Современные представления о механизмах возникновения и прогрессирования атеросклероза
Содержание
- Основные гипотезы инициации атеросклеротического повреждения
- Формирование атеросклеротического повреждения
- Прогрессирование атеросклеротического процесса
- Фазы атеротромбоза
- Патофизиологические механизмы формирования осложненной атеромы
- Основные виды атером
- Понятие угрожаемой и высокорисковой атеромы
- Идентификация угрожаемой атеромы
- Васкулярное ремоделирование, ассоциированное с атеросклеротическим поражением сосуда
- Иммунопатология атеросклероза
- Основные биомаркеры провоспалительной активации и их клиническое значение
- Роль провоспалительных биомаркеров как предикторов высокого кардиоваскулярного риска
- СРП
- Фибриноген
- Циркулирующий ингибитор активатора плазминогена
- Солюбилизированные лиганды CD40
- Миелоидзависимый протеин 8/14
- Адипонектин
- ИЛ-18
- ММП-9
- Дендритические клетки стенки артерий
- Белки теплового шока (heat shock proteins)
- Литература
В последние годы представления об атеросклерозе подверглись действительно революционным изменениям (Libby P., Theroux P., 2005). Первоначальные знания о локальном характере этого процесса, ассоциированном с окклюзионными поражениями артерий, приводящими к ишемическим изменениям органов и тканей, не отражали всей полноты проблемы (Hansson G.K., Libby P., 2006). В частности, было установлено, что атеротромботические осложнения, такие как инфаркт миокарда или мозговой ишемический инсульт, не всегда являются непосредственным результатом формирования критического стеноза в зависимой артерии. Многие исследователи пришли к заключению о том, что практически в половине случаев инфаркт миокарда возникает у лиц, не переносивших до этого каких-либо ишемических событий. Более того, внезапная смерть зачастую является первым и единственным кардиоваскулярным эпизодом. В этой связи попытки стратификации пациентов в группу высокого риска манифестации атеротромбоза выглядят вполне обоснованными и своевременными (Libby P., Ridker P.M., 1999; 2004).
Традиционно, патофизиология атеротромбоза тесно согласуется с липидной теорией (Libby P., 2006), поскольку существует достаточно большой объем научных данных, подтверждающих существование прямой связи между плазменной концентрацией общего ХС и ХС ЛПНП с одной стороны и частотой кардиоваскулярных событий в популяции — с другой (Tabas I., 2002). Понимание важной роли субинтимального накопления недоокисленных модифицированных липидов, тесно ассоциированного с генетически обусловленным дефектом трансмембранной регуляции их транспорта, в формировании атеромы существенным образом стимулировало прогресс клинической липидологии, который, в конечном итоге, обусловил успешное завершение целого ряда крупных эпидемиологических и рандомизированных клинических исследований (Libby P. et al., 2000). Результаты последних привели к созданию программ первичной и вторичной профилактики возникновения кардиоваскулярных событий на основе достижения контроля над гипер- и дислипидемией (Castelli W.P., 1996). В настоящее время выработаны и созданы клинические рекомендации, посвященные превенции и лечению нарушений липидного обмена у пациентов с высоким кардиоваскулярным риском, в которых мониторинг плазменных концентраций общего ХС, ХС ЛПНП, ХС ЛПВП, ТГ, апо-А/апо-В рассматривается как неотъемлемая часть эффективной стратегии улучшения клинических исходов (Steinberg D., 2004; 2005a; b; 2006a; b).
С другой стороны, на протяжении последних лет интерес исследователей стали вызывать процессы локального воспаления в области атеромы, инициируемые процессами модификации ХС ЛПНП и их ремнантов. Последние рассматриваются в качестве одного из важнейших механизмов не только формирования атеромы, но и возникновения атеротромбоза (Glass C.K., Witztum J.L., 2001). Установлено, что многие факторы риска атеротромбоза, такие как артериальная гипертензия, курение, употребление алкоголя, сахарный диабет, ожирение, способны потенцировать накопление модифицированных молекул ХС ЛПНП и индуцировать локальный воспалительный процесс (Libby P., Aikawa M., 2002).
Таким образом, провоспалительная активация, косвенно вовлекающая в патологический процесс иммунологические механизмы, не только может быть рассмотрена как еще один фактор риска формирования и прогрессирования атеросклероза, но и как модулятор, связывающий между собой процессы нарушения липидного обмена, депозицию ремнантных липопротеидов, асептическое воспаление в субинтиме и угрожаемых участках атеромы, а также нарушения функции эндотелия, коагуляционного каскада и различные виды межклеточной кооперации.
Настоящая глава посвящена обсуждению основных патофизиологических механизмов, лежащих в основе формирования и прогрессирования атеросклероза.
Основные гипотезы инициации атеросклеротического повреждения
К середине XIX в. сформировались две основные теории, объясняющие механизм возникновения атеросклероза: инкрустационная (incrustation hypothesis) гипотеза, сформулированная Рокитанским (Rokitansky) в 1852 г., и липидная теория, предложенная Вирховым (Virchow) в 1856 г. (Fuster V. et al., 1992). Обе гипотезы рассматривали аккумуляцию липидов, ассоциированную с локальным накоплением фибрина и экспансией коллагенового внеклеточного матрикса, как начальный этап в формировании атеросклеротического поражения артерий. Вместе с тем, именно Вирхов, используя термин «деформирующий эндартериит», впервые предложил рассматривать инфильтрацию липидами субинтимы артерий как ответ на повреждение (response-to-injury hypothesis) вследствие воспаления задолго до того, как прямые доказательства справедливости его представлений были подтверждены классическими исследованиями R. Ross и соавторов (Ross R., Glomset J.A., 1976; Ross R., 1999).
К настоящему времени ретенция липопротеидов, ассоциированная с нарушением их транспорта, рассматривается в неразрывной связи с провоспалительной активацией, нарушением кооперации иммунокомпетентных клеток, особенно на ранних стадиях атерогенеза (Williams K.J., Tabas I., 1995). Более того, установлено, что асептическое воспаление не только играет важную роль в механизмах формирования атеромы, но и принимает непосредственное участие в возникновении так называемого феномена усталости покрышки и атеротромботических осложнений (во многом индуцированных ею) (Moreno P.R. et al., 1994; Falk E. et al., 1995; Libby P., 1995; 2002a; b; Davies M.J., 1996). Поэтому в настоящее время более принято говорить об атеротромбозе, чем об атеросклерозе вообще, поскольку первый термин значительно лучше определяет этиологическую и патогенетическую исключительность описываемых событий.
Атеротромбоз является системным патологическим процессом, преимущественно локализованным в интимо-медиальном сегменте артерий эластического или мышечно-эластического типов (аорта, ветви брахиоцефального ствола, коронарные и бедренные артерии, некоторые периферические артерии), главным компонентом которого является атеросклеротическая бляшка (атерома) (Stary H.C., 1992; Stary H.C. et al., 1994; 1995; Schwartz S.M. et al., 1995; Daugherty A. et al., 1997; Daugherty A., Rateri D.L., 2002; Libby P. et al., 2002). Последняя состоит как из клеточных (макрофаги и пенистые клетки преимущественно моноцитарного происхождения, Т-лимфоциты, гладкомышечные клетки), так и неклеточных элементов ядра (кристаллы ХС, эстерифицированный ХС, фосфолипиды) и покрышки (коллаген, протеогликаны, фибронектин, эластин) с возможными фибринными и тромботическими наложениями на ее внешней поверхности (рис. 7.1).
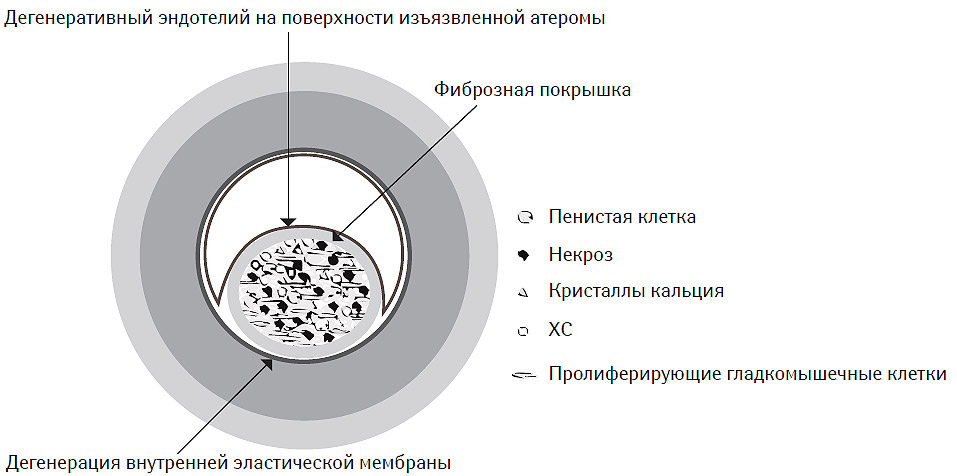
Рис. 7.1. Структура атеромы
Таблица 7.1 Номенклатура и дефиниции морфологических особенностей атером, ассоциированных с формированием атеротромбоза и острого коронарного синдрома (модифицировано из работы Schaar J.A. et al., 2004)
| Название атеромы | Дефиниция |
| Атерома, ответственная за повреждение (Culprit lesion) | Атерома, осложненная тромбозом, распространенным в пределах люминальной пластинки сосудистой стенки, ответственная за возникновение инфаркта миокарда, внезапной сердечной смерти, нестабильной стенокардии |
| Эрозированная атерома (Eroded plaque) | Атерома, обогащенная гладкомышечными клетками, протеогликанами, обычно не имеющая дополнительных дефектов покрышки, кроме отсутствия эндотелиоцитов на последней, сопровождающаяся формированием дисфункции эндотелия выше и ниже места поражения |
| Атерома высокого риска (High-risk), угрожаемая (Vulnerable) атерома, атерома, осложненная пристеночным тромбозом (Thrombosis-prone plaque) | Термины используются как синонимы при описании атеромы, сопровождающейся высоким риском возникновения пристеночного тромбоза и быстрого прогрессирования стенозирования сосуда |
| Воспалительно-измененная фиброатерома с тонкой покрышкой (Inflamed thin-cap fibroatheroma — TCFA) | Атерома с истонченной покрышкой, обогащенная липидами и клетками воспалительного происхождения, с некрозом ядра. Термин часто используется для описания угрожаемой по возникновению тромбоза атеромы |
| Кальцифицированная атерома (Plaque with a calcified nodule) | Атерома с отсутствием эндотелиоцитов на покрышке, с включением кристаллов кальция, является результатом эволюции фиброзной атеромы, рассматривается как вариант атеромы высокого риска |
| Атерома с тромбозом (Thrombosed plaque) | Атерома, ассоциированная с формированием тромба как с распространением в направлении люминальной пластинки сосудистой стенки, так и с протрузией в просвет сосуда с последующим его стенозированием |
| Разорвавшаяся атерома (Ruptured plaque) | Рассматривается как одна из основных причин тромбоза вследствие разрушения фиброзной покрышки и обнажения тромбогенного липидного ядра |
Указанные компоненты, представленные в различных соотношениях в атеромах, обусловливают гетерогенность последних. Кроме того, особенности состава атеромы могут оказывать определенное влияние на морфологические характеристики медиа и адвентиции (Moreno P.R. et al., 2002), включая vasa vasorum сосудистой стенки (Williams J.K., Heistad D.D., 1996; Ware J.A., 2001; Kolodgie F.D. et al., 2003; Moreno P.R. et al., 2004).
Формирование атеросклеротического повреждения
Согласно современным представлениям инфильтрация интимы артерий СЖК с образованием жировых полосок уже может сопровождаться провоспалительной активацией. Так, СЖК, инкорпорированные в экстрацеллюлярный матрикс интимы посредством связи с протеогликанами, способствуют ослаблению антиоксидантных качеств эндотелия и создают благоприятные условия для оксидативной модификации ЛПНП (Williams K.J., Tabas I., 1998; Kruth H.S., 2002; Skalen K. et al., 2002). Последние представляют собой своеобразную смесь окисленных протеинов и липидов, находящихся на различных этапах модификации молекул. Модифицированные таким образом липопротеиды являются активными индукторами асептического воспалительного процесса (Miller Y.I. et al., 2003).
В норме эндотелиоциты высокорезистентны к адгезии лейкоцитов. Провоспалительные стимулы, такие как высококалорийная диета, обогащенная насыщенными жирами, гиперхолестеринемия, гипертриглицеридемия, курение, инсулинорезистентность, ожирение, являются триггерами экспрессии молекул клеточной адгезии (VCAM-1), которые обеспечивают фиксацию на эндотелиоцитах циркулирующих в крови моноцитов и лимфоцитов (Cybulsky M.I., Gimbrone M.A. Jr., 1991; Li H. et al., 1993; Cybulsky M.I. et al., 2001). Интересно, что формирующиеся в области разветвлений и бифуркаций артерий атеромы способствуют возникновению турбулентного кровотока, который повышает способность эндотелия к экспрессии VCAM-1 и редуцирует эндогенную продукцию оксида азота (Jongstra-Bilen J. et al., 2006).
Хемоаттрактантные факторы, которые включают, прежде всего, моноцитарный хемоаттрактантный протеин-1, продуцируются сосудистой стенкой в ответ на инфильтрацию модифицированными ЛПНП и способствуют миграции в субэндотелий моноцитов, замыкая порочный круг (Boring L. et al., 1998; Gu L. et al., 1998). Моноциты кооперируются с эндотелиоцитами и способствуют индукции активности матричной металлопротеиназы-9 (MMP-9), обеспечивающей последующее проникновение лейкоцитов через базальную мембрану сосуда (Amorino G.P., Hoover R.L., 1998). В пределах интимального и субинтимального пространства моноцит трасформируется в макрофаг под действием макрофагального колониестимулирующего фактора (МКСФ), экспрессия которого увеличивается вследствие накопления окисленных ЛПНП (Rajavashisth T.B. et al., 1990; Swirski F.K. et al., 2007). МКСФ повышает экспрессию на поверхности мембраны макрофага специфических scavenger-рецепторов, поглощающих модифицированные ЛПНП посредством эндоцитоза. Аккумуляция эстерифицированных молекул ХС ЛПНП в цитоплазме макрофагов способствует конвертации их в так называемые пенистые клетки. Этот этап знаменует формирование одной из наиболее ранних стадий атерогенеза. Параллельно макрофаги пролиферируют, что обеспечивает постоянно возрастающую секрецию провоспалительных цитокинов и факторов роста, таких как ФНО-α и ИЛ-1β (Swirski F.K. et al., 2007; Tacke F. et al., 2007).
T-клетки также могут играть существенную роль в атерогенезе, способствуя вовлечению в воспалительный процесс хемокин-индуцированного протеина-10, монокин- индуцированного интерферона (МИИ) и ряда хемоаттрактантов (Mach F. et al., 1999). CD4+ субтип лимфоцитов, презентирующих фрагменты антигенов в непосредственной связи с молекулами II типа HLA, могут также быть активированы модифицированными молекулами ХС ЛПНП (Stemme S. et al., 1995). Установлено, что атерома может содержать цитокины (ИЛ-4, ИЛ-10, ИЛ-12, ИФ-γ), способствующие активации T1- хелперов и дифференциации их в T1-эффекторы (Robertson A.K., Hansson G.K., 2006). Последние выступают в роли амплификаторов для многих провоспалительных цитокинов, таких как МИИ и CD40-лиганд (CD40L, CD154), участвующих в процессах интенсификации роста атеромы.
Важную роль в регуляции процессов атерогенеза играет адипонектин, продуцирующийся непосредственно адипоцитами и проявляющий высокий антиатерогенный и антивоспалительный потенциал (Okamoto Y. et al., 2006). Адипонектин способен оказывать многочисленные биологические эффекты как аутокринно/паракринного, так и центрального характера. Установлена его способность повышать чувствительность тканей к инсулину, снижать плазменную концентрацию ТГ и объем депонированного нейтрального жира, а также повышать уровень ХС ЛПВП (Matsuzawa Y., 2006). Кроме того, доказана его способность индуцировать катаболизм липопротеидов посредством активации липопротеинлипазы (Lara-Castro C. et al., 2007). Адипонектин оказывает благоприятный эффект в отношении функционирования эндотелиоцитов, а также способен снижать экспрессию VCAM-1 и интенсивность инфильтрации моноцитами интимы, редуцировать экспрессию scavenger-рецепторов на макрофагах и продукцию ФНО-α (Fantuzzi G., Mazzone T., 2007).
Прогрессирование атеросклеротического процесса
Макрофаги и T-клетки обычно инфильтрируют плечевую зону сформировавшейся атеромы, обеспечивая ее последующий рост. Установлено, что макрофаги атеромы рекрутируются из моноцитов крови посредством связывания на поверхности покрышки с молекулами клеточной адгезии (молекула внутриклеточной адгезии-1, васкулярная молекула клеточной адгезии-1). Фиксированные на эндотелиоцитах покрышки моноциты путем активной миграции проникают в интиму. Этот процесс в значительной мере опосредуется хемоаттрактантными протеинами, такими как моноцитарная хемоаттрактантная молекула-1, обеспечивающими процесс проникновения моноцитов в сосудистую стенку. Локально депонированные моноциты поглощают и аккумулируют липиды, трансформируясь в так называемые пенистые клетки. Последние принимают непосредственное участие в аутопаракринной регуляции лейкоцитарной инфильтрации атеромы, накоплении гладкомышечных клеток, миграции эндотелиоцитов, неоваскуляризации сосудистой стенки, продукции прокоагулянтных факторов, аполипопротеина Е, медиаторов воспаления и ряда цитокинов. Полагают, что пенистые клетки могут играть центральную роль в эволюции липидной полоски как начального звена формирования атеросклеротического поражения сосуда в осложненную атерому за счет модуляции процессов аккумуляции гладкомышечных клеток, повышения образования внеклеточного матрикса, формирования центрального ядра, содержащего свободные внеклеточные липиды. Причем между размерами липидного ядра, его тромбогенным потенциалом, интенсивностью апоптоза макрофагов и степенью нестабильности атеромы имеется отчетливая прямая корреляционная взаимосвязь. Не случайно Р. Libbi образно называл нестабильную атерому «кладбищем» макрофагов.
Кульминацией этих процессов является разрыв фиброзной покрышки атеромы, обнажение липидного ядра и формирование тромба, последующий рост которого обусловливает прогрессирующее стенозирование сосуда вплоть до его окклюзии (рис. 7.2).
В качестве ключевого критического компонента, обеспечивающего резистентность атеромы к дестабилизации, рассматриваются морфологические особенности ее покрышки, зависящие, в частности, от интенсивности синтеза коллагена и ряда других структурных компонентов внеклеточного матрикса. В этом аспекте процессы, оказывающие непосредственное влияние в отношении синтеза и деградации макромолекул последнего, особенно связанные с продукцией коллагена гладкомышечными клетками, могут иметь принципиальное значение для обеспечения стабильности покрышки атеромы (рис. 7.3).

Рис. 7.2. Принципиальные механизмы формирования атеромы

Рис. 7.3. Основные механизмы, обеспечивающие стабилизацию атеромы
Стабильность покрышки обеспечивается преимущественно за счет аккумуляции фиброзной ткани. Общий же объем атеромы тесно зависит от интенсивности продукции внеклеточного матрикса гладкомышечными клетками и накопления свободных липидов (Raines E.W., Ferri N., 2005). Вследствие высвобождения тромбоцитарного фактора роста из активированных макрофагов и эндотелиоцитов гладкомышечные клетки мигрируют из медиа в интиму, где участвуют в процессах деградации экстрацеллюлярного матрикса посредством продукции MMP-9 и других протеиназ. Установлена возможность эволюции моноцитов в гладкомышечные клетки непосредственно в интиме (Mason D.P. et al., 1999). В итоге, именно в интиме под влиянием различных факторов роста гладкомышечные клетки пролиферируют, секретируют различные матричные протеины, включая интерстициальный коллаген. Этот процесс является основной причиной эволюции обогащенной липидами нестабильной атеромы в стабильную фиброзную кальцифицированную атеросклеротическую бляшку, формирующую фиксированный стеноз.
Установлено, что в атероме увеличивается экспрессия ИЛ-18 и особенно его рецептора ИЛ-18R/β (Gerdes N. et al., 2002). Основным источником ИЛ-18 являются мононуклеарные фагоциты, тогда как эндотелиоциты, гладкомышечные клетки и макрофаги экспрессируют в основном ИЛ-18R/β. Особенно важно, что ИЛ-18 рассматривается как сигнальный мессенджер для привлечения (VCAM-1) хемокинов (ИЛ-8), цитокинов (ИЛ-6) и MMP-1/-9/-13. Кроме того, ИЛ-18, особенно в комбинации с ИЛ-12, ответственен за индукцию и регуляцию экспрессии большинства провоспалительных цитокинов, участвующих в атерогенезе. Так, ИЛ-18 активирует МИИ не только в T-клетках, но и в макрофагах, гладкомышечных клетках (Okamura H. et al., 1995). Многими исследователями этот процесс рассматривается как доказательство существования паракринной регуляции атерогенеза за счет модуляции провоспалительной активности (Gerdes N. et al., 2002).
Еще одним из патофизиологических механизмов, лежащих в основе формирования и прогрессирования атеромы, является неоваскуляризация артерий (Moulton K.S. et al., 1999). Предполагается, что vasa vasorum могут создавать дополнительные условия для инфильтрации стенки артерий лейкоцитами (Moulton K.S. et al., 2003). Более того, нарушение кровообращения в vasa vasorum вследствие тромбоза или других причин может стать причиной локального кровоизлияния в область атеромы, обеспечивая ее рост или возникновение пристеночного тромбоза. Кроме того, локальное кровоизлияние за счет высвобождения тромбина, а также XII и II фактора свертывания крови способствует активации тромбоцитов, гладкомышечных клеток, моноцитов и эндотелиоцитов (Croce K., Libby P., 2007), с последующей продукцией провоспалительных медиаторов, цитокинов (CD40L, RANTES) и фактора, ингибирующего миграцию макрофагов. Указанные молекулы создают благоприятные условия для активации тромбообразования по внешнему пути и возникновения осложненной атеромы (Libby P., Simon D.I., 2001). Установлено, что и тромбоциты могут играть важную роль в продукции провоспалительных медиаторов, таких как тромбоцитарный фактор роста, способствующий непосредственной инкорпорации лейкоцитов в состав атеромы, миелоидзависимый протеин-8/14 и CD40L. Последний экспрессируется практически на всех типах клеток, вовлеченных в атерогенез: гладкомышечных клетках, макрофагах/моноцитах, эндотелиоцитах, Т-клетках, тромбоцитах (Mach F. et al., 1997). CD40 обеспечивает связь между экспрессией молекул клеточной адгезии и секрецией ряда цитокинов и MMP, вовлекаемых в процессы деградации внеклеточного матрикса (Mach F. et al., 1997; Schonbeck U. et al., 1997; Schonbeck U. et al., 1999). Установлено, что CD40-лиганд обладает высоким тромбогенным потенциалом (Bavendiek U. et al., 2002) и вовлекает активированные макрофаги и гладкомышечные клетки в коагуляционный каскад (Mach F. et al., 1998). С другой стороны, получены экспериментальные подтверждения того факта, что ингибирование активности CD40 способствует реверсии процессов атерогенеза на ранних этапах (Mach F. et al., 1998) и ограничения прогрессирования на поздних (Schonbeck U. et al., 2000a; b; Croce K., Libby P., 2007).
Таким образом, именно макрофаги ответственны за регуляцию последовательности смены фаз клеточной инфильтрации атеромы, результатом которой являются нарушения резистентности покрышки к провоспалительным, механическим или иным факторам, что приводит к ее разрыву, часто в угрожаемой плечевой области, кровоизлиянию (из vasa vasorum или просвета сосуда) или эрозированию.
Фазы атеротромбоза
В соответствии с критериями, предложенными Американской ассоциацией сердца (American Heart Association Committee on Vascular Lesions), в эволюции атеротромбоза выделяют пять основных фаз (Stary H.C. et al., 1995; Stary H.C., 2000).
Фаза 1 (ранняя). Обычно определяется у лиц молодого возраста и характеризуется тремя последовательными типами повреждения сосудистой стенки. I тип обусловлен накоплением в интиме и субинтиме сосуда макрофагов, поглощающих липиды с последующей трансформацией в пенистые клетки. Привлечение в зону инфильтрации гладкомышечных клеток, продуцирующих коллаген, составляющий основной компонент внеклеточных депозитов, характеризует наступление II типа поражения сосуда. В дальнейшем вокруг зоны клеточной инфильтрации развивается фиброз, формируются липидное ядро и фиброзная покрышка атеромы (III тип). Уже на этапе формирования I типа повреждения моноцитарные фагоциты, проникая в стенку артерий между эндотелиоцитами, способствуют инициализации воспалительной реакции в зоне субинтимы за счет повышения продукции провоспалительных цитокинов и оксидации ЛПНП, что негативно отражается на продукции оксида азота и способствует формированию дисфункции эндотелия. Повышение экспрессии молекул клеточной адгезии на мембранах эндотелиоцитов, опосредованных локальной провоспалительной активацией, замыкает порочный круг интернационализации клеточной субинтимальной инфильтрации. Необходимо отметить, что инфильтрация и аккумуляция ЛПНП в субинтиме артерий зависит не только от активности депонированных макрофагов, но может модулироваться взаимоотношением апо-В-липопротеина с матриксными протеинами сосудистой стенки. Последние в основном представлены протеогликанами, гликозаминогликанами, коллагенами и фибронектином (Khalil M.F. et al., 2004). Протеогликаны компартментализированы на участке между базальной мембраной с эндотелиоцитами и внутренней эластической пластинкой. Полагают, что образование взаимосвязи между окисленными ЛПНП, инфильтрирующими стенку артерий, и протеогликанами является одной из ключевых стадий развития атерогенеза, поскольку этот процесс в значительной мере препятствует ретенции липидов с помощью ЛПВП, способствует модификации ЛПНП и их аккумуляции (Shah P.K. et al., 2001a; b; Zhang Y. et al., 2003; Barter P., 2004; Barter P.J. et al., 2004). Важную роль в этих процессах играют скавенджер-рецепторы SR-A и CD-36, экспрессия которых на макрофагах повышается в ответ на поглощение ими окисленных ЛПНП. Результатом этого взаимодействия является активация факторов роста, таких как ядерный фактор транскрипции kB, способствующий потенциации хемоаттрактантного цикла привлечения гладкомышечных клеток в зону макрофагальной инфильтрации. Именно этот процесс в дальнейшем обеспечивает формирование атеромы за счет продукции гладкомышечными клетками внеклеточного коллагенового каркаса вокруг зоны аккумуляции липидов, что и приводит к возникновению покрышки атеромы.
Фаза 2 (развернутая). В эту фазу отчетливо прослеживается наличие сформированной атеромы, которая не всегда обусловливает стенотическое поражение сосуда. Покрышка атеромы может быть целостной или нарушенной с элементами деэндотелизации и истончения, формирования фиссур, особенно в плечевой области, участков инфильтрации клетками, имеющими воспалительное происхождение (макрофаги, Т-лимфоциты). Морфологически эти атеромы могут относиться к двум основным типам. Для первого характерно формирование крупного липидного ядра, содержащего свободные липиды, с истонченной покрышкой, а для второго — умеренная липидная внеклеточная аккумуляция при наличии фиброзной покрышки.
Фаза 3 (формирование уязвимой атеромы). Эта фаза характеризуется появлением осложненной атеромы, ассоциированной с разрывом или эрозированием покрышки, а также формированием интрамурального или внутрисосудистого тромбоза, обычно не приводящего к окклюзии. Часто этот процесс протекает субклинически или может способствовать возникновению стенокардии (Davies M.J., 1996). В настоящее время рассматриваются две основные причины, приводящие к разрыву атеромы: влияние физических факторов, способствующих появлению так называемой механической усталости покрышки и воспалительной активации, опосредованной аккумуляцией липидов и клеточной инфильтрацией (рис. 7.4). Необходимо отметить, что оба процесса фактически неразрывно связаны между собой. Вместе с тем, основной причиной, повлекшей за собой разделение обоих механизмов возникновения разрыва атеромы, стали сведения о вовлечении различных участков атеромы в процесс образования фиссуры. Так, при воздействии гемодинамических или физических факторов разрыв атеромы чаще происходит в плечевой области проксимальнее участка стенозирования, тогда как при других причинах разрыв локализуется в зоне верхушки атеромы на участках деэндотелизации интимы.
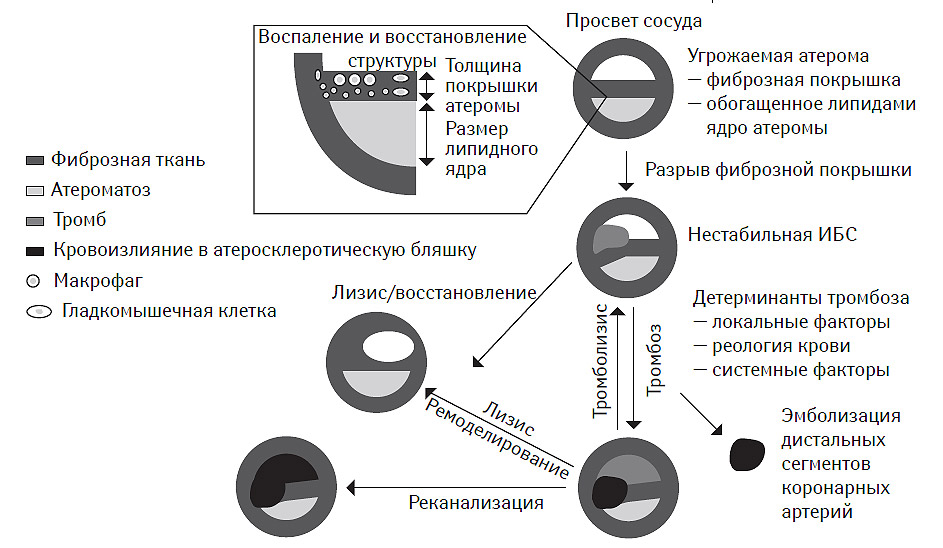
Рис. 7.4. Основные причины и механизмы реализации разрыва атеромы
Фаза 4 (возникновение осложненной атеромы). На этой стадии возникает окклюзия просвета сосуда сформированным тромбом, что приводит к появлению острого коронарного синдрома или инфаркта миокарда. В некоторых случаях течение процесса может быть бессимптомным (Canto J.G. et al., 2000; Sheifer S.E. et al., 2001). Необходимо отметить, что в подавляющем большинстве случаев острого коронарного синдрома связан с окклюзирующим тромбозом на фоне атеромы, не обтурирующей просвет сосуда. Вместе с тем, в 30% случаев возможно возникновение интраваскулярного тромбоза на поверхности атеромы, формирующей суб- или критический стеноз артерии (Falk E. et al., 1995).
Фаза 5 (формирование фиксированного стеноза сосуда). В этот период формируется или кальцифицированная атерома, или атеромы с жесткой фиброзной покрышкой, способствующие фиксированному стенозированию сосуда. Клинически эта фаза обычно соответствует стенокардии напряжения (Pohl T. et al., 2001; Werner G.S. et al., 2001). Установлено, что кальцификация является атрибутом взаимодействия гидроксиапатитов с внеклеточным органическим матриксом, преимущественно представленным молекулами коллагена I типа. Депонирование коллагенассоциированных кристаллов инициирует минерализацию везикул, расположенных в матриксе атеромы, что приводит, в свою очередь, к формированию кальцифицированной атеромы. Согласно современным представлениям минерализация атеромы является активным регулируемым процессом, напоминающим таковой в костной ткани, в котором важную модулирующую роль играют остеопонтин, остеонектин, остеопротегерин, матриксный протеин Gla. Последние экспрессируются в клетках адвентиции, макрофагах, гладкомышечных клетках медиа и атеромы (O’Brien E.R. et al., 1994; Takemoto M. et al., 2000). Доказана способность остеопонтина к индукции клеточной адгезии, стимуляции таксиса гладкомышечных клеток и макрофагов (Ikeda T. et al., 1993). Причем кальцификация покрышки атеромы может быть зарегистрирована уже на ранних этапах ее формирования (Liaw L. et al., 1994; Liaw L. et al., 1997). Предполагают, что диффузная минерализация фиброзной покрышки без очагового накопления кристаллов кальция является морфологической основой жесткой атеромы.
Патофизиологические механизмы формирования осложненной атеромы
Традиционно разрыв атеромы с формированием пристеночного тромбоза рассматривается как одно из наиболее неблагоприятных осложнений атерогенеза. Именно с атеротромбозом связана клиническая манифестация большинства кардиоваскулярных событий. Однако в большинстве случаев острый тромбоз не приводит к критическому стенозу или окклюзии, что априори создает известные сложности для интерпретации рутинно проведенных количественных ангиограмм (Hackett D. et al., 1988). Предполагается, что именно провоспалительная активация, а не собственно степень стенозирования, и является тем фактором, который обеспечивает тяжесть последующих ишемических повреждений тканей (Libby P., 2001). Гистологически в сайтах разрыва покрышки атеромы концентрируется наибольшее количество гладкомышечных клеток и макрофагов, от которых зависит интенсивность секреции провоспалительных медиаторов. С другой стороны, некоторые ИЛ (ИЛ-1), CD40 и интерферон способствуют не только деградации внеклеточного матрикса, но и формированию так называемой усталости покрышки за счет снижения накопления коллагена (Amento E.P. et al., 1991) и аккумуляции MMP-9 (Galis Z.S. et al., 1994; Gough P.J. et al., 2006). Это приводит к фрагментации покрышки, чаще всего в плечевой зоне, (Sukhova G.K. et al., 1999) и формированию осложненной атеромы (Morishige K. et al., 2003). Более того, рядом исследований было установлено, что локальная избыточная экспрессия (overexpression) MMP-9 способствует интраваскулярному тромбозу посредством высвобождения тканевого фактора свертывания (Morishige K. et al., 2003).
Таким образом, серьезные кардиоваскулярные события обычно возникают вследствие непосредственного контакта содержимого атеромы с пристеночной кровью (Libby P., 2001). Это взаимодействие инициирует формирование тромба, который не всегда приводит к критическому стенозу/окклюзии, хотя и достаточно часто заканчивается фатальными последствиями. Предполагается, что сопутствующая активация фибринолиза приводит к ранней реканализации зависимой артерии и это не означает, что окклюзирующий тромбоз не имел место вообще (Vaughan D.E., 2005). Более того, провоспалительная активация способствует повышению плазменной концентрации циркулирующего ингибитора плазменного активатора и фибриногена, что пролонгирует существование тромбоза (Naghavi M. et al., 2003а; b; Libby P., Theroux P., 2005).
Основные виды атером
К основным видам атером относят липидные полоски, неосложненные и осложненные атеросклеротические бляшки. Липидные полоски представляют собой локально депонированные в субинтиме липиды, содержащие нагруженные липидами макрофаги (пенистые клетки) и гладкомышечные клетки. Результатом эволюции липидной полоски является фиброзная атерома, которая отличается от последней наличием вступающей в просвет сосуда относительно жесткой фиброзной покрышки и сформированного липидного ядра, содержащего свободные липиды. Атерома, ассоциированная с формированием тромба, кальцификации или кровоизлияния в липидное ядро рассматривается как осложненная.
Понятие угрожаемой и высокорисковой атеромы
Полагают, что прогрессирование атеросклероза осуществляется преимущественно за счет экспансии липидного ядра и аккумуляции пенистых клеток на его периферии в непосредственной близости от так называемой покрышки атеромы. При этом ее жесткость, обусловленная накоплением коллагена, особенно в плечевой области, рассматривается как основной фактор, определяющий способность атеромы к разрыву (Burke A.P. et al., 1997). Так, при аутопсии больных с ИБС, умерших внезапно, разрыв атеромы ассоциировался с повышением соотношения общий ХС/ХС ЛПВП, тогда как такие известные факторы риска кардиоваскулярной смерти, как курение и артериальная гипертензия не продемонстрировали устойчивой корреляции с риском возникновения угрожаемой (vulnerable plaque) атеромы.
Несмотря на то, что разрыв атеромы является одной из основных причин возникновения атеротромбоза, почти в 30–40% локализацию уязвимой атеромы установить не удается. Результаты исследования A. Farb и соавторов (1996) показали, что только у 22 из 55 больных, умерших внезапно вследствие коронарного атеротромбоза, удалось идентифицировать поверхностно-эрозированную атерому, обогащенную протеогликанами с большим содержанием гладкомышечных клеток. При этом локализация атеромы соответствовала участку ишемии и некроза. У 28 пациентов структура атеромы не рассматривалась как потенциально угрожаемая разрыву. Авторы пришли к заключению, что уязвимые атеромы обычно не подвержены выраженной кальцификации, не приводят к формированию гемодинамически значимых стенотических поражений артерий, менее инфильтрированы макрофагами, а также чаще идентифицируются у женщин в пост- и перименопаузальный период, чем у мужчин того же возраста.
Необходимо отметить, что покрышка атером, подвергшихся разрыву, обычно умеренно утолщена или истончена (в среднем 23±19 µм) и в 95% случаев имеет абсолютную толщину <64 µм. Кроме того, подобные атеромы богато инфильтрированы макрофагами и Т-лимфоцитами, а липидное ядро обогащено эстерифицированым ХС (Burke A.P. et al., 1997). Достаточно часто выявляют новообразование сосудов из vasa vasorum, прорастающих в интиму атеромы со стороны адвентиции сосуда.
Несколько реже (приблизительно в 30% случаев) при проведении аутопсий отмечают несколько иной вариант атером, обычно идентифицирующихся как эрозированные атеросклеротические бляшки. Для последних характерно образование тромбоза на поверхности покрышки, отсутствие эндотелиальной выстилки при сохранении толщины последней, липидное ядро может отсутствовать вообще или обогащено протеогликанами, обычно не определяется выраженная инфильтрация воспалительными клетками. Подобные изменения часто выявляют при проведении рентгеноконтрастной ангиографии у пациентов с острым коронарным синдромом с элевацией сегмента ST на ЭКГ до возникновения указанного события в инфарктзависимой артерии. Причем подобные атеромы не создают гемодинамически значимого стеноза и часто локализуются либо в проксимальных сегментах крупных коронарных артерий, либо в участках их бифуркаций. Кроме того, по данным гистологических исследований, эрозированные атеромы — достаточно часто определяют у женщин в возрасте моложе 50 лет, умерших внезапно вследствие коронарных причин (Schoenhagen P. et al., 2000; Lakoske S.G. et al., 2007; Detrano R. et al., 2008; Ambrose J.A., Srikanth S., 2010).
Таким образом, к настоящему времени удалось идентифицировать как минимум два типа потенциально угрожаемых атером: так называемая воспалительно-измененная фиброатерома с истонченной покрышкой и эрозированная атерома (Libby P., 2005).
Необходимо отметить, что оба типа атеросклеротических бляшек существенно отличаются друг от друга не только излюбленной локализацией в коронарных артериях, но и морфологически. В то же время полагают, что процессы разрыва и эрозирования покрышки атеромы обусловлены двумя принципиально различными механизмами, обычно завершающимися формированием тромба и окклюзии сосуда. В этом контексте термины атерома высокого риска (high-risk) и угрожаемая (vulnerable) атерома обычно употребляются как синонимы, поскольку описывают риск возникновения атеротромбоза (Schaar J.A. et al., 2003). В то же время в клинической практике широко применяются сугубо морфологические термины, такие как эрозированная атерома, атерома, ответственная за повреждение, воспалительно-измененная фиброатерома с тонкой покрышкой (inflamed thin-cap fibroatheroma — ITCFA), кальцифицированная атерома, атерома с тромбозом, которые характеризуют различные стадии процесса дестабилизации атеросклеротической бляшки (Libby P., 2005; Detrano R. et al., 2008). Тем не менее, неоднозначность в понимании и множественность сочетаемых характеристик указанных дефиниций создавали некоторые проблемы в описании особенностей патологического процесса, что потребовало разработки стандартизированной концепции, касающейся формирования представлений о наличии четко определенных критериев для так называемых осложненных атером (Naghavi M. et al., 2006). Наиболее удачно эту задачу удалось выполнить коллективу экспертов J.A. Schaar, J.E. Muller, E. Falk и соавторам (2004), результаты работы которых представлены в табл. 7.1.
Вместе с тем, существует и иной подход, позволяющий классифицировать типы атеросклеротического поражения, среди которых атерома не является единственной формой нарушения структуры и архитектоники сосудистой стенки (Virmani R. et al., 2000). При этом в качестве основных признаков, позволяющих идентифицировать высокорисковую атерому является интрамуральный или окклюзирующий тромбоз (табл. 7.2).
Таблица 7.2 Главные типы поражения сосудистой стенки при атеросклерозе (модифицировано из работы Virmani R. et al., 2000)
| Характер поражения сосуда | Гистопатологическое описание | Наличие тромбоза |
| Неатеросклеротическое поражение интимы | ||
| Утолщение интимы | Нормальная аккумуляция ГМК при отсутствии накопления липидов, макрофагов/пенистых клеток, сохраняется принципиальная возможность регресса | Отсутствует |
| Ксантомы или липидные полоски интимы | Субэндотелиальное накопление пенистых клеток без формирования некротического липидного ядра или фиброзной капсулы с возможностью спонтанного регресса | Отсутствует |
| Прогрессирующее атеросклеротическое поражение | ||
| Патологическое утолщение интимы без эрозий | ГМК расположены в обогащенном протеогликанами матриксе вокруг зоны экстрацеллюлярной аккумуляции свободных липидов без участков некроза | Отсутствует |
| Патологическое утолщение интимы с эрозиями | Интрамуральный или иногда окклюзирующий | |
| Атерома с фиброзной капсулой без эрозии | Хорошо сформированное некротическое ядро с покрывающей его фиброзной капсулой без эрозии | Отсутствует |
| Атерома с фиброзной капсулой с эрозией | Формирование эрозии на поверхности фиброзной капсулы | Интрамуральный или иногда окклюзирующий |
| Тонкокапсульная атерома | Истонченная фиброзная капсула атеромы инфильтрирована макрофагами и лимфоцитами, редко выявляют ГМК, некротический характер липидного ядра | Отсутствует, иногда отмечают кровоизлияние внутри атеромы |
| Тонкокапсульная атерома с разрывом | Фиброатерома с разорванной покрышкой, тромбоз на поверхности люминальной пластинки сосудистой стенки, покрывающий некротическое липидное ядро | Тромбоз обычно носит окклюзирующий характер |
| Кальцифицированная атерома | Разрыв покрышки с кальцификацией и формированием фиброкальциноза | Тромбоз обычно неокклюзирующий |
| Фиброкальцифицированная атерома | Атерома, обогащенная коллагеном, обычно формирующая клинически значимое стенотическое поражение артерий, содержит обширную зону кальцифицированных элементов (кристаллов), окруженную клетками воспалительного происхождения, некротическое липидное ядро может сохраняться | Тромбоз обычно отсутствует |
ГМК — гладкомышечные клетки.
Таким образом, общая концепция формирования атеротромбоза преимущественно основана на исключительной роли атеромы с эрозированной или разорванной покрышкой, презентирующей тромбогенное содержимое некротического липидного ядра, что приводит к формированию интрамурального или окклюзирующего тромбоза. Последний рассматривается как атрибут процессов про- и антикоагулянтного гемостаза, модулированного, в частности, тканевым тромбопластином липидного ядра. В результате этих процессов наблюдается появление клинических эквивалентов атеротромбоза. Восстановление целостности фиброзной покрышки атеромы, с одной стороны, способствует ее росту, кальцификации и формированию фиксированного клинически значимого стеноза, а с другой — снижает риск тромбообразования (рис. 7.5). Вместе с тем, эта концепция, несмотря на ее высочайшую прогностическую ценность, не позволяет идентифицировать атерому, определяющую клинические исходы при прогрессировании атеротромбоза. Все это стало основанием для внедрения в клиническую практику понятия «уязвимая» или «угрожаемая» атерома.

Рис. 7.5. Современная концепция прогрессирования атеротромбоза ОКС — острый коронарный синдром.
Идентификация угрожаемой атеромы
Теоретически идентификация угрожаемой атеромы предоставляет возможность для проведения локальной терапии или более агрессивного системного подхода с использованием статинов, антиагрегантов и блокаторов ренин-ангиотензиновой системы в популяции пациентов высокого риска. Сегодня предпринимаются попытки для внедрения в клиническую практику методов инвазивной или неинвазивной визуализации атером с истонченной покрышкой. Вместе с тем, измерение толщины покрышки фиброатеромы не является единственной возможностью для последующей идентификации угрожаемой атеромы, поэтому применяемая процедура должна предполагать визуализацию некротически измененного липидного ядра, участков кальцификации, vasa vasorum. Этим условиям удовлетворяет ряд методов, представленных в табл. 7.3. Однако необходимо отметить, что все перечисленные подходы базируются на допущении о том, что угрожаемая атерома имеет такие характеристики, как и осложненная атеросклеротическая бляшка. Вместе с тем, есть сведения о том, что это предположение не всегда выполняется (см. табл. 7.1). Еще одним лимитирующим фактором для широкого внедрения вышеперечисленных методов визуализации в рутинную клиническую практику является исходно неизвестная локализация угрожаемой атеромы, что вызывает необходимость расширения объема и количества процедур, негативно отражающихся на общей стоимости исследования (Ambrose J.A., Srikanth S., 2010).
С другой стороны, не существует согласованного мнения по поводу того, какие именно морфологические характеристики атеромы следует рассмотреть в качестве достаточного основания для идентификации последней как угрожаемой (Ambrose J.A., 2008). На рис. 7.6 представлены две принципиальные возможности для эволюции стабильной атеромы в осложненную через формирование угрожаемой бляшки. Во всех случаях выявляют наличие большого липидного ядра, тонкой покрышки, инфильтрированной клетками воспалительного происхождения, как правило, Т-лимфоцитами и макрофагами. Вместе с тем, появление некротических изменений в толще липидного ядра возможно далеко не всегда, а воспалительная инфильтрация проявляет высокую степень вариабельности. Многие эксперты полагают, что таким образом мы имеем возможность наблюдать атеромы «в процессе», то есть на разных стадиях формирования угрожаемой атеросклеротической бляшки. Кроме того, при проведении спиральной контрастной компьютерной томографии оказалось, что в ряде случаев покрышка фиброатеромы не сохраняет свою целостность, а является эрозированной или разорванной в нескольких местах, причем зачастую без существенного изменения ее толщины и интенсивности субкапсулярной инфильтрации. Вполне возможно, что большинство угрожаемых атером уже на субклиническом этапе могут иметь эрозированную покрышку или осложняться тромбозом без выраженной протрузии в просвет сосуда (Ambrose J.A., 2008). С другой стороны, около трети всех пациентов с острым коронарным синдромом обычно имеют более одной атеромы, характеристики которых могут быть приняты во внимание как потенциально угрожаемые для возникновения последующего атеротромбоза. Поскольку до настоящего времени отсутствуют доступные методы проведения локальной терапии, попытки документирования угрожаемых атером хотя и выглядят как достаточно научно-обоснованные, но являются малопривлекательными с практической точки зрения (Naghavi M. et al., 2003a; b; Libby P., 2005). Системная терапия, включающая применение статинов, антиагрегантов, ингибиторов ангиотензинпревращающего фермента (иАПФ), иногда антикоагулянтов или ω-3 ненасыщенных жирных кислот в высоких дозах, не требует обязательной идентификации типа и локализации атеромы высокого риска и позволяет минимизировать затраты системы здравоохранения при приемлемом уровне эффективности и безопасности. В настоящее время рекомендованным является комплексный подход к идентификации пациентов группы высокого риска на основании традиционных шкал оценки риска, популяционных критериев, данных дополнительных методов исследования и ряда биологических маркеров (Khot U.N. et al., 2003; Naghavi M. et al., 2003a; b) (табл. 7.4). Вероятно, скрининг и агрессивное локальное лечение «угрожаемых» пациентов является подходом будущего.
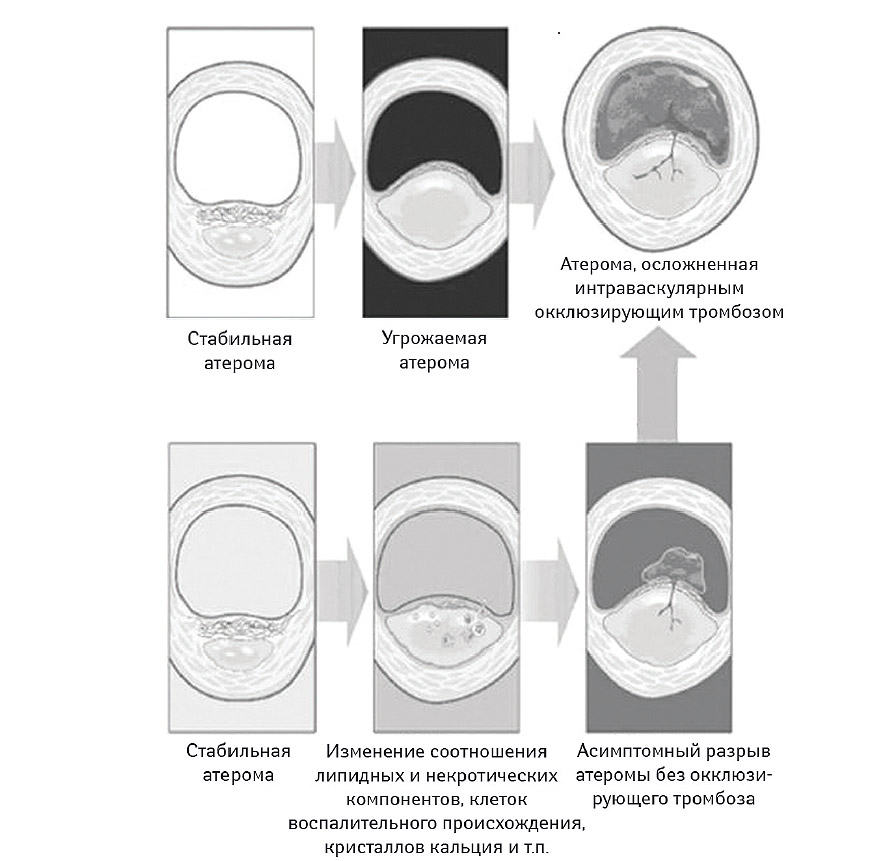
Рис. 7.6. Принципиальные морфологические характеристки угрожаемой атеромы (модифицировано с изменениями из работы Schoenhagen P. et al., 2001)
Таблица 7.3 Методы визуализации угрожаемой атеромы
| № | Основные методы исследования |
| 1 | Ангиоскопия |
| 2 | Интраваскулярная эхо- и допплерография (IVUS — intravascular ultrasound — технология) |
| 3 | Методы виртуальной гистологии/молекулярные исследования |
| 4 | Термография |
| 5 | Визуализация vasa vasorum с помощью контрастной спиральной компьютерной ангиогафии |
| 6 | ЯМР-томография (инвазивная и неинвазивная) |
| 7 | Позитронная эмиссионная томография |
| 8 | Спиральная компьютерная томография |
| 9 | Оптическая когерентная томография |
Таблица 7.4 Методы, использующиеся для косвенной оценки риска возникновения угрожаемых атером в рутинной клинической практике (модифицирована из работы Ambrose J.A., Srikanth S., 2010)
| Название | Характеристика |
| Традиционные системы оценки риска | Framingham Risk Score |
| European Society Risk Score | |
| Prospective Cardiovascular Munster Study Risk Score | |
| Маркеры индивидуального высокого кардиоваскулярного риска | Гипертрофия левого желудочка |
| Гиперлипидемия | |
| Снижение плазменной концентрации ХС ЛПВП | |
| Апо-В100 и другие качественные нарушения структуры аполипопротеинов | |
| Микроальбуминурия | |
| Ожирение | |
| Метаболический синдром/сахарный диабет 2-го типа | |
| Сниженная физическая активность | |
| Эректильная дисфункция | |
| Низкий социально-экономический статус | |
| Маркеры атеросклеротического процесса | Утолщение комплекса интимы-медиа общей сонной артерии |
| Документированная дисфункция эндотелия | |
| Жесткость сосудистой стенки | |
| Снижение плече-лодыжечного индекса | |
| Доказательства ИБС (ЭКГ-критерии, результаты коронароангиографии) | |
| Методы неинвазивной визуализации | ЯМР-томография |
| Позитронная эмиссионная томография | |
| Спиральная компьютерная томография (64 среза) | |
| Биомаркеры | Уровень гомоцистеина |
| Уровень фибриногена | |
| Уровень цистацита-С | |
| Уровень тропонина-I | |
| Уровень мозгового натрийуретического пептида (МНУП) | |
| Уровень липопротеинассоциированной фосфолипазы А2 | |
| Уровень СРП | |
| Генетические маркеры | Точечный нуклеотидный полиморфизм |
Васкулярное ремоделирование, ассоциированное с атеросклеротическим поражением сосуда
Формирование атеросклеротической бляшки часто рассматривается в аспекте сопутствующего васкулярного ремоделирования. Принято выделять как минимум два типа последнего: экспансивный и констриктивный (рис. 7.7). На сегодня установлено, что констриктивный вариант васкулярного ремоделирования в значительной мере обусловливает возникновение фиксированного стеноза артерии, а также является компонентом ряда осложнений ангиопластики (de Smet B.J.G.L. et al., 1998), в том числе и стентинга, таких как рестеноз или подострый тромбоз стента (Ozaki Y. et al., 1996).
Напротив, экспансивный вариант рассматривается как благоприятный процесс, возможно имеющий компенсаторный характер, повышающий демпфирующие свойства сосудистой стенки, способствующий сохранению ламинарного характера кровотока и препятствующий дальнейшему увеличению жесткости сосуда (Hermiller J.B. et al., 1993; Pasterkamp G. et al., 1995).
В исследованиях in vivo с использованием внутрисосудистой эхо-допплерографии было установлено, что ремоделирование артерий, ассоциированное с атеросклеротическим поражением, может иметь двоякий характер (рис. 7.8). С одной стороны, существуют варианты этого процесса, при которых площадь наружной эластической мембраны стенки сосуда может увеличиваться за счет смещения ее липидным ядром проксимальнее и дистальнее локализации атеромы. Этот тип ремоделирования принято называть позитивным или адекватным. С другой стороны, в некоторых случаях в области атеромы со стороны адвентиции сосуда формируется «провал» сосудистой стенки, что приводит к уменьшению площади наружной эластической мембраны. Это так называемый негативный или неадекватный тип васкулярного ремоделирования (McPherson D.D. et al., 1991; Ge J. et al., 1993; Hermiller J.B. et al., 1993; Pasterkamp G. et al., 1995; Nishioka T. et al., 1996). Причем в обоих случаях отмечают стенозирование сосуда атеромой.
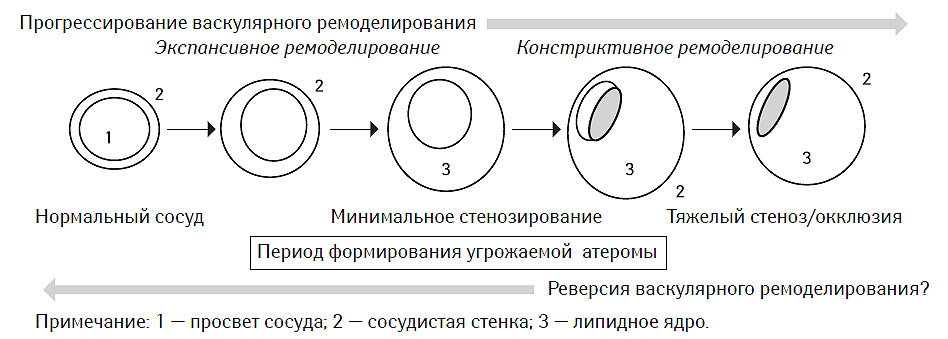
Рис. 7.7. Эволюция васкулярного ремоделирования, ассоциированного с формированием угрожаемой атеромы и последующей окклюзией сосуда (модифицировано с изменениями из работы Schoenhagen P. et al., 2001)
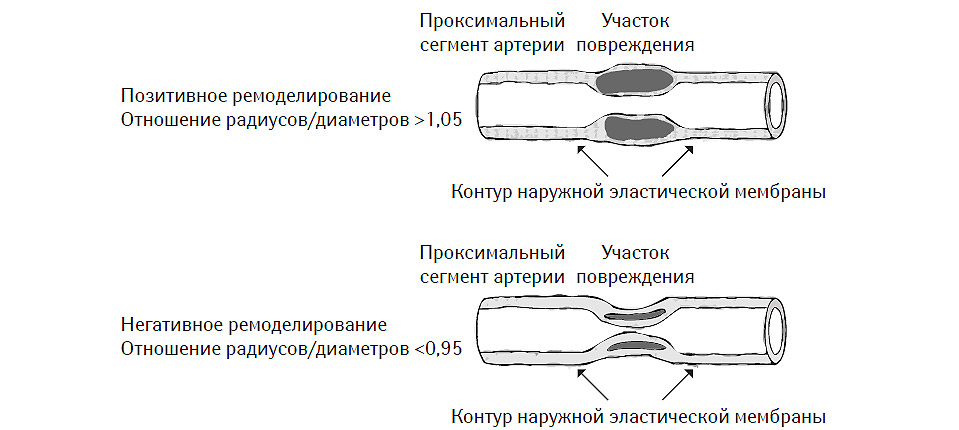
Рис. 7.8. Позитивный и негативный тип васкулярного ремоделирования (модифицировано с изменениями из работы Schoenhagen P. et al., 2001)
При проведении внутрисосудистого ультразвукового исследования наружная эластическая мембрана обычно хорошо лоцируется, а измерение ее площади в области атеромы дает возможность четко верифицировать характер и тип васкулярного ремоделирования. На рис. 7.9 приведен образец изображения, полученный с помощью интраваскулярного допплерографического исследования (Intravascular ultrasound — IVUS) левой нисходящей коронарной артерии у пациента с ангиографически интактными коронарными артериями. Темная и светлая стрелки указывают на позицию катетера и область эксцентрического ремоделирования, обусловленную некальцифицированной атеромой, соответственно. Наружной пунктирной линией обозначена наружная эластическая мембрана, а внутренней — люминальная пластинка.

Рис. 7.9. Образец изображения, полученный с помощью интраваскулярного допплерографического исследования
Собственно, термины «позитивный» и «негативный» тип ремоделирования являются атрибутами следующего математического отношения: площадь наружной эластической мембраны непосредственно над участком атеромы/площадь наружной эластической мембраны над проксимально расположенным интактным участком. В первом или втором случаях соответственно результат этого отношения больше или меньше единицы. Именно эти особенности и были приняты во внимание S. Glagov и соавторами (1987), выполнившими пионерские работы в этом направлении. Впоследствии оказалось, что описанные ими типы васкулярного ремоделирования свойственны в большей степени периферическим артериям, чем коронарным, хотя и выявляются при исследовании крупных коронарных артерий, в основном ствола левой или правой артерии.
Необходимо отметить, что достаточно часто (57%) у пациентов с ангиографически интактными коронарными артериями при проведении внутрисосудистого ультразвукового исследования выявляли адекватный вариант васкулярного ремоделирования, тогда как неадекватный отмечают значительно реже. По данным G.S. Mintz и соавторов (1997), неадекватный тип васкулярного ремоделирования выявляют приблизительно у 15% пациентов с документированной ИБС. В то же время стало известно, что позитивное васкулярное ремоделирование чаще отмечается в проксимальных сегментах крупных и средних коронарных артерий, зачастую — у пациентов с острым коронарным синдромом и инфарктом миокарда, ассоциируется с формированием нестабильной атеромы и рассматривается как предиктор неблагоприятного клинического исхода, что повышает диагностическую значимость идентификации подобных изменений (Gerber T.C. et al., 1994; von Birgelen C. et al., 2001; Ward M.R. et al., 2001; Prati F. et al., 2003).
Механизмы, опосредующие формирование обоих типов васкулярного ремоделирования сходы между собой, тогда как причины, приводящие к столь существенным различиям, до конца не определены (Libby P. et al., 2002a; b; Gurfinkel E. et al., 2009). Предполагается, что значительную роль в этом процессе могут играть генетические факторы, определяющие экспрессию ряда сигнальных молекул (трансформирующий фактор роста-β, тромбоцитарный фактор роста, CD40-лиганд) и ферментных систем (металлопротеиназы, капсазы), принимающих участие в продукции и деградации внеклеточного коллагенового матрикса (Brasselet C. et al., 2005; Hellings W.E. et al., 2008). Предпринимались попытки сопоставления експансивного/конструктивного вариантов васкулярного ремоделирования с адекватным/неадекватным типом последнего (рис. 7.10).

Рис. 7.10. Сопоставление различных вариантов васкулярного ремоделирования (модифицировано с изменениями из работы Schoenhagen P. et al., 2001)
При этом оказалось, что экспансивный вариант ремоделирования часто выявляют на ранних стадиях атерогенеза, он ассоциируется с формированием фиброзной атеромы с большим липидным ядром, смещающим наружную эластическую мембрану и формирующим адекватный тип ремоделирования. Вместе с тем, именно такие атеромы чаще всего и подвергаются эрозированию и разрыву, что не может рассматриваться как благоприятный или позитивный в клиническом отношении вариант ремоделирования сосуда. Напротив, констриктивный тип ремоделирования чаще всего опосредован атеромой с жесткой покрышкой, редко подвергаемой дестабилизации и сопровождающейся неадекватным вариантом ремоделирования сосуда, хотя в клиническом смысле это далеко не так. Двойственность представлений о клинической ценности вариантов ремоделирования артерий при атеросклерозе привели к формированию концепции об ассоциации стабильной атеромы с негативным вариантом ремоделирования, тогда как позитивный вариант ремоделирования, напротив, рассматривается в качестве предиктора высокого риска атеротромботических событий (рис. 7.11).
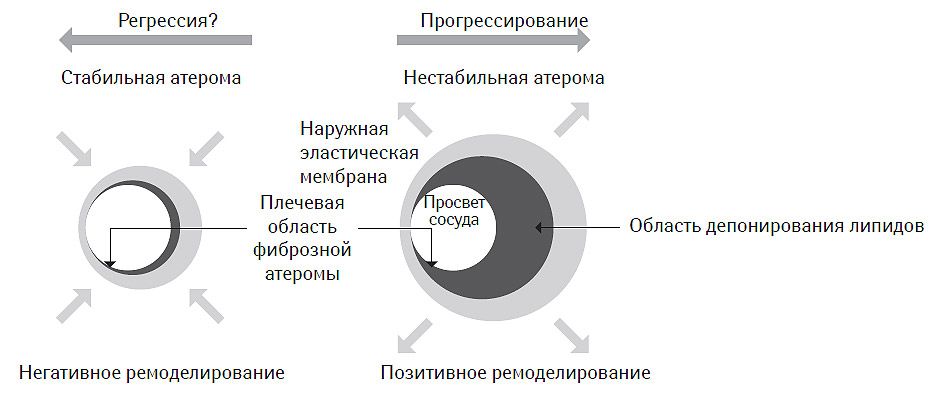
Рис. 7.11. Клиническая ценность вариантов ремоделирования артерий при атеросклерозе
Таким образом, появление доступной техники для проведения внутрисосудистой эхолокации благоприятно отразилось на возможности индивидуализации оценки риска манифестации атеротромботических событий с учетом особенностей ремоделирования стенозированного сосуда.
Иммунопатология атеросклероза
Элементы воспалительной реакции присутствуют практически на всех стадиях атерогенеза. Установлено, что в формировании атеромы принимают участие не только эндотелиоциты, гладкомышечные клетки, но и потенциально антигенпрезентирующие клетки: мононуклеарные фагоциты и Т-лимфоциты. Моноциты с помощью адгезивных молекул, экспрессирующихся на поверхности эндотелиоцитов, проникают в субинтиму, где после поглощения ЛПНП превращаются в пенистые клетки. Последние привлекают гладкомышечные клетки, которые являются главным источником продукции коллагена, формирующего покрышку атеромы. При этом основная роль в регуляции синтеза и деградации внеклеточного матрикса принадлежит именно Т-лимфоцитам. Синтезируемый последними γ-ИФ супрессирует продукцию коллагена. Контактно или посредством растворимых медиаторов Т-лимфоциты активируют макрофаги, которые продуцируют металлопротеиназы, участвующие в деградации покрышки атеромы. Установлено, что на ранних стадиях атерогенеза аккумулированные в интиме артерий Т-лимфоциты способны экспрессировать рецептор, распознающий окисленные формы ЛПНП и идентифицированный как νβ-6. Предполагается, что фрагменты окисленных ЛПНП могут являться индукторами активации Th-1-зависимых процессов локально в стенке артерий еще до формирования атеромы. Кроме того, оказалось, что у лиц с высоким риском возникновения атеротромботических событий в плазме крови часто выявляется клон Т-лимфоцитов, экспрессирующий рецепторы к окисленным ЛПНП, а также циркулирующие антитела к последним.
Таким образом, активные воспалительные процессы в интиме и субинтиме артерий способствуют избыточному разрушению и снижению синтеза коллагена, что предрасполагает к формированию феномена механической «усталости» покрышки атеромы, повышает риск ее разрыва и возникновения тромбоза как внутри некротического липидного ядра, так и в просвете сосуда. Кроме того, в настоящее время провоспалительная активация рассматривается как один из факторов, приводящих к дисбалансу между тромбозом и фибринолизом при формировании осложненной атеромы (Kohler H.P., Grant P.J., 2000; Eren M. et al., 2002). При этом уровень продукции провоспалительных цитокинов связывается с общей величиной тромбогенного потенциала плазмы крови (Hansson G.K., 2005; Luster A.D. et al., 2005; Maxfield F.R., Tabas I., 2005; Charo I.F., Ransohoff R.M., 2006; Hotamisligil G.S., 2006; Tedgui A., Mallat Z., 2006; van Gaal L.F. et al., 2006).
Основные биомаркеры провоспалительной активации и их клиническое значение
К наиболее важным биомаркерам провоспалительной активации относят VCAM-1; ФНО-α, ИЛ-1, ИЛ-18; металлопротеиназы, такие как MMП-9; месенджерные цитокины, такие как ИЛ-6; продукты тромбоцитарной активации (лиганд CD40, миелоидзависимый протеин 8/14); адипонектин; CРП, фибриноген и циркулирующий ингибитор плазменного активатора фибринолиза (ЦИПАФ).
Наиболее изученным и известным биомаркером, без сомнения, является СРП, уровень которого тесно ассоциируется с риском возникновения кардиоваскулярных событий (Ridker P.M., 2003a; b; Danesh J. et al., 2004; Blankenberg S., Yusuf S., 2006; Kinlay S., Egido J., 2006). Вместе с тем, линейная взаимосвязь между кардиоваскулярным риском и плазменной концентрацией СРП возникает только при достаточно высоких уровнях последней (>10 мг/дл), что предполагает проведение дополнительных исследований в отношении возможной контаминации образцов или наличия инфекционного процесса (Pasceri V. et al., 2000; Zwaka T.P. et al., 2001). Кроме того, последние исследования in vivo позволили предположить, что СРП может продуцироваться не только в гепатоцитах, но и в гладкомышечных клетках и не только в ответ на стимуляцию ИЛ-1β. При этом СРП активно вовлекается в процессы атеротромбоза, включая дисрегуляцию фибринолиза посредством повышения экспрессии и активности ЦИПАФ (Devaraj S. et al., 2003; Calabro P. et al., 2003), а также в формирование дисфункции эндотелия артерий (Jabs W.J. et al., 2003; Singh P. et al., 2007). В этой связи мониторирование плазменного пула СРП приносит бесспорно существенную клиническую пользу.
Другие острофазовые реактанты, такие как фибриноген и циркулирующий ингибитор плазменного активатора фибринолиза, имеют существенно меньшую диагностическую ценность, чем СРП. Адипонектины, в отличие от фибриногена и циркулирующего ингибитора плазменного активатора фибринолиза, имеют не столь вариабельную плазменную концентрацию и более пригодны для рутинного измерения. Другие медиаторы, такие как ИЛ-1 и ИЛ-6 имеют очень короткий период полужизни, что создает серьезные проблемы для внедрения их мониторинга в клиническую практику. Растворимые VCAM-1 не обладают способностью прогнозировать риск возникновения серьезных кардиоваскулярных событий, например инфаркта миокарда, у здоровых мужчин (de Lemos J.A. et al., 2000). Тем не менее, роль VCAM-1 как предиктора высокого сосудистого риска была продемонстрирована в экспериментальной модели (Li H. et al., 1993; Gerszten R.E. et al., 1996; Gerszten R.E., et al., 1998; Kawakami A. et al., 2006).
Таким образом, существует настоятельная необходимость в поиске новых маркеров провоспалительной активации, отвечающих всем требованиям теории тестов.
Роль провоспалительных биомаркеров как предикторов высокого кардиоваскулярного риска
В настоящее время в соответствии с требованиями Adult Treatment Panel III для скрининга и мониторинга суммарного кардиоваскулярного риска используются, в частности, абсолютные величины плазменных концентраций ряда липидных фракций: ХС ЛПНП, ХС ЛПВП, ТГ, общего ХС (Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults, 2001). Вместе с тем, величина общего ХС плазмы крови плохо коррелирует с частотой кардиоваскулярных событий в популяции (Ridker P.M. et al., 2002). В этой связи в качестве дополнительных кандидатов на роль биомаркеров, способных повысить предсказующую ценность анализа липидного профиля плазмы крови, рассматриваются молекулы, отражающие активность воспалительной активности, оксидантного стресса, тромбогенного потенциала (Libby P., 2002a; b; Libby P. et al., 2002; Tsimikas S. et al., 2006).
Необходимо отметить, что рекомендации о расчете именно глобального кардиоваскулярного риска призваны улучшить стратификацию пациентов, оптимизировать подходы к их лечению и профилактике, и, тем самым, снизить вероятность наступления неблагоприятного исхода (Greenland P. et al., 2003). В тоже время дополнительный анализ уровня провоспалительной активности плазмы крови создает предпосылки для еще более точной оценки суммарного кардиоваскулярного риска (Libby P., Ridker P.M., 1999). К сожалению, использование таких диагностических инструментов, как анализ плазменной концентрации СРП, солюбилизированных лигандов CD40, адипонектина, ИЛ-18 и MMП-9 обосновано только в специально спланированных научных исследованиях, тогда как в рутинной клинической практике не установлены дополнительные преимущества этих исследований перед стандартными методами.
СРП
Результатами многочисленных клинических исследований показано, что СРП является независимым фактором высокого кардиоваскулярного риска, включая риск инфаркта миокарда, инсульта и внезапной сердечной смерти (Kuller L.H. et al., 1996; Ridker P.M. et al., 1997; Ridker P.M. et al., 2000; Koenig W. et al., 2004; Pai J.K. et al., 2004). Зависимость между этими факторами фактически линейная. Учитывая это, многие исследователи склонны стратифицировать больных с высокими уровнями СРП в группы высокого риска даже при наличии целевых уровней ХС ЛПНП в крови (Ridker P.M. et al., 2002). Мониторинг плазменного пула СРП существенным образом повышает прогностическую ценность не только ХС ЛПНП, но и всех остальных показателей Фремингемской шкалы риска (Ridker P.M. et al., 2002; Koenig W. et al., 2004). Установлено, что фактически все дефинизирующие компоненты метаболического синдрома тесно коррелируют с плазменной концентрацией СРП (Ridker P.M. et al., 2002; Ridker P.M. et al., 2003a; b). Эти данные позволили использовать СРП как один из компонентов шкалы риска Рейно (Reynolds risk score) для глобальной оценки кардиоваскулярного риска у женщин (Ridker P.M. et al., 2007). Алгоритм добавления оценки СРП во Фремингемскую шкалу риска способствует более точной оценке риска более чем в 50% случаев у лиц, ранее отнесенных в когорту «низкорисковых» пациентов. В этой связи Centers for Disease Control and Prevention and the American Heart Association настоятельно рекомендует рутинно использовать уровень СРП в качестве маркера кардиовасулярного риска (Pearson T.A. et al., 2003), особенно у пациентов, не имеющих клинических признаков кардиоваскулярных заболеваний или с рассчитанной промежуточной величиной риска. В клинической практике плазменный уровень СРП <1 мг/л рассматривается как показатель низкого риска, а уровень >3 мг/л — высокого. Необходимо отметить, что после внедрения статинов в клиническую практику их способность редуцировать плазменную концентрацию СРП на 20–30% стали рассматривать как одну из целей терапии (Ridker P.M. et al., 1999). Причем статины способствуют снижению СРП в плазме крови независимо от их гиполипидемического эффекта (Ridker P.M., 2003a; b), что позитивно отражается на ближайшем и отдаленном прогнозе (Ridker P.M. et al., 2001a; b). Так, в рандомизированном клиническом исследовании PROVE-IT TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 trial) было установлено, что максимальную вероятность выживания при одинаковом снижении ХС ЛПНП под влиянием статина отмечают у пациентов с острым коронарным синдромом с наиболее низким пулом СРП (Ridker P.M. et al., 2005). В рандомизированном клиническом исследовании REVERSAL (The Reversal of Atherosclerosis with Aggressive Lipid Lowering) снижение СРП плазмы крови благоприятно отражалось на реверсии атеросклеротического поражения артерий (Nissen S.E. et al., 2005).
Таким образом, мониторинг плазменной концентрации СРП оказывается весьма полезным инструментом в оценке не только вероятности выживания, но и эффективности модуляции кардиоваскулярного риска.
Фибриноген
В многочисленных эпидемиологических и рандомизированных клинических исследованиях было установлено, что плазменная концентрация фибриногена может отражать риск возникновения инфаркта миокарда и инсульта (Wilhelmsen L. et al., 1984; Kannel W.B. et al., 1987; Ma J. et al., 1999). Сопоставление прогностической ценности уровня СРП и фибриногена показало преимущества первого в отношении величины прогностической ценности возникновения любых кардиоваскулярных событий (Ridker P.M. et al., 2001a; b). Между тем, существует достаточно тесная корреляция между плазменными концентрациями СРП и фибриногена в общей популяции, что дает основания использовать оба фактора для оценки 10-летнего кардиоваскулярного риска (Mora S. et al., 2006).
Циркулирующий ингибитор активатора плазминогена
Циркулирующий ингибитор активатора плазминогена (ЦИАП) имеет период полужизни около 6 мин, что создает трудности для его качественной детекции. Однако многие кардиоваскулярные факторы риска, в том числе генетические (Dawson S. et al., 1991; Festa A. et al., 2003), метаболические (Juhan-Vague I., Alessi M.C., 1997), нейрогуморальные (Brown N.J. et al., 1998), могут оказывать непосредственное влияние на продукцию ЦИАП. При этом избыточный уровень ЦИАП тесно ассоциируется с высоким риском первого инфаркта миокарда у мужчин и у женщин (Thogersen A.M. et al., 1998). Необходимо отметить, что в настоящее время уже четко установлено, что хроническая блокада ренин-ангиотензиновой системы, в частности с помощью иАПФ, позволяет добиться достоверного и значительного снижения плазменной концентрации ЦИАП (Vaughan D.E. et al., 1997; Brown N.J. et al., 2002). Исследователи полагают, что этот эффект может в некоторой степени объяснять АПФ-независимые влияния иАПФ на выживаемость. В целом, использование оценки уровня ЦИАП вне Фремингемской шкалы риска или шкалы SCORE является дискутабельным.
Солюбилизированные лиганды CD40
Концентрация солюбилизированных лигандов CD40 (sCD40L) в плазме крови детектируется с большой вариабельностью (Varo N. et al., 2006; Weber M. et al., 2006). Вместе с тем, установлено, что у больных с метаболическим синдромом плазменный уровень CD40 существенно выше, чем в здоровой популяции (Lee W.L. et al., 2006). Тем не менее, результаты Dallas Heart Study показали, что sCD40L не способны помочь в идентификации субклинического атеросклеротического процесса у лиц в общей популяции (de Lemos J.A. et al., 2005). У больных с ангиографически документированной ИБС показана тесная ассоциация между уровнем sCD40L и наличием атеромы, стеноза коронарной артерии (Tayebjee M.H. et al., 2005). Аналогичные данные получены и для пациентов с документированным стенозом сонных артерий (Blake G.J. et al., 2003). Более того, в исследовании Women’s Health Study была установлена взаимосвязь между концентрацией sCD40L в плазме крови и величиной кардиоваскулярного риска среди здоровых женщин (Schonbeck U. et al., 2001). При этом использование для расчета риска пула sCD40L приводило к повышению расчетного значения относительного риска возникновения любых кардиоваскулярных событий в 2 раза (Schonbeck U. et al., 2001). Даже у лиц с асимптомным стенозом сонных артерий элевация sCD40L в плазме крови сопровождалась снижением вероятности выживания (Novo S. et al., 2005). Кроме того, повышение плазменной концентрации sCD40L детектируется и у пациентов, находящихся на гемодиализе, что рассматривается как доказательство наличия у них высокого риска фатальных и нефатальных кардиоваскулярных событий (относительный риск (ОР)=6,8) (Hocher B. et al., 2007). Некоторые исследователи полагают, что у пациентов с повышенным уровнем sCD40L могут отмечать повышение выживаемости от назначения ингибиторов IIb/IIIa рецепторов (Heeschen C. et al., 2003) и статинов. Последние позволяют добиться почти 48% редукции плазменного пула sCD40L (Kinlay S. et al., 2004).
Миелоидзависимый протеин 8/14
Установлено, что элевация миелоидзависимого протеина-8/14 у здоровых лиц является хорошим прогностическим признаком высокого риска возникновения кардиоваскулярных событий (Healy A.M. et al., 2006). Причем в этой когорте лиц повышение относительного риска манифестации сердечно-сосудистых заболеваний не зависит от наличия других факторов риска, в том числе СРП и гиперлипидемии.
Адипонектин
Концентрация адипонектина, повышающего чувствительность тканей к инсулину, оказывается значительно сниженной у пациентов с артериальной гипертензией, сахарным диабетом 2-го типа, ожирением по сравнению со здоровой популяцией (Matsuzawa Y., 2006). В экспериментальных и клинических исследованиях была установлена тесная негативная взаимосвязь между плазменным пулом адипонектина и компонентами метаболического синдрома, в том числе и ожирением (Salmenniemi U. et al., 2004). Кроме того, у мужчин плазменное содержание адипонектина существенно ниже, чем у пременопаузальных женщин. Снижение концентрации адипонектина ассоциируется с инсулинорезистентностью, висцеральным ожирением, наличием компонентов метаболического синдрома, ИБС, артериальной гипертензией, сахарным диабетом 2-го типа (Patel D.A. et al., 2006). Установлено, что у пременопаузальных женщин с ожирением (индекс массы тела >30 кг/м2), но без артериальной гипертензии, сахарного диабета 2-го типа и гиперлипидемии, уменьшение массы тела не менее чем на 10% в результате диетических ограничений сопровождается восстановлением плазменной концентрации адипонектина до уровня, близкого к нормальному (Esposito K. et al., 2003). Таким образом, оказалось, что содержание адипонектина в плазме крови является чрезвычайно вариабельным и, в то же время, может отражать метаболический статус пациента. В условиях 5-летнего проспективного исследования в когорте лиц без сахарного диабета, сниженная концентрация адипонектина была хорошим прогностическим признаком возникновения впервые выявленной артериальной гипертензии (Chow W.S. et al., 2007). По данным B. Iglseder и соавторов (2005) концентрация адипонектина у мужчин среднего возраста хорошо коррелирует с утолщением интимо-медиального сегмента общей сонной артерии. Аналогичные данные были получены R.P. Dullaart и соавторами (2007) у больных с сахарным диабетом 2-го типа. Кроме того, исследователи выявили устойчивую негативную корреляционную взаимосвязь между содержанием адипонектина и концентрацией СРП. Концентрация адипонектина также снижается у пациентов с ангиографически документированной ИБС независимо от их возраста и пола (Rothenbacher D. et al., 2005) и этнической принадлежности (Lu G. et al., 2007). Более того, плазменное содержание адипонектина негативно коррелирует с тяжестью атеросклеротических поражений коронарных артерий (Otsuka F. et al., 2006). В исследовании Health Professionals Follow-up Study (n=18 225 мужчин в возрасте 40–75 лет без верифицированных кардиоваскулярных заболеваний с периодом наблюдения 6 лет) впервые было установлено, что уровень адипонектина является хорошим и мощным прогностическим фактором возникновения первого инфаркта миокарда (Pischon T. et al., 2004). Прогностическое значение сниженной концентрации адипонектина в отношении возникновения любых фатальных и нефатальных кардиоваскулярных заболеваний сохраняется у пациентов с сахарным диабетом 2-го типа, с и без нарушений липидного профиля плазмы крови, а также у больных с изолированным снижением ХС ЛПВП без документированных сердечно-сосудистых заболеваний (Koenig W. et al., 2006; Lim S. et al., 2006) и у пациентов с хроническими заболеваниями почек, особенно на их конечных стадиях (Zoccali C. et al., 2002).
Вместе с тем, в течение последних лет появились некоторые сомнения относительно ассоциации плазменного содержания адипонектина и величины 10-летнего кардиоваскулярного риска. Так, в условиях 20-летнего наблюдения G.A. Laughlin и соавторы (2007) не выявили непосредственной взаимосвязи между плазменной концентрацией адипонектина и риском возникновения фатальных кардиоваскулярных событий. Интересно, что в еще одном проспективном 6-летнем наблюдении (n=4046 мужчин в возрасте 60–79 лет) было установлено, что высокая концентрация адипонектина сопровождается повышением общей смертности и кардиоваскулярной летальности в случае предшествующего наличия у больного документированного кардиоваскулярного заболевания (Wannamethee S.G. et al., 2007). Эти результаты, являясь бесспорно, неоднозначными, диктуют настоятельную необходимость проведения специальных исследований в этом направлении.
Интересно, что ген-промотор адипонектина содержит элементы, отвечающие на активацию рецепторов PPAR (peroxisome proliferator-activated receptor). Получены данные о том, что такие известные стимуляторы PPAR, как фибраты и безафибрат, в частности, способны повышать плазменную концентрацию адипонектина (Hiuge A. et al., 2007). По мнению многих исследователей, высокий уровень этого пептида в крови негативно коррелирует с риском возникновения впервые выявленного сахарного диабета, а мониторинг содержания адипонектина в крови может быть использован как способ контроля за изменением кардиоваскулярного риска (Hiuge A. et al., 2007). Кроме того, оказалось, что у пациентов без верифицированного сахарного диабета 2-го типа с низким уровнем ХС ЛПВП глитазоны (розиглитазон) также достоверно увеличивают плазменное содержание адипонектина без существенного изменения уровня ХС ЛПВП (Samaha F.F. et al., 2006). Другой представитель класса тиазолидиндионов — пиоглитазон — в комбинации с симвастатином продемонстрировал аналогичный эффект у лиц без сахарного диабета с высоким кардиоваскулярным риском (Forst T. et al., 2007).
Таким образом, возможность детекции и мониторирования плазменного пула адипонектина выглядит достаточно привлекательно с точки зрения перспективы контроля за модификацией кардиоваскулярного риска в процессе лечения.
ИЛ-18
Преадипоциты человека на стадии дифференциации обладают способностью к секреции ИЛ-18, что поддерживает гипотезу об их участии в процессах формирования провоспалительной активации при ожирении и сахарном диабете (Skurk T. et al., 2005). Установлено, что интенсивность освобождения ИЛ-18 из адипоцитов у больных с ожирением более чем в 3 раза превышает уровень здоровых лиц (Skurk T. et al., 2005). Повышение концентрации ИЛ-18 в плазме крови ассоциируется со значительным повышением риска манифестации сахарного диабета у мужчин и женщин любого возраста, причем это влияние не зависит от наличия других традиционных факторов риска (Thorand B. et al., 2005). Кроме того, уровень ИЛ-18 в плазме крови позитивно коррелирует с риском возникновения и количеством компонентов метаболического синдрома (Hung J. et al., 2005).
В настоящее время анализ плазменного содержания ИЛ-18 не рассматривается в качестве валидного теста для скрининга наличия атеросклеротического поражения артерий в общей популяции (Chapman C.M. et al., 2006; Zirlik A. et al., 2007). Однако его роль как предиктора возникновения кардиоваскулярных событий продолжает активно изучаться. Так, в испытании AtheroGene study плазменное содержание ИЛ-18 позитивно коррелировало с риском наступления смерти от кардиоваскулярных причин у больных с документированной ИБС (Blankenberg S. et al., 2002). В этой когорте больных концентрация ИЛ-18 на уровне верхнего квартиля сопровождалась трехкратным повышением кардиоваскулярного риска по сравнению с первым квартилем. Более того, в исследовании PRIME (Prospective Epidemiological Study of Myocardial Infarction) были получены данные о наличии независимой взаимосвязи между плазменной концентрацией ИЛ-18 и риском будущих кардиоваскулярных событий в мужской популяции среднего возраста (Blankenberg S. et al., 2003). Эти данные свидетельствуют о принципиальной возможности детекции плазменного пула ИЛ-18 для получения дополнительной информации о величине кардиоваскулярного риска в когортах пациентов без документированных сердечно-сосудистых заболеваний.
ММП-9
Установлено, что жесткость стенки аорты является независимым предиктором наступления кардиоваскулярных событий. Предполагается, что деградация эластина в сосудистой стенке, ассоциированная с увеличением ММП-9, и обеспечивает повышение ее жесткости, особенно у пациентов с изолированной систолической артериальной гипертензией (McEniery Yasmin C.M. et al., 2005). Показано, что уровень ММП-9 обычно повышен и у больных с документированной ИБС (Tayebjee M.H. et al., 2005). При этом в этой когорте лиц отмечена позитивная взаимосвязь между ММП-9 и ХС ЛПНП (McEniery Yasmin C.M. et al., 2005). Кроме того, у пациентов с острым коронарным синдромом плазменный пул ММП-9 обычно превышает нормальные значения более чем в 3 раза (Kai H. et al., 1998; Inokubo Y. et al., 2001). Это позволяет предполагать участие ММП-9 в формировании осложненной атеромы (Kai H. et al., 1998). В проспективном исследовании с медианой наблюдения 4,4 года было установлено, что среди 50% больных со стенозом сонных артерий плазменный уровень ММП-9 в 2 раза выше популяционного и тесно ассоциируется с риском развития ипсилатерального мозгового инсульта (Eldrup N. et al., 2006). При этом абсолютный риск кардиоваскулярных событий в когорте пациентов с документированными стенозами сонных артерий колебался от 34 до 17% в зависимости от высокого или низкого плазменного уровня ММП-9 соответственно (Blankenberg S. et al., 2003). Таким образом, ММП-9 в настоящее время рассматривается как весьма перспективный в отношении мониторирования провоспалительный фактор кардиоваскулярного риска. Хотя концентрации некоторых циркулирующих провоспалительных факторов и ассоциируются с повышенным кардиоваскулярным риском, не все они пригодны для рутинного клинического применения и, в частности, для мониторинга. СРП оказывается особенно привлекательным благодаря простоте выполнения теста, хотя не все исследователи признают наличие дополнительной пользы от его оценки. Дополнительные биомаркеры: адипонектин, растворимый лиганд CD40, ИЛ-18 и MMП-9 продемонстрировали свою диагностическую ценность только в научных исследованиях, хотя и могут вносить дополнительный вклад в глобальную оценку риска по Фремингемской шкале. Однако их применение ограничивается серьезными проблемами в стандартизации проведения теста.
Таким образом, требуются дополнительные исследования для более четкого формулирования полезности оценки провоспалительных биомаркеров в контексте стратификации больных в группы высокого кардиоваскулярного риска.
Дендритические клетки стенки артерий
Дендритические клетки представляют собой разновидность мононуклеарных фагоцитов (Abbas A.K. et al., 2000). Их сеть присутствует в слизистой оболочке желудочно-кишечного тракта, трахеи, бронхов, эпидермисе. Выявленные в стенке артерий дендритические клетки имеют сходные фенотип и численность с дендритическими клетками эпидермиса кожи — клетками Лангерганса. Функция последних заключается в фиксации белковых антигенов и транспорте их к дренирующим лимфатическим узлам, где происходит распознавание антигенов Т-лифоцитами. В отличие от клеток Ларгенганса, дендритические клетки стенки артерий способны к презентации антигенной детерминанты, принимая тем самым непосредственное участие в возникновении локального иммунного ответа. Интересно, что наибольшее скопление дендритических клеток в стенке артерий выявлено на участках бифуркаций или турбулунтного кровотока. Предполагают, что именно дендритические клетки могут связывать между собой такие разнородные процессы, как напряжение сдвига на эндотелии и иммунный ответ, играющие важную роль в формировании и прогрессировании атеросклероза.
Белки теплового шока (heat shock proteins)
В последнее время интерес к белкам теплового шока (heat shock proteins — HSP) особенно высок. Последние представляют собой семейство протеинов, включающих 25 молекул с различной молекулярной массой. Основная биологическая роль HSP сводится к предотвращению агрегации внутриклеточных белков с конформационными изменениями, развивающимися при длительном стрессе, гипертермии, инфекционных и аутоиммунных заболеваниях. Кроме того, HSP препятствует агрегации белковых молекул в процессе их синтеза и транспорта, а также ускоряет восстановление нарушенной структуры протеинов. Один из представителей этого семейства HSP60 у всех млекопитающих является важным антигеном при хронических воспалительных заболеваниях и, возможно, при атеросклерозе. Установлено, что хламидийный белок теплового шока HSP65 более чем на 50% идентичен HSP60 человека. В экспериментальных условиях было показано, что после проведения иммунизации HSP65 у животных развивался атеросклероз независимо от наличия гиперлипидемии. Причем этот процесс может быть подвергнут регуляции путем введения специфических Т-лимфоцитов. В последующем оказалось, что предрасположенность к атерогенезу может быть передана с помощью антител и Т-лимфоцитов от иммунизированных HSP60 животных. Причем в составе IgG исследователями были идентифицированы антиэндотелиальные антитела. Интересно, что одним из мессенджеров, обусловливающих фиксацию последних, является гликопротеин β2-GPI, экспрессирующийся на поверхности мембран эндотелиоцитов, а также входящий в состав ЛПВП, ЛПОНП, хиломикронов, липидного ядра атеромы. Установлено, что β2-GPI активирует эндотелиоциты, способствуя продукции эндотелина-1, а также принимает непосредственное участие в модуляции внутреннего пути активации процессов коагуляции. Последнее достигается преимущественно за счет повышения активности факторов XII и Xa, а также путем активации АДФ-индуцированной агрегации тромбоцитов и ингибирования протеина G.
Существует достаточно интересная концепция аутоиммунного патогенеза атеросклероза, предложенная G. Wick и соавторами (2001), согласно которой атеросклероз является своеобразной платой за иммунитет против микробного HSP60. Необходимо отметить, что последний более чем на 50% идентичен человеческому, что создает возможность для перекрестного реагирования антител против микробного HSP60 с собственными HSP60. Причем ряд факторов способен индуцировать повышение экспрессии HSP60 на поверхности эндотелиоцитов. К таковым относятся некоторые цитокины, липополисахариды бактериальной стенки, вирусы, гемодинамические силы, оксиленные ЛПНП. По мнению G. Wick и соавторов (2001), наиболее важное значение в инициации атеросклеротического поражения сосудов приобретают гемодинамические силы и окисленные ЛПНП. Концепция G. Wick и соавторов (2001) основана на том, что в плазме крови большинства людей выявляют антитела и специфичные Т-лимфоциты к микробному HSP60. При наличии иммунитета против HSP60 и воздействии указанных выше атерогенных факторов антимикробные антитела способны перекрестно реагировать с собственными HSP60, экспрессированными на поверхности мембран эндотелиоцитов. Все это, в конечном итоге, приводит к возникновению первой воспалительной стадии атеросклероза, а в дальнейшем — и к формированию атеромы (рис. 7.12).

Рис. 7.12. Механизмы реализации гуморального и клеточного иммунного ответа на HSP60 в соответствии с концепцией G. Wick (2001)
У части людей не обнаруживается перекрестная реакция антител к микробному HSP60 с собственным протеином. В этих случаях даже при наличии проатерогенных факторов и иммунитета против микробного HSP60 атеросклероз не развивается. Таким образом, комплемент-активирующие антитела к HSP60 вполне могут оказаться одним из новых семейных факторов высокого кардиоваскулярного риска.
В целом, роль цитокинов в модуляции процессов атерогенеза является несомненной. Однако большинство исследователей рассматривают цитокиновую активацию в качестве своеобразного мессенджера межклеточной кооперации, обусловливающей локальный воспалительный процесс в атероме и субинтиме, что отражается на прогрессировании заболевания. Принципиальная схема регуляции активности атеросклеротической бляшки с вовлечением иммунологических механизмов представлена на рис. 7.13. С одной стороны, субпопуляция Тh-1 (T-helper-1) выделяет ряд провоспалительных цитокинов, в том числе γ-ИФ, ИЛ-1, ФНО-α. Последние активируют эндотелиоциты, макрофаги, а также способствуют накоплению свободных радикалов, индуцируют продукцию протеиназ, повышают протромботическую активность, поддерживая воспалительный процесс в субинтиме артерий. С другой стороны, Тh-2-клетки продуцируют медиаторы с антивоспалительными свойствами, в том числе ИЛ-4, ИЛ-10, тканевой фактор роста-β, которые способствуют пролиферации гладкомышечных клеток, продукции коллагена, снижению степени деградации внеклеточного коллагенового матрикса за счет супрессии активности маталлопротеиназ, что способствует интенсификации процессов репарации. Баланс между этими двумя процессами в значительной мере определяет степень активности атеромы и ее «угрожаемость» в отношении возникновения атеротромботических событий.

Рис. 7.13. Регуляция активности атеросклеротической бляшки с вовлечением иммунологических механизмов
ГМК — гладкомышечные клетки.
В заключение необходимо отметить, что несмотря на серьезное продвижение в понимании природы и эволюции атеротромбоза, достигнутое за последние десятилетия, многие механизмы его инициации, регуляции и прогрессирования до сих пор требуют пристального внимания исследователей. До сих пор не предложены методы корректной оценки стадии процесса, индивидуального риска пациента, основанного на идентификации угрожаемой атеромы, что приводит к необходимости использования популяционных критериев для формирования стратегии первичной и вторичной профилактики кардиоваскулярных событий.
Литература
- Abbas A.K. et al. (2000) Cellular and Molecular Immunology. Philadelphia, PA: W.B. Saunders.
- Ambrose J.A. (2008) In search of the «vulnerable plaque». Can it be localized and will focal regional therapy ever be an option for cardiac prevention? J. Am. Coll. Cardiol., 51: 1539–1542.
- Ambrose J.A., Srikanth S. (2010) Vulnerable plaques and patients: improving prediction of future coronary events. Am. J. Med., 123: 10–16.
- Amento E.P., Ehsani N., Palmer H. et al. (1991) Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler. Thromb., 11: 1223–1230.
- Amorino G.P., Hoover R.L. (1998) Interactions of monocytic cells with human endothelial cells stimulate monocytic metalloproteinase production. Am. J. Pathol., 152: 199–207.
- Barter P. (2004) HDLa recipe for longevity. Atheroscler. Suppl., 5: 25–31.
- Barter P.J., Nicholls S., Rye K.A. et al. (2004) Antiinflammatory properties of HDL Circ. Res., 95: 764–772.
- Bavendiek U., Libby P., Kilbride M. et al. (2002) Induction of tissue factor expression in human endothelial cells by CD40 ligand is mediated via activator protein 1, nuclear factor kappa B, and Egr-1. J. Biol. Chem., 277: 25032–25039.
- Blake G.J., Ostfeld R.J., Yucel E.K. et al. (2003) Soluble CD40 ligand levels indicate lipid accumulation in carotid atheroma: an in vivo study with high-resolution MRI. Arterioscler. Thromb. Vasc. Biol., 23: e11–e14.
- Blankenberg S., Luc G., Ducimetiere P. et al. (2003) Interleukin-18 and the risk of coronary heart disease in European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation, 108: 2453–2459.
- Blankenberg S., Rupprecht H.J., Poirier O. et al. (2003) Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation, 107: 1579–1585.
- Blankenberg S., Tiret L., Bickel C. et al. (2002) Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation, 106: 24–30.
- Blankenberg S., Yusuf S. (2006) The inflammatory hypothesis: any progress in risk stratification and therapeutic targets? Circulation, 114: 1557–1560.
- Boring L., Gosling J., Cleary M. et al. (1998) Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature, 394: 894–897.
- Brasselet C., Durand E., Addad F. et al. (2005) Collagen and elastin cross-linking: a mechanism of constrictive remodeling after arterial injury Am. J. Physiol. Heart Circ. Physiol., 289(5): H2228–H2233.
- Brown N.J., Abbas A., Byrne D. (2002) Comparative effects of estrogen and angiotensin-converting enzyme inhibition on plasminogen activator inhibitor-1 in healthy postmenopausal women. Circulation, 105: 304–309.
- Brown N.J., Agirbasli M.A., Williams G.H. et al. (1998) Effect of activation and inhibition of the renin-angiotensin system on plasma PAI-1. Hypertension, 32: 965–971.
- Burke A.P., Farb A., Malcom G.T. et al. (1997) Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med., 336: 1276–1282.
- Calabro P., Willerson J.T., Yeh E.T. (2003) Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation, 108: 1930–1932.
- Canto J.G., Shlipak M.G., Rogers W.J. et al. (2000) Prevalence, clinical characteristics, and mortality among patients with myocardial infarction presenting without chest pain JAMA, 283: 3223–3229.
- Castelli W.P. (1996) Lipids, risk factors and ischaemic heart disease. Atherosclerosis, 124, Suppl.: S1–S9.
- Chapman C.M., McQuillan B.M., Beilby J.P. et al. (2006) Interleukin-18 levels are not associated with subclinical carotid atherosclerosis in a community population: the Perth Carotid Ultrasound Disease Assessment Study (CUDAS). Atherosclerosis, 189: 414–419.
- Charo I.F., Ransohoff R.M. (2006) The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med., 354: 610–621.
- Chow W.S., Cheung B.M., Tso A.W. et al. (2007) Hypoadiponectinemia as a predictor for the development of hypertension: a 5-year prospective study. Hypertension, 49: 1455–1461.
- Croce K., Libby P. (2007) Intertwining of thrombosis and inflammation in atherosclerosis. Curr. Opin. Hematol., 14: 55–61.
- Cybulsky M.I., Iiyama K., Li H. et al. (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest., 107: 1255–1262.
- Cybulsky M.I., Gimbrone M.A. Jr. (1991) Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science, 251: 788–791.
- Danesh J., Wheeler J.G., Hirschfield G.M. et al. (2004) C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N. Engl. J. Med., 350: 1387–1397.
- Daugherty A., Pure E., Delfel-Butteiger D. et al. (1997) The effects of total lymphocyte deficiency on the extent of atherosclerosis in apolipoprotein E-/- mice. J. Clin. Invest., 100: 1575–1580.
- Daugherty A., Rateri D.L. (2002) T lymphocytes in atherosclerosisthe yin-yang of Th1 and Th2 influence on lesion formation. Circ. Res., 90: 1039–1040.
- Davies M.J. (1996) Stability and instability: two faces of coronary atherosclerosis. The Paul Dudley White Lecture 1995. Circulation, 94: 2013–2020.
- Dawson S., Hamsten A., Wiman B. et al. (1991) Genetic variation at the plasminogen activator inhibitor-1 locus is associated with altered levels of plasma plasminogen activator inhibitor-1 activity. Arterioscler. Thromb., 11: 183–190.
- de Lemos J.A., Hennekens C.H., Ridker P.M. (2000) Plasma concentration of soluble vascular cell adhesion molecule-1 and subsequent cardiovascular risk. J. Am. Coll. Cardiol., 36: 423–426.
- de Lemos J.A., Zirlik A., Schonbeck U. et al. (2005) Associations between soluble CD40 ligand, atherosclerosis risk factors, and subclinical atherosclerosis: results from the Dallas Heart Study. Arterioscler. Thromb. Vasc. Biol., 25: 2192–2196.
- de Smet B.J.G.L., Pasterkamp G., van der Helm Y.J. et al. (1998) The relation between de novo atherosclerotic remodeling and angioplasty-induced remodeling in an atherosclerotic yucatan micropig model. Arterioscl. Thromb. Vasc. Biol., 188: 702–707.
- Detrano R., Guerci A.D., Carr J.J. et al. (2008) Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med., 358: 1336–1345.
- Devaraj S., Xu D.Y., Jialal I. (2003) C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation, 107: 398–404.
- Dullaart R.P., de Vries R., van Tol A. et al. (2007) Lower plasma adiponectin is a marker of increased intima-media thickness associated with type 2 diabetes mellitus and with male gender. Eur. J. Endocrinol., 156: 387–394.
- Eldrup N., Gronholdt M.L., Sillesen H. et al. (2006) Elevated matrix metalloproteinase-9 associated with stroke or cardiovascular death in patients with carotid stenosis. Circulation, 114: 1847–1854.
- Eren M., Painter C.A., Atkinson J.B. et al. (2002) Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation, 106: 491–496.
- Esposito K., Pontillo A., Di Palo C. et al. (2003) Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA, 289: 1799–1804.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (2001) Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA, 285(19): 2486–2497.
- Falk E., Shah P.K., Fuster V. (1995) Coronary plaque disruption. Circulation, 92: 657–671.
- Fantuzzi G., Mazzone T. (2007) Adipose tissue and atherosclerosis: exploring the connection. Arterioscler. Thromb. Vasc. Biol., 27: 996–1003.
- Farb A., Burke A.P., Tang A.L. et al. (1996) Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation, 93: 1354–1363.
- Festa A., D’Agostino R. Jr., Rich S.S. et al. (2003) Promoter (4G/5G) plasminogen activator inhibitor-1 genotype and plasminogen activator inhibitor-1 levels in blacks, Hispanics, and non-Hispanic whites: the Insulin Resistance Atherosclerosis Study. Circulation, 107: 2422–2427.
- Forst T., Pfutzner A., Lubben G. et al. (2007) Effect of simvastatin and/or pioglitazone on insulin resistance, insulin secretion, adiponectin, and proinsulin levels in nondiabetic patients at cardiovascular risk – the PIOSTAT Study. Metabolism, 56: 491–496.
- Fuster V., Badimon L., Badimon J.J. et al. (1992) The pathogenesis of coronary artery disease and the acute coronary syndromes (1). N. Engl. J. Med., 326(4): 242–250.
- Fuster V., Badimon L., Badimon J.J. et al. (1992) The pathogenesis of coronary artery disease and the acute coronary syndromes (2). N. Engl. J. Med., 326(5): 310–318.
- Galis Z.S., Sukhova G.K., Lark M.W. et al. (1994) Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest., 94: 2493–2503.
- Ge J., Erbel R., Zamoran J. et al. (1993) Coronary artery remodeling in atherosclerotic disease. Coron. Artery. Dis., 4: 981–986.
- Gerber T.C., Erbel R., Gorge G. et al. (1994) Extent of atherosclerosis and remodeling of the left main coronary artery determined by intravascular ultrasound. Am. J. Cardiol., 73: 666–671.
- Gerdes N., Sukhova G.K., Libby P. (2002) Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J. Exp. Med., 195: 245–257.
- Gerszten R.E., Lim Y.C., Ding H.T. et al. (1998) Adhesion of monocytes to vascular cell adhesion molecule-1-transduced human endothelial cells: implications for atherogenesis. Circ. Res., 82: 871–878.
- Gerszten R.E., Luscinskas F.W., Ding H.T. et al. (1996) Adhesion of memory lymphocytes to vascular cell adhesion molecule-1-transduced human vascular endothelial cells under simulated physiological flow conditions in vitro. Circ. Res., 79: 1205–1215.
- Glagov S., Weisenberg E., Zarins C.K. et al. (1987) Compensatory enlargement of human atherosclerotic coronary arteries. N. Engl. J. Med., 316: 1371–1375.
- Glass C.K., Witztum J.L. (2001) Atherosclerosis: the road ahead. Cell., 104: 503–516.
- Gough P.J., Gomez I.G., Wille P.T. et al. (2006) Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Invest., 116: 59–69.
- Greenland P., Knoll M.D., Stamler J. et al. (2003) Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. JAMA, 290: 891–897.
- Gu L., Okada Y., Clinton S.K. et al. (1998) Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell., 2: 275–281.
- Gurfinkel E., Vigliano C., Janavel J.V. (2009) Presence of vulnerable coronary plaques in middle-aged individuals who suffered a brain death. Eur. Heart J., 30(23): 2845–2853.
- Hackett D., Davies G., Maseri A. (1988) Pre-existing coronary stenoses in patients with first myocardial infarction are not necessarily severe. Eur. Heart J., 9: 1317–1323.
- Hansson G.K. (2005) Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med., 352: 1685–1695.
- Hansson G.K., Libby P. (2006) The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol., 6: 508–519.
- Healy A.M., Pickard M.D., Pradhan A.D. et al. (2006) Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation, 113: 2278–2284.
- Heeschen C., Dimmeler S., Hamm C.W. et al. (2003) Soluble CD40 ligand in acute coronary syndromes. N. Engl. J. Med., 348: 1104–1111.
- Hellings W.E., Moll F.L., De Vries J.-P.P.M. et al. (2008) Atherosclerotic Plaque Composition and Occurrence of Restenosis After Carotid Endarterectomy. JAMA, 299(5): 547–554.
- Hermiller J.B., Tenaglia A.N., Kisslo K.B. et al. (1993) In vivo validation of compensatory enlargement of atherosclerotic coronary arteries. Am. J. Cardiol., 71: 665–668.
- Hiuge A., Tenenbaum A., Maeda N. et al. (2007) Effects of peroxisome proliferator-activated receptor ligands, bezafibrate and fenofibrate, on adiponectin level. Arterioscler. Thromb. Vasc. Biol., 27: 635–641.
- Hocher B., Liefeldt L., Quaschning T. et al. (2007) Soluble CD154 is a unique predictor of nonfatal and fatal atherothrombotic events in patients who have end-stage renal disease and are on hemodialysis. J. Am. Soc. Nephrol., 18: 1323–1330.
- Hotamisligil G.S. (2006) Inflammation and metabolic disorders. Nature (Lond.), 444: 860–867.
- Hung J., McQuillan B.M., Chapman C.M. et al. (2005) Elevated interleukin-18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler. Thromb. Vasc. Biol., 25: 1268–1273.
- Iglseder B., Mackevics V., Stadlmayer A. et al. (2005) Plasma adiponectin levels and sonographic phenotypes of subclinical carotid artery atherosclerosis: data from the SAPHIR Study. Stroke, 36: 2577–2582.
- Ikeda T., Shirasawa T., Esaki Y. et al. (1993) Osteopontin mRNA is expressed by smooth muscle-derived foam cells in human atherosclerotic lesions of the aorta J. Clin. Invest., 92: 2814–2820.
- Inokubo Y., Hanada H., Ishizaka H. et al. (2001) Plasma levels of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 are increased in the coronary circulation in patients with acute coronary syndrome. Am. Heart J., 141: 211–217.
- Jabs W.J., Theissing E., Nitschke M. et al. (2003) Local generation of C-reactive protein in diseased coronary artery venous bypass grafts and normal vascular tissue. Circulation, 108: 1428–1431.
- Jongstra-Bilen J., Haidari M., Zhu S.N. et al. (2006) Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med., 203: 2073–2083.
- Juhan-Vague I., Alessi M.C. (1997) PAI-1, obesity, insulin resistance and risk of cardiovascular events. Thromb. Haemost., 78: 656–660.
- Kai H., Ikeda H., Yasukawa H. et al. (1998) Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J. Am. Coll. Cardiol., 32: 368–372.
- Kannel W.B., Wolf P.A., Castelli W.P. et al. (1987) Fibrinogen and risk of cardiovascular disease: the Framingham Study. JAMA, 258: 1183–1186.
- Kawakami A., Aikawa M., Alcaide P. et al. (2006) Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation, 114: 681–687.
- Khalil M.F., Wagner W.D., Goldberg I.J. (2004) Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis Arterioscler. Thromb. Vasc. Biol., 24: 2211–2218.
- Khot U.N., Khot M.B., Baizer C.T. et al. (2003) Prevalence of conventional risk factors in patients with coronary heart disease. JAMA, 290: 898–904.
- Kinlay S., Egido J. (2006) Inflammatory biomarkers in stable atherosclerosis. Am. J. Cardiol., 98: 2P–8P.
- Kinlay S., Schwartz G.G., Olsson A.G. et al. (2004) Effect of atorvastatin on risk of recurrent cardiovascular events after an acute coronary syndrome associated with high soluble CD40 ligand in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study. Circulation, 110: 386–391.
- Koenig W., Khuseyinova N., Baumert J. et al. (2006) Serum concentrations of adiponectin and risk of type 2 diabetes mellitus and coronary heart disease in apparently healthy middle-aged men: results from the 18-year follow-up of a large cohort from southern Germany. J. Am. Coll. Cardiol., 48: 1369–1377.
- Koenig W., Lowel H., Baumert J. et al. (2004) C-reactive protein modulates risk prediction based on the Framingham Score: implications for future risk assessment: results from a large cohort study in southern Germany. Circulation, 109: 1349–1353.
- Kohler H.P., Grant P.J. (2000) Plasminogen-activator inhibitor type 1 and coronary artery disease. N. Engl. J. Med., 342: 1792–1801.
- Kolodgie F.D., Gold H.K., Burke A.P. et al. (2003) Intraplaque hemorrhage and progression of coronary atheroma N. Engl. J. Med., 349: 2316–2325.
- Kruth H.S. (2002) Sequestration of aggregated low-density lipoproteins by macrophages. Curr. Opin. Lipidol., 13: 483–488.
- Kuller L.H., Tracy R.P., Shaten J. et al. (1996) Relation of C-reactive protein and coronary heart disease in the MRFIT nested case-control study. Multiple Risk Factor Intervention Trial. Am. J. Epidemiol., 144: 537–547.
- Lakoske S.G., Greenland P., Wong N. et al. (2007) Coronary artery calcium scores and risk for cardiovascular events in women classified as «low risk» based on Framingham Risk Score. Arch. Intern. Med., 167: 2437–2442.
- Lara-Castro C., Fu Y., Chung B.H. et al. (2007) Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease. Curr. Opin. Lipidol., 18: 263–270.
- Laughlin G.A., Barrett-Connor E., May S. et al. (2007) Association of adiponectin with coronary heart disease and mortality: the Rancho Bernardo study. Am. J. Epidemiol., 165: 164–174.
- Lee W.L., Lee W.J., Chen Y.T. et al. (2006) The presence of metabolic syndrome is independently associated with elevated serum CD40 ligand and disease severity in patients with symptomatic coronary artery disease. Metabolism, 55: 1029–1034.
- Li H., Cybulsky M.I., Gimbrone M.A. et al. (1993) An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler. Thromb., 13: 197–204.
- Li H., Cybulsky M.I., Gimbrone M.A. Jr. et al. (1993) Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am. J. Pathol., 143: 1551–1559.
- Liaw L., Almeida M., Hart C.E. et al. (1994) Osteopontin promotes vascular cell adhesion and spreading and is chemotactic for smooth muscle cells in vitro. Circ. Res., 74: 214–224.
- Liaw L., Lombardi D.M., Almeida M.M. et al. (1997) Neutralizing antibodies directed against osteopontin inhibit rat carotid neointimal thickening after endothelial denudation Arterioscler. Thromb. Vasc. Biol., 17: 188–193.
- Libby P. (1995) Molecular bases of the acute coronary syndromes Circulation, 91: 2844–2850.
- Libby P. (2001) Current concepts of the pathogenesis of the acute coronary syndromes. Circulation, 104: 365–372.
- Libby P. (2002) Atherosclerosis: the new view. Sci. Am., 286: 46–55.
- Libby P. (2002) Inflammation in atherosclerosis. Nature, 420: 868–874.
- Libby P. (2005) Act local, act global: inflammation and the multiplicity of «vulnerable» coronary plaques. J. Am. Coll. Cardiol., 45: 1600–1602.
- Libby P. (2006) Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr., 83: 456S–460S.
- Libby P., Aikawa M. (2002) Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat. Med., 8: 1257–1262.
- Libby P., Ridker P.M. (1999) Novel inflammatory markers of coronary risk: theory versus practice. Circulation, 100: 1148–1150.
- Libby P., Ridker P.M. (2004) Inflammation and atherosclerosis: role of C-reactive protein in risk assessment. Am. J. Med., 116(Suppl. 6A): 9S–16S.
- Libby P., Ridker P.M. (2006) Inflammation and atherothrombosis: from population biology and bench research to clinical practice. J. Am. Coll. Cardiol., 48: A33–A46.
- Libby P., Simon D.I. (2001) Inflammation and thrombosis: the clot thickens. Circulation, 103: 1718–1720.
- Libby P., Theroux P. (2005) Pathophysiology of coronary artery disease. Circulation, 111: 3481–3488.
- Libby P., Aikawa M., Schonbeck U. (2000) Cholesterol and atherosclerosis. Biochim. Biophys. Acta., 1529: 299–309.
- Libby P., Ridker P.M., Maseri A. (2002) Inflammation and atherosclerosis. Circulation, 105: 1135–1143.
- Lim S., Koo B.K., Cho S.W. et al. (2006) Association of adiponectin and resistin with cardiovascular events in Korean patients with type 2 diabetes: the Korean atherosclerosis study (KAS) A 42-month prospective study. Atherosclerosis, 54: 1232.
- Lu G., Chiem A., Anuurad E. et al. (2007) Adiponectin levels are associated with coronary artery disease across Caucasian and African-American ethnicity. Transl. Res., 149: 317–323.
- Luster A.D., Alon R., von Andrian U.H. (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol., 6: 1182–1190.
- Ma J., Hennekens C.H., Ridker P.M. et al. (1999) A prospective study of fibrinogen and risk of myocardial infarction in the Physicians’ Health Study. J. Am. Coll. Cardiol., 33: 1347–1352.
- Mach F., Sauty A., Iarossi A.S. et al. (1999) Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J. Clin. Invest., 104: 1041–1050.
- Mach F., Schonbeck U., Bonnefoy J.Y. et al. (1997) Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation, 96: 396–399.
- Mach F., Schonbeck U., Sukhova G.K. et al. (1997) Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl Acad. Sci. USA, 94: 1931–1936.
- Mach F., Schonbeck U., Sukhova G.K. et al. (1998) Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature (Lond.), 394: 200–203.
- Mason D.P., Kenagy R.D., Hasenstab D. et al. (1999) Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ. Res., 85: 1179–1185.
- Matsuzawa Y. (2006) Therapy insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med., 3: 35–42.
- Maxfield F.R., Tabas I. (2005) Role of cholesterol and lipid organization in disease. Nature, 438: 612–621.
- McEniery Yasmin C.M., Wallace S., Dakham Z. et al. (2005) Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler. Thromb. Vasc. Biol., 25: 372.
- McPherson D.D., Sirna S.J., Hiratzka L.F. et al. (1991) Coronary arterial remodeling studied by high-frequency epicardial echocardiography. J. Am. Coll. Cardiol., 17: 79–86.
- Miller Y.I., Chang M.K., Binder C.J. et al. (2003) Oxidized low density lipoprotein and innate immune receptors. Curr. Opin. Lipidol., 14: 437–445.
- Mintz G.S., Kent K.M., Pichard A.D. et al. (1997) Contribution of inadequate arterial remodeling to the development of focal coronary artery stenoses. Circulation, 95: 1791–1798.
- Mora S., Rifai N., Buring J.E. et al. (2006) Additive value of immunoassay-measured fibrinogen and high-sensitivity C-reactive protein levels for predicting incident cardiovascular events. Circulation, 114: 381–387.
- Moreno P.R., Falk E., Palacios I.F. et al. (1994) Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation, 90: 775–778.
- Moreno P.R., Purushothaman K.R., Fuster V. et al. (2004) Tunica media and plaque neovascularization is increased in ruptured atherosclerotic lesions of human aortaimplications for plaque vulnerability. Circulation, 110: 2032–2038.
- Moreno P.R., Purushothaman K.R., Fuster V. et al. (2002) Intimomedial interface damage and adventitial inflammation is increased beneath disrupted atherosclerosis in the aortaimplications for plaque vulnerability. Circulation, 105: 2504–2511.
- Morishige K., Shimokawa H., Matsumoto Y. et al. (2003) Overexpression of matrix metalloproteinase-9 promotes intravascular thrombus formation in porcine coronary arteries in vivo. Cardiovasc. Res., 57: 572–585.
- Moulton K.S., Heller E., Konerding M.A. et al. (1999) Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation, 99: 1726–1732.
- Moulton K.S., Vakili K., Zurakowski D. et al. (2003) Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl Acad. Sci. USA, 100: 4736–4741.
- Naghavi M., Falk E., Hecht H.S. et al. (2006) From vulnerable plaque to vulnerable patient- Part III: Executive summary of the Screening for Heart Attack Prevention and Education (SHAPE) task force report. Am. J. Cardiol., 98(Suppl.): 2H–15H.
- Naghavi M., Libby P., Falk E. et al. (2003а) From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation, 108: 1664–1672.
- Naghavi M., Libby P., Falk E. et al. (2003b) From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part II. Circulation, 108: 1772–1778.
- Nishioka T., Luo H., Eigler N.L. et al. (1996) Contribution of inadequate compensatory enlargement to development of human artery stenosis. J. Am. Coll. Cardiol., 27: 1571–1576.
- Nissen S.E., Tuzcu E.M., Schoenhagen P. et al. (2005) Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N. Engl. J. Med., 352: 29–38.
- Noji Y., Kajinami K., Kawashiri M.A. et al. (2001) Circulating matrix metalloproteinases and their inhibitors in premature coronary atherosclerosis. Clin. Chem. Lab. Med., 39: 380–384.
- Novo S., Basili S., Tantillo R. et al. (2005) Soluble CD40L and cardiovascular risk in asymptomatic low-grade carotid stenosis. Stroke, 36: 673–675.
- O’Brien E.R., Garvin M.R., Stewart D.K. et al. (1994) Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques Arterioscler. Thromb., 14: 1648–1656.
- Okamoto Y., Kihara S., Funahashi T. et al. (2006) Adiponectin: a key adipocytokine in metabolic syndrome. Clin. Sci., 110: 267–278.
- Okamura H., Tsutsi H., Komatsu T. et al. (1995) Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (Lond.), 378: 88–91.
- Otsuka F., Sugiyama S., Kojima S. et al. (2006) Plasma adiponectin levels are associated with coronary lesion complexity in men with coronary artery disease. J. Am. Coll. Cardiol., 48: 1155–1162.
- Ozaki Y., Violaris A.G., de Feyter P. et al. (1996) Role of underlying vascular remodeling mode in the mechanism of acute luminal gain and late restenosis after balloon angioplasty (BA) and directional coronary atherectomy (DCA). Circulation, 94(Suppl.): I–134.
- Pai J.K., Pischon T., Ma J. et al. (2004) Inflammatory markers and the risk of coronary heart disease in men and women. N. Engl. J. Med., 351: 2599–2610.
- Pasceri V., Willerson J.T., Yeh E.T. (2000) Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation, 102: 2165–2168.
- Pasterkamp G., Wensing P.J.W., Post M.J. et al. (1995) Paradoxical arterial wall shrinkage contributes to luminal narrowing of human atherosclerotic femoral arteries. Circulation, 91: 1444–1449.
- Patel D.A., Srinivasan S.R., Xu J.H. et al. (2006) Adiponectin and its correlates of cardiovascular risk in young adults: the Bogalusa Heart Study. Metabolism, 55: 1551–1557.
- Pearson T.A., Mensah G.A., Alexander R.W. et al. (2003) Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation, 107: 499–511.
- Pischon T., Girman C.J., Hotamisligil G.S. et al. (2004) Plasma adiponectin levels and risk of myocardial infarction in men. JAMA, 291: 1730–1737.
- Pohl T., Seiler C., Billinger M. et al. (2001) Frequency distribution of collateral flow and factors influencing collateral channel development. Functional collateral channel measurement in 450 patients with coronary artery disease J. Am. Coll. Cardiol., 38: 1872–1878.
- Prati F., Arbustini E., Labellarte A. et al. (2003) Eccentric atherosclerotic plaques with positive remodelling have a pericardial distribution: apermissive role of epicardial fat? A three dimensional intravascular ultrasound study of left anterior descending artery lesions. Eur. Heart J., 24: 329–336.
- Raines E.W., Ferri N. (2005) Thematic review series: the immune system and atherogenesis, cytokines affecting endothelial and smooth muscle cells in vascular disease. J. Lipid. Res., 46: 1081–1092.
- Rajavashisth T.B., Andalibi A., Territo M.C. et al. (1990) Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature, 344: 254–257.
- Ridker P.M. (2003a) Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation, 107: 363–369.
- Ridker P.M. (2003b) Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: rationale and design of the JUPITER trial. Circulation, 108: 2292–2297.
- Ridker P.M., Buring J.E., Cook N.R. et al. (2003) C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation, 107: 391–397.
- Ridker P.M., Buring J.E., Rifai N. et al. (2007) Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds ris k score. JAMA, 297: 611–619.
- Ridker P.M., Cannon C.P., Morrow D. et al. (2005) C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med., 352: 20–28.
- Ridker P.M., Cushman M., Stampfer M.J. et al. (1997) Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med., 336: 973–979.
- Ridker P.M., Hennekens C.H., Buring J.E. et al. (2000) C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med., 342: 836–843.
- Ridker P.M., Rifai N., Clearfield M. et al. (2001a) Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Engl. J. Med., 344: 1959–1965.
- Ridker P.M., Rifai N., Pfeffer M.A. et al. (1999) Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation, 100: 230–235.
- Ridker P.M., Rifai N., Rose L. et al. (2002) Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med., 347: 1557–1565.
- Ridker P.M., Stampfer M.J., Rifai N. (2001b) Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA, 285: 2481–2485.
- Robertson A.K., Hansson G.K. (2006) T cells in atherogenesis: for better or for worse?. Arterioscler. Thromb. Vasc. Biol., 26: 2421–2432.
- Ross R. (1999) Atherosclerosis — an inflammatory disease. N. Engl. J. Med., 340: 115–126.
- Ross R., Glomset J.A. (1976) The pathogenesis of atherosclerosis (first of two parts). N. Engl. J. Med., 295: 369–377.
- Ross R., Glomset J.A. (1976) The pathogenesis of atherosclerosis (second of two parts). N. Engl. J. Med., 295: 420–425.
- Rothenbacher D., Brenner H., Marz W. et al. (2005) Adiponectin, risk of coronary heart disease and correlations with cardiovascular risk markers. Eur. Heart J., 26: 1640–1646.
- Salmenniemi U., Ruotsalainen E., Pihlajamaki J. et al. (2004) Multiple abnormalities in glucose and energy metabolism and coordinated changes in levels of adiponectin, cytokines, and adhesion molecules in subjects with metabolic syndrome. Circulation, 110: 3842–3848.
- Samaha F.F., Szapary P.O., Iqbal N. et al. (2006) Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high-density lipoprotein cholesterol and metabolic syndrome. Arterioscler. Thromb. Vasc. Biol., 26: 624–630.
- Schaar J.A., Muller J.E., Falk E. et al. (2004) Terminology for high-risk and vulnerable coronary artery plaques. Report of a meeting on the vulnerable plaque, June 17 and 18, 2003, Santorini, Greece. Eur. Heart J., 25(12): 1077–1082
- Schoenhagen P., Zinda K.M., Kapadia S.R. et al. (2000) Extent and direction of arterial remodeling in stable versus unstable coronary syndrome: an intravascular ultrasound study. Circulation, 101: 604–610.
- Schoenhagen P., Ziada K.M., Vince D.G. et al. (2001) Arterial remodeling and coronary artery disease: the concept of “dilated” versus “obstructive” coronary atherosclerosis. J. Am. Coll. Cardiol., 38(2): 297–306.
- Schonbeck U., Mach F., Sukhova G.K. et al. (2000a) CD40 ligation induces tissue factor expression in human vascular smooth muscle cells. Am. J. Pathol., 156: 7–14.
- Schonbeck U., Mach F., Sukhova G.K. et al. (1999) Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J. Exp. Med., 189: 843–853.
- Schonbeck U., Mach F., Sukhova G.K. et al. (1997) Regulation of matrix metalloproteinase expression in human vascular smooth muscle cells by T lymphocytes: a role for CD40 signaling in plaque rupture? Circ. Res., 81: 448–454.
- Schonbeck U., Sukhova G.K., Shimizu K. et al. (2000b) Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc. Natl Acad. Sci. USA, 97: 7458–7463.
- Schonbeck U., Varo N., Libby P. et al. (2001) Soluble CD40L and cardiovascular risk in women. Circulation, 104: 2266–2268.
- Schwartz S.M., deBlois D., O’Brien E.R. (1995) The intima. Soil for atherosclerosis and restenosis. Circ. Res., 77: 445–465.
- Shah P.K., Kaul S., Nilsson J. et al. (2001a) Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteinsan idea whose time for testing is coming, part I. Circulation, 104: 2376–2383.
- Shah P.K., Kaul S., Nilsson J.et al. (2001b) Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteinsan idea whose time for testing is coming, part II. Circulation, 104: 2498–2502.
- Sheifer S.E., Manolio T.A., Gersh B.J. (2001) Unrecognized myocardial infarction. Ann. Intern. Med., 135: 801–811.
- Singh P., Hoffmann M., Wolk R. et al. (2007) Leptin induces C-reactive protein expression in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol., 27: e302–e307.
- Skalen K., Gustafsson M., Rydberg E.K. et al. (2002) Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature (Lond.), 417: 750–754.
- Skurk T., Kolb H., Muller-Scholze S. et al. (2005) The proatherogenic cytokine interleukin-18 is secreted by human adipocytes. Eur. J. Endocrinol., 152: 863–868.
- Stary H.C. (1992) Composition and classification of human atherosclerotic lesions. Virchows Arch., 421: 277–290.
- Stary H.C. (2000) Natural history and histologic classification of atherosclerotic lesionsan update. Arterioscler. Thromb. Vasc. Biol., 20: 1177–1178.
- Stary H.C., Chandler A.B., Dinsmore R.E. et al. (1995) A definition of advanced types of atherosclerotic lesions and a histologic classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association Circulation, 92: 1355–1374.
- Stary H.C., Chandler A.B., Glagov S. et al. (1994) A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association Circulation, 89: 2462–2478.
- Steinberg D. (2006a) The pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part IV: the 1984 coronary primary prevention trial ends it — almost. J. Lipid. Res., 47: 1–14.
- Steinberg D. (2004) Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part I. J. Lipid. Res., 45: 1583–1593.
- Steinberg D. (2005a) Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: part II: the early evidence linking hypercholesterolemia to coronary disease in humans. J. Lipid. Res., 46: 179–190.
- Steinberg D. (2005b) Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part III: mechanistically defining the role of hyperlipidemia. J. Lipid. Res., 46: 2037–51.
- Steinberg D. (2006b) Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. J. Lipid. Res., 47: 1339–51.
- Stemme S., Faber B., Holm J. et al. (1995) T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl Acad. Sci. USA, 92: 3893–3897.
- Sukhova G.K., Schonbeck U., Rabkin E. et al. (1999) Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation, 99: 2503–2509.
- Swirski F.K., Libby P., Aikawa E. et al. (2007) Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest., 117: 195–205.
- Tabas I. (2002) Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest., 110: 905–911.
- Tacke F., Alvarez D., Kaplan T.J. et al. (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest., 117: 185–194.
- Takemoto M., Yokote K., Nishimura M. et al. (2000) Enhanced expression of osteopontin in human diabetic artery and analysis of its functional role in accelerated atherogenesis Arterioscler. Thromb. Vasc. Biol., 20: 624–628.
- Tayebjee M.H., Lip G.Y., Tan K.T. et al. (2005) Plasma matrix metalloproteinase-9, tissue inhibitor of metalloproteinase-2, and CD40 ligand levels in patients with stable coronary artery disease. Am. J. Cardiol., 96: 339–345.
- Tedgui A., Mallat Z. (2006) Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev., 86: 515–581.
- Thogersen A.M., Jansson J.H., Boman K. et al. (1998) High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: evidence for the fibrinolytic system as an independent primary risk factor. Circulation, 98: 2241–2247.
- Thorand B., Kolb H., Baumert J. et al. (2005) Elevated levels of interleukin-18 predict the development of type 2 diabetes: results from the MONICA/KORA Augsburg Study, 1984–2002. Diabetes, 54: 2932–2938.
- Tsimikas S., Willerson J.T., Ridker P.M. (2006) C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J. Am. Coll. Cardiol., 47: C19–C31.
- Van Gaal L.F., Mertens I.L., De Block C.E. (2006) Mechanisms linking obesity with cardiovascular disease. Nature, 444: 875–880.
- Varo N., Nuzzo R., Natal C. et al. (2006) Influence of pre-analytical and analytical factors on soluble CD40L measurements. Clin. Sci., 111: 341–347.
- Vaughan D.E. (2005) PAI-1 and atherothrombosis. J. Thromb. Haemost., 3: 1879–1883.
- Vaughan D.E., Rouleau J.L., Ridker P.M. et al. (1997) Effects of ramipril on plasma fibrinolytic balance in patients with acute anterior myocardial infarction. HEART Study Investigators. Circulation, 96: 442–447.
- Virmani R., Kolodgie F.D., Burke A.P. et al. (2000) Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol., 20: 1262–1275.
- von Birgelen C., Klinkhart W., Mintz G.S. et al. (2001) Plaque distribution and vascular remodeling of ruptured and nonruptured coronary plaques in the same vessel: an intravascular ultrasound study in vivo. J. Am. Coll. Cardiol., 37: 1864–1870.
- Wannamethee S.G., Whincup P.H., Lennon L. et al. (2007) Circulating adiponectin levels and mortality in elderly men with and without cardiovascular disease and heart failure. Arch. Intern. Med., 167: 1510–1517.
- Ward M.R., Jeremias A., Hibi K. et al. (2001) The influence of plaque orientation (pericardial or myocardial) on coronary arterial remodelling. Atherosclerosis, 154: 179–183.
- Ware J.A. (2001) Too many vessels? Not enough? The wrong kind? The VEGF debate continues. Nat. Med., 7: 403–404.
- Weber M., Rabenau B., Stanisch M. et al. (2006) Influence of sample type and storage conditions on soluble CD40 ligand assessment. Clin. Chem., 52: 888–891.
- Werner G.S., Ferrari M., Betge S. et al. (2001) Collateral function in chronic total coronary occlusions is related to regional myocardial function and duration of occlusion Circulation, 104: 2784–2790.
- Wick G., Perschinka H., Millonig G. (2001) Atherosclerosis as an autoimmune disease: an update. Trends Immunol., 22(12): 665–669.
- Wilhelmsen L., Svardsudd K., Korsan-Bengtsen K. et al. (1984) Fibrinogen as a risk factor for stroke and myocardial infarction. N. Engl. J. Med., 311: 501–505.
- Williams J.K., Heistad D.D. (1996) The vasa vasorum of the arteries. J. Mal. Vasc., 21(Suppl. C): 266–269.
- Williams K.J., Tabas I. (1995) The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol., 15: 551–561.
- Williams K.J., Tabas I. (1998) The response-to-retention hypothesis of atherogenesis reinforced. Curr. Opin. Lipidol., 9: 471–474.
- Zhang Y., Zanotti I., Reilly M.P. et al. (2003) Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation, 108: 661–663.
- Zirlik A., Abdullah S.M., Gerdes N. et al. (2007) Interleukin-18, the metabolic syndrome, and subclinical atherosclerosis: results from the Dallas Heart Study. Arterioscler Thromb. Vasc. Biol.
- Zoccali C., Mallamaci F., Tripepi G. et al. (2002) Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. J. Am. Soc. Nephrol., 13: 134–141.
- Zwaka T.P., Hombach V., Torzewski J. (2001) C-reactive protein-mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation, 103: 1194–1197.