмена с высоким выделением антраниловой кислоты и аутоиммунный механизм с блокадой эритропоэза в нормальном костном мозгу лейкоцитами пациента с анемией Блекфана — Даймонда (60% всех пациентов благоприятно реагируют на лечение глюкокортикоидами).
Развивается у детей грудного возраста или даже у новорожденных. В периферической крови нормо- или гиперхромная нормоцитарная анемия с отсутствием ретикулоцитов без признаков гемолиза. В костном мозгу значительно уменьшено количество эритробластов. У 30% пациентов выявляют отставание в росте, микроцефалию, страбизм, аномалии почек, сердца, гипогонадизм, сколиоз, аномалии костей.
Видимо, существуют переходные формы анемии Фанкони и анемии Блекфана — Даймонда. Но истинная анемия Блекфана — Даймонда протекает без хромосомных аномалий и пороков развития.
Истинный приобретенный эритробластофтиз — заболевание, возникающее очень редко — при опухолях тимуса. В крови выявляют цитотоксические антитела к эритробластам. Преходящие эритобластопении, часто субклинические, сопровождают острые инфекции.
Почечные анемии в ДД имеют большое практическое значение. Абсолютной корреляции между степенью почечной недостаточности и выраженностью анемии нет, хотя наиболее значимые анемии наблюдаются при хронических процессах, особенно при высоком уровне креатинина и мочевины. Анемия, как правило, нормоцитарная. Основная причина — дефицит эритропоэтина и непостоянно выраженное снижение длительности жизни эритроцитов.
Анемии при белковом голодании известны более из экспериментов над животными. У людей всегда в условиях голодания на первый план выдвигается дефицит железа, фолиевой кислоты и витамина В12.
Анемии при эндокринопатиях известны при поражении щитовидной железы (тиреопривные) и гипофиза. Щитовидная железа активирует обмен железа и образование эритропоэтина, поэтому при гипотиреозе возможны нормоцитарные анемии, хотя описаны наблюдения микро- и макроцитарных анемий. Вероятно, эти анемии отражают снижение потребности тканей в кислороде. Отметим, что сродство гемоглобина к кислороду у таких больных повышено. Аналогичный механизм анемий имеет значение и при недостаточности передней доли гипофиза.
Анемии при хронических заболеваниях нормоцитарные с тенденцией к микроцитарным. Они являются следствием недостатка эритропоэтина, нарушения доставки железа к эритробластам, уменьшения продолжительности жизни эритроцитов. В противоположность истинным железодефицитным состояниям уровень трансферрина в сыворотке крови нормален или даже понижен, а ферритан в сыворотке крови в норме или повышен. То есть нарушается распределение железа. Железо, освобождающееся из эритроцитов, остается блокированным и не поступает в транспортную систему. Для ДД можно провести пункцию костного мозга. При железодефицитных анемиях интерстициальное железо отсутствует, а при обсуждаемой ситуации его много.
Оптимальный способ лечения пациентов с анемией при хронических болезнях — курация основного заболевания. Препараты железа, как правило, неэффективны. Лечение эритропоэтином дорогостоящее.
Панцитопении диагностируются, если наряду с уменьшением количества эритроцитов отмечают гранулоцитопению (число нейтрофильных гранулоцитов <1000/мм3) и тромбоцитопению (<150 000/мм3). Но если в анализах крови выявляется уменьшение количества тромбо- и гранулоцитов, а уровень гемоглобина соответсвует нижней границе нормы, то все равно нет оснований отказываться от диагноза «панцитопения». Потому что этот диагноз может иметь решающее значение для тактики обследования и лечения. Необходимо отметить, что хотя мы употребили по отношению к панцитопении термин «диагноз», это состояние является не диагнозом в узком смысле слова, а лабораторным синдромом и требует расшифровки. Определяющим в ДД панцитопений чаще всего оказывается изучение биоптата костного мозга. Панцитопению отмечают при следующих состояниях:
- иммунопатологические состояния (СКВ);
- инфекции, в первую очередь милиарный туберкулез;
- дефицит витамина В12 или фолиевой кислоты;
- лучевые, лекарственные, токсические поражения;
- остеопетроз;
- гиперспленизм;
- пароксизмальная ночная гемоглобинурия;
- апластические анемии приобретенные и конституциональные;
- миелодиспластический синдром;
- опухолевая инфильтрация костного мозга.
Нарушения пролиферации при инфильтрации костного мозга эритропоэтически неактивными клетками более известны при лимфомах и лейкозах (см. соответствующие разделы), но в практике выявляют и при метастазирующих опухолях. Типичные опухолевые анемии микроцитарные, уровень железа и трансферрина в сыворотке крови снижен, ферритин повышен. Но диффузная метастатическая инфильтрация может проявиться выраженнейшим лейкоцитозом до 5ОООО/мм3, что требует ДД с лейкемиями и лейкемоидными реакциями крови.
Диагноз «метастатическое поражение костного мозга» основывается на:
- рентгеновских методах диагностики, при этом ищут первичный очаг (пиелография, маммография ит. д.);
- сцинтиграфии (выявление очагов в скелете);
- пункции костного мозга, где выявляют опухолевые клетки;
- определении активности щелочной и кислой фосфатазы. Повышение кислой фосфатазы у мужчины требует сосредоточиться на исключении рака простаты. Щелочная фосфатаза повышается при холестазе (чему свойственно повышение лейцина-минопептидазы), матастазах, а также при болезни Педжета, остеомаляции и гиперпаратиреоидизме.
Парапротеинемии характеризуются патологическим увеличением количества плазматических и лимфоидных клеток в костном мозгу или в ЛУ, которые продуцируют моноклональные антитела. Выделяют три формы парапротеинемий:
А. Идиопатическая, в большинстве доброкачественная. При этом варианте нет доказательств поражения лимфоплазмоцитарной системы, нет клинических проявлений заболевания. Кроме того, доброкачественной парапротеинемии не свойственны анемия, гиперкальциемия, остеолиз, парапротеинурия. Уровень парапротеинов в крови не превышает 10 г/л, что не подавляет продукцию иммуноглобулинов. В пунктате костного мозга плазматических клеток не более 15%. У приблизительно 1–2% людей в возрасте старше 70 лет выявляют доброкачественную парапротеинемию.
Б. Факультативная парапротеинемия возникает при целом ряде гемобластозов: волосатоклеточном лейкозе, хроническом лимфолейкозе, злокачественных лимфомах, а также при карциномах, инфекционных заболеваниях и болезнях печени.
В. Облигатная парапротеинемия при болезни холодовых аглютининов, макроглобулинемии Вальденстрема, множественной миеломе.
Парапротеины, соответственно физиологически предшествующим им иммуноглобулинам, подразделяются на 5 классов: IgA, IgM, IgD, IgG, IgE. Каждый парапротеин, как и иммуноглобулин, состоит из 4 цепей: двух легких (ню или дельта типы) с молекулярной массой около 25 000 и двух тяжелых с молекулярной массой 50 000–60 000. Выявление парапротеинов осуществляется с помощью электрофореза. При этом парапротеины выявляются как узкая полоска (так называемый градиент М) в области альфа, бета или гамма-глобулинов. Иммуноэлектрофорез позволяет выявить природу легких или тяжелых цепей.
Макроглобулинемия Вальденстрема. Возникает нечасто. Поражает в основном людей не младше 50 лет. На первый план выдвигаются такие общие симптомы, как утомляемость, снижение работоспособности, частые инфекционные заболевания. Практически постоянно возникают носовые и желудочно-кишечные кровотечения, ЛУ увеличиваются. Гистологически отмечается картина иммуноцитомы, так называемой лимфомы низкой степени злокачественной. То есть макроглобулинемия Вальденстрема представляет собой В-клеточный вариант особой формы неходжкинской лимфомы типа иммуноцитомы, причем только 20% опухоли этого типа продуцируют парапротеин. Гиперпротеинемия вызывает значительное повышение вязкости крови, что приводит к резким нарушениям тканевой гемоциркуляции. Характерно изменение глазного дна по типу fundus paraproteinaemicus с дилатацией вен сетчатки и ретинальными кровоизлияниями. В противоположность множественной миеломе никогда не развивается остеопороз. У большинства (80–85%) пациентов развивается анемия с одновременным уменьшением количества лейко- и тромбоцитов, гипергамма-глобулинемия и значительно повышенная СОЭ. При электрофорезе выявляют патогномоничные М-парапротеины. Костный мозг инфильтрирован малыми лимфоидными клетками. При PAS-реакции они выявляют маленькие капли (виноградинки). Рядом с лимфоидными клетками выявляют плазматические. Очень характерно для этого заболевания увеличение тканевых мастоцитов.
Множественная миелома — относительно частое заболевание с распространенностью до 3:100 000 населения. Начало хроническое. Отмечаются общие симптомы утомляемости, неспецифической ревмаподобной боли, что служит причиной первоначально ошибочных диагнозов. В таких ситуациях только развивающаяся в последующем интенсивная ночная боль в костях и даже их переломы заставляют проводить полноценное радиологическое и биохимическое обследование. Рентгенологически выявляют два варианта поражения костей. Чаще это диффузный остеопороз, особенно отчетливый в позвонках, что может привести к их компрессии. При втором варианте выявляют штампованные остеолитические дефекты костей, особенно в плоских костях черепа. Практически никогда не увеличиваются печень, селезенка и ЛУ. На первый план выходят симптомы почечной недостаточности за счет инфильтрации коры плазмоцитами. Большое значение имеет повреждение канальцев белком Бенса — Джонса. Типично развитие полинейропатий.
Очень редко выявляют экстрамедуллярную плазмоцитому или унилокулярную миелому в одном костном регионе. Несецернирующую множественную миелому диагностируют в 2% случаев всех миелом, она характеризуется сохранением парапротеинов в плазматических клетках без выхода патологического белка в кровь. Диагноз этой формы миеломы устанавливается только на основании изучения костного мозга и флюоресцентного подтверждения внутриклеточного парапротеионоза. Парапротеинурию типа Бенса — Джонса отмечают в 40–60% случаев миелом, но выделение только легких цепей — крайне редко. Диагностика таких состояний чрезвычайно сложна, поскольку электрофорез белков в сыворотке крови на бумаге и СОЭ не изменяются. Но повреждение почечных канальцев быстро приводит к почечной недостаточности. В противоположность этому состоянию при болезни тяжелых цепей парапротеин не содержит легких цепей. Описаны несколько типов этого варианта парапротеиноза: тип гамма-цепей (болезнь Франклина), альфа- и мю-цепи. Гамма-тип характеризуется гепатоспленомегалией и лимфаденопатией, частыми интеркурентными инфекциями.
При альфа-типе развивается синдром мальабсорбции. Мю-тип напоминает гамма-болезнь.
При дополнительных исследованиях миеломы характеризуются существенно повышенной СОЭ, особенно в случае так называемой чистой миеломы Бенса — Джонса. В мазках почти всегда анемия. В связи с наличием холодовых аглютининов эритроциты седиментируются по типу монетных столбиков. В дебюте заболевания число лейкоцитов и тромбоцитов не изменяется, в динамике — уменьшается. В биохимических анализах — гиперкальцемия и гиперфосфатемия. Общий белок сыворотки крови значительно повышен, при электрофорезе определяется увеличение моноклональных иммуноглобулинов с формированием типичного М-градиента. Иммуноэлектрофорез позволяет идентифицировать иммуноглобулин. Криоглобулины при множественной миеломе выявляют нечасто. Но если они присутствуют, то способны вызвать тяжелые нарушения микроциркуляции. Парапротеины синтезируются плазматическими клетками, поэтому в костном мозгу выявляется значительное (более 15%) содержание плазматических клеток. Это могут быть морфологически нормальные клетки, но отмечаются и клетки с несколькими ядрами или с наличием множества вакуолей (Расселовские тельца). В ДД-оценке феномена плазматизации костного мозга необходимо принимать во внимание заболевания печени, почек, хронические инфекции или ревматические заболевания. Парапротеин Бенса — Джонса в моче может быть достаточно просто выявлен классической температурной пробой. При нагревании мочи до 50–60 °C белок выпадает в осадок в виде рыхлого облака, при дальнейшем нагревании до 90–95 °C растворяется. После охлаждения осадок опять появляется. Эта проба недостаточно чувствительна и в современных условиях должна быть заменена на электрофорез мочи (иммунный или на бумаге). Прогноз миелом плохой, даже при интенсивной своевременной цитостатической терапии ожидаемая продолжительность жизни не превышает 4 лет.
Амилоидоз — один из вариантов парапротеинозов. Может быть первичным, генетически детерминированным, и вторичным, представляя собой исход хронических нагноительных заболеваний (туберкулез, бронхоэктатическая болезнь), некоторых опухолей, хронического артрита (особенно его системных форм) и периодической болезни. Но даже при вторичных формах амилоидоза нельзя исключить генетическую предрасположенность к его формированию. Большое значение для установления диагноза имеет эхография (эхоГ), регистрирующая повышение ригидности миокарда, инфильтративную кардиомиопатию, увеличение размеров почек, печени и селезенки с утратой ими типичной архитектоники.
Развитие анемии идет по принципу вытеснения костного мозга патологическими отложениями, поражением почек со снижением продукции эритропоэтина, развитием синдрома мальабсорбции.